

Abstract
Methylation alterations of Jagged1 and Notch1 genes have been reported in non-tumor lesions and a few cancers. However, methylation profiles of Jagged1 promoter and Notch1 exon25 in breast cancer and matched normal tissue and the association of methylation with clinicopathological characteristics still remain unclear. To explore the potential effects of aberrant DNA methylation of Jagged1 and Notch1 on occurrence and progression of breast cancer, we detected the quantitative DNA methylation of Jagged1 and Notch1 in 73 breast cancer (BC) and 20 adjacent normal breast tissues (ANBT) by using MassARRAY spectrometry. The methylation level of overall and majority individual CpG sites of the two genes were synergistically significantly lower in BC than in ANBT. The overall hypomethylation of the two genes, particularly of Jagged1 CpG_8.9.10 and Notch1 CpG_14.15.16 in primary tumors, were markedly associated with lymph node metastasis, advanced stage and high grade. The protein expressions of the both genes were examined by immunohistochemical staining in same cohorts. The expression was significantly inverse correlation with methylation. The two proteins in primary tumor were synergistically up-regulated and dramatically related to lymph node metastasis, advanced stage and high grade. Our findings suggest that the synergetic hypomethylation of Jagged1 and Notch1 genes, especially of Jagged1 CpG_8.9.10 and Notch1 CpG_14.15.16, may involve tumorigenesis and development of breast cancer. The negative relationship between methylation and expression indicates methylation role for expression regulation. The synergetic overexpression of the two proteins further indicates the effects on occurrence and progression of breast cancer.
Keywords: Jagged1, Notch1, methylation, expression, occurrence, progression, breast cancer
Introduction
Breast cancer is one of the most common malignancies among women [1]. To date, the mechanisms of occurrence and prognosis of breast cancer is not fully elucidated. The Notch signaling plays crucial role during the development of adenocarcinoma of murine mammary glands [2]. Human Notch pathway family consists of five ligands (Jagged1/2, Delta-like ligand 1/3/4) and four Notch receptors (Notch1, 2, 3 and 4). The activation of the Notch signaling requires a cell to cell contact that involves the physical interaction between the transmembrane Notch receptor and its ligand [3,4]. In our previous study, we have observed the high expression of Notch1 protein is associated with tumorigenesis, metastasis and poor prognosis of patients with breast cancer [5,6]. It has been reported that increased expression of Jagged1 correlates with frequent recurrence and severe malignancy of breast cancer [7,8], and the synergistic elevation of Jagged1 and Notch1 expression affects the overall survival of breast cancer patients [9].
Jagged1 and Notch1 gene expressions are governed by complex genetic and epigenetic alterations. Epigenetic processes such as DNA methylation may control the meticulous spatial and temporal expression of the Notch1 or Jagged1 gene [10-12]. A few studies have showed the Jagged1 promoter hypermethylation occurring in acute lymphoblastic leukemia [13] and neural cell differentiation in mouse [14]. However, Jagged1 methylation status in breast cancer is still not reported so far. For Notch1 expression regulation, researchers have showed that Notch1 methylation on promoter region regulates its expression in muscle cells and its diseases [15-17], asthma [18], systemic lupus erythematosus [19], hepatic stellate cells [20], mantle cell lymphoma [21], and oral squamous cell carcinomas [22]. DNA methylation can occur on promoter or non-promoter (exon) region [23,24]. Our previous study [25] has shown that Notch1 methylation on exon25 is obviously decreased in invasive carcinoma compared with those in ductal carcinoma in situ, atypical ductal hyperplasia and usual ductal hyperplasia, and hypomethylation corresponds with overexpression. Nevertheless, methylation alteration of Notch1 exon25 in carcinoma and matched normal tissues and the role of aberrant methylation in progression of breast cancer still remain unclear.
To investigate whether aberrant methylation of Jagged1 and Notch1 genes affect on occurrence and progression of breast cancer, we detected the methylation profiles of Jagged1 promoter and Notch1 exon25 in matched pairs of breast carcinoma and adjacent non-cancer tissue by using quantitative DNA methylation analysis (MassARRAY spectrometry), and followed the evaluating for the association of methylation status with clinicopathological characteristics as well as the association of methylation alteration with expression level.
Materials and methods
Patients and tissues
A total of 73 consecutive breast cancer (BC) patients with complete clinicopathological data were collected from the First Affiliated Hospital of Shihezi University School of Medicine from January 2008 to June 2009. Of these, 20 patients had adjacent normal breast tissue (ANBT). None of the patients received therapy before sample collection. Patients underwent modified radical mastectomy and the acquired breast tissues were fixed in 10% neutral formalin and embedded in paraffin. Clinical and pathological information (Table S1) were available and reviewed. The clinical stage was based on TNM staging system (American Joint Committee on Cancer classification) [26]. The histological type and grade was according to WHO classification [27]. The staining results o estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) were independently evaluated by two experienced pathologists without prior knowledge of clinical information. ER and PR expression were considered as positive when there was ≥1 % of nuclear staining [28]. Membranous immunostaining for HER2 was scored on a scale of 0 to 3+. Tumors with immunohistochemical scores of 3+ or with a ≥2.2-fold increase in HER2 gene amplification as determined by FISH (fluorescence in situ hybridization) were considered to be positive for HER2. The matched normal breast tissues were collected at least 4 cm away from the tumor site. The study was approved by the Ethics Committee of the above-mentioned hospital and all patients signed informed consent forms.
MALDI-TOF-MS based DNA methylation analysis
High-throughput methylation detection was performed on the MassARRAY system (SEQUENOM, Inc. San Diego, CA) applying the Mass CLEAVETM (SEQUENOM, Inc. San Diego, CA) biochemistry and MALDI-TOF mass spectrometry (Bruker SEQUENOM) [29,30].
DNA extraction and bisulfite treatment
The portions of samples that consisted of at least 70% cancer cells or normal luminal cells in each of the 73 formalin-fixed paraffin-embedded BC and 20 ANBT were used. Genomic DNA was isolated by using tissue DNA extraction kit (Qiagen Inc., Valencia, CA, USA) [31]. Following manufacturer’s instructions, genomic DNA bisulfite conversion was achieved by using EZ-96 Bisulfite Kit (Zymo Research, Orange County, CA), which converts native cytosine (“C”) nucleotides into uracil (“U”), but the 5-methyl-protected cytosine residues remain as “C”, and the resulting artificial sequence is conserved during PCR amplification. The PCR was performed as follows: 95°C for 30 sec, 50°C for 15 min, repeating these 2 steps for 20 cycles.
Primer design and PCR tagging for EpiTYPER assay
The sequence of CpG islands on Jagged1 and Notch1 genes was identified by using UCSC genome browser (http://genome.ucsc.edu/). Using EpiDesigner software (http://epidesigner.com), we designed the primers covering the region with maximum CpG sites on the Jagged1 and Notch1 genes (Table 1). The forward primer was tagged with 10-mer sequence to balance the PCR primer length, and T7 promoter (31 bp) was added to the reverse primer to facilitate the conversion of double stranded PCR product into single stranded RNA with simultaneous second level of amplification. The PCR conditions were one cycle, 94°C for 4 min; 45 cycles, 94°C for 20 sec, Ta for 30 sec (Table 1), 72°C for 1 min; one cycle, 72°C for 3 min. After the round of PCR amplification, 2 μl PCR products was used for in vitro transcription.
Table 1.
Primer sequence, length, and PCR conditions for methylation analysis
| Gene | Primer* | Sequence (5’→3’) | Length | Ta# (°C) | Amplified Length (bp) |
|---|---|---|---|---|---|
| Jagged1 | tag-FW | aggaagagagAAAATTTTTTTGGAGTTAGGTTTGT | 10+25 | 56 | 314 |
| T7-RV | cagtaatacgactcactatagggagaaggctAACCCATCTCTTACCACCCAA | 31+21 | |||
| Notch1 | tag-FW | aggaagagagTTTTTTTAGGGGTTATTGAAGTTGA | 10+25 | 56 | 282 |
| T7-RV | cagtaatacgactcactatagggagaaggctCCCCTACTACAACCAAAAAACCTAT | 31+25 |
FW, forward; RV, reverse.
Ta, annealing temperature.
In vitro transcription and T-cleavage assay
Unincorporated dNTPs in PCR products were removed by adding 0.3 units of shrimp alkaline phosphatase (SAP, SEQUENOM, Inc. San Diego, CA) and 1.7 μl ddH2O. The reaction mixture was incubated at 37°C for 20 minutes and the SAP was heat inactivated for 5 minutes at 85°C. After the SAP treatment, 5 ml T Cleavage Transcription/RNase Cocktail (Epicentre, Madison, WI) containing 0.89 μl 5× T7 polymerase buffer, 0.24 μl T cleavage mix, 3.14 mM dithiothreitol, 22 U T7 RNA and DNA polymerase, 0.09 mg/ml RNase A, and a total of 2 μl of the product of PCR/SAP reactions was mixed and incubated at 37°C for 3 hours for in vitro transcription and RNase A digestion.
Mass spectrometry
We robotically dispensed 15 nl of the RNase A treated product onto silicon chips preloaded with matrix (SpectroCHIP; SEQUENOM). The mass spectra data were collected using a MassARRAY Compact MALDI-TOF (SEQUENOM, Inc. San Diego, CA), and spectra’s methylation proportions were generated by Epityper software v.1.0 (SEQUENOM, Inc. San Diego, CA).
Immunohistochemistry (IHC)
The paraffin-embedded breast tissues were cut into 4-μm thick sections and were deparaffinized in xylene and rehydrated in graded alcohol. The microwave antigen-retrieval procedure in sodium citrate buffer (pH 6.0) was performed and endogenous peroxidase activity was blocked by H2O2. Subsequently, the slides were incubated with anti-Jagged1 (1:300, sc-8303, Santa Cruz, CA) or anti-Notch1 (1:50, ab44986, Abcam , Cambriadge, MA) over night. Following secondary antibody incubation, the product visualization was performed with diaminobenzidine (DAB) substrate chromogen. Human lung tumor tissue was served as positive control and PBS replaced primary antibody in the negative control. All slides were assessed independently by two pathologists. The proportion scores were given as a percentage of cells with positive cytoplasmic staining on a scale of 0 to 4 (0: 0-5%, 1: 6-25%, 2: 26-50%, 3: 51-75%, and 4: 76-100% positive cells). The absolute intensity of cytoplasmic staining were based on a scale of 0 to 3 (0: no staining, 1: heterogeneous staining, 2: homogenous staining, and 3: intense homogenous staining). Percentages and intensities of positive cells were then multiplied to generate immunoreactivity score (IS) for each case. The staining results were divided into 2 categories based on IS: < (0, 1, 2, and 3) which was considered as low expression, while ≥4 (4, 6, 8, 9, and 12) was defined as high expression.
Statistical analysis
Data analyses were performed by using the SPSS software package version 17.0. Quantitative DNA methylation profiles of Jagged1 and Notch1 gene were compared between cancer and normal tissue by using two-way hierarchical cluster analysis. The statistical difference of methylation quantity of overall or each CpG site between cancer and normal tissues was identified by t-test or Mann-Whitney-U-Test. The receiver operating characteristic (ROC) curve was used to calculate the methylation sensitivity and specificity. The correlation of methylation or the correlation of expression between Jagged1 and Notch1 gene was analyzed by Spearman’s rank test. The relationship between methylation and clinicopathological parameters or between methylation and expression in overall or each CpG site was evaluated by Mann-Whitney-U-Test or Kruskal-Wallis H test. The comparison of proteins expression between cancer and normal tissues and the association of expression with clinicopathological features were detected by Chi-square test or Fisher’s Exact Test. All statistic analyses were two-sided and P<0.05 was considered to be statistically significant.
Results
Methylation profiles of Jagged1 and Notch1 genes in breast cancer and normal tissues
The DNA methylation were detected on the region from -1,396 to -1083 bp (314 bp) relative to transcription start site (TSS) on Jagged1 promoter and on the region from +40,075 to +40,356 bp (282 bp) relative to TSS on Notch1 exon25 (Figure S1) because of CpG cluster-rich (>20 sites) [32]. For Jagged1 gene, the amplicon region covered 28 CpG sites and actually covered 28 CpG sites. Fifteen of 28 CpG sites could be analyzed and the 15 CpG sites included 6 single sites and 9 composite sites. For Notch1 gene, the amplicon region covered 21 CpG sites then actually covered 19 CpG sites, and 13 of 19 CpG sites were able to be analyzed and were divided into 8 single sites and 5 composite sites (Table S2).
The methylation status of Jagged1 gene in BC and ANBT was showed in Figure 1. The CpG methylation levels could be identified by color with low methylation highlighted in yellow and high methylation in red (Figure 1A). The overall methylation was markedly lower in BC (mean methylation rate 11.97%) than ANBT (mean methylation rate 20.17%) (Figure 1B). Over 86% CpG sites methylation value were less than 20% (median methylation rate 11%) in BC group, and 41% CpG sites showed increased methylation with more than 20% value compared with median 18% in ANBT. The methylation level of all individual CpG site was lower in BC than ANBT (Figure 1C). Particularly, the methylation levels of 10 CpG sites (CpG-2, CpG-6, CpG_7, CpG_11.12, CpG_13, CpG_16.17, CpG_20.21.22, CpG_23.24.25, CpG_26, and CpG_27.28) were significantly lower in BC than ANBT group (Table S3).
Figure 1.

Methylation profiles of Jagged1 gene in breast cancer (BC) and matched adjacent normal breast tissue (ANBT). A. Two-way hierarchical cluster analysis of methylation alteration. B. Comparison of overall methylation status. C. Methylation level of 15 individual CpG.
Consistent with Jagged1 methylation profile in cancer, Notch1 gene displayed lower methylation in cancer (yellow clusters) than normal tissues (red clusters) (Figure 2A), and the alteration was provided with statistical significance in BC (mean methylation rate 14.57%) and ANBT group (mean methylation rate 41.66%) (Figure 2B). Moreover, in cancer group, the methylation values of 85% CpG sites were less than 20% (median methylation rate 15.07%), then in normal group, 42% CpG sites were greater than 45% compared to median value of 43.69%. In addition, all 13 individual CpG site exhibited significantly decreased methylation level in BC (Figure 2C, Table S4).
Figure 2.

Methylation profiles of Notch1 gene in breast cancer (BC) and matched adjacent normal breast tissue (ANBT). A. Two-way hierarchical cluster analysis of methylation alteration. B. Comparison of overall methylation status. C. Methylation level of 13 individual CpG.
Furthermore, the receiver operating characteristic (ROC) analysis was performed to compare the methylation status of Jagged1 and Notch1 gene in BC. The sensitivity and specificity of the two genes were obviously high in cancer, respectively (Figure 3). The association analysis of methylation frequency in the two genes revealed a significantly positive relationship in BC but not markedly association in ANBT. (Figure S2).
Figure 3.
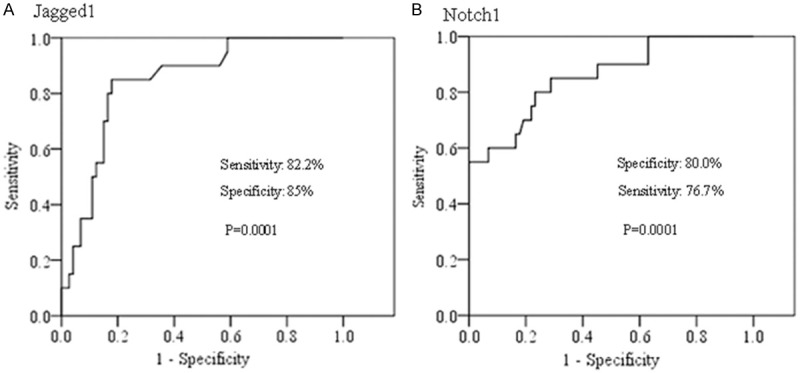
ROC analysis for methylation status of Jagged1 (A) and Notch1 (B) genes in BC samples.
Relationship between methylation alteration of Jagged1 and Notch1 genes and clinicopathological characteristics in breast cancer patients
The correlation between Jagged1 gene methylation and clinicopathological features of patients was displayed in Table 2. The overall methylation frequency in primary cancer was significantly lower with lymph node metastasis (p=0.000), advanced stage (p=0.000) and high grade groups (p=0.000). CpG_8.9.10 was significantly lower methylation degree in primary tumor with lymph node metastasis (p=0.002). The hypomethylation of CpG_8.9.10 and CpG_23.24.25 were markedly associated with advanced stage (p=0.001, p=0.038). The lower methylation status of CpG_11.12 was showed in high grade group (p=0.002). To note, CpG_8.9.10 hypomethylation in primary cancer was statistically related not only to lymph node metastasis but also to advanced stage. There were no significant methylation alterations in luminal subtype (ER+, PR+, HER2-) and HER2 overexpression subtype (ER-, PR-, HER2+) breast cancer.
Table 2.
Comparison between Jagged1 gene methylation and patient clinicopathological parameters
| Parameters | N | Jagged1 (Mean methylation %) | |||
|---|---|---|---|---|---|
|
| |||||
| Overall | CpG_8.9.10 | CpG_11.12 | CpG_23.24.25 | ||
| Lymph node metastasis | |||||
| - (LNN) | 39 | 13.15* | 9.92* | 11.72 | 11.64 |
| + (LNP) | 34 | 10.81 | 5.70 | 9.03 | 9.30 |
| TNM stage | |||||
| I | 10 | 15.19* | 12.1* | 12.50 | 13.8* |
| II | 29 | 12.47 | 9.26 | 11.62 | 10.85 |
| III | 34 | 10.42 | 4.96 | 8.41 | 9.07 |
| Histological grade | |||||
| 1 | 8 | 16.02* | 9.00 | 19.88* | 15.38 |
| 2 | 57 | 12.03 | 7.77 | 10.75 | 10.71 |
| 3 | 8 | 9.58 | 7.25 | 6.38 | 10.50 |
| Receptor status | |||||
| ER(+)/PR(+) Her2(-) | 52 | 12.63 | 6.40 | 10.80 | 10.20 |
| ER(-) PR(-) Her2(+) | 12 | 11.44 | 5.28 | 8.28 | 9.65 |
ER: estrogen receptor; PR: progesterone receptor; HER2: human epidermal growth factor receptor 2.
P<0.001.
The association of Notch1 methylation with clinicopathological parameters was presented in Table 3. The overall hypomethylation in primary tumor significantly related to lymph node metastasis (p=0.000), advanced stage (p=0.000), high grade (p=0.000), and HER2 overexpression subtype breast cancer (p=0.000). The low methylation of CpG_4.5, CpG_10.11 and CpG_14.15.16 in primary cancer were markedly associated with lymph node metastasis (p=0.008, p=0.013, and p=0.000). The CpG_14.15.16 and CpG_18 methylation significantly decreased in advanced stage group (p=0.003 and p=0.011). CpG_1.2 and CpG_12.13 were hypomethylated with high grade (p=0.005 and p=0.001). The CpG_3, CpG_8 and CpG_14.15.16 methylation significantly decreased in HER2 overexpression subtype breast cancer (p=0.011, p=0.013 and p=0.009). Interestingly, CpG_14.15.16 hypomethylation in primary tumor was a specific feature for lymph node metastasis, advanced stage, and HER2 overexpression subtype breast cancer.
Table 3.
Comparison between Notch1 gene methylation and patient clinicopathological parameters
| Parameters | N | Notch1 (Mean methylation %) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Oveall | CpG_1.2 | CpG_3 | CpG_4.5 | CpG_8 | CpG_10.11 | CpG_12.13 | CpG_14.15.16 | CpG_18 | ||
| Lymph node metastasis | ||||||||||
| - (LNN) | 39 | 15.24* | 11.85 | 21.80 | 21.00* | 21.14 | 7.13* | 16.46 | 11.97* | 15.71 |
| + (LNP) | 34 | 12.02 | 7.09 | 14.61 | 15.68 | 19.20 | 3.36 | 14.31 | 6.24 | 14.48 |
| TNM stage | ||||||||||
| I | 10 | 19.94* | 13.40 | 18.60 | 22.90 | 19.00 | 7.40 | 16.70 | 16.80* | 24.80* |
| II | 29 | 13.57 | 8.82 | 15.82 | 20.21 | 18.07 | 5.67 | 15.38 | 9.82 | 17.00 |
| III | 34 | 12.37 | 7.48 | 13.25 | 19.45 | 17.23 | 3.61 | 13.92 | 7.86 | 13.96 |
| Histological grade | ||||||||||
| 1 | 8 | 17.69* | 17.88* | 18.38 | 28.50 | 19.75 | 7.75 | 28.25* | 12.00 | 17.25 |
| 2 | 57 | 13.26 | 8.70 | 14.64 | 19.61 | 19.12 | 5.09 | 15.20 | 9.21 | 14.91 |
| 3 | 8 | 12.36 | 5.38 | 11.50 | 21.38 | 24.29 | 2.50 | 12.25 | 8.63 | 14.71 |
| Receptor status | ||||||||||
| ER(+)/PR(+) Her2(-) | 52 | 20.33* | 11.33 | 24.69* | 22.20 | 23.67* | 6.17 | 21.00 | 17.64* | 26.17 |
| ER(-) PR(-) Her2(+) | 12 | 14.09 | 9.83 | 18.60 | 20.77 | 16.71 | 6.10 | 20.91 | 8.27 | 24.27 |
P<0.05.
Impacts of DNA methylation in Jagged1 and Notch1 genes on protein expression, following the association of expression with clinicopathological features in breast cancer
We detected Jagged1 expression by immunohistochemistry staining in same cohorts (Figure 4A). The results showed the inverse correlation between methylation and expression in BC, especially the hypomethylation of overall and CpG_1, CpG_3.4.5, CpG_8.9.10, CpG_11.12, CpG_14.15 were significantly associated with high expression (p=0.000, p=0.033, p=0.043, p=0.039, p=0.018, and p=0.008) (Table 4). The expression level of Jagged1 was markedly higher in BC than in ANBT (Figure 5A) and was presented mainly in high expression status in cancer (42/73, 57.5%). The high expression of Jagged1 in primary cancer significantly related to lymph node metastasis, advanced stage and high grade (Table 5).
Figure 4.
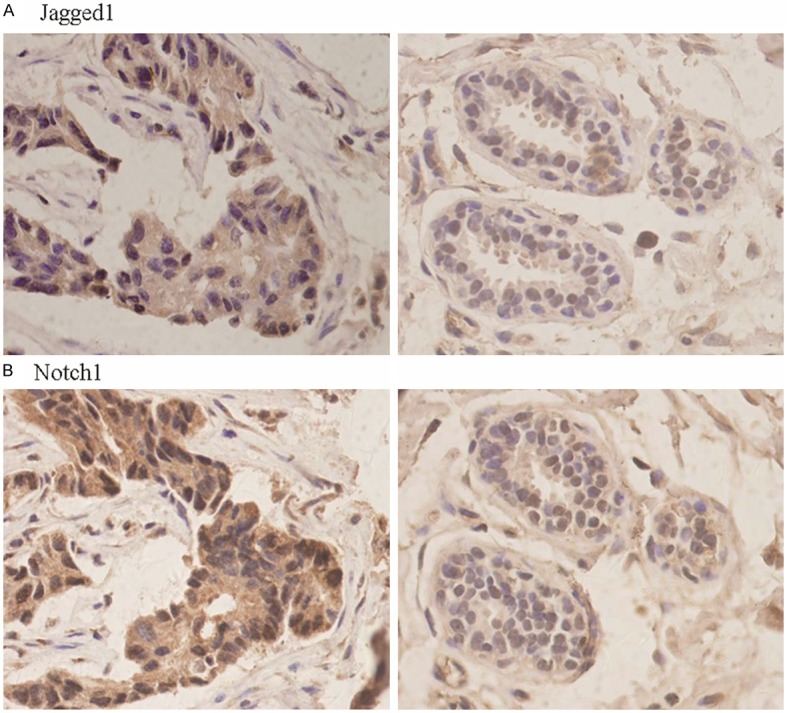
Expression of Jagged1 protein (A) in BC (left) and ANBT (right) and Notch1 protein (B) in BC (left) and ANBT (right). Note immunohistochemical strong staining in cytoplasm of cancer cells then weak staining in cytoplasm of normal epithelial cells (original magnification ×400).
Table 4.
Association of methyaltion with expression of Jagged1 and Notch1 genes in BC
| Jagged1 expression | Jagged1 (Mean methylation %) | Notch1 expression | Notch1 (Mean methylation %) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||||||||||||
| N | Overall | CpG_1 | CpG_3.4.5 | CpG_8.9.10 | CpG_11.12 | CpG_14.15 | N | Overall | CpG_3 | CpG_4.5 | CpG_8 | CpG_9 | CpG_12.13 | CpG_14.15.16 | CpG_17 | CpG_19 | CpG_20 | ||
| High | 42 | 10.71* | 6.12* | 19.61* | 6.36* | 7.50* | 10.32* | High | 52 | 11.54* | 13.81* | 16.39* | 11.40* | 6.95* | 13.39* | 7.93* | 11.59* | 12.56* | 19.56* |
| Low | 31 | 13.06 | 11.36 | 24.35 | 9.36 | 10.92 | 13.97 | Low | 21 | 20.42 | 29.93 | 31.31 | 25.77 | 18 | 24.21 | 15.07 | 22.86 | 18.29 | 23.23 |
P<0.05.
Figure 5.
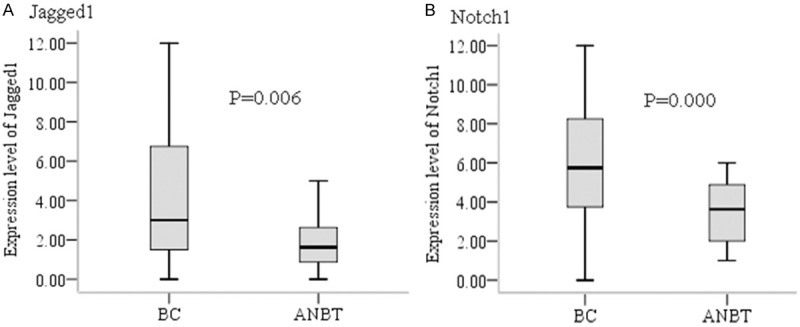
Comparison of Jagged1 (A) and Notch1 (B) expression in BC and ANBT.
Table 5.
Association of expression of Jagged1 and Notch1 with clinicopathological variables
| Parameters | N | Jagged1 expression | Notch1 expression | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| High n (%) | Low n (%) | χ2 | P | High n (%) | Low n (%) | χ2 | P | ||
| Lymph node metastasis | |||||||||
| - (LNN) | 39 | 22 (56.4%) | 17 (43.6%) | 5.665 | 0.023* | 24 (61.5%) | 15 (38.5%) | 5.153 | 0.023* |
| + (LNP) | 34 | 28 (82.4%) | 6 (17.6%) | 29 (85.3%) | 5 (14.7%) | ||||
| TNM stage | |||||||||
| I | 10 | 3 (30.0%) | 7 (70.0%) | 6.475 | 0.041* | 7 (70.0%) | 3 (30.0%) | 6.772 | 0.034* |
| II | 29 | 16 (55.2%) | 13 (44.8%) | 15 (51.7%) | 14 (48.3%) | ||||
| III | 34 | 25 (73.5%) | 9 (26.5%) | 28 (82.4%) | 6 (17.6%) | ||||
| Histological grade | |||||||||
| 1 | 8 | 3 (37.5%) | 5 (62.5%) | 7.107 | 0.021* | 3 (37.5%) | 5 (62.5%) | 6.783 | 0.040* |
| 2 | 57 | 15 (26.3%) | 42 (73.7%) | 22 (38.6%) | 35 (61.4%) | ||||
| 3 | 8 | 6 (75.0%) | 2 (25.0%) | 7 (87.5%) | 1 (12.5%) | ||||
| Receptor status | |||||||||
| ER(+)/PR(+) Her2(-) | 52 | 37 (71.2%) | 15 (28.8%) | 0.071 | 0.789 | 26 (50.0%) | 26 (50.0%) | 4.402 | 0.036* |
| ER(-) PR(-) Her2(+) | 12 | 9 (75.0%) | 3 (25.0%) | 10 (83.3%) | 2 (16.7%) | ||||
ER: estrogen receptor; PR: progesterone receptor; HER2: human epidermal growth factor receptor 2.
P<0.05.
We also examined Notch1 protein expression (Figure 4B). Similarly, Notch1 gene methylation was negative associated with expression, specially, the methylation level of overall and CpG_3, CpG_4.5, CpG_8, CpG_9, CpG_12.13, CpG_14.15.16, CpG_17, CpG_19, CpG_20 was remarkably reduced (p=0.000, p=0.008, p=0.015, p=0.000, p=0.001, p=0.008, p=0.010, p=0.001, p=0.033, and p=0.036) in high expression BC samples (Table 4). The expression level of Notch1 was dramatically increased in BC (Figure 5B) and mainly in high expression status in cancer (52/73, 71.2%). The high expression of Notch1 in primary tumor was positive correlation with lymph node metastasis, advanced stage, high grade and HER2 overexpression type breast cancer (Table 5).
The correlation between Jagged1 expression and Notch1 expression were analyzed in BC and ANBT groups. Our data revealed the expression of the two proteins was statistically positive association in BC and ANBT samples (Figure S3).
Discussion
Aberrant DNA methylation contributes to tumorigenesis and progression of numerous malignant tumors [33-36]. Breast cancer is a genetic and epigenetic disease, and several studies have indicated the association of gene abnormal methylation with carcinogenesis and development of breast cancer [37-40]. In this study, we investigated DNA methylation status of ligand Jagged1 and receptor Notch1 of Notch pathway in BC and ANBT. To our knowledge, this study provides the first analysis of DNA methylation on Jagged1 promoter and Notch1 exon25 in breast carcinoma.
We first detected methylation levels of Jagged1 and Notch1 genes in paired BC and ANBT. These results showed significant hypomethylation of overall and 10 of 15 CpG sites on Jagged1 promoter in tumor tissue, which indicated the methylation heterogeneity in cancer and normal tissue, and suggested a potential role of hypomethylation facilitating to breast carcinogenesis. Our results were in contrast to published data that hypermethylated Jagged1 occurred in B cell acute lymphoblastic leukemia [13]. This discrepancy can be attributed to difference in cancer cell type and sample size. Similar to Jagged1, the overall as well as all 13 individual CpG sites on Notch1 exon25 had significantly lower methylation in cancer than in normal tissue, which implied that Notch1 hypomethylation can be used as a biomarker for occurrence of breast cancer. These findings were strongly supported by our previous data of decreased methylation of Notch1 gene in carcinoma but elevated methylation in premalignant lesion tissues [25], and also were consistent with other observations of Notch1 hypomethylation in mantle cell lymphoma [21] and oral squamous cell carcinomas [22]. The high sensitivity and specificity of hypomethylation in cancer further revealed abnormal methylation of Jagged1 and Notch1 genes may be valuable markers for breast cancer diagnosis. Additionally, a significantly positive relationship of methylation between Jagged1 and Notch1 gene implied a possible synergetic effect of the both genes on breast carcinogenesis. Although our investigation has paved the way for studying Jagged1 and Notch1 gene methylation by using MALDI-TOF MS technology, the study is limited by small sample size, therefore warrants further validation.
Furthermore, we wanted to determine whether methylation alterations of Jagged1 and Notch1 genes can predict breast cancer progression. The association of DNA methylation status with clinicopathological characteristics was investigated. The overall hypomethylation of Jagged1 and Notch1 genes in primary tumors significantly correlated with lymph node metastasis, advanced stage and high grade, which suggested the aberrant methylation may be important predictors for breast carcinoma development. The results consistent with our previous conclusion that hypomethylated Notch1 gene can be a poor prognosis biomarker [25]. The individual CpG site, specially, Jagged1 CpG_8.9.10 was dramatically decreased methylation in primary cancers with lymph node metastasis and advanced stage, and the low methylation of Notch1 CpG_14.15.16 in primary tumors was also observed in lymph node metastasis, advanced stage and HER2 overexpression subtype breast cancer. The findings indicated that hypomethylation on some individual CpG sites especially on Jagged1 CpG_8.9.10 and Notch1 CpG_14.15.16 was specific feature for cancer progression and may provide an evidence of the heterogeneity of individual CpG site with clinicopathological characteristics. Although the aberrant methylation of the two genes influenced clinical biological features of breast cancer, but the study did not delve into the molecular role of DNA methylation, so we will need further to study this phenomenon in cancer cell lines and animal models. Nevertheless, we first show the hypomethylation from overall to specific individual CpG site of Jagged1 and Notch1 genes in primary cancer is associated with breast cancer progression, particularly associated with lymph node metastasis and advanced stage.
In addition, we explored the aberrant DNA methylation impacting on protein expression in same cohorts. For Jagged1 gene, the methylation of overall and individual CpG, specifically CpG_8.9.10, was markedly inversely proportional to the protein expression, which indicated the low methylation may result in elevated expression and CpG_8.9.10 hypomethylation may be one of crucial causes of dysfunction of Jagged1. Additional, Jagged1 was higher expression in cancer than in normal tissue and the increased expression in primary tumors was positively related to lymph node metastasis, advanced stage and high grade, which were similar to above-mentioned findings of hypomethylation in cancer and the relationship between hypomethylation and clinicopathological variables. These data further confirmed the important role of Jagged1 gene in occurrence and progression of breast cancer. For Notch1 protein, we found the significant correlation between high expression and low methylation of overall and each CpG sites, particularly Notch1 CpG_14.15.16, suggesting that hypomethylation was one of the reasons for aberrant high expression and the hypomethylation of CpG_14.15.16 especially may be important for expression regulation of Notch1 protein. Our results was consistent with other reports that DNA methylation on Notch1 promoter correlated with transcriptional silencing in mantle cell lymphoma [21] and non-tumor samples [15], however, the relationship on Notch1 exox25 in our finding needed further being verified. When the exact mechanism was unknown, the losing Notch1 exox25 methylation may be an immediate cause of Notch1 expression. Moreover, we observed the Notch1 overexpression in cancer and Notch1 overexpression in primary cancer significantly related to lymph node metastasis, advanced stage, high grade and HER2 overexpression subtype, which was a keeping with the hypomethylation in cancer and the correlation between hypomethylation and clinicopathological features. These findings further indicated Notch1 facilitating to tumorigenesis and development of breast cancer. In addition, the expression of Jagged1 and Notch1 had significantly positive relationship in cancer, in accordance with the relationship of methylation status in the two genes, which also confirmed the two genes cooperatively effecting on carcinogenesis of breast tissue.
In summary, we present the first evidence that ligand Jagged1 promoter region and receptor Notch1 exon25 region are synergistically hypomethylated and signifies carcinogenesis and development of breast cancer. DNA hypomethylation of the two genes may play important role in regulating overexpression. The high expression of the two proteins further confirms the role of Jagged1 and Notch1 gene in promoting occurrence and progression of breast carcinoma.
Acknowledgements
This work was supported in part by the Natural Science Foundation of China (No. 81260301) and The First Affiliated Hospital of Shihezi University School.
Disclosure of conflicts of interest
The authors declare that they have no completing interests.
Abbreviations
- BC
breast cancer
- ANBT
adjacent normal breast tissues
- MALDI-TOF MS
matrix assisted laser desorption/ionization time-of-flight mass spectrometry
- ER
estrogen receptor
- PR
progesterone receptor
- HER2
human epidermal growth factor receptor 2
- FISH
fluorescence in situ hybridization
- IHC
immunohistochemistry
- IS
immunoreactivity score
- ROC
receiver operating characteristic
- TSS
transcription start site
Supporting Information
References
- 1.Wang YC, Wei LJ, Liu JT, Li SX, Wang QS. Comparison of cancer incidence between China and the USA. Cancer Biol Med. 2012;9:128–32. doi: 10.3969/j.issn.2095-3941.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kiaris H, Politi K, Grimm LM, Szabolcs M, Fisher P, Efstratiadis A, Artavanis-Tsakonas S. Modulation of notch signaling elicits signature tumors and inhibits hras1-induced oncogenesis in the mouse mammary epithelium. Am J Pathol. 2004;165:695–705. doi: 10.1016/S0002-9440(10)63333-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Artavanis T, sakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–32. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 4.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–89. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 5.Cao YW, Wan GX, Sun JP, Cui XB, Hu JM, Liang WH, Zheng YQ, Li WQ, Li F. Implications of the Notch1-Snail/Slug-epithelial to mesenchymal transition axis for lymph node metastasis in infiltrating ductal carcinoma. Kaohsiung J Med Sci. 2015;31:70–6. doi: 10.1016/j.kjms.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Cao YW, Li WQ, Wan GX, Li YX, Du XM, Li YC, Li F. Correlation and prognostic value of SIRT1 and Notch1 signaling in breast cancer. J Exp Clin Cancer Res. 2014;33:97. doi: 10.1186/s13046-014-0097-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickson BC, Mulligan AM, Zhang H, Lockwood G, O’Malley FP, Egan SE, Reedijk M. High level JAG1 mRNA and protein predict poor outcome in breast cancer. Mod Pathol. 2007;20:685–93. doi: 10.1038/modpathol.3800785. [DOI] [PubMed] [Google Scholar]
- 8.Reedijk M, Pinnaduwage D, Dickson BC, Mulligan AM, Zhang H, Bull SB, O’Malley FP, Egan SE, Andrulis IL. JAG1 expression is associated with a basal phenotype and recurrence in lymph node-negative breast cancer. Breast Cancer Res Treat. 2008;111:439–48. doi: 10.1007/s10549-007-9805-3. [DOI] [PubMed] [Google Scholar]
- 9.Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, Lockwood G, Egan SE. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65:8530–7. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- 10.Costello JF, Plass C. Methylation matters. J Med Genet. 2001;38:285–303. doi: 10.1136/jmg.38.5.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–66. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 12.Piazzi G, Fini L, Selgrad M, Garcia M, Daoud Y, Wex T, Malfertheiner P, Gasbarrini A, Romano M, Meyer RL, Genta RM, Fox JG, Boland CR, Bazzoli F, Ricciardiello L. Epigenetic regulation of Delta-Like1 controls Notch1 activation in gastric cancer. Oncotarget. 2011;2:1291–301. doi: 10.18632/oncotarget.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuang SQ, Fang Z, Zweidler-McKay PA, Yang H, Wei Y, Gonzalez-Cervantes EA, Boumber Y, Garcia-Manero G. Epigenetic inactivation of Notch-Hes pathway in human B-cell acute lymphoblastic leukemia. PLoS One. 2013;8:e61807. doi: 10.1371/journal.pone.0061807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cortese R, Lewin J, Bäckdahl L, Krispin M, Wasserkort R, Eckhardt F, Beck S. Genome-wide screen for differential DNA methylation associated with neural cell differentiation in mouse. PLoS One. 2011;6:e26002. doi: 10.1371/journal.pone.0026002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terragni J, Zhang G, Sun Z, Pradhan S, Song L, Crawford GE, Lacey M, Ehrlich M. Notch signaling genes: Myogenic DNA hypomethylation and 5-hydroxymethylcytosine. Epigenetics. 2014;9:842–50. doi: 10.4161/epi.28597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felician G, Collesi C, Lusic M, Martinelli V, Ferro MD, Zentilin L, Zacchigna S, Giacca M. Epigenetic modification at Notch responsive promoters blunts efficacy of inducing Notch pathway reactivation after myocardial infarction. Circ Res. 2014;115:636–49. doi: 10.1161/CIRCRESAHA.115.304517. [DOI] [PubMed] [Google Scholar]
- 17.Acharyya S, Sharma SM, Cheng AS, Ladner KJ, He W, Kline W, Wang H, Ostrowski MC, Huang TH, Guttridge DC. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA mehtylation mediated repression: implications in duchenne muscular dystrophy. PLoS One. 2010;5:e12479. doi: 10.1371/journal.pone.0012479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui ZL, Gu W, Ding T, Peng XH, Chen X, Luan CY, Han RC, Xu WG, Guo XJ. Histone modifications of Notch1 promoter affect lung CD4+ T cell differentiation in asthmatic rats. Int J Immunopathol Pharmacol. 2013;26:371–81. doi: 10.1177/039463201302600210. [DOI] [PubMed] [Google Scholar]
- 19.Rauen T, Grammatikos AP, Hedrich CM, Floege J, Tenbrock K, Ohl K, Kyttaris VC, Tsokos GC. cAMP-responsive element modulatorα(CREMα) contributes to decreased Notch-1 expression in T cells from patients with active systemic lupus erythematosus (SLE) J Biol Chem. 2012;287:42525–32. doi: 10.1074/jbc.M112.425371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reister S, Kordes C, Sawitza I, Haussinger D. The epigenetic regulation of stem cell factor in hepatic stellate cells. Stem Cells Dev. 2011;20:1687–99. doi: 10.1089/scd.2010.0418. [DOI] [PubMed] [Google Scholar]
- 21.Leshchenko VV, Kuo PY, Shaknovich R, Yang DT, Gellen T, Petrich A, Yu Y, Remache Y, Weniger MA, Rafiq S, Suh KS, Goy A, Wilson W, Verma A, Braunschweig I, Muthusamy N, Kahl BS, Byrd JC, Wiestner A, Melnick A, Parekh S. Genome wide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood. 2010;116:1025–34. doi: 10.1182/blood-2009-12-257485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao S, Krogdahl A, Eiberg H, Liu CJ, Sørensen JA. LOH at chromosome 9q34.3 and the Notch1 gene methylation are less involved in oral squamous cell carinomas. J Oral Pathol Med. 2007;36:173–6. doi: 10.1111/j.1600-0714.2007.00520.x. [DOI] [PubMed] [Google Scholar]
- 23.Tang Y, Liu C, Wang X, Liu D, Ingvarsson S, Chen H. Demethylation of the region around exon2 of MLH1 gene in gastrointestinal cancer. Anticancer Res. 2012;32:4861–4. [PubMed] [Google Scholar]
- 24.Uekusa S, Kawashima H, Sugito K, Yoshizawa S, Shinojima Y, Igarashi J, Ghosh S, Wang X, Fujiwara K, Ikeda T, Koshinaga T, Soma M, Nagase H. Nr4a3, a possible oncogenic factor for neuroblastoma associated with CpG methylation within the third exon. Int J Oncol. 2014;44:1669–77. doi: 10.3892/ijo.2014.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang N, Sun ZZ, Li F, Cao YW, Zhao CX, Liang WH, Sun HP, Li HA, Fu XG. Detection and clinical significance of Notch1 methylation in breast cancer and intraductal proliferative breast lesions. Zhonghua Bing Li Xue Za Zhi. 2011;40:324–9. [PubMed] [Google Scholar]
- 26.Edge SB. AJCC Cancer Staging Manual. In: Edge SB, editor. American Joint Committee on Cancer. 7th ed. New York Inc; 2010. [DOI] [PubMed] [Google Scholar]
- 27.Lakhani SR. International Agency for Research on Cancer. WHO Classification of Tumours of the Breast. In: Lakhani SR, editor. Lyon WHO Classification of Tumours. Lyon: World Health Organization; 2012. [Google Scholar]
- 28.Fujiwara S, Hung M, Yamamoto-Ibusuk CM, Yamamoto Y, Yamamoto S, Tomiguchi M, Takeshita T, Hayashi M, Sueta A, Iwase H. The localization of HER4 intracellular domain and expression of its alternately-spliced isoforms have prognostic significance in ER+ HER2- breast cancer. Oncotarget. 2014;5:3919–30. doi: 10.18632/oncotarget.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, Cantor CR, Field JK, van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A. 2005;102:15785–90. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radpour R, Haghighi MM, Fan AX, Torbati PM, Hahn S, Holzgreve W, Zhong XY. High-throughput hacking of the methylation patterns in breast cancer by in vitro transcription and thymidine-specific cleavage mass array on MALDI-TOF silico-chip. Mol Cancer Res. 2008;6:1702–9. doi: 10.1158/1541-7786.MCR-08-0262. [DOI] [PubMed] [Google Scholar]
- 31.Cui X, Zhao Z, Liu D, Guo T, Li S, Hu J, Liu C, Yang L, Cao Y, Jiang J, Liang W, Liu W, Li S, Wang L, Wang L, Gu W, Wu C, Chen Y, Li F. Inactivation of micR-34a by aberrant CpG methylation in Kazakh patients with esophageal carcinoma. J Exp Clin Cancer Res. 2014;33:20. doi: 10.1186/1756-9966-33-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellido ML, Radpour R, Lapaire O, De Bie I, Hösli I, Bitzer J, Hmadcha A, Zhong XY, Holzgreve W. MALDI-TOF mass array analysis of RASSF1A and SERPINB5 methylation patterns in human placenta and plasma. Biol Reprod. 2010;82:745–50. doi: 10.1095/biolreprod.109.082271. [DOI] [PubMed] [Google Scholar]
- 33.Van Breda SG, Claessen SM, Lo K, van Herwijnen M, Brauers KJ, Lisanti S, Theunissen DH, Jennen DG, Gaj S, de Kok TM, Kleinjans JC. Epigenetic Mechanisms underlying arsenic-associated lung carcinogenesis. Arch Toxicol. 2014 doi: 10.1007/s00204-014-1351-2. [DOI] [PubMed] [Google Scholar]
- 34.Brait M, Maldonado L, Noordhuis MG, Begum S, Loyo M, Poeta ML, Barbosa A, Fazio VM, Angioli R, Rabitti C, Marchionni L, de Graeff P, van der Zee AG, Wisman GB, Sidransky D, Hoque MO. Association of Promoter Methylation of VGF and PGP9.5 with Ovarian Cancer Progression. PLoS One. 2013;8:e70878. doi: 10.1371/journal.pone.0070878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ganapathi SK, Beggs AD, Hodgson SV, Kumar D. Expression and DNA methylation of TNF, IFNG and FOXP3 in colorectal cancer and their prognostic significance. Br J Cancer. 2014;111:1581–9. doi: 10.1038/bjc.2014.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu Y, Feng MX, Yu J, Ma MZ, Liu XJ, Li J, Yang XM, Wang YH, Zhang YL, Ao JP, Xue F, Qin W, Gu J, Xia Q, Zhang ZG. DNA methylation-mediated silencing of matricellular protein dermatopontin promotes hepatocellular carcinoma metastasis by α3β1 integrin-Rho GTPase signaling. Oncotarget. 2014;5:6701–15. doi: 10.18632/oncotarget.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cowling VH. Enhanced mRNA cap methylation increases cyclin D1 expression and promoters cell transformation. Oncogene. 2010;29:930–936. doi: 10.1038/onc.2009.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tommasi S, Karm DL, Wu X, Yen Y, Pfeifer GP. Methylation of homeobox genes is a frequent and early epigenetic event in breast cancer. Breast Cancer Res. 2009;11:R14. doi: 10.1186/bcr2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shargh SA, Sakizli M, Khalaj V, Movafagh A, Yazdi H, Hagigatjou E, Sayad A, Mansouri N, Mortazavi-Tabatabaei SA, Khorram Khorshid HR. Down regulation of E-cadherin expression in breast cancer by promoter hypermethylation and its relation with progression and prognosis of tumor. Med Oncol. 2014;31:250. doi: 10.1007/s12032-014-0250-y. [DOI] [PubMed] [Google Scholar]
- 40.Moarii M, Pinheiro A, Sigal-Zafrani B, Fourquet A, Caly M, Servant N, Stoven V, Vert JP, Reyal F. Epigenomic alteration in breast carcinoma from primary tumor to locoreginal recurrences. PLoS One. 2014;9:e103986. doi: 10.1371/journal.pone.0103986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.