

Abstract
Hallmarks of cancer cells comprise altered glucose metabolism (aerobic glycolysis) and differences in DNA damage response (DDR). Glucose transporters (GLUT), glycolytic enzymes such as hexokinase (HK) and metabolic pathways (e.g. PI3K/Akt/mTor) have been shown to be upregulated in multiple myeloma and other cancer cell lines. Here we have investigated the effects of clinically used inhibitors of topoisomerases, of DDR and of the PI3K/Akt/mTor pathway on glucose metabolism and on cell survival in multiple myeloma cells. The effects of DNA damaging topoisomerase inhibitors (doxorubicin, etoposide, topotecan), non-DNA damaging agents (bortezomib, vincristine) as well as of molecular inhibitors of DNA damage related kinases PIKKs (KU55933 [ATM], NU7026 [DNA-PKCs]) and PI3K/Akt/mTor signaling (BEZ235 [PI3K/mTor], MK-2206 [Akt]) were analyzed 24 hours after treatment of OPM-2 multiple myeloma cells. For this purpose we monitored [18F]-FDG uptake, cell viability using an ATP assay and expression of GLUT-1, hexokinase II (HKII), cleaved caspase-3 and cleaved PARP via Western-blotting. All topoisomerase inhibitors used could upregulate expression of GLUT-1 and HKII in OPM-2 cells, resulting in elevated [18F]-FDG uptake and promotion of cell survival. In contrast, bortezomib and vincristine induced a decline in [18F]-FDG uptake combined with early induction of apoptosis. Combination treatment with topoisomerase inhibitors and molecular inhibitors of PIKK and PI3K could reverse elevated [18F]-FDG uptake, as observed after application of topoisomerase inhibitors only, and aggravate induction of apoptosis. Thus, elevated glucose consumption in OPM-2 cells can be reversed by targeting both DDR and PI3K/Akt/mTOR signaling, thus providing a promising strategy in the treatment of cancer.
Keywords: Glucose metabolism, DNA damage response, topoisomerase inhibitors, apoptosis, PI3K/Akt/mTor pathway, cell survival, cancer treatment
Introduction
The DNA damage response (DDR) is essential to genomic integrity. It subsumes a great variety of interwoven pathways that respond to all different types of DNA lesions via the regulation of kinase activities. Defects in DDR can result in carcinogenesis and promote rapid tumor growth [1]. While minor damages to DNA are efficiently repaired by the cellular base and nucleotide excision repair systems, more serious lesions such as DNA double-strand breaks (DSB) induce two major mechanisms of DDR: homologous recombination (HR) and non-homologous end joining (NHEJ) [2]. While HR aims at reconstructing DNA structure by resecting the lesion and copying the deleted information from the sister chromatid, NHEJ is error-prone as it simply ligates two ends of nearby DNA fragments [3]. HR and NHEJ are initiated by a family known as the phosphatidylinositol 3-kinase related kinases (PIKKs) which include ATM (ataxia telangiectasia mutated protein), ATM-Rad3-related (ATR) and the DNA-dependent protein kinase catalytic subunit (DNA-PKCs) [4]. The PIKKs are the first responders to DNA damage and act through phosphorylation of scaffolding proteins and downstream kinases such as p53, H2AX, and Chk2 [5].
Topoisomerase inhibitors like etoposide and doxorubicin are known to trigger the DNA damage response via activation of ATM due to efficient induction of DSB [6]. Inhibition of topoisomerases (subtypes I and II) suppresses relaxation of supercoiled DNA during replication and transcription [7]. Defects in DSB repair may increase efficacy of DNA damaging agents: cancer cells with impaired NHEJ have been shown to preferentially respond to treatment with topoisomerase II inhibitors with high sensitivity [8]. In contrast, cells with a high activity of DNA-PK have been shown to develop resistance to treatment with etoposide and doxorubicin [9,10].
Moreover, DNA repair is an energy-consuming process that utilizes various ATP-dependent chromatin-remodeling complexes which are not fully characterized yet [11]. Besides, members of the structural maintenance of chromosome (SMC) protein family hydrolyze ATP in order to recognize and reorganize damaged DNA [12].
Thus, repair of DNA damage requires an increased uptake of glucose via the cell membrane to generate ATP. Cancer cells are known to generate ATP by aerobic glycolysis (Warburg effect), i.e. the conversion of glucose into lactate, even in the presence of oxygen [13]. Correspondingly, many cancer cells are characterized by highly active upstream regulators of metabolic signaling. PI3K/Akt is known to promote the switch towards aerobic glycolysis by stimulating the expression of glucose transporters on the cell surface [14] and the expression of glycolytic enzymes in the cytoplasm of cancer cells [15,16]. Akt also controls the activity of the ‘mechanistic target of rapamycin’ (mTor) pathway. The mTor-complex consists of mTORC1 and mTORC2 which have different regulating functions in cell proliferation and protein synthesis [17]. The expression and activation of PI3K/Akt in different multiple myeloma cell lines and particularly in OPM-2 cells has been reported before [18,19]. Evidence is arising that Akt is also involved in the repair of genotoxic damage. Akt was shown to respond to DNA double-strand breaks (DSB) in a DNA-PK- or ATM/ATR-dependent manner and to actively stimulate the repair of DNA-DSB by NHEJ [20]. Akt may therefore be the pivot point in connecting the DNA damage response to aerobic glycolysis.
Elevated activities of glycolytic enzymes and upregulated expression of glucose transporters are prerequisites for the Warburg effect of tumor cells. This accounts especially for GLUT-1 whose up-regulation has been shown for several types of cancer [21-23] and whose expression levels have been connected to malignancy and metastasis [24]. GLUT-1 activity has been shown to correlate inversely with the survival of cancer patients [25,26]. Following uptake via glucose transporters, glucose is phosphorylated by subtypes of the key glycolytic enzyme hexokinase (HK). Particularly hexokinase II (HKII) is considered to play a fundamental role in the promotion of aerobic glycolysis and cell survival [27,28]. HKII has been shown to be constitutively over-expressed in various multiple myeloma cell lines [19]. As HKII is bound to the outer membrane of mitochondria in cancer cells, it is not inhibited by its own cytosolic product glucose-6-phosphate and gains access to the ATP that is generated in mitochondria coupling oxidative phosphorylation to the glycolytic pathway [29]. The feature of HKII to phosphorylate, but not to metabolize 2-deoxy-D-glucose is widely utilized in visualization and assessment of therapy response in cancerous lesions via [18F]-FDG PET [30,31].
Nevertheless, the molecular connection between the activation of DDR by chemotherapeutics and the resultant changes in glucose metabolism and cell survival of cancer cells has not been studied extensively so far. Only few studies could demonstrate a relation between the changes in bioenergetic response pathways in apoptotic cell death and the down-/up-regulation of glucose transporter genes and glycolytic enzymes after treatment with genotoxic compounds [32,33].
The purpose of this study was to further elucidate the influence of DDR inducing topoisomerase inhibitors (doxorubicin, etoposide, topotecan) on glucose metabolism and cell survival in multiple myeloma cells (OPM-2). Furthermore, we investigated the effects of the chemotherapeutic, non-DNA damaging drugs bortezomib and vincristine. In summary we could show correlations between [18F]-FDG uptake in OPM-2 cells and expression levels of GLUT-1 and HKII. We propose that OPM-2 cells react differently to chemotherapy with DNA damaging agents with regard to glucose consumption and promotion of cell survival. At low concentrations of the drugs, we observed promotion of cell survival. This may hold implications for the understanding of resistance to genotoxic stress. Moreover, we could show that targeting of DNA damage response with molecular inhibitors of ATM, DNA-PKCs and PI3K/Akt/mTor signaling is effective in reversing these effects and in synergistically inducing apoptosis (schematic representation see Figure 1).
Figure 1.
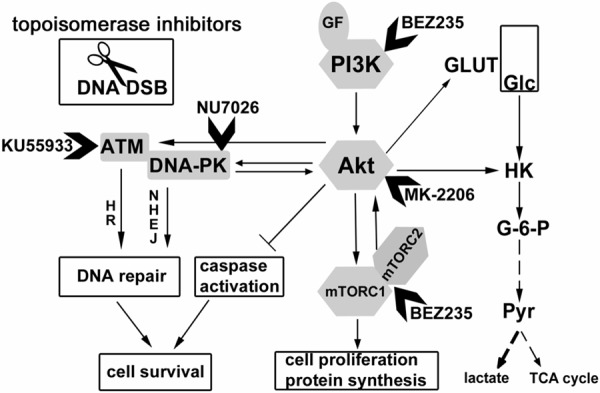
Signaling network in regulation of glucose uptake and DNA repair in cancer cells. This scheme displays prominent features of glycolysis, PI3K/Akt/mTor signaling and DNA damage response. It represents an idea how these pathways might be connected, and how cell metabolism might respond to genotoxic stress (20, 34). Activation and inhibition are represented by arrows and bars, respectively. ATM Ataxia telangiectasia mutated, DNA-PK DNA-dependent protein kinase, HK hexokinase, DSB double-strand break, GF growth factor, Glc glucose, GLUT glucose transporter, G-6-P glucose-6-phosphate, HR homologous recombination, NHEJ non-homologous end joining, mTOR mammalian target of rapamycin, PI3K phosphatidylinositol-4,5-bisphosphate 3-kinase, Pyr pyruvate, TCA cycle tricarboxylic acid cycle.
Materials and methods
Cell culture and treatment
The multiple myeloma cell line OPM-2 (obtained from T. Dechow, Technische Universität München) was cultured in RPMI 1640 medium (Biochrom) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin and 1% L-glutamine (Biochrom). OPM-2 cells were cultivated at 37°C in a humidified atmosphere with 5% CO2. OPM-2 cells were treated with dilution series of the proteasome inhibitor bortezomib (5 nM-5.12 μM), the mitotic inhibitor vincristine (0.625 nM-0.64 μM) and the topoisomerase inhibitors doxorubicin (5 nM-51.2 µM), etoposide (0.5 μM-1.024 mM), topotecan (25 nM-25.6 µM) (all obtained from pharmacy of Technische Universität München). The dual PI3K/mTor inhibitor BEZ235 (200 nM), the ATM inhibitor KU55933 (10 µM), the Akt inhibitor MK-2206 (1 µM) and the DNA-PKC inhibitor NU7026 (10 µM) were all dissolved in DMSO prior to application (all supplied by SelleckChem).
Determination of glucose consumption via [18F]-FDG uptake in OPM-2 cells
For evaluation of chemotherapeutic effects on glucose metabolism, [18F]-FDG uptake was measured in vitro. 2 × 105 OPM-2 cells/ml were seeded in 24-well plates (Greiner) as quadruple with different concentrations of inhibitors and incubated for 24 h in RPMI1640. After removal of culture medium, OPM-2 cells were incubated with 370 kBq of [18F]-FDG per sample in PBS for 30 min. Subsequently, cells were washed three times with cold PBS (4°C) and centrifuged at 1,300 rpm. Counts per minute of cell pellets (standards and blank values included) were measured using a gamma counter (1480 Wizard, Wallac, Finnland). The results were related to the number of viable cells which were determined using an automated cell counter (Countess®, Life Technologies, Germany). [18F]-FDG uptake was plotted as fold of the untreated control.
Cell viability analysis of OPM-2 cells subject to treatment with inhibitors
Cell viability was assessed using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega). With this test the percentage of metabolically active cells is determined based on the quantification of ATP. 2 × 104 cells/100 µl were seeded in quadruple in 96-well plates (Greiner) and incubated for 24 h with increasing concentrations of various inhibitors (see above) or left untreated. Subsequently cells were incubated for 10 min with 100 µl of CellGlo®-substrate and the assay was quantified by measuring luminescence in a microplate reader (Mithras LB 940, Berthold Technologies, Germany). Cell viability was calculated relative to untreated controls (set to 100 %).
Western blot analysis
OPM-2 cells (1 × 106 per 75 cm2 culture flask) were incubated for 24 h with different inhibitors at various concentrations (see above). Cells were washed with cold PBS (4°C) and lysed in an appropriate buffer (50 mM Tris, pH 7.5; 250 mM NaCl; 0.1% Triton X-100; 1 mM EDTA; 50 mM NaF; protease inhibitor cocktail [Roche, Germany]) for 30 min on ice. Lysates were centrifuged at 14,800 rpm for 20 min at 4°C. Pellets were discarded and protein concentrations of the supernatants were determined using the BCA protein assay kit (Pierce, Germany). Twenty µg of the lysates each were subjected to SDS-Page and subsequently blotted onto a PVDF membrane. For subsequent incubation of blocked (TBS-T/5% nonfat dry milk) PVDF membranes the following antibodies were used: cleaved caspase-3 (1:2,500), PARP (1:5,000) (#9664, #9542 Cell Signaling Technologies), hexokinase II (1:5,000) (AB3279, Millipore) and GLUT-1 (1:200) (clone SP168, Biomol). Except for GLUT-1 (5% BSA, 1 h, RT) all antibodies were incubated overnight at 4°C in 5% nonfat dry milk. Subsequent incubation with anti-IgG (mouse and rabbit) horseradish peroxidase-conjugated secondary antibodies (1:10,000 in TBS-T/5% nonfat dry milk) (Millipore, Germany) was done for 1 h at RT. Protein-antibody complexes were detected using the SuperSignal West Pico Chemiluminescent Substrate (ThermoScientific, Germany). Images were generated via the ChemiDoc™ XRS+ System (Bio-Rad, Germany). All experiments were done in triplicate.
Statistics
Statistical analysis was performed using SPSS (version 19, IBM, Ehningen, Germany). For calculation an independent students t-test was performed and the results were regarded as statistically significant (*) if a significance level < 0.05 was achieved.
Results
Treatment with DNA damaging drugs results in elevated [18F]-FDG uptake in OPM-2 cells
We first examined the effects of the topoisomerase inhibitors doxorubicin, etoposide and topotecan on [18F]-FDG uptake at 24 h after start of incubation with different concentrations of the inhibitors (Figure 2A-C). Treatment with doxorubicin increased the [18F]-FDG uptake compared to the untreated control at 0.1 µM-1.6 µM with a maximum increase at 0.4 µM (+42.7%) (Figure 2A). Etoposide induced a significant rise of [18F]-FDG uptake at 2 µM-32 µM, peaking at 2 µM (+98.7%) (Figure 2B). Topotecan triggered increased [18F]-FDG uptake at 0.05 µM-1.6 µM. At 0.2 µM, the [18F]-FDG uptake was 2.3-fold higher than that of the untreated control (Figure 2C). In summary, elevation of [18F]-FDG uptake in OPM-2 cells by topoisomerase inhibitors was comparatively high after treatment with comparatively low concentrations. However, the concentrations that induced the highest increase were different for the different inhibitors. At higher concentrations, the application of all three topoisomerase inhibitors resulted in suppressed [18F]-FDG uptake.
Figure 2.
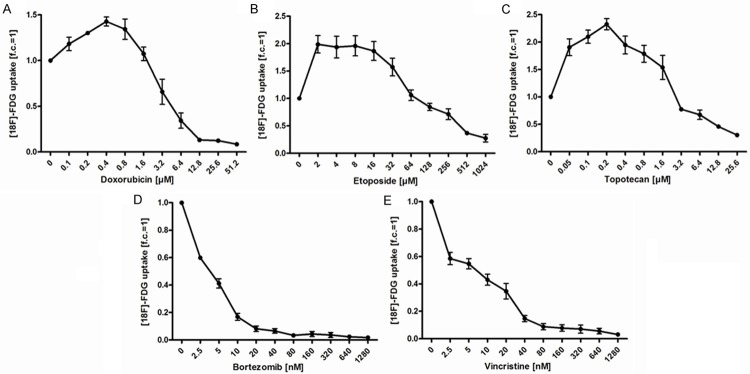
[18F]-FDG uptake in OPM-2 cells is regulated differentially by DNA-damaging topoisomerase inhibitors (A-C) and non-DNA damaging compounds (D, E). OPM-2 cells were left untreated (0) or treated with increasing concentrations of doxorubicin (A), etoposide (B), topotecan (C), bortezomib (D) and vincristine (E) for 24 h. After incubation with [18F]-FDG (370 kBq/ml) for 30 min, counts per minute (CPM), representing [18F]-FDG uptake, were measured in the cell pellets after centrifugation and related to the number of viable cells. Treatment with topoisomerase inhibitors (A-C) resulted in an initial increase of [18F]-FDG uptake, while both bortezomib and vincristine exclusively caused a decrease of [18F]-FDG uptake. All values are expressed as multiple or fraction of untreated controls (fold of control, f.c.), being set to 1. Data show means ± SEM (n = 4).
In contrast, the non-DNA damaging drugs bortezomib and vincristine did not increase [18F]-FDG uptake at any concentrations tested. Application of both compounds led to an immediate decline in [18F]-FDG uptake by more than half and finally resulted in total suppression of [18F]-FDG uptake in OPM-2 cells (Figure 2D, 2E). [18F]-FDG uptake in OPM-2 cells was also reduced by co-treatment with bortezomib/doxorubicin and vincristine/etoposide (data not shown).
Increased [18F]-FDG uptake corresponds to improved survival of OPM-2 cells after treatment with DNA damaging drugs
Given the increase in [18F]-FDG uptake following application of topoisomerase inhibitors (Figure 2A-C), we investigated whether these changes were associated with differences in cell survival in OPM-2 cells at the indicated concentrations. Doxorubicin insignificantly increased cell viability of OPM-2 cells at concentrations of 0.05 μM to 1.6 µM (Figure 3A). The IC50 of doxorubicin was determined as 2.1 µM (95% CI: 1.9-2.3 µM). At 3.2 µM, the number of viable cells dropped significantly. Etoposide did not turn out as a potent inducer of cell death in OPM-2 cells at all analyzed concentrations (Figure 3B). At the highest concentration applied (1.024 mM), the cell viability was still 28% of that of the untreated control. The IC50 of etoposide in OPM-2 cells was determined as 131.2 µM (95% CI: 105.7-162.8 µM). Treatment with topotecan had a negligible effect on viability from 0.025 to 0.4 µM (Figure 3C). At concentrations from 0.8 to 25.6 μM, the number of viable cells decreased significantly with an IC50 of 1.01 µM (95% CI: 1.0-1.2 µM). Overall, treatment with topoisomerase inhibitors required comparatively high concentrations to affect cell viability. At lower concentrations a significant effect on cell survival could not be observed. Moreover, increased uptake of [18F]-FDG could not be correlated with cell survival (Figure 3A-C). Consequently, we examined if similar effects could be observed after treatment with non-DNA damaging drugs.
Figure 3.
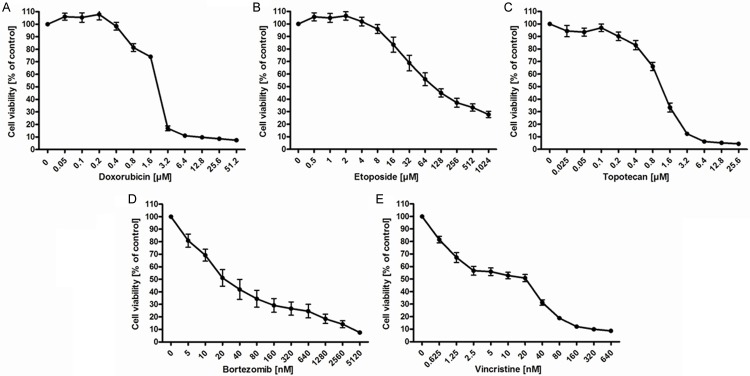
Viability of OPM-2 cells after treatment with DNA-damaging topoisomerase inhibitors (A-C) and non-DNA damaging compounds (D, E). Viability of OPM-2 cells was analyzed using the CellTiter-Glo® luminescent cell viability assay 24 h after treatment with increasing concentrations of doxorubicin (A), etoposide (B), topotecan (C), bortezomib (D) and vincristine (E). Untreated controls (0) were set to 100%. Data represent means ± SEM of at least 4 independent experiments.
Treatment of OPM-2 cells with bortezomib resulted in a dose-dependent, almost linear decrease of viability with an IC50 at 34.4 nM (95% CI: 24.1-49.1 nM) (Figure 3D). Treatment with vincristine also led to a strong decrease in viability at concentrations of up to 640 nM (Figure 3E). The IC50 was determined as 8.7 nM (95% CI: 7.3-10.2 nM). Thus, non-DNA damaging drugs potently affected cell survival in OPM-2 cells clearly inducing a dose-dependent decrease in viability already at the lowest concentrations applied (Figure 3D, 3E). Furthermore, we could demonstrate a strict correlation between [18F]-FDG uptake and cell viability.
High expression levels of GLUT-1 and HKII after genotoxic exposure correlate to inhibition of apoptosis
To elucidate the molecular mechanisms by which low concentrations of topoisomerase inhibitors trigger an increased [18F]-FDG uptake, OPM-2 cells were analyzed by immunoblotting. We found that the glycolytic enzymes are regulated differently following application of DNA damaging and non-DNA damaging compounds. Untreated OPM-2 cells exhibited a constitutive expression of GLUT-1 and HKII. The topoisomerase inhibitors doxorubicin, etoposide and topotecan induced a slight decrease in the expression levels of GLUT-1, notably at the highest concentrations applied (Figure 4A-C). In contrast, protein levels of HKII steadily decreased in a dose-dependent way, particularly after treatment with doxorubicin and topotecan. Noticeably, levels of cleaved caspase-3 and cleaved PARP concomitantly increased with down-regulation of HKII and almost stable expression of GLUT-1 (Figure 4A-C).
Figure 4.
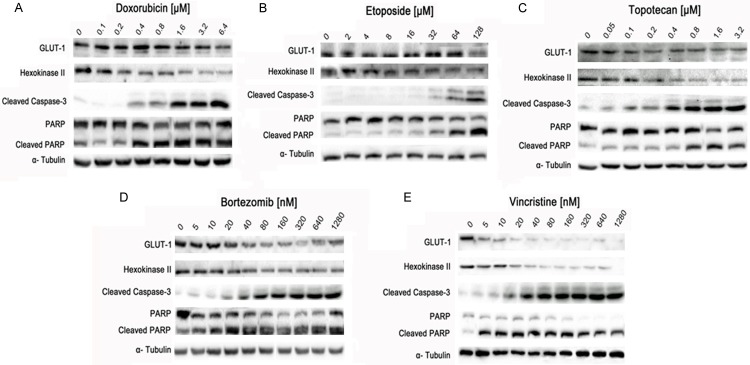
Effects of DNA-damaging topoisomerase inhibitors (A-C) and non-DNA damaging compounds (D, E) on expression of enzymes involved in glucose metabolism and apoptosis. OPM-2 cells were left untreated (0) or treated for 24 h with increasing concentrations of doxorubicin (A), etoposide (B) topotecan (C), bortezomib (D) and vincristine (E). Cell lysates were subjected to SDS-PAGE and immunoblotting using antibodies against GLUT-1, HKII, cleaved PARP and cleaved caspase-3. An anti-tubulin antibody was used as loading control. The effects of bortezomib (D) and vincristine (E) on down-regulation of GLUT-1 and HKII as well as on up-regulation of cleaved PARP and cleaved caspase-3 were more prominent than of doxorubicin (A), etoposide (B) and topotecan (C). Data were confirmed at least three times in independent experiments.
These findings suggest that OPM-2 cells were able to maintain their anabolic drive in terms of elevated ATP levels. Exemplarily, slightly decreased expression of HKII and almost steady expression of GLUT-1 during etoposide treatment might trigger delayed appearance of apoptotic markers (cleaved caspase 3 and cleaved PARP) compared to treatment with doxorubicin and topotecan (Figure 4B, compare to Figure 4A and 4C).
In comparison, GLUT-1 and HKII expression slightly decreased dose-dependently under treatment with bortezomib and steadily decreased upon incubation with vincristine. Correspondingly, induction of apoptosis, as visualized via cleavage of caspase-3 and PARP, increased with decreasing expression of GLUT-1 and HKII (Figure 4D, 4E). In summary, non-DNA damaging drugs more effectively induced apoptosis compared to the topoisomerase inhibitors. The quantification results of the Western blot signals are shown in the supplementary data.
PIKK/PI3K inhibitors and DNA damaging drugs act synergistically in the reduction of glucose metabolism
Given these findings, we tried to further increase the rate of cells undergoing apoptosis and to knockdown glucose consumption in OPM-2 cells. For that we analyzed the effects of KU55933 and NU7026, inhibitors of the DNA damage response related kinase ATM and DNA-PKCs, respectively, as well as the Akt-inhibitor MK-2206 and the dual PI3K/mTor inhibitor BEZ235. These inhibitors were also combined with topoisomerase inhibitors at concentrations that had triggered maximum [18F]-FDG uptake in previous experiments (doxorubicin [0.4 µM], etoposide [8 µM] and topotecan [0.2 µM]).
KU55933 (10 µM), MK-2206 (1 µM) and NU7026 (10 µM) applied separately induced only insignificant changes in [18F]-FDG uptake compared to controls (Figures 5B-D, 6B-D and 7B-D). Besides, expression of HKII was not altered strikingly by these inhibitors compared to untreated controls. Expression of GLUT-1 was slightly suppressed particularly by MK-2206 and NU7026. In contrast, BEZ235 (200 nM) demonstrated a distinct inhibitory effect on [18F]-FDG uptake compared to the untreated (RPMI) controls. As well, BEZ235 induced a clear down-regulation of HKII (Figures 5A, 6A and 7A).
Figure 5.
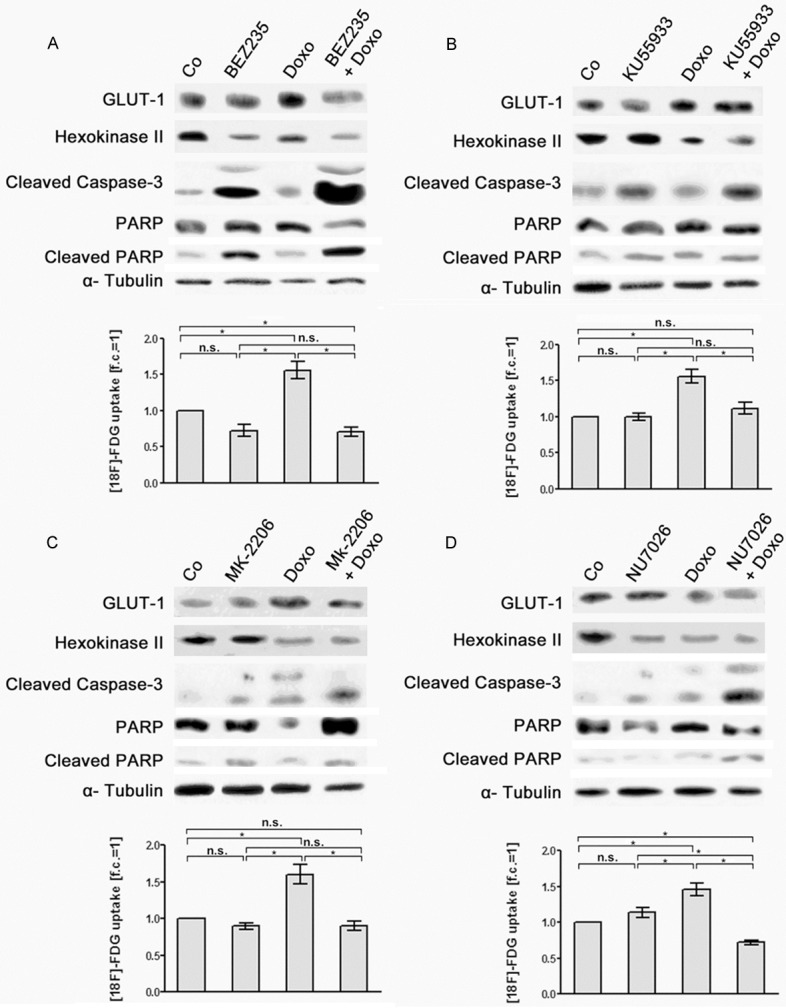
Effects of doxorubicin, of PIKK/PI3K inhibitors and of combination treatment with doxorubicin and PIKK/PI3K inhibitors on [18F]-FDG uptake and expression of glycolytic enzymes and apoptotic marker proteins in OPM-2 cells. OPM-2 cells were left untreated (Co), treated for 24 h with doxorubicin (0.4 µM) (Doxo, A-D), with (A) BEZ235 (200 nM), (B) KU55933 (10 µM), (C) MK-2206 (1 µM) and (D) NU7026 (10 µM) as a single treatment, respectively, or subjected to a combination treatment with doxorubicin and the different PIKK/PI3K inhibitors (A-D). Subsequently, [18F]-FDG uptake of the cells was quantified (means ± SEM; n ≥ 3; fold of control, f.c.) and expression of GLUT-1, HKII, cleaved caspase-3 and cleaved PARP was monitored in corresponding cell lysates by Western blot analysis. Significances of differences in [18F]-FDG uptake were calculated using the students t-test. n.s. not significant; *significant.
Figure 6.
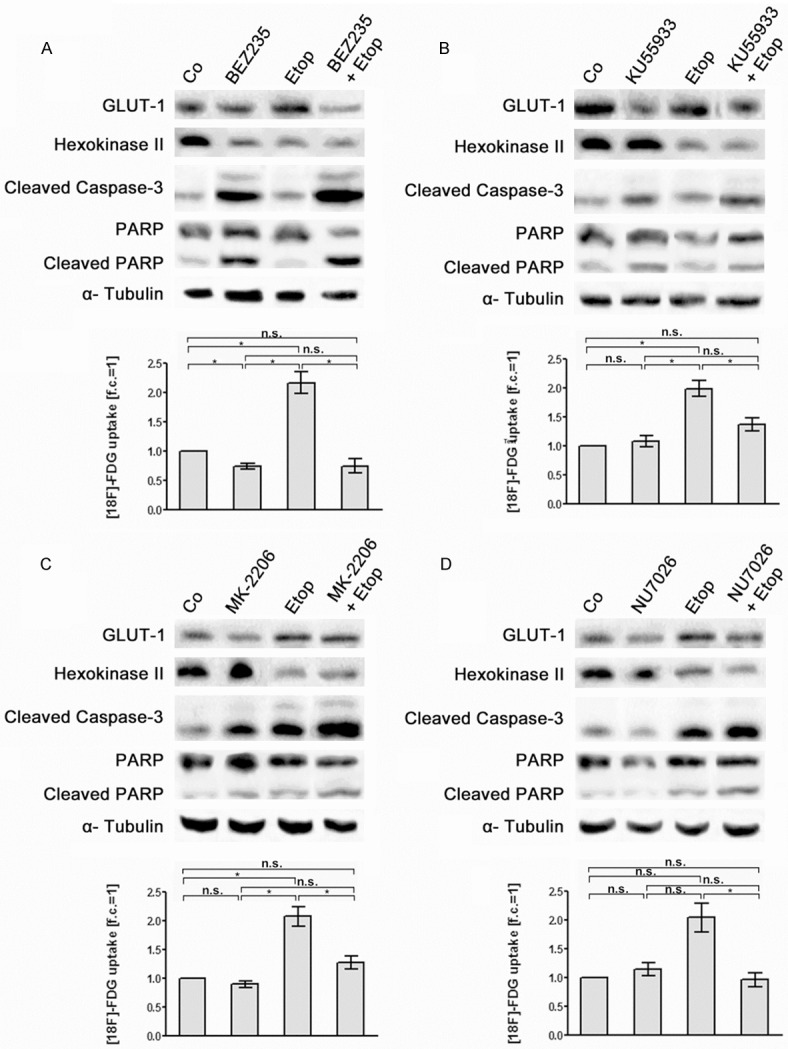
Effects of etoposide, of PIKK /PI3K inhibitors and of combination treatment with etoposide and PIKK /PI3K inhibitors on [18F]-FDG uptake and expression of glycolytic enzymes and apoptotic marker proteins in OPM-2 cells. OPM-2 cells were left untreated (Co), treated for 24 h with etoposide (8 µM) (Etop, A-D), with (A) BEZ235 (200 nM), (B) KU55933 (10 µM), (C) MK-2206 (1 µM) and (D) NU7026 (10 µM) as a single treatment, respectively, or subjected to a combination treatment with etoposide and the different PIKK/PI3K inhibitors (A-D). Subsequently, [18F]-FDG uptake of the cells was quantified (means ± SEM; n ≥ 3; fold of control, f.c.) and expression of GLUT-1, HKII, cleaved caspase-3 and cleaved PARP was monitored in corresponding cell lysates by Western blot analysis. Significances of differences in [18F]-FDG uptake were calculated using the students t-test. n.s. not significant; *significant.
Figure 7.
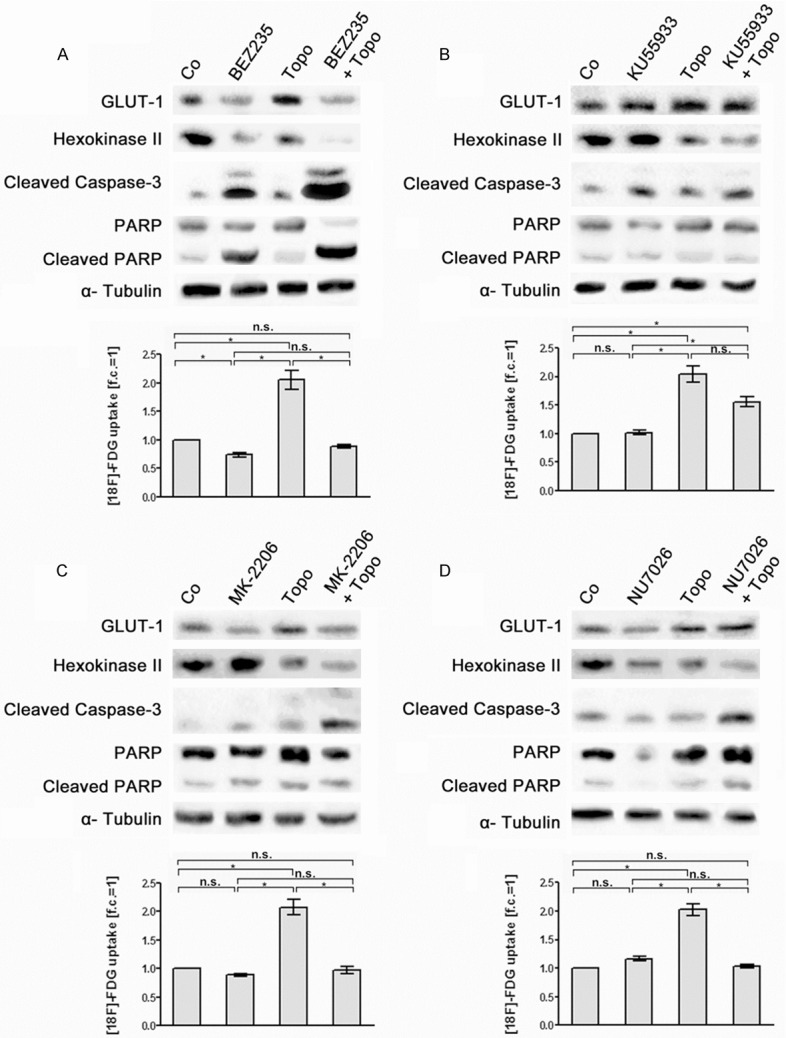
Effects of topotecan, of PIKK /PI3K inhibitors and of combination treatment with topotecan and PIKK /PI3K inhibitors on [18F]-FDG uptake and expression of glycolytic enzymes and apoptotic marker proteins in OPM-2 cells. OPM-2 cells were left untreated (Co), treated for 24 h with topotecan (0.2 µM) (Topo, A-D), with (A) BEZ235 (200 nM), (B) KU55933 (10 µM), (C) MK-2206 (1 µM) and (D) NU7026 (10 µM) as a single treatment, respectively, or subjected to a combination treatment with topotecan and the different PIKK/PI3K inhibitors (A-D). Subsequently, [18F]-FDG uptake of the cells was quantified (means ± SEM; n ≥ 3; fold of control, f.c.) and expression of GLUT-1, HKII, cleaved caspase-3 and cleaved PARP was monitored in corresponding cell lysates by Western blot analysis. Significances of differences in [18F]-FDG uptake were calculated using the students t-test. n.s. not significant; *significant.
Co-treatment of OPM-2 cells with BEZ235 and the topoisomerase inhibitors doxorubicin, etoposide and topotecan resulted in a decrease of [18F]-FDG uptake similar to treatment with BEZ235 alone. Moreover, combined treatment with BEZ235 and the three topoisomerase inhibitors more efficiently suppressed GLUT-1 and HKII expression than BEZ235 alone (Figures 5A, 6A and 7A). Co-treatment with KU55933, MK-2206 and NU7026 plus topoisomerase inhibitors did not significantly alter [18F]-FDG uptake in OPM-2 cells compared to treatment with KU55933, MK-2206 and NU7026 only. However, co-treatment was more effective in down-regulation of HKII than with KU55933, MK-2206 and NU7026 alone (Figures 5B-D, 6B-D and 7B-D). For example, co-treatment with MK-2206 plus topotecan (Figure 7C) as well as with NU7026 plus etoposide (Figure 6D) remarkably decreased HKII expression.
The expression of the apoptotic markers cleaved caspase 3 and cleaved PARP-1 in OPM-2 cells was clearly increased by co-treatment with BEZ235 plus the topoisomerase inhibitors doxorubicin, etoposide and topodecan compared to treatment with BEZ235 alone (Figures 5A, 6A and 7A). For all the other combined treatments analyzed intensity of apoptotic markers was not markedly increased compared to the treatments with KU55933, MK-2206 and NU7026 alone (Figures 5B-D, 6B-D and 7B-D). The quantification results of the Western blot signals are shown in the supplementary data.
Discussion
Cancer cells are in need of a high metabolic rate as aerobic glycolysis (Warburg effect) only provides about 4 mol ATP/mol glucose [34]. Many types of tumor cells have been shown to encounter the enhanced demand of glucose by increased levels of GLUT-1 and HKII [35-38]. Several studies have discussed the potential of targeting this bioenergetic pathway in different tumor cells, especially as intact glucose metabolism is associated with apoptosis resistance and long-term survival [39,40]. Because many anti-cancer drugs induce DNA-damage triggered apoptosis [41], detailed knowledge of the mechanisms in response to DNA damage is crucial. Some evidence has been provided that DNA-damaging drugs such as etoposide, vincristine, cisplatin, camptothecine, and doxoribicin regulate the activity of GLUT genes and glucose metabolism [32,33]. In this study, we have investigated the linkage between genotoxic damage, glucose metabolism and cell survival and provide new findings on targets of the PI3K/Akt/mTor pathway in multiple myeloma cells.
Essentially, the treatment of OPM-2 cells with different chemotherapeutic agents exhibited two ways of regulation of glucose metabolism, depending on the method of action of the applied drugs. The DNA-damage inducing topoisomerase inhibitors doxorubicin, etoposide and topotecan increased [18F]-FDG uptake in OPM-2 cells with delayed occurrence of apoptosis. In contrast, treatment of OPM-2 cells with the non-DNA damaging proteasome inhibitor bortezomib and the mitotic inhibitor vincristine resulted in decreased [18F]-FDG uptake combined with reduced cell survival.
Topoisomerase inhibitors are known to cause DNA double-strand breaks which are repaired via ATP-dependent mechanisms [11,12]. Although the inhibitors of topoisomerase II, doxorubicin and etoposide, as well as the inhibitor of topoisomerase I, topotecan, actually differ in the way of targeting DNA, they produced very similar changes in glucose metabolism, probably due to similar patterns of DNA-damage in OPM-2 cells. Our results indicate that OPM-2 cells increase uptake and metabolism of glucose in response to DNA-damage via a dose-dependent up-regulation of GLUT-1 and HKII protein levels. Most probably, OPM-2 cells raise production of ATP in the glycolytic pathway as well as of nucleotides in the pentose phosphate pathway [32]. If OPM-2 cells are not able to increase efficacy of aerobic glycolysis they are obviously killed more efficiently.
The significance of effective glucose uptake by glucose-transporter proteins for cancer progression has been demonstrated in various studies. Growth factor IL-3-dependent pro-B-cells have been shown to delay apoptosis subject to overexpression of GLUT-1, even after withdrawal of the growth factor [42]. More recently, it has been proven that treatment of HeLa cells with doxorubicin and etoposide induces down-regulation of GLUT-3, but not of GLUT-1. This emphasizes the difference in suppression of GLUT-1 and GLUT-3 genes with respect to the damaging agent [33]. As well, the investigated species (e.g. man or mouse) and the enzyme composition of a specific cell type seem to play an important role in prediction of the cellular response to DNA damage. In a study of Zhou et al. a strong suppression of GLUT-1 and GLUT-3 protein levels in hematopoetic precursor cells of murine fetal liver was described after genotoxic exposure (etoposide, cisplatin, γ-radiation) together with decreased glycolysis and oxygen consumption [32].
While GLUT-1 expression in OPM-2 cells was rather unaffected by topoisomerase inhibitors, decreasing ATP levels in the cells - as measured via the cell proliferation assay - strongly correlated with reduced HKII protein levels. This is in accordance with findings in which the most widely used HKII-inhibitor 3-bromopyruvate (3BrPA) suppressed ATP production and induced apoptosis in HKII-overexpressing multiple myeloma cells [19]. A related study highlighted that inhibition of glycolysis by 3BrPA could resensitize multiple myeloma cells to doxorubicin treatment by inactivation of ATP-dependent efflux pumps (ABC transporter family) [43]. As these transporters have often been associated with chemoresistance in anti-cancer therapy [44], elevated ATP levels after treatment with topoisomerase inhibitors in OPM-2 cells might contribute to the observed anti-apoptotic effects.
Moreover, we could prove the potential of targeting glycolysis and DNA damage response with inhibitors (i) of the PI3K/Akt/mTor pathway (MK-2206, BEZ235), (ii) of ATM (KU55933) and (iii) of DNA-PK (NU7026). The combined application of each of these inhibitors with one of the topoisomerase inhibitors each (doxorubicin, etoposide or topotecan) reversed the up-regulation of [18F]-FDG uptake as observed after treatment with the respective topoisomerase inhibitors only (Figures 5, 6 and 7). Of the inhibitors investigated, only BEZ235 effectively reduced [18F]-FDG uptake when applied as a single substance. This corresponds to previous findings on quantification of [18F]-FDG uptake in 3D tumor spheroids prepared from EMT6 (mouse mammary) and FaDu (human nasopharyngeal) cells [45]. BEZ235 induces of dual inhibition of PI3K and mTORC1/2. Furthermore, BEZ235 has been shown to inhibit the kinases ATM and DNA-PKCs, both of which are involved in DNA damage response (DDR), in glioblastoma cells more potently than the known specific inhibitors KU55933 and NU7026, thereby blocking both non-homologous end joining (NHEJ) and homologous recombination (HR) [46]. Effective induction of apoptosis by BEZ235 has also been described in multiple myeloma cells [18,47], but none of these studies was related to changes in the expression of GLUT-1 and HKII. As shown in our study, combined treatment of OPM-2 cells with BEZ235 and any of the topoisomerase inhibitors used (doxorubicin, etoposide or topotecan), triggered down-regulation of GLUT-1 expression and a drastic increase of the apoptotic markers cleaved caspase-3 and cleaved PARP-1. Such changes were not observed using the inhibitors KU55933, MK-2206 and NU7026 in combination with the topoisomerase inhibitors (Figures 5-7). Evidence is growing that dual inhibition of DDR kinases and mTORC1 is necessary to facilitate apoptosis in cancers [48]. Apoptosis induction in multiple myeloma cells has also been shown to be dependent on inhibition of both PI3K and mTor. PI3K feedback loops and mTor activation downstream of the MAP kinase pathway were able to inhibit drugs that target Akt or the mTor-complex individually [49].
Notably, co-treatment with topoisomerase inhibitors and molecular inhibitors of both PIKK and PI3K similarly decreased HKII levels and activated cleavage of caspase-3 (Figures 5, 6 and 7) which suggests a point of intersection between metabolic pathways and DNA repair pathways. As shown previously the PI3K inhibitor LY294002 reduced HKII levels and lactate production in various multiple myeloma cells [19]. It is known that PI3K/Akt stimulates the coupling of HKII to a voltage-dependent anion channel (VDAC) on the mitochondrial membrane thereby improving cell viability [50]. Therefore, we suppose that in our study administration of topoisomerase inhibitors and PIKK/PI3K inhibitors led to the disruption of HKII and contributed to changes in glycolysis and cell survival in OPM-2 cells as described previously for hepatocellular carcinoma [29].
The effect of the inhibitors KU55933, MK-2206 and NU7026 of DDR kinases in combination with the topoisomerase inhibitors doxorubicin, etoposide and topotecan on apoptosis in OPM-2 cells was evident, but not as potent as with the dual PI3K/mTor inhibitor BEZ235. This might be explained by the dependence of the activity of the DDR kinases on the status of tumor suppressor genes, the enzymatic configuration of DNA repair or the cell-cycle phase of the tumor cells. In previous studies, the ATM inhibitor KU55933 was shown to increase etoposide-induced DNA damage only in highly proliferating leukemic cells, but not in resting cells [51]. As well, ATM inhibition was shown to sensitize cancer cells to DNA damaging drugs only in p53-deficient cells, while drug resistance was evident in p53-proficient cells. In the same study it was shown that ATM-deficient cells with defective HR were more vulnerable to doxorubicin induced DSBs after concurrent suppression of DNA-PKCs triggering NHEJ [52]. Our data showed that the DNA-PK inhibitor NU7026 effectively reversed elevated [18F]-FDG uptake in combination with the administered topoisomerase poisons. As DNA-PK is thought to play an important role in the stimulation of Akt (protein kinase B), known to promote cell proliferation, after induction of DNA double strand breaks [53], the development of drugs that inhibit both DNA-PK and PI3K seems to be a very attractive option for treatment of multiple myeloma cells.
In this study we demonstrated that targeting the genomic integrity and DNA damage response in OPM-2 cells via topoisomerase inhibitors is closely linked to a modulation of the glucose metabolism. We presume that over-expression of GLUT-1 and HKII might result in resistance against these genotoxic agents by boosting aerobic glycolysis and the production of ATP which finally will increase DNA repair. As we have observed, DNA damage-induced apoptosis in OPM-2 cells was strictly correlated with a decreasing activity of HKII.
Obviously, the mechanism by which antineoplastic drugs affect the glycolytic status of cancer cells seems to be a key component of tumor sensitivity in chemotherapy. Because the molecular inhibitors of DDR kinases and PI3K/Akt/mTor signaling administered in this study (particularly BEZ235) have been shown to disrupt the glucose supply and therefore to eradicate OPM-2 cells, we see a promising therapeutic potential in dual targeting of these pathways.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, SFB 824).
Disclosure of conflict of interest
None.
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finlay MR, Griffin RJ. Modulation of DNA repair by pharmacological inhibitors of the PIKK protein kinase family. Bioorg Med Chem Lett. 2012;22:5352–5359. doi: 10.1016/j.bmcl.2012.06.053. [DOI] [PubMed] [Google Scholar]
- 3.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 4.Furgason JM, Bahassi el M. Targeting DNA repair mechanisms in cancer. Pharmacol Ther. 2013;137:298–308. doi: 10.1016/j.pharmthera.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Woods D, Turchi JJ. Chemotherapy induced DNA damage response: convergence of drugs and pathways. Cancer Biol Ther. 2013;14:379–389. doi: 10.4161/cbt.23761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 7.Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caldecott K, Banks G, Jeggo P. DNA double-strand break repair pathways and cellular tolerance to inhibitors of topoisomerase II. Cancer Res. 1990;50:5778–5783. [PubMed] [Google Scholar]
- 9.Shen H, Schultz M, Kruh GD, Tew KD. Increased expression of DNA-dependent protein kinase confers resistance to adriamycin. Biochim Biophys Acta. 1998;1381:131–138. doi: 10.1016/s0304-4165(98)00020-8. [DOI] [PubMed] [Google Scholar]
- 10.Hansen LT, Lundin C, Helleday T, Poulsen HS, Sorensen CS, Petersen LN, Spang-Thomsen M. DNA repair rate and etoposide (VP16) resistance of tumor cell subpopulations derived from a single human small cell lung cancer. Lung Cancer. 2003;40:157–164. doi: 10.1016/s0169-5002(03)00026-6. [DOI] [PubMed] [Google Scholar]
- 11.Lans H, Marteijn JA, Vermeulen W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenetics Chromatin. 2012;5:4. doi: 10.1186/1756-8935-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinoshita E, van der Linden E, Sanchez H, Wyman C. RAD50, an SMC family member with multiple roles in DNA break repair: how does ATP affect function? Chromosome Res. 2009;17:277–288. doi: 10.1007/s10577-008-9018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 14.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 16.Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene. 2005;24:4165–4173. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- 17.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 18.Baumann P, Mandl-Weber S, Oduncu F, Schmidmaier R. The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Exp Cell Res. 2009;315:485–497. doi: 10.1016/j.yexcr.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Nakano A, Miki H, Nakamura S, Harada T, Oda A, Amou H, Fujii S, Kagawa K, Takeuchi K, Ozaki S, Matsumoto T, Abe M. Up-regulation of hexokinaseII in myeloma cells: targeting myeloma cells with 3-bromopyruvate. J Bioenerg Biomembr. 2012;44:31–38. doi: 10.1007/s10863-012-9412-9. [DOI] [PubMed] [Google Scholar]
- 20.Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: a double-edged sword in cell proliferation and genome stability. J Oncol. 2012;2012:951724. doi: 10.1155/2012/951724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamamoto T, Seino Y, Fukumoto H, Koh G, Yano H, Inagaki N, Yamada Y, Inoue K, Manabe T, Imura H. Over-expression of facilitative glucose transporter genes in human cancer. Biochem Biophys Res Commun. 1990;170:223–230. doi: 10.1016/0006-291x(90)91263-r. [DOI] [PubMed] [Google Scholar]
- 22.Binder C, Binder L, Marx D, Schauer A, Hiddemann W. Deregulated simultaneous expression of multiple glucose transporter isoforms in malignant cells and tissues. Anticancer Res. 1997;17:4299–4304. [PubMed] [Google Scholar]
- 23.Smith TA. Facilitative glucose transporter expression in human cancer tissue. Br J Biomed Sci. 1999;56:285–292. [PubMed] [Google Scholar]
- 24.Tateishi U, Yamaguchi U, Seki K, Terauchi T, Arai Y, Hasegawa T. Glut-1 expression and enhanced glucose metabolism are associated with tumour grade in bone and soft tissue sarcomas: a prospective evaluation by [18F] fluorodeoxyglucose positron emission tomography. Eur J Nucl Med Mol Imaging. 2006;33:683–691. doi: 10.1007/s00259-005-0044-8. [DOI] [PubMed] [Google Scholar]
- 25.Tsukioka M, Matsumoto Y, Noriyuki M, Yoshida C, Nobeyama H, Yoshida H, Yasui T, Sumi T, Honda K, Ishiko O. Expression of glucose transporters in epithelial ovarian carcinoma: correlation with clinical characteristics and tumor angiogenesis. Oncol Rep. 2007;18:361–367. [PubMed] [Google Scholar]
- 26.Mori Y, Tsukinoki K, Yasuda M, Miyazawa M, Kaneko A, Watanabe Y. Glucose transporter type 1 expression are associated with poor prognosis in patients with salivary gland tumors. Oral Oncol. 2007;43:563–569. doi: 10.1016/j.oraloncology.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 27.Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta. 2002;1555:14–20. doi: 10.1016/s0005-2728(02)00248-7. [DOI] [PubMed] [Google Scholar]
- 28.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 29.Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19:17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czernin J, Phelps ME. Positron emission tomography scanning: current and future applications. Annu Rev Med. 2002;53:89–112. doi: 10.1146/annurev.med.53.082901.104028. [DOI] [PubMed] [Google Scholar]
- 31.Weber WA. Assessing tumor response to therapy. J Nucl Med. 2009;50:1S–10S. doi: 10.2967/jnumed.108.057174. [DOI] [PubMed] [Google Scholar]
- 32.Zhou R, Vander Heiden MG, Rudin CM. Genotoxic exposure is associated with alterations in glucose uptake and metabolism. Cancer Res. 2002;62:3515–3520. [PubMed] [Google Scholar]
- 33.Watanabe M, Naraba H, Sakyo T, Kitagawa T. DNA damage-induced modulation of GLUT3 expression is mediated through p53-independent extracellular signal-regulated kinase signaling in HeLa cells. Mol Cancer Res. 2010;8:1547–1557. doi: 10.1158/1541-7786.MCR-10-0011. [DOI] [PubMed] [Google Scholar]
- 34.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown RS, Wahl RL. Overexpression of Glut-1 glucose transporter in human breast cancer. An immunohistochemical study. Cancer. 1993;72:2979–2985. doi: 10.1002/1097-0142(19931115)72:10<2979::aid-cncr2820721020>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 36.Reske SN, Grillenberger KG, Glatting G, Port M, Hildebrandt M, Gansauge F, Beger HG. Overexpression of glucose transporter 1 and increased FDG uptake in pancreatic carcinoma. J Nucl Med. 1997;38:1344–1348. [PubMed] [Google Scholar]
- 37.Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005;202:654–662. doi: 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- 38.Adams V, Kempf W, Hassam S, Briner J. Determination of hexokinase isoenzyme I and II composition by RT-PCR: increased hexokinase isoenzyme II in human renal cell carcinoma. Biochem Mol Med. 1995;54:53–58. doi: 10.1006/bmme.1995.1008. [DOI] [PubMed] [Google Scholar]
- 39.Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol. 2008;10:1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA Jr, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaina B. DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem Pharmacol. 2003;66:1547–1554. doi: 10.1016/s0006-2952(03)00510-0. [DOI] [PubMed] [Google Scholar]
- 42.Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol. 2001;21:5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakano A, Tsuji D, Miki H, Cui Q, El Sayed SM, Ikegame A, Oda A, Amou H, Nakamura S, Harada T, Fujii S, Kagawa K, Takeuchi K, Sakai A, Ozaki S, Okano K, Nakamura T, Itoh K, Matsumoto T, Abe M. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS One. 2011;6:e27222. doi: 10.1371/journal.pone.0027222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer. 2010;10:147–156. doi: 10.1038/nrc2789. [DOI] [PubMed] [Google Scholar]
- 45.Kelly CJ, Hussien K, Muschel RJ. 3D tumour spheroids as a model to assess the suitability of [18F] FDG-PET as an early indicator of response to PI3K inhibition. Nucl Med Biol. 2012;39:986–992. doi: 10.1016/j.nucmedbio.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 46.Mukherjee B, Tomimatsu N, Amancherla K, Camacho CV, Pichamoorthy N, Burma S. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia. 2012;14:34–43. doi: 10.1593/neo.111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McMillin DW, Ooi M, Delmore J, Negri J, Hayden P, Mitsiades N, Jakubikova J, Maira SM, Garcia-Echeverria C, Schlossman R, Munshi NC, Richardson PG, Anderson KC, Mitsiades CS. Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer Res. 2009;69:5835–5842. doi: 10.1158/0008-5472.CAN-08-4285. [DOI] [PubMed] [Google Scholar]
- 48.Muller A, Zang C, Chumduri C, Dorken B, Daniel PT, Scholz CW. Concurrent inhibition of PI3K and mTORC1/mTORC2 overcomes resistance to rapamycin induced apoptosis by down-regulation of Mcl-1 in mantle cell lymphoma. Int J Cancer. 2013;133:1813–1824. doi: 10.1002/ijc.28206. [DOI] [PubMed] [Google Scholar]
- 49.Stengel C, Cheung CW, Quinn J, Yong K, Khwaja A. Optimal induction of myeloma cell death requires dual blockade of phosphoinositide 3-kinase and mTOR signalling and is determined by translocation subtype. Leukemia. 2012;26:1761–1770. doi: 10.1038/leu.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korwek Z, Sewastianik T, Bielak-Zmijewska A, Mosieniak G, Alster O, Moreno-Villanueva M, Burkle A, Sikora E. Inhibition of ATM blocks the etoposide-induced DNA damage response and apoptosis of resting human T cells. DNA Repair (Amst) 2012;11:864–873. doi: 10.1016/j.dnarep.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 52.Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;23:1895–1909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30:203–213. doi: 10.1016/j.molcel.2008.02.024. [DOI] [PubMed] [Google Scholar]