

Abstract
Perillyl alcohol (POH) is a naturally occurring dietary monoterpene isolated from the essential oils of lavender, peppermint, and other plants. Medical interest in this compound was generated by research findings showing that POH was able to inhibit the growth of tumor cells in cell culture and exert cancer preventive and therapeutic activity in a variety of animal tumor models. Based on this promising preclinical work, POH was formulated in soft gelatine capsules and orally administered to cancer patients several times a day on a continuous basis. However, such clinical trials in humans yielded disappointing results, also because the large number of capsules that had to be swallowed caused hard-to-tolerate intestinal side effects, causing many patients to withdraw from treatment due to unrelenting nausea, fatigue, and vomiting. As a result, efforts to treat cancer patients with oral POH were abandoned and did not enter clinical practice. Intriguingly, clinical trials in Brazil have explored intranasal POH delivery as an alternative to circumvent the toxic limitations of oral administration. In these trials, patients with recurrent malignant gliomas were given comparatively small doses of POH via simple inhalation through the nose. Results from these studies show this type of long-term, daily chemotherapy to be well tolerated and effective. In this review, we will present the vicissitudes of POH’s evaluation as an anticancer agent, and its most recent success in therapy of patients with malignant brain tumors.
Keywords: Monoterpene, intranasal drug delivery, inhalation drug delivery, glioblastoma
Background
Perillyl alcohol (POH; IUPAC name: [4-(prop-1-en-2-yl)cyclohex-1-en-1-yl] methanol) and its precursor limonene are naturally occurring monocyclic terpenes derived from the mevalonate pathway in plants. POH is a constituent of caraway, lavender and lilac oil, cherries, cranberries, sage, spearmint, peppermint, celery seeds, and certain other plants [1]. D-Limonene (1-methyl-4-(1-methylethenyl)-cyclohexene) is the predominant constituent of peel oil from citrus fruits and the essential oils of caraway, and an important ingredient of the flavor and aroma profiles of anise, black pepper, cinnamon, coriander, ginger, lavender, mint, nutmeg, rosemary, sage, thyme, and some other herbs [1]. It is metabolized to POH via hydroxylation by cytochrome P450-type enzymes, and this catalytic process has been documented in a variety of microbes and in microsomal preparations from plants [2,3]. Recently, limonene production and subsequent conversion to POH was achieved after engineering E. coli with a heterologous mevalonate pathway and limonene synthase, coupled with a cytochrome P450 enzyme specifically hydroxylating limonene to produce POH [4]. Chemical synthesis of POH, in four steps from commercially available limonene oxide, has been accomplished as well [5].
Humans and other mammals produce neither limonene nor POH, but do harbor P450 liver enzymes for the oxidation of limonene to POH and other metabolic products. For instance, liver microsomes of humans, mice, rats, guinea pigs, rabbits, dogs, and monkeys readily produce POH after addition of limonene as a substrate [6,7]. In humans, dogs, and rats, POH was shown to be rapidly metabolized to perillyl aldehyde (perillaldehyde), perillic acid, and cis- and trans-dihydroperillic acids, followed by glucuronidation and excretion primarily in the urine and to a lesser extent in bile [8-12] (Figure 1).
Figure 1.
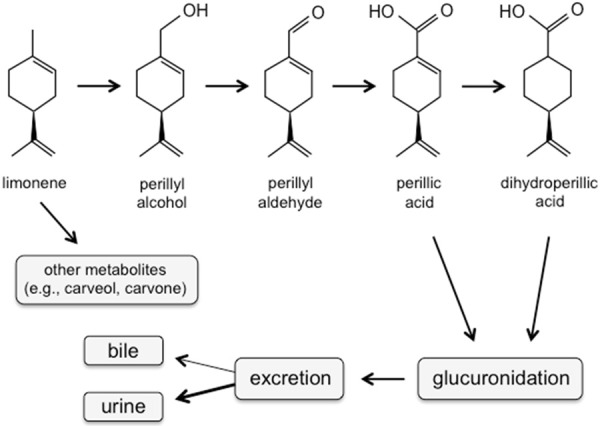
Major components of POH metabolism in mammals. POH, either derived from limonene via hydroxylation by P450 liver enzymes, or administered directly into the patient or other mammal, undergoes stepwise oxidation to its metabolic end products, followed by glucuronidation by UDP-glucuronyltransferase and subsequent excretion. (Shown are the (篓C)-(S)-enantiomers of the stereoisomeric molecules).
Traditionally, limonene and POH have a number of manufacturing and household uses and are common ingredients in cleaning products, cosmetics, and as fragrance in toiletries [12,13]. They are permitted by the U.S. Food and Drug Administration (FDA) as food additives, primarily for the purpose as flavoring agents [14]. Most relevant for the following review, limonene and POH both have attracted attention from the medical community, based on their anticancer activity in a number of preclinical models (e.g., [15,16]), with POH generally displaying greater activity than limonene [17-20]. In the following, we will present the vicissitudes of POH’s evaluation as an anticancer agent, and its most recent success in therapy of patients with malignant brain tumors.
Chemopreventive activity
POH and D-limonene have revealed chemopreventive activity in preclinical animal models of breast, colon, lung, pancreas, and skin cancer [21,22]. For instance, dietary POH at 1 or 2 g/kg greatly reduced the incidence and multiplicity of invasive adenocarcinomas of the colon of rats injected with the carcinogen azoxymethane (AOM) [23]. Similarly, when POH was injected intraperitoneally at a dose of 75 mg/kg three times per week in mice, it significantly reduced lung tumor formation triggered by simultaneous injection of carcinogenic NNK (4-(methyl-nitrosoamino)-1-(3-pyridyl)-1-butanone) [24,25]. Similar studies indicated preventive activity of POH in a hamster pancreatic cancer model [26].
In contrast to the above studies, chemopreventive activity of POH could not be established in rat models of esophageal [27] or hepatic carcinogenesis [28]. In the latter model, rats were treated with a single dose of N-nitrosomorpholine (NNM), followed by 1 g/kg daily POH in the diet. After several months, animals treated with POH failed to show fewer neoplastic liver foci as compared to rats that had not received dietary POH, indicating a lack of detectable chemopreventive effect of the monoterpene in the early stages of rat hepatocarcinogenesis [28]. Within the context of other studies that instead had demonstrated positive effects of POH [29,30], the authors suggested that POH perhaps might act differently in the early and late stages of carcinogenesis. Overall, the potential of POH and other dietary phytochemicals for chemoprevention of hepatocarcinogenesis needs to be explored further, possibly beginning with the phase 0 approach [22,31].
In mouse skin tumor models, POH (10 mM) applied topically to the ears and shaved dorsal surface significantly inhibited tumor development in response to exposure to ultraviolet B radiation [32] or painting of the skin of TPras mice with DMBA (dimethylbenz[a]anthracene) [33]. POH was also effective in the classic two-stage skin carcinogenesis model, where tumor initiation with DMBA is followed by tumor promotion with TPA (12-O-tetradecanoylphorbol-13-acetate) [34]. The activity of topical POH in these preclinical studies encouraged the design of a Phase 1 study in participants with normal-appearing skin, which showed that a cream-based topical formulation of POH [35] was well tolerated at a dose of 0.76% (w/w) without severe cutaneous toxicities, systemic toxicities, or histopathological abnormalities [36]. The purpose of a subsequent randomized, placebo-controlled, double-blind Phase 2a study was to determine whether POH cream, applied twice daily to the forearms for three months, could reverse actinic (sun) damage. Although a modest effect of POH in sun-damaged skin could be detected, the overall outcome was unimpressive, and the authors proposed that improved delivery to the skin might be necessary [37].
Chemotherapeutic activity in vivo
Several studies in preclinical animal tumor models have characterized POH as a powerful chemotherapeutic agent against different cancer types, including pancreatic, breast, liver, and brain cancers. For example, a diet mixed with 2-4% (1.2-2.4 g/kg per day) POH resulted in significant reduction of tumor growth in hamsters injected with pancreatic carcinoma cells, including complete regression in 20% of the animals [38]. At the same time, there was no observable toxicity in H & E-stained sections of the liver, kidney, and normal pancreas. Subsequent studies with human pancreatic cancer cells implanted into nude mice further confirmed the therapeutic activity of POH against this tumor type [39].
In studies with rats, 2.5% dietary POH resulted in regression of 81% of small mammary carcinomas and 75% of advanced mammary carcinomas initiated by 7,12-dimethylbenz(a)anthracene (DMBA) [18]. In a mouse model with orthotopically transplanted human breast carcinoma cells, POH at a dose of 75 mg/kg was injected intraperitoneally three times a week for 6 weeks, which resulted in suppression of primary tumor growth and inhibition of metastatic spread to regional lymph nodes [40]. The ability of POH to block metastatic spread was also confirmed in the chorioallantoic membrane (CAM) model with the use of the C6 rat glioma cell line [41].
POH also revealed significant potency against diethylnitrosoamine (DEN)-induced liver tumors in rats. Two weeks after the removal of DEN, the animals were placed on a diet containing 2% POH for 19 weeks. Compared to control animals that had not received POH, the liver weight of POH-treated rats was 10-fold less, with substantially smaller tumor tissue present [30]. POH also exerted cancer therapeutic activity after intranasal delivery, where mice with intracranially xenografted glioblastoma cells received 0.76 or 1.9 mg/kg POH into alternating nostrils every other day. Both POH-treated groups of animals survived significantly longer than control animals treated with vehicle only [42]. Similar to the other in vivo studies, histopathological analysis failed to reveal apparent pathological abnormalities in POH-treated animals.
Antiangiogenic activity
The potent cancer therapeutic activity of POH documented in various preclinical tumor models appears to result from its dual impact on the tumor cells per se, as well as the endothelial cells forming the tumor vasculature. On one hand, inhibitory potency of POH against cultured tumor cells has been described in a number of studies (e.g., [43-50]); on the other hand, interference of POH with the process of angiogenesis has been documented as well.
POH was able to induce apoptosis of endothelial cells, and prevented new blood vessel growth in the chicken CAM assay and in a Matrigel model of endothelial tubule formation [51,52]. As well, it differentially modulated the release of two important angiogenic regulators, vascular endothelial growth factor (VEGF) from tumor cells and angiopoietin 2 (Ang2) from endothelial cells, resulting in suppression of neovascularization and induction of vessel regression. In related studies, it was confirmed that POH decreased the production of proangiogenic growth factors, such as VEGF and interleukin-8 (IL-8), in cultured glioblastoma cells [42].
Phase I trials of oral POH
In seven phase I clinical trials, POH was administered orally to cancer patients with advanced and refractory malignancy. POH was given in divided doses ranging from 2,400 to 16,200 mg per day (equivalent to approximately 40-270 mg/kg). Treatment duration varied with each patient, but was generally between 2 and 9 months. Nausea was cited as a common side effect, along with other gastrointestinal toxicities such as vomiting, eructation, and satiety, which became dose limiting in several of these trials.
Howard Bailey’s group at the University of Wisconsin Comprehensive Cancer Center initiated four of these phase I trials. The first one [53] involved 18 patients with advanced malignancies for whom no effective standard therapy was available. POH was formulated in soft gelatin capsules containing 250 mg POH and 250 mg soybean oil, which were administered p.o. on a continuous three-times-a-day basis. The dose-escalation scheme started with 800 mg/m2/dose in four patients and increased up to 2,400 mg/m2/dose in seven patients. About half the patients in each group remained on therapy for ≥ 3 months. The main toxicities, which appeared to be dose related, were gastrointestinal (GI) and included nausea and vomiting, anorexia, unpleasant taste, satiety, and eructation, and grade 1-2 fatigue was also noted [53]. Two heavily pre-treated ovarian cancer patients experienced reversible ≥ grade 3 granulocytopenia. Disease stabilization for ≥ 6 months was seen, although not objective tumor response was noted.
The subsequent phase I study from this group [9] increased continuous POH dosing from three to four times a day. Sixteen patients with refractory malignancies received gelatin capsules at 800, 1,200, and 1,600 mg/m2/dose four times a day, and several of these patients remained on this schedule for ≥ 3 months, with one patient in the highest-dose group remaining for ≥ 24 months. As before, the predominant toxicities seen were GI-related and fatigue. No significant problems with myelosuppression were seen. Grade 1 leukopenia and neutropenia were observed in several patients, but this did not appear to be drug related. Grade 1 anemia and thrombocytopenia were seen in one patient at the lowest dose level. No hepatic, renal, or neurological toxicities thought to be related to the drug were seen. Overall, POH appeared to be better tolerated when taken in a fed state as opposed to fasting, and it was concluded that the maximum tolerated dose of POH given continuously four times a day was 1,200 mg/m2/dose [9]. Several patients presented with stable disease for ≥ 6 months, and one patient with metastatic colon cancer experienced a near-complete response of > 2 years duration.
To avoid the large amounts of ingested soybean oil, the following phase I trial from the Bailey group [54] used a new formulation with 700 mg capsules containing 650 mg POH, in an effort to improve POH dose and metabolite concentration. A total of 19 patients with refractory solid malignancies received escalating dose levels/dose of 1,350 mg, 2,025 mg, 2,700 mg, 3,375 mg, or 4,050 mg, administered orally four times a day in a 28-day cycle. Within the first four dose levels, no dose-limiting toxicity occurred, but at the highest dose level one patient (out of 6) experienced grade 3 vomiting. Overall, as before, GI toxicities predominated, with nausea and vomiting in 63% of patients (12/19). The same proportion of patients (12/19) experienced heartburn and indigestion, primarily grade 1. Although the side effects were mild in nature, three patients withdrew from treatment, citing intolerable GI toxicity. The authors concluded that this reformulation of POH appeared to represent an improvement upon prior formulation, by reducing the number of capsules ingested and the degree of GI toxicity per dose, where a dose of 2,050 mg administered four times a day was easily tolerated [54].
The fourth phase I trial by the Bailey group, performed in collaboration with a team at the University of Iowa Holden Comprehensive Cancer Center [55], tested the original 500 mg capsules (250 mg POH and 250 mg soybean oil) in an interrupted 28-day schedule, consisting of 14 continuous days of treatment followed by 14 rest days, for up to three cycles. The rationale was to examine whether an interrupted administration schedule could possibly lead to increased tolerability with reduced severity of POH side effects. POH was administered orally to 20 patients four times a day at doses between 1,200 and 2,000 mg/m2/dose. As before, the most common toxicities were nausea, GI distress, and fatigue. Other toxicities noted were hypokalemia and one incidence of pancreatitis. Due to these toxicities, four patients declined further treatment during or after the second cycle. No objective responses were observed, and the authors concluded that an interrupted administration schedule of POH did not reveal significant advantages over continuous dosing schedules, as the associated toxicities of drug treatment did not seem to lessen as compared to continuous daily dosing [55].
A similar cycle of 14 days on/14 days off was also investigated at the Fox Chase Center in Philadelphia, PA, where 17 patients received POH at 1,600, 2,100, and 2,800 mg/m2/dose three times a day [8]. They observed increasing GI toxicity starting at the initial dose of 1,600 mg/m2/dose, and dose-limiting nausea and fatigue at the highest dose. Grade 1-2 hypokalemia was common at 2,100 and 2,800 mg/m2/dose. In comparison, this three-times-daily dosing of POH seemed better tolerated than the four-times-daily dosing reported by Bailey et al., 2004, which was also given on a 14 days on/14 days off cycle [55].
A phase I study performed by a team at the Yale Cancer Center in New Haven, CT, had 21 patients treated with POH orally three times a day on a continuous schedule [56]. The average number of days that patients remained on the study was 48 (range 11-172). Soft gelatin capsules (250 mg POH, 250 mg soybean oil) were used, with a starting dose of 600 mg/m2/dose on an empty stomach, with escalation to 2,800 mg/m2/dose, where fatigue and nausea became dose limiting. Reversible neutropenia occurred in a small minority of the patients. Central nervous system (CNS) toxicities, manifested as mild disorientation, loss of balance, and impaired ability to concentrate, were observed in 11 patients; one patient on the highest dose level developed slurred speech. All CNS toxicities resolved upon withdrawal of the drug. Stabilization of disease was observed in one of the 16 patients evaluable for response [56].
Finally, a phase I study performed by a team at Memorial Sloan-Kettering Cancer Center in New York [57] investigated POH delivered orally as soft gelatin capsules, four times daily, on a continuous schedule at doses ranging from 1,200 to 2,800 mg/m2/dose. The median time on study was 4 weeks. The dose-limiting toxicities in this trial were nausea and vomiting, encountered in all patients at the highest dose level. Overall, there were no objective tumor responses. Five patients continued on the study for 2 months or more with stable disease, including one patient with stage IV non-small-cell lung cancer metastatic to lung and lymph nodes, who remained on the study for 13 months [57].
The maximum tolerated dose (MTD) of POH in this latter study by Azzoli et al. ([57] was determined to be 8,400 mg/m2 per day [57], which was higher than what was observed in other four-times-a-day dosing trials, such as the ones by Ripple et al. [9] and Bailey et al. [55] that reported MTDs of 4,800 and 6,400 mg/m2 per day, respectively. In comparison, the three-times-a-day treatment schedule used by Hudes et al. [8] and Murren et al. [56] resulted in an MTD of 6,300 mg/m2 per day.
The variable MTDs of POH presumably are attributable to the nonspecific and subjective GI side effects of the drug, where considerable interpatient variability was noted, and where measures to ameliorate the toxicity were of little help for some and no help for others. In fact, the unremitting nature of drug side effects was significant enough to result in several subjects declining to participate further in the trials. Another consistently noted issue with oral dosing of POH was the number of capsules required (15-20 capsules four times a day at higher doses). A different formulation with more concentrated capsules used by Morgan-Meadows et al. [54] presented an improvement and resulted in a high, tolerated dose of 8,200 mg per day, although it did not appear to offer any metabolite pharmacokinetic advantage.
Phase II trials of oral POH
The above-mentioned phase I studies demonstrated that POH was reasonably well tolerated, with the exception of mild to moderate toxicities, most commonly gastrointestinal symptoms and fatigue. Doses up to 2,400 mg/m2 p.o. three times a day did not reach the MTD; however, a dose of 1,200 mg/m2 p.o. four times a day was recommended for phase II trials that examined perillyl alcohol in patients with advanced ovarian cancer [58], metastatic colorectal cancer [59], androgen-independent prostate cancer [60], and treatment-refractory metastatic breast cancer [61]. In two of these trials, patients tolerating the initial dose were dose-escalated to 1,500 mg/m2 [61] and 1,600 mg/m2 [59].
The results of all four studies were disappointing, with the uniform conclusion that this POH treatment regimen did not exhibit objective clinical antitumor activity. As well, tolerance to this regimen was poor, which contributed to suspension of enrollment short of planned accrual, or early withdrawal of patients from therapy due to the unpleasant experience. In some cases, even grade 2 toxicity was not tolerable when it was chronic and unremitting, which also limited the ability to escalate the dose beyond 1,200 mg/m2 [58-61].
Intranasal delivery of POH
While efforts are underway to create high-dose formulations of oral POH that might reduce GI toxicities, an alternative delivery method was explored in clinical trials performed in Brazil. In these trials, POH was administered via nasal inhalation to patients with recurrent malignant glioma (see below). The rationale behind intranasal delivery of POH to brain cancer patients included the idea that direct nose-to-brain transport might support increased drug access to the intracranial tumor site. Other advantages of nasal drug uptake are based on the highly vascularized epithelium of nasal mucosa and its high total blood flow, its large surface area, and its lower enzyme levels as compared to gastrointestinal tract and liver. In general, these features enable easy accessibility, rapid drug absorption that avoids first-pass hepatic metabolism, altogether resulting in enhanced bioavailability and quick onset of drug action [62-66].
Intranasal drug delivery is a rapidly developing field that seeks to deliver a wide range of therapeutic agents to target a variety of medical conditions from rhinitis to neurological disorders such as Alzheimer’s, migraine, or schizophrenia [62,65,67,68]. However, the exact drug transport mechanisms and processes involved are incompletely understood [69]. For instance, while the determination of drug concentrations in the brain after intranasal delivery can be achieved via measurements of cerebrospinal fluid (CSF), it is substantially more difficult to establish the tracks of drug movement between nose and brain, as there appear to exist several direct and indirect pathways that can be utilized by drugs to reach the CSF. These pathways include direct brain entry via the olfactory pathway or the trigeminal nerve pathway, or indirect routes via the blood vasculature and lymphatic system [63,68,70,71].
Several existing and emerging nasal delivery devices and dispersion technologies are explored for optimal nasal delivery and clinical performance, including conventional inhalers, mechanical spray pumps, gas driven or electrically powered nebulizers, and a variety of powder devices [72,73]. For example, ViaNaseTM, a hand-held electronic atomizer developed by Kurve Technology [74], allows for control of droplet size, velocity and trajectories, and enables targeted deposition of pharmaceuticals to the entire nasal cavity, including the olfactory regions and the paranasal sinuses [75,76].
In the meantime, clinical trials in Brazil already have demonstrated therapeutic efficacy and easy tolerability of POH when administered four times a day via nasal inhalation. In an initial phase I/II study [77], 37 patients with recurrent malignant glioma received 0.3% v/v POH (55 mg) per dose, totaling 220 mg per day. It was reported that no patient presented with signs of toxicity, inclusive of 4 patients with more than 1 year of daily POH treatment, and no dose reduction or drug discontinuation was required for any of the study participants. At the same time, this novel delivery strategy led to an increase in progression-free survival and decrease in tumor size in several patients [77].
Two follow-up reports [78,79] presented data on the long-term outcome of POH intranasal delivery to a cohort of 198 patients, including 155 with recurrent glioblastoma multiforme (GBM), 27 with grade III astrocytoma (AA), and 16 with anaplastic oligodendroglioma (AO). All patients were only under palliative symptomatic treatment because they had failed current standard of care for malignant glioma recurrence. For therapy of these patients, POH was diluted in mineral water with pH above 7 (in an attempt to alkalinize the acidity of peritumoral edema) and was administered in a common nebulizer by intranasal inhalation four times a day. The initial individual doses were 67 mg qid (268 mg total per day), which were escalated up to 133 mg (533 mg total). Clinical toxicity and overall survival following treatment were compared with tumor size, topography, extent of peritumoral edema, and histological classification (see representative examples in Figure 2).
Figure 2.
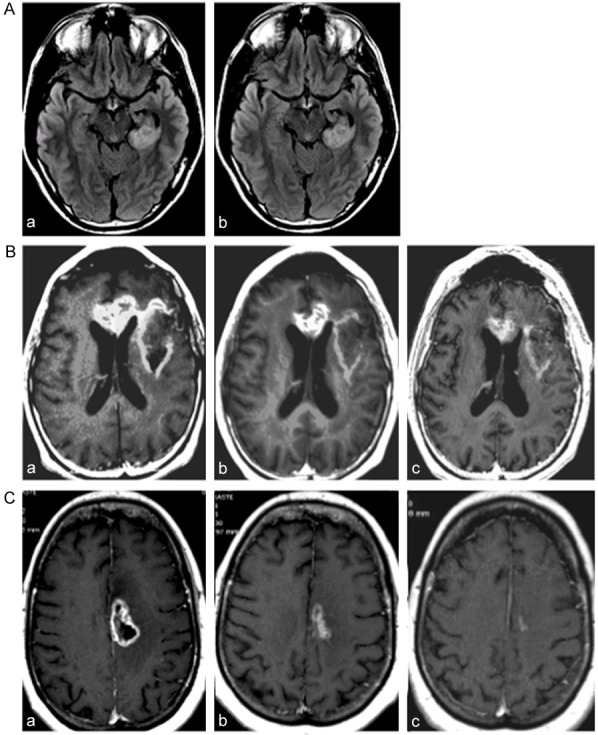
Representative MRIs of POH-treated patients. (A) MRIs of patient with astrocytoma grade II under treatment with four times daily intranasal POH at 66.7 mg/dose (266.8 mg/day). Note persistent tumor size in initial MRI (a), and after 3 years (b) of continuous treatment. (B) MRIs of patient with recurrent glioblastoma multiforme under treatment with same intranasal POH schedule (66.7 mg qid). Note mild reduction of tumor size from initial MRI (a) to 2 years (b) and 4 years (c) of daily treatment. (C) MRIs of patient with recurrent glioblastoma multiforme under treatment with same intranasal POH schedule (66.7 mg qid) in combination with temozolomide. Note mild reduction of tumor size from initial MRI (a) to 2 years (b) and 3 years (c) of daily POH treatment.
It was noted that adhesion to the protocol was high (> 95%). At the highest dose, POH occasionally caused nose soreness and in rare instances nose bleed. After 4 years under continuous, exclusive POH treatment, 19% of patients still remain in clinical remission, while drug side effects were almost non-existent. It was concluded that long-term POH inhalation therapy is a safe and non-invasive strategy with efficacy against recurrent malignant glioma [78,79].
Pharmacokinetics
Pharmacokinetic studies of oral perillyl alcohol have been challenging due to high inter- and intrapatient variability. The two main metabolites in humans were identified as perillic acid (PA) and dihydroperillic acid (DHPA), which were present in a ratio similar to that observed in dog studies [80], and minor metabolites included perillaldehyde. In contrast, the parent drug POH was not detectable in the plasma. Peak levels were noted 2-3 hours post-ingestion for PA and 3-5 hours post-ingestion for DHPA [8,9,53-57]. Metabolite half-lives measured from 1-4 hours for each metabolite [9,53-55], although one study reported a half-life for PA of less than one hour [56]. Consistently, there was no evidence of drug accumulation in the blood with time, supporting the necessity for frequent dosing. POH, PA, and DHPA were detectable in the urine of patients at higher dose levels. Independent of the administered dose, about 9-10% of the total dose was recovered in the first 24 hours, with PA as the major component, DHPA as a lesser component, and only a very small fraction (less than 1%) as POH [9,53,55].
The single-dose AUC0-6 h values for both PA and DHPA were similar to those seen on a TID or QID schedule at comparable doses, suggesting more consistent exposure to higher circulating metabolite levels on the more frequent dosing schedule [9]. Metabolite levels seen in random samples from rats at dietary levels of POH shown to be effective at inducing tumor regression were 390-480 µM for PA and 110-230 µM for DHPA [18]. In comparison, in patients the peak levels (Cmax) of PA were similar after patients received 1,200 mg/m2/dose, and reached even higher values (600 and 774 µM) after treatment with 2,000 or 2,800 mg/m2/dose, respectively [8,9,55]. However, Cmax for DHPA remained well below the range measured in rats, even at a human dose of 2,800 mg/m2, possibly because of known species differences in metabolism [7-9,55].
Taking the drug in a fed vs. fasting state did not have a substantial impact on metabolite levels (AUC) within any given dose level [9,53], although fasting patients appeared to reach Cmax somewhat faster [57], and in most cases had slightly higher 24-h AUC and Cmax at 1,200 mg/m2 and 1,600 mg/m2 dose levels, although not at 2,000 mg/m2/dose [55]. However, high inter- and intrapatient variability in all trials of oral POH allowed few conclusions regarding the relationship of AUC and toxicity.
Measuring POH turnover and metabolite levels represents a critical component of POH clinical studies, also because metabolites themselves may exert pharmacological activity. Several in vitro studies, for instance, have demonstrated tumor cell killing by perillyl aldehyde (perillaldehyde) [11,20,81]. In one study with rat PC12 pheochromocytoma cells [11], perillyl aldehyde exerted stronger effects than POH and caused apoptosis at 200 µM. In comparison, POH required 500 µM for the same outcome, whereas perillic acid was inactive in these assays. In related studies with murine B16 melanoma cells [20], the IC50 for growth inhibition by perillyl aldehyde and POH was 120 µM and 250 µM, respectively. In contrast to these previous studies, another study [81] with human carcinoma cell lines demonstrated that POH exerted stronger apoptosis-inducing potency than perillyl aldehyde. Thus, while cell-type specific responses might influence the overall potency of POH as compared to its metabolites, these studies nonetheless revealed cytotoxic potency of perillyl aldehyde.
Cellular targets and mechanism of action
The mechanisms of action of monoterpenes are not clearly defined. Several investigators have suggested cellular effects, such as G1 block causing cytostasis or differentiation, induction of apoptosis, or aggravation of endoplasmic reticulum (ER) stress [29,30,42,43,49,82]. Biochemical effects, such as alterations in mevalonate metabolism and inhibition of isoprenylation, might be involved as well [83-86]. More specifically, POH might target key components of signal transduction pathways, such as the Ras oncoprotein [87-89], transforming growth factor beta (TGFβ) receptor [29], nuclear factor kappa B (NF-κB) [90], c-fos and c-jun proto-oncogenes [91], or components of the cell cycle machinery [44,46,47,49,82] and appears to inhibit certain cellular enzymes, such as telomerase [92,93] and sodium/potassium adenosine triphosphatase (Na/K-ATPase) [94]. The chemopreventive effects of POH may be related to induction of phase I and phase II liver enzymes, resulting in carcinogen detoxification [95,96]. However, the contribution of all of these components to the biological and clinical impact of POH treatment remains to be fully characterized.
Role of Ras as a proposed target of POH
Early studies on POH function suggested that POH might act as an inhibitor of farnesyl-protein transferase (FPTase) and geranylgeranyl-protein transferases (GGPTases) [17,84-86]. Such interference with the mevalonate pathway generated substantial excitement, because posttranslational prenylation had been recognized as a critical modification of Ras oncoproteins, single-unit GTPases of molecular weight 21 (p21). Ras genes are well established as the most frequently mutated oncogenes in human cancer, and posttranslational farnesylation or geranylation is required for normal activity, as well as transforming function, of all three Ras protein isoforms (H-Ras, K-Ras, N-Ras) [97,98]. It was therefore postulated that POH might exert its anticancer effects via inhibition of Ras activity, resulting from the blockage of the proteins’ posttranslational modifications [99,100].
However, a number of experimental observations contradict the above model and greatly minimize a role, if any, of Ras proteins in POH-mediated anticancer effects. For instance, in vitro enzyme experiments revealed that POH concentrations required to inhibit FPTase and GGPTase activity were generally higher than those that impacted cell proliferation and survival [85,101,102]. As well, the observed decrease in farnesylated Ras levels upon treatment of cells with high concentrations of POH did not correlate with greater levels of cytosolic Ras, but rather appeared to be a consequence of decreased de novo synthesis of the protein [102,103]. While one study [104] did not detect lower activity of Ras downstream targets, such as p42/44 MAP kinase/ERK or collagenase promoter, another study [105] did find inhibition of MAPK/ERK, as well as upstream MAP kinase kinase (MEK), by POH. Inhibition of MEK and MAPK/ERK closely correlated with cell growth inhibition by POH, but was determined to be entirely independent of any involvement of Ras [105].
In an effort to gauge the impact of POH on Ras activity in patients, Hudes et al. [8] used PBMCs as an easily accessible surrogate tissue from six patients receiving 2,800 mg/m2/dose three times daily. Samples from day 1 (pre-treatment) and days 8 and 15 were compared for p21 Ras expression levels, but no consistent changes were found. These authors also treated cultured MCF-7 breast carcinoma and DU145 prostate carcinoma cells with the POH metabolites PA and DHPA at concentrations that exceeded those achieved in patient plasma after POH treatment; yet, no change in p21 Ras expression or its isoprenylation status could be detected [8], indicating that growth inhibition of these tumor cells was not related to altered Ras activity, consistent with in vitro results by others [103]. A lack of Ras processing in peripheral blood cells was also documented in a separate study with patients receiving a four-times-a-day schedule of POH at 1,200 mg/m2/dose [55]. Hudes et al. concluded that “Ras function may be neither a relevant target for POH nor a suitable intermediate end point in the dose range tolerated by humans” [8].
Altogether, a significant involvement of Ras protein in mediating the anticancer effects of POH seems rather unlikely, as the majority of more recent studies are not supportive of this model. In the meantime, a large number of additional POH targets have emerged, and many of them seem reasonable candidates for mediating the antiproliferative effects of POH. However, the validation of their precise roles in these events remains to be established.
Summary
The naturally occurring monoterpene POH exerts cytotoxic effects when added to tumor cells in culture. Early on, it was surmised that the key mechanism by which POH triggers cell death was through the inhibition of the Ras oncoprotein; however, later studies could not convincingly establish this model. Subsequently, a growing number of cellular targets of POH were identified. While the precise contribution of each of these additional components remains to be established, it appears that POH might cause tumor cell death through pleiotropic effects impacting a variety of cellular functions.
When investigated in vivo, POH exerted convincing therapeutic activity in different animal tumor models. However, in clinical trials that followed, POH initially did not fulfill its promise as a novel cancer therapeutic when it was administered orally to patients with different types of neoplasms. Among the challenges of these trials was the observation that the ingestion of large gram dosages of POH caused intestinal toxicities, causing many patients to withdraw from treatment due to unrelenting nausea, fatigue, and vomiting. As a result, oral POH was abandoned and did not enter clinical practice.
Clinical trials in Brazil spearheaded an alternative mode of POH delivery to patients. Here, sub-gram daily quantities of POH were administered through nasal inhalation to recurrent glioma patients, who previously had become unresponsive to standard cancer therapeutic regimens and faced dismal prognosis. Intriguingly, these studies not only demonstrated clinical activity of POH, but also revealed that long-term intranasal inhalation of the compound was very well tolerated over several years of daily use. In the United States, the FDA (Food and Drug Administration) has accepted IND (investigational new drug) filing of NEO100, a highly purified form of POH, and clinical trials to treat recurrent glioblastoma patients with this compound are anticipated to begin sometime during the year 2015.
Acknowledgements
Work in the authors’ labs was supported by the Hale Family Research Fund and Sounder Foundation (to TCC), and the California Breast Cancer Research Program (to AHS).
Disclosure of conflict of interest
TCC and CODF are founders and stakeholders of NeOnc Technologies, Woodland Hills, California, USA. AHS declares no conflict of interest.
References
- 1.Crowell PL, Elson CE. Isoprenoids, Health and Disease. In: Wildman REC, editor. Neutraceuticals and Functional Foods. Boca Raton, FL: CRC Press, LLC; 2001. [Google Scholar]
- 2.Duetz WA, Bouwmeester H, van Beilen JB, Witholt B. Biotransformation of limonene by bacteria, fungi, yeasts, and plants. Appl Microbiol Biotechnol. 2003;61:269–277. doi: 10.1007/s00253-003-1221-y. [DOI] [PubMed] [Google Scholar]
- 3.Lupien S, Karp F, Ponnamperuma K, Wildung M, Croteau R. Cytochrome P450 limonene hydroxylases of Mentha species. Drug Metabol Drug Interact. 1995;12:245–260. doi: 10.1515/dmdi.1995.12.3-4.245. [DOI] [PubMed] [Google Scholar]
- 4.Alonso-Gutierrez J, Chan R, Batth TS, Adams PD, Keasling JD, Petzold CJ, Lee TS. Metabolic engineering of Escherichia coli for limonene and perillyl alcohol production. Metab Eng. 2013;19:33–41. doi: 10.1016/j.ymben.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Geoghegan K, Evans P. Synthesis of (+)-perillyl alcohol from (+)-limonene. Tetrahedron Lett. 2014;55:1431–1433. [Google Scholar]
- 6.Miyazawa M, Shindo M, Shimada T. Metabolism of (+)- and (-)-limonenes to respective carveols and perillyl alcohols by CYP2C9 and CYP2C19 in human liver microsomes. Drug Metab Dispos. 2002;30:602–607. doi: 10.1124/dmd.30.5.602. [DOI] [PubMed] [Google Scholar]
- 7.Shimada T, Shindo M, Miyazawa M. Species differences in the metabolism of (+)- and (-)-limonenes and their metabolites, carveols and carvones, by cytochrome P450 enzymes in liver microsomes of mice, rats, guinea pigs, rabbits, dogs, monkeys, and humans. Drug Metab Pharmacokinet. 2002;17:507–515. doi: 10.2133/dmpk.17.507. [DOI] [PubMed] [Google Scholar]
- 8.Hudes GR, Szarka CE, Adams A, Ranganathan S, McCauley RA, Weiner LM, Langer CJ, Litwin S, Yeslow G, Halberr T, Qian M, Gallo JM. Phase I pharmacokinetic trial of perillyl alcohol (NSC 641066) in patients with refractory solid malignancies. Clin Cancer Res. 2000;6:3071–3080. [PubMed] [Google Scholar]
- 9.Ripple GH, Gould MN, Arzoomanian RZ, Alberti D, Feierabend C, Simon K, Binger K, Tutsch KD, Pomplun M, Wahamaki A, Marnocha R, Wilding G, Bailey HH. Phase I clinical and pharmacokinetic study of perillyl alcohol administered four times a day. Clin Cancer Res. 2000;6:390–396. [PubMed] [Google Scholar]
- 10.Zhang Z, Chen H, Chan KK, Budd T, Ganapathi R. Gas chromatographic-mass spectrometric analysis of perillyl alcohol and metabolites in plasma. J Chromatogr B Biomed Sci Appl. 1999;728:85–95. doi: 10.1016/s0378-4347(99)00065-1. [DOI] [PubMed] [Google Scholar]
- 11.Boon PJ, van der Boon D, Mulder GJ. Cytotoxicity and biotransformation of the anticancer drug perillyl alcohol in PC12 cells and in the rat. Toxicol Appl Pharmacol. 2000;167:55–62. doi: 10.1006/taap.2000.8988. [DOI] [PubMed] [Google Scholar]
- 12.Bhatia SP, McGinty D, Letizia CS, Api AM. Fragrance material review on p-mentha-1,8-dien-7-ol. Food Chem Toxicol. 2008;46(Suppl 11):S197–200. doi: 10.1016/j.fct.2008.06.071. [DOI] [PubMed] [Google Scholar]
- 13.Laszlo P. Citrus: A History. Chicago, Illinois: University of Chicago Press; 2007. [Google Scholar]
- 14.U.S. Food and Drug Administration, 21CFR172.515, Code of Federal Regulations Title 21, April 1, 2014. Part 172: Food Additives Permitted for Direct Addition to Food for Human Consumption. [Google Scholar]
- 15.Crowell PL, Gould MN. Chemoprevention and therapy of cancer by d-limonene. Crit Rev Oncog. 1994;5:1–22. doi: 10.1615/critrevoncog.v5.i1.10. [DOI] [PubMed] [Google Scholar]
- 16.Crowell PL. Prevention and therapy of cancer by dietary monoterpenes. J Nutr. 1999;129:775S–778S. doi: 10.1093/jn/129.3.775S. [DOI] [PubMed] [Google Scholar]
- 17.Crowell PL, Ren Z, Lin S, Vedejs E, Gould MN. Structure-activity relationships among monoterpene inhibitors of protein isoprenylation and cell proliferation. Biochem Pharmacol. 1994;47:1405–1415. doi: 10.1016/0006-2952(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 18.Haag JD, Gould MN. Mammary carcinoma regression induced by perillyl alcohol, a hydroxylated analog of limonene. Cancer Chemother Pharmacol. 1994;34:477–483. doi: 10.1007/BF00685658. [DOI] [PubMed] [Google Scholar]
- 19.Haag JD, Lindstrom MJ, Gould MN. Limonene-induced regression of mammary carcinomas. Cancer Res. 1992;52:4021–4026. [PubMed] [Google Scholar]
- 20.He L, Mo H, Hadisusilo S, Qureshi AA, Elson CE. Isoprenoids suppress the growth of murine B16 melanomas in vitro and in vivo. J Nutr. 1997;127:668–674. doi: 10.1093/jn/127.5.668. [DOI] [PubMed] [Google Scholar]
- 21.Crowell PL. Monoterpenes in breast cancer chemoprevention. Breast Cancer Res Treat. 1997;46:191–197. doi: 10.1023/a:1005939806591. [DOI] [PubMed] [Google Scholar]
- 22.Ong TP, Cardozo MT, de Conti A, Moreno FS. Chemoprevention of hepatocarcinogenesis with dietary isoprenic derivatives: cellular and molecular aspects. Curr Cancer Drug Targets. 2012;12:1173–1190. doi: 10.2174/156800912803987986. [DOI] [PubMed] [Google Scholar]
- 23.Reddy BS, Wang CX, Samaha H, Lubet R, Steele VE, Kelloff GJ, Rao CV. Chemoprevention of colon carcinogenesis by dietary perillyl alcohol. Cancer Res. 1997;57:420–425. [PubMed] [Google Scholar]
- 24.Lantry LE, Zhang Z, Crist KA, Wang Y, Hara M, Zeeck A, Lubet RA, You M. Chemopreventive efficacy of promising farnesyltransferase inhibitors. Exp Lung Res. 2000;26:773–790. doi: 10.1080/01902140150216819. [DOI] [PubMed] [Google Scholar]
- 25.Lantry LE, Zhang Z, Gao F, Crist KA, Wang Y, Kelloff GJ, Lubet RA, You M. Chemopreventive effect of perillyl alcohol on 4-(methylni-trosamino)-1-(3-pyridyl)-1-butanone induced tumorigenesis in (C3H/HeJ X A/J)F1 mouse lung. J Cell Biochem Suppl. 1997;27:20–25. doi: 10.1002/(sici)1097-4644(1997)27+<20::aid-jcb6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 26.Burke YD, Ayoubi AS, Werner SR, McFarland BC, Heilman DK, Ruggeri BA, Crowell PL. Effects of the isoprenoids perillyl alcohol and farnesol on apoptosis biomarkers in pancreatic cancer chemoprevention. Anticancer Res. 2002;22:3127–3134. [PubMed] [Google Scholar]
- 27.Liston BW, Nines R, Carlton PS, Gupta A, Aziz R, Frankel W, Stoner GD. Perillyl alcohol as a chemopreventive agent in N-nitrosome-thylbenzylamine-induced rat esophageal tumorigenesis. Cancer Res. 2003;63:2399–2403. [PubMed] [Google Scholar]
- 28.Low-Baselli A, Huber WW, Kafer M, Bukowska K, Schulte-Hermann R, Grasl-Kraupp B. Failure to demonstrate chemoprevention by the monoterpene perillyl alcohol during early rat hepatocarcinogenesis: a cautionary note. Carcinogenesis. 2000;21:1869–1877. doi: 10.1093/carcin/21.10.1869. [DOI] [PubMed] [Google Scholar]
- 29.Ariazi EA, Satomi Y, Ellis MJ, Haag JD, Shi W, Sattler CA, Gould MN. Activation of the transforming growth factor beta signaling pathway and induction of cytostasis and apoptosis in mammary carcinomas treated with the anticancer agent perillyl alcohol. Cancer Res. 1999;59:1917–1928. [PubMed] [Google Scholar]
- 30.Mills JJ, Chari RS, Boyer IJ, Gould MN, Jirtle RL. Induction of apoptosis in liver tumors by the monoterpene perillyl alcohol. Cancer Res. 1995;55:979–983. [PubMed] [Google Scholar]
- 31.Bishayee A, Thoppil RJ, Waghray A, Kruse JA, Novotny NA, Darvesh AS. Dietary phytochemicals in the chemoprevention and treatment of hepatocellular carcinoma: in vivo evidence, molecular targets, and clinical relevance. Curr Cancer Drug Targets. 2012;12:1191–1232. [PubMed] [Google Scholar]
- 32.Barthelman M, Chen W, Gensler HL, Huang C, Dong Z, Bowden GT. Inhibitory effects of perillyl alcohol on UVB-induced murine skin cancer and AP-1 transactivation. Cancer Res. 1998;58:711–716. [PubMed] [Google Scholar]
- 33.Lluria-Prevatt M, Morreale J, Gregus J, Alberts DS, Kaper F, Giaccia A, Powell MB. Effects of perillyl alcohol on melanoma in the TPras mouse model. Cancer Epidemiol Biomarkers Prev. 2002;11:573–579. [PubMed] [Google Scholar]
- 34.Chaudhary SC, Alam MS, Siddiqui MS, Athar M. Perillyl alcohol attenuates Ras-ERK signaling to inhibit murine skin inflammation and tumorigenesis. Chem Biol Interact. 2009;179:145–153. doi: 10.1016/j.cbi.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 35.Gupta A, Myrdal PB. Development of a perillyl alcohol topical cream formulation. Int J Pharm. 2004;269:373–383. doi: 10.1016/j.ijpharm.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 36.Stratton SP, Saboda KL, Myrdal PB, Gupta A, McKenzie NE, Brooks C, Salasche SJ, Warneke JA, Ranger-Moore J, Bozzo PD, Blanchard J, Einspahr JG, Dorr RT, Levine N, Alberts DS. Phase 1 study of topical perillyl alcohol cream for chemoprevention of skin cancer. Nutr Cancer. 2008;60:325–330. doi: 10.1080/01635580701840391. [DOI] [PubMed] [Google Scholar]
- 37.Stratton SP, Alberts DS, Einspahr JG, Sagerman PM, Warneke JA, Curiel-Lewandrowski C, Myrdal PB, Karlage KL, Nickoloff BJ, Brooks C, Saboda K, Yozwiak ML, Krutzsch MF, Hu C, Lluria-Prevatt M, Dong Z, Bowden GT, Bartels PH. A phase 2a study of topical perillyl alcohol cream for chemoprevention of skin cancer. Cancer Prev Res (Phila) 2010;3:160–169. doi: 10.1158/1940-6207.CAPR-09-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stark MJ, Burke YD, McKinzie JH, Ayoubi AS, Crowell PL. Chemotherapy of pancreatic cancer with the monoterpene perillyl alcohol. Cancer Lett. 1995;96:15–21. doi: 10.1016/0304-3835(95)03912-g. [DOI] [PubMed] [Google Scholar]
- 39.Lebedeva IV, Su ZZ, Vozhilla N, Chatman L, Sarkar D, Dent P, Athar M, Fisher PB. Chemoprevention by perillyl alcohol coupled with viral gene therapy reduces pancreatic cancer pathogenesis. Mol Cancer Ther. 2008;7:2042–2050. doi: 10.1158/1535-7163.MCT-08-0245. [DOI] [PubMed] [Google Scholar]
- 40.Yuri T, Danbara N, Tsujita-Kyutoku M, Kiyozuka Y, Senzaki H, Shikata N, Kanzaki H, Tsubura A. Perillyl alcohol inhibits human breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat. 2004;84:251–260. doi: 10.1023/B:BREA.0000019966.97011.4d. [DOI] [PubMed] [Google Scholar]
- 41.Teruszkin Balassiano I, Alves de Paulo S, Henriques Silva N, Curie Cabral M, Gibaldi D, Bozza M, Orlando da Fonseca C, Da Gloria da Costa Carvalho M. Effects of perillyl alcohol in glial C6 cell line in vitro and anti-metastatic activity in chorioallantoic membrane model. Int J Mol Med. 2002;10:785–788. doi: 10.3892/ijmm.10.6.785. [DOI] [PubMed] [Google Scholar]
- 42.Cho HY, Wang W, Jhaveri N, Torres S, Tseng J, Leong MN, Lee DJ, Goldkorn A, Xu T, Petasis NA, Louie SG, Schonthal AH, Hofman FM, Chen TC. Perillyl alcohol for the treatment of temozolomide-resistant gliomas. Mol Cancer Ther. 2012;11:2462–2472. doi: 10.1158/1535-7163.MCT-12-0321. [DOI] [PubMed] [Google Scholar]
- 43.Clark SS. Perillyl alcohol induces c-Myc-dependent apoptosis in Bcr/Abl-transformed leukemia cells. Oncology. 2006;70:13–18. doi: 10.1159/000091181. [DOI] [PubMed] [Google Scholar]
- 44.Koyama M, Sowa Y, Hitomi T, Iizumi Y, Watanabe M, Taniguchi T, Ichikawa M, Sakai T. Perillyl alcohol causes G1 arrest through p15(INK4b) and p21(WAF1/Cip1) induction. Oncol Rep. 2013;29:779–784. doi: 10.3892/or.2012.2167. [DOI] [PubMed] [Google Scholar]
- 45.Lebedeva IV, Su ZZ, Vozhilla N, Chatman L, Sarkar D, Dent P, Athar M, Fisher PB. Mechanism of in vitro pancreatic cancer cell growth inhibition by melanoma differentiation-associated gene-7/interleukin-24 and perillyl alcohol. Cancer Res. 2008;68:7439–7447. doi: 10.1158/0008-5472.CAN-08-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiseman DA, Werner SR, Crowell PL. Cell cycle arrest by the isoprenoids perillyl alcohol, geraniol, and farnesol is mediated by p21(Cip1) and p27(Kip1) in human pancreatic adenocarcinoma cells. J Pharmacol Exp Ther. 2007;320:1163–1170. doi: 10.1124/jpet.106.111666. [DOI] [PubMed] [Google Scholar]
- 47.Yeruva L, Pierre KJ, Elegbede A, Wang RC, Carper SW. Perillyl alcohol and perillic acid induced cell cycle arrest and apoptosis in non small cell lung cancer cells. Cancer Lett. 2007;257:216–226. doi: 10.1016/j.canlet.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 48.Fernandes J, da Fonseca CO, Teixeira A, Gattass CR. Perillyl alcohol induces apoptosis in human glioblastoma multiforme cells. Oncol Rep. 2005;13:943–947. [PubMed] [Google Scholar]
- 49.Shi W, Gould MN. Induction of cytostasis in mammary carcinoma cells treated with the anticancer agent perillyl alcohol. Carcinogenesis. 2002;23:131–142. doi: 10.1093/carcin/23.1.131. [DOI] [PubMed] [Google Scholar]
- 50.Sahin MB, Perman SM, Jenkins G, Clark SS. Perillyl alcohol selectively induces G0/G1 arrest and apoptosis in Bcr/Abl-transformed myeloid cell lines. Leukemia. 1999;13:1581–1591. doi: 10.1038/sj.leu.2401536. [DOI] [PubMed] [Google Scholar]
- 51.Loutrari H, Hatziapostolou M, Skouridou V, Papadimitriou E, Roussos C, Kolisis FN, Papapetropoulos A. Perillyl alcohol is an angiogenesis inhibitor. J Pharmacol Exp Ther. 2004;311:568–575. doi: 10.1124/jpet.104.070516. [DOI] [PubMed] [Google Scholar]
- 52.d’Alessio PA, Mirshahi M, Bisson JF, Bene MC. Skin repair properties of d-Limonene and perillyl alcohol in murine models. Antiinflamm Antiallergy Agents Med Chem. 2014;13:29–35. doi: 10.2174/18715230113126660021. [DOI] [PubMed] [Google Scholar]
- 53.Ripple GH, Gould MN, Stewart JA, Tutsch KD, Arzoomanian RZ, Alberti D, Feierabend C, Pomplun M, Wilding G, Bailey HH. Phase I clinical trial of perillyl alcohol administered daily. Clin Cancer Res. 1998;4:1159–1164. [PubMed] [Google Scholar]
- 54.Morgan-Meadows S, Dubey S, Gould M, Tutsch K, Marnocha R, Arzoomanin R, Alberti D, Binger K, Feierabend C, Volkman J, Ellingen S, Black S, Pomplun M, Wilding G, Bailey H. Phase I trial of perillyl alcohol administered four times daily continuously. Cancer Chemother Pharmacol. 2003;52:361–366. doi: 10.1007/s00280-003-0684-y. [DOI] [PubMed] [Google Scholar]
- 55.Bailey HH, Wilding G, Tutsch KD, Arzoomanian RZ, Alberti D, Feierabend C, Simon K, Marnocha R, Holstein SA, Stewart J, Lewis KA, Hohl RJ. A phase I trial of perillyl alcohol administered four times daily for 14 days out of 28 days. Cancer Chemother Pharmacol. 2004;54:368–376. doi: 10.1007/s00280-004-0788-z. [DOI] [PubMed] [Google Scholar]
- 56.Murren JR, Pizzorno G, DiStasio SA, McKeon A, Peccerillo K, Gollerkari A, McMurray W, Burtness BA, Rutherford T, Li X, Ho PT, Sartorelli A. Phase I study of perillyl alcohol in patients with refractory malignancies. Cancer Biol Ther. 2002;1:130–135. doi: 10.4161/cbt.57. [DOI] [PubMed] [Google Scholar]
- 57.Azzoli CG, Miller VA, Ng KK, Krug LM, Spriggs DR, Tong WP, Riedel ER, Kris MG. A phase I trial of perillyl alcohol in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2003;51:493–498. doi: 10.1007/s00280-003-0599-7. [DOI] [PubMed] [Google Scholar]
- 58.Bailey HH, Levy D, Harris LS, Schink JC, Foss F, Beatty P, Wadler S. A phase II trial of daily perillyl alcohol in patients with advanced ovarian cancer: Eastern Cooperative Oncology Group Study E2E96. Gynecol Oncol. 2002;85:464–468. doi: 10.1006/gyno.2002.6647. [DOI] [PubMed] [Google Scholar]
- 59.Meadows SM, Mulkerin D, Berlin J, Bailey H, Kolesar J, Warren D, Thomas JP. Phase II trial of perillyl alcohol in patients with metastatic colorectal cancer. Int J Gastrointest Cancer. 2002;32:125–128. doi: 10.1385/IJGC:32:2-3:125. [DOI] [PubMed] [Google Scholar]
- 60.Liu G, Oettel K, Bailey H, Ummersen LV, Tutsch K, Staab MJ, Horvath D, Alberti D, Arzoomanian R, Rezazadeh H, McGovern J, Robinson E, DeMets D, Wilding G. Phase II trial of perillyl alcohol (NSC 641066) administered daily in patients with metastatic androgen independent prostate cancer. Invest New Drugs. 2003;21:367–372. doi: 10.1023/a:1025437115182. [DOI] [PubMed] [Google Scholar]
- 61.Bailey HH, Attia S, Love RR, Fass T, Chappell R, Tutsch K, Harris L, Jumonville A, Hansen R, Shapiro GR, Stewart JA. Phase II trial of daily oral perillyl alcohol (NSC 641066) in treatment-refractory metastatic breast cancer. Cancer Chemother Pharmacol. 2008;62:149–157. doi: 10.1007/s00280-007-0585-6. [DOI] [PubMed] [Google Scholar]
- 62.Illum L. Nasal drug delivery - recent developments and future prospects. J Control Release. 2012;161:254–263. doi: 10.1016/j.jconrel.2012.01.024. [DOI] [PubMed] [Google Scholar]
- 63.Gizurarson S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr Drug Deliv. 2012;9:566–582. doi: 10.2174/156720112803529828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolfe TR, Bernstone T. Intranasal drug delivery: an alternative to intravenous administration in selected emergency cases. J Emerg Nurs. 2004;30:141–147. doi: 10.1016/j.jen.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 65.Bitter C, Suter-Zimmermann K, Surber C. Nasal drug delivery in humans. Curr Probl Dermatol. 2011;40:20–35. doi: 10.1159/000321044. [DOI] [PubMed] [Google Scholar]
- 66.Ugwoke MI, Agu RU, Verbeke N, Kinget R. Nasal mucoadhesive drug delivery: background, applications, trends and future perspectives. Adv Drug Deliv Rev. 2005;57:1640–1665. doi: 10.1016/j.addr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 67.Gomez D, Martinez JA, Hanson LR, Frey WH 2nd, Toth CC. Intranasal treatment of neurodegenerative diseases and stroke. Front Biosci (Schol Ed) 2012;4:74–89. doi: 10.2741/252. [DOI] [PubMed] [Google Scholar]
- 68.Lochhead JJ, Thorne RG. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev. 2012;64:614–628. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 69.Merkus FW, van den Berg MP. Can nasal drug delivery bypass the blood-brain barrier?: questioning the direct transport theory. Drugs R D. 2007;8:133–144. doi: 10.2165/00126839-200708030-00001. [DOI] [PubMed] [Google Scholar]
- 70.Pardeshi CV, Belgamwar VS. Direct nose to brain drug delivery via integrated nerve pathways bypassing the blood-brain barrier: an excellent platform for brain targeting. Expert Opin Drug Deliv. 2013;10:957–972. doi: 10.1517/17425247.2013.790887. [DOI] [PubMed] [Google Scholar]
- 71.Dhuria SV, Hanson LR, Frey WH 2nd. Intranasal delivery to the central nervous system:mechanisms and experimental considerations. J Pharm Sci. 2010;99:1654–1673. doi: 10.1002/jps.21924. [DOI] [PubMed] [Google Scholar]
- 72.Djupesland PG. Nasal drug delivery devices: characteristics and performance in a clinical perspective-a review. Drug Deliv Transl Res. 2013;3:42–62. doi: 10.1007/s13346-012-0108-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Djupesland PG, Messina JC, Mahmoud RA. The nasal approach to delivering treatment for brain diseases: an anatomic, physiologic, and delivery technology overview. Ther Deliv. 2014;5:709–733. doi: 10.4155/tde.14.41. [DOI] [PubMed] [Google Scholar]
- 74. www.kurve.com.
- 75.Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, Fishel MA, Plymate SR, Breitner JC, DeGroodt W, Mehta P, Craft S. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2008;70:440–448. doi: 10.1212/01.WNL.0000265401.62434.36. [DOI] [PubMed] [Google Scholar]
- 77.da Fonseca CO, Schwartsmann G, Fischer J, Nagel J, Futuro D, Quirico-Santos T, Gattass CR. Preliminary results from a phase I/II study of perillyl alcohol intranasal administration in adults with recurrent malignant gliomas. Surg Neurol. 2008;70:259–266. doi: 10.1016/j.surneu.2007.07.040. [DOI] [PubMed] [Google Scholar]
- 78.da Fonseca CO, Simao M, Lins IR, Caetano RO, Futuro D, Quirico-Santos T. Efficacy of monoterpene perillyl alcohol upon survival rate of patients with recurrent glioblastoma. J Cancer Res Clin Oncol. 2011;137:287–293. doi: 10.1007/s00432-010-0873-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.da Fonseca CO, Teixeira RM, Silva JC, DE Saldanha DA Gama Fischer J, Meirelles OC, Landeiro JA, Quirico-Santos T. Long-term outcome in patients with recurrent malignant glioma treated with perillyl alcohol inhalation. Anticancer Res. 2013;33:5625–5631. [PubMed] [Google Scholar]
- 80.Phillips LR, Malspeis L, Supko JG. Pharmacokinetics of active drug metabolites after oral administration of perillyl alcohol, an investigational antineoplastic agent, to the dog. Drug Metab Dispos. 1995;23:676–680. [PubMed] [Google Scholar]
- 81.Elegbede JA, Flores R, Wang RC. Perillyl alcohol and perillaldehyde induced cell cycle arrest and cell death in BroTo and A549 cells cultured in vitro. Life Sci. 2003;73:2831–2840. doi: 10.1016/s0024-3205(03)00701-x. [DOI] [PubMed] [Google Scholar]
- 82.Bardon S, Picard K, Martel P. Monoterpenes inhibit cell growth, cell cycle progression, and cyclin D1 gene expression in human breast cancer cell lines. Nutr Cancer. 1998;32:1–7. doi: 10.1080/01635589809514708. [DOI] [PubMed] [Google Scholar]
- 83.Ren Z, Gould MN. Inhibition of ubiquinone and cholesterol synthesis by the monoterpene perillyl alcohol. Cancer Lett. 1994;76:185–190. doi: 10.1016/0304-3835(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 84.Crowell PL, Chang RR, Ren ZB, Elson CE, Gould MN. Selective inhibition of isoprenylation of 21-26-kDa proteins by the anticarcinogen d-limonene and its metabolites. J Biol Chem. 1991;266:17679–17685. [PubMed] [Google Scholar]
- 85.Gelb MH, Tamanoi F, Yokoyama K, Ghomashchi F, Esson K, Gould MN. The inhibition of protein prenyltransferases by oxygenated metabolites of limonene and perillyl alcohol. Cancer Lett. 1995;91:169–175. doi: 10.1016/0304-3835(95)03747-k. [DOI] [PubMed] [Google Scholar]
- 86.Ren Z, Elson CE, Gould MN. Inhibition of type I and type II geranylgeranyl-protein transferases by the monoterpene perillyl alcohol in NIH3T3 cells. Biochem Pharmacol. 1997;54:113–120. doi: 10.1016/s0006-2952(97)00151-2. [DOI] [PubMed] [Google Scholar]
- 87.Stayrook KR, McKinzie JH, Barbhaiya LH, Crowell PL. Effects of the antitumor agent perillyl alcohol on H-Ras vs. K-Ras farnesylation and signal transduction in pancreatic cells. Anticancer Res. 1998;18:823–828. [PubMed] [Google Scholar]
- 88.Hohl RJ, Lewis K. Differential effects of monoterpenes and lovastatin on RAS processing. J Biol Chem. 1995;270:17508–17512. doi: 10.1074/jbc.270.29.17508. [DOI] [PubMed] [Google Scholar]
- 89.Cerda SR, Wilkinson J 4th, Thorgeirsdottir S, Broitman SA. R-(+)-perillyl alcohol-induced cell cycle changes, altered actin cytoskeleton, and decreased ras and p34(cdc2) expression in colonic adenocarcinoma SW480 cells. J Nutr Biochem. 1999;10:19–30. doi: 10.1016/s0955-2863(98)00078-3. [DOI] [PubMed] [Google Scholar]
- 90.Berchtold CM, Chen KS, Miyamoto S, Gould MN. Perillyl alcohol inhibits a calcium-dependent constitutive nuclear factor-kappaB pathway. Cancer Res. 2005;65:8558–8566. doi: 10.1158/0008-5472.CAN-04-4072. [DOI] [PubMed] [Google Scholar]
- 91.Satomi Y, Miyamoto S, Gould MN. Induction of AP-1 activity by perillyl alcohol in breast cancer cells. Carcinogenesis. 1999;20:1957–1961. doi: 10.1093/carcin/20.10.1957. [DOI] [PubMed] [Google Scholar]
- 92.Sundin T, Peffley D, Hentosh P. eIF4E-Overexpression imparts perillyl alcohol and rapamycin-mediated regulation of telomerase reverse transcriptase. Exp Cell Res. 2013;319:2103–2112. doi: 10.1016/j.yexcr.2013.05.029. [DOI] [PubMed] [Google Scholar]
- 93.Sundin T, Peffley DM, Gauthier D, Hentosh P. The isoprenoid perillyl alcohol inhibits telomerase activity in prostate cancer cells. Biochimie. 2012;94:2639–2648. doi: 10.1016/j.biochi.2012.07.028. [DOI] [PubMed] [Google Scholar]
- 94.Garcia DG, Amorim LM, de Castro Faria MV, Freire AS, Santelli RE, Da Fonseca CO, Quirico-Santos T, Burth P. The anticancer drug perillyl alcohol is a Na/K-ATPase inhibitor. Mol Cell Biochem. 2010;345:29–34. doi: 10.1007/s11010-010-0556-9. [DOI] [PubMed] [Google Scholar]
- 95.Maltzman TH, Christou M, Gould MN, Jefcoate CR. Effects of monoterpenoids on in vivo DMBA-DNA adduct formation and on phase I hepatic metabolizing enzymes. Carcinogenesis. 1991;12:2081–2087. doi: 10.1093/carcin/12.11.2081. [DOI] [PubMed] [Google Scholar]
- 96.Austin CA, Shephard EA, Pike SF, Rabin BR, Phillips IR. The effect of terpenoid compounds on cytochrome P-450 levels in rat liver. Biochem Pharmacol. 1988;37:2223–2229. doi: 10.1016/0006-2952(88)90585-0. [DOI] [PubMed] [Google Scholar]
- 97.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3:1787–1808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging Ras Back in the Ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 99.Gould MN. Prevention and therapy of mammary cancer by monoterpenes. J Cell Biochem Suppl. 1995;22:139–144. doi: 10.1002/jcb.240590818. [DOI] [PubMed] [Google Scholar]
- 100.da Fonseca CO, Linden R, Futuro D, Gattass CR, Quirico-Santos T. Ras pathway activation in gliomas: a strategic target for intranasal administration of perillyl alcohol. Arch Immunol Ther Exp (Warsz) 2008;56:267–276. doi: 10.1007/s00005-008-0027-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hardcastle IR, Rowlands MG, Barber AM, Grimshaw RM, Mohan MK, Nutley BP, Jarman M. Inhibition of protein prenylation by metabolites of limonene. Biochem Pharmacol. 1999;57:801–809. doi: 10.1016/s0006-2952(98)00349-9. [DOI] [PubMed] [Google Scholar]
- 102.Holstein SA, Hohl RJ. Monoterpene regulation of Ras and Ras-related protein expression. J Lipid Res. 2003;44:1209–1215. doi: 10.1194/jlr.M300057-JLR200. [DOI] [PubMed] [Google Scholar]
- 103.Ruch RJ, Sigler K. Growth inhibition of rat liver epithelial tumor cells by monoterpenes does not involve Ras plasma membrane association. Carcinogenesis. 1994;15:787–789. doi: 10.1093/carcin/15.4.787. [DOI] [PubMed] [Google Scholar]
- 104.Karlson J, Borg-Karlson AK, Unelius R, Shoshan MC, Wilking N, Ringborg U, Linder S. Inhibition of tumor cell growth by monoterpenes in vitro: evidence of a Ras-independent mechanism of action. Anticancer Drugs. 1996;7:422–429. doi: 10.1097/00001813-199606000-00008. [DOI] [PubMed] [Google Scholar]
- 105.Clark SS, Zhong L, Filiault D, Perman S, Ren Z, Gould M, Yang X. Anti-leukemia effect of perillyl alcohol in Bcr/Abl-transformed cells indirectly inhibits signaling through Mek in a Ras- and Raf-independent fashion. Clin Cancer Res. 2003;9:4494–4504. [PubMed] [Google Scholar]