

Abstract
The inflammatory tumor microenvironment has been identified to play a pivotal role in tumor development and metastasis. Tumor necrosis factor-α (TNF-α) is one of the key cytokines that regulate the inflammatory processes in tumor promotion. In the current study, we treated three oral squamous cell carcinoma (OSCC) cell lines with TNF-α to study its role in inflammation-induced tumor progression. Here we show that TNF-α induces stabilization of the transcriptional repressor Snail and activates NF-κB pathway in the three OSCC cell lines. These activities resulted in the increased motility and invasiveness of three OSCC cell lines. In addition, upon dealing with TNF-α for the indicated time, three OSCC cell lines underwent epithelial-to-mesenchymal transition (EMT), in which they presented a fibroblast-like phenotype and had a decreased expression of epithelial marker (E-cadherin) and an increased expression of mesenchymal marker (vimentin). We further demonstrated that TNF-α can up-regulate the expression of Id2 while inducing an EMT in oral cancer cells. Finally, we showed that Id2 interacted with Snail which may constrain Snail-dependent suppression of E-cadherin. In conclusion, our study indicates that TNF-α induces Snail stabilization is dependent on the activation of NF-κB pathway and results in increasing cell invasion and migration in OSCC cells. Id2 may contribute to regulate the function of Snail during TNF-α-mediated EMT in OSCC. These findings have significant implications for inflammation-induced tumor promotion in OSCC.
Keywords: Epithelial-to-mesenchymal transition, Id2, oral squamous cell carcinoma, Snail, tumor necrosis factor-α
Introduction
Mounting evidence has demonstrated that the inflammatory tumor microenvironment contributes a pivotal role to tumor development and metastasis [1-4]. The inflammatory tumor microenvironment is largely orchestrated by immune cells and their secretory cytokines and/or chemokines, which facilitate extracellular matrix breakdown, angiogenesis, tissue remodeling and promote tumor cell motility [5]. In particular, tumor necrosis factor-α (TNF-α) is one of the key cytokines that regulate the inflammatory processes in tumor promotion, which is mainly derived from activated macrophages infiltrated in tumor. Despite high-dose of TNF-α used as a cytotoxic agent, increasing evidences have provided that chronic and low level of TNF-α can acquire a tumorigenic feature. TNF-α can activate the major inflammatory response NF-κB pathway, which facilitates to mediate many critical processes of tumor progression [6,7]. Recently, studies revealed that TNF-α could induce epithelial-mesenchymal transition (EMT) and further promote invasion and metastasis in breast cancer, renal cell carcinoma cells and prostate cancer [8-10]. In addition, it has been reported that TNF-α dominates genotypes were considered as independent predictors for early, advanced and overall oral squamous cell carcinoma (OSCC) stages [11]. However, there is little known about the role of TNF-α in OSCC progression.
EMT is defined as the conversion of epithelial cells to mesenchymal cells with migratory and invasive capabilities during embryonic development, tissue remolding, wound healing and tumor metastasis [12-14]. During this transition, epithelial cells lose apico-basal polarity and cell-cell interactions, acquire mesenchymal and migratory behavior [13,15,16]. One hallmark of EMT is the loss of E-cadherin, which is thought to be a functional suppressor of invasion during carcinoma progression [15,17]. Currently, several transcription factors has emerged as fundamental regulators for the potent repression of E-cadherin, such as the Snail (Snail1 and Slug), ZEB (ZEB1 and ZEB2) and basic helix-loop-helix (bHLH: Twist, E12/E47) families [18].
Snail was first identified in Drosophila as the zinc-finger transcriptional repressor, which can bind to E-boxes of the E-cadherin promoter [17]. Snail is known as a central mediator of EMT and can strongly induce EMT by repressing the transcription of E-cadherin in tumor progression [19-21]. In addition, overexpression of Snail correlates with tumor grade, tumor recurrence and cancer cell invasion [22]. Recent study has demonstrated that TNF-α stabilizes Snail dependent on NF-κB activation and triggers EMT [23]. However, the role of TNF-α inducing EMT in OSCC has not been studied yet and the underlying molecular mechanism remains undefined.
Inhibitor of DNA-binding-2 (Id2) is a member of the Id proteins that belongs to the HLH protein family. The Id protein functions by binding to specific transcription factors and preventing their dimerization and DNA binding. Id2 has been shown to be important in regulating cellular differentiation, proliferation, development and tumorigenesis [24-28]. In tumor, Id2 plays two opposite roles in the same or different types of cells depending on extracellular signals and microenvironments [26,27]. Over-expression of Id2 has been shown to be associated with tumors progression in pancreatic cancer, neuroblastoma, and lung cancer [29-31]. In contrast, studies have demonstrated that over-expression of Id2 blocks EMT induced by TGF-β to have an anti-oncogenic potential in breast cancer and hepatocellular carcinoma [32-35]. However, the expression pattern and functional mechanism of Id2 in OSCC is rarely investigated.
Based on the above studies, we explored the role of TNF-α in OSCC progression. Here we show that TNF-α induces Snail stabilization through activation of NF-κB pathway, and further promotes the invasive and migratory activities in OSCC. In addition, TNF-α can up-regulate the expression of Id2 while inducing an EMT in oral cancer cells. Finally, we also demonstrate that Snail interacts with Id2, which may have an effect on the suppression of E-cadherin mediated by Snail and regulate TNF-α-induced EMT in OSCC.
Materials and methods
Cell culture
Three human OSCC cell lines (SCC4, SCC9 and SCC25) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in a mixture of Dulbecco’s Modified Eagle’s medium and Ham’s F12 medium (1:1) (Invitrogen, Burlington, Ontario, Canada) supplemented with 400 ng/ml hydrocortisone (Sigma-Aldrich, St Louis, MO, USA), penicillin (100 U/ml), streptomycin (100 μg/ml) (Invitrogen) and 10% fetal bovine serum (FBS, Invitrogen). After serum starvation for 16 to 18 h, tumor cells (2×105/ml) were treated with or without TNF-α (10 ng/ml) (Peprotech, Rocky Hill, NJ, USA) for the indicated time.
Real-timeRT-PCR
Total mRNA of OSCC cells was extracted after treatment of TNF-α (10 ng/ml) for 0 h, 0.5 h, 1h, 2 h, 4 h, 8 h, 24 h, 48 h and 72 h using the TRIzol reagent (Invitrogen). For cDNA synthesis, mRNA was reverse-transcribed into cDNA using the 5×PrimeScript RT Master Mix (TaKaRa) at 37°C for 15 min and 85°C for 5 s according to the manufacturer’s protocol. Gene expression was quantified by real-time quantitative PCR using 2×SYBR Premix Ex Taq (TaKaRa) with a 7300 ABI Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) under the conditions of 95°C for 30 s, 95°C for 5 s, and 60°C for 31 s for 40 cycles. The relative gene expression was calculated using the 2(-ΔΔCT) method at least three independent experiments. Briefly, the resultant mRNA was normalized to its own GAPDH. The primers utilized for the real-time RT-PCR were as followed: GAPDH (5’-GAAGGTGAAGGTCGGAGTC-3’, 5’-GAGATGGTGATGGGATTTC-3’), E-cadherin (5’-TACACTGCCCAGGAGCCAGA-3’, 5’-TGGCACCAGTGTCCGGATTA-3’), vimentin (5’-TGAGTACCGGAGACAGGTGCAG-3’, 5’-TAGCAGCTTCAACGGCAAAGTTC-3’), Snail (5’-GACCACTATGCCGCGCTCTT-3’, 5’-TCGCTGTAGTTAGGCTTCCGATT-3’), ID2 (5’-CATCTTGGACCTGCAGATCG, 5’-ATGAACACCGCTTATTCAGC-3’).
Western blotting
The cells were lysed using RIPA lysis buffer (Beyotime) after treated with TNF-α (10 ng/ml) for 0 h, 0.5 h, 1 h, 2 h, 4 h, 8 h, 24 h, 48 h, 72 h. Total protein (30 μg) from each sample was separated on the SDS-PAGE and then transferred into PVDF membranes (Millipore, Billerica, MA, USA), which were blocked with 5% nonfat milk in PBST for 2 h at room temperature, then the membranes were incubated with primary antibodies at 4°C overnight. The antibodies were used as following: anti-β-actin (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-E-cadherin (1:1000, Cell Signaling Technology, Danvers, MA, USA), anti-vimentin (1:500, Santa Cruz Biotechnology), anti-Snail (1:500, Abcam, Cambridge, MA, USA), anti-Id2 (1:500, Abcam). Then the membranes were incubated with HRP-conjugated secondary antibodies (1:1000, R&D Systems, Minneapolis, MN, USA) for 2 h at room temperature. Immunoreactive material was visualized using the Immun-Star WesternC Kit (Bio-Rad, Hercules, CA, USA) products and bands were detected via ImageQuantLAS4000 (GE, Fairfield , CT, USA).
Transwell invasion assay
According to the manufacturer’s procedures, the polycarbonate filters (8 μm pore size, costar, Lowell, MA, USA) were pre-coated with Matrigel Basement Membrane Matrix (BD Biosciences, MA, USA) diluted with serum-free medium (1:3) for 30 min at 37°C. The cells (3×105/ml) were resuspended with 200 μl serum-free medium inoculated in the upper chamber while 500 μl medium containing 10% FBS with or without TNF-α (10 ng/ml) was placed in the lower chamber. The plates were placed at 37°C in 5% CO2 for 24 h. The chambers were fixed with 4% paraformaldehyde (PFA) and stained with 0.1% crystal violet (Beyotime) for 30 min. Then non-invasive cells were carefully wiped, and the invasive cells presenting on the lower surface of the upper chamber were counted and captured under the microscope at ×100 magnification (10 random fields per chamber).
Wound healing assay
For the wound healing assay, the cells were allowed to grow to 90% confluence and then wounded by scratching with a pipette tip in the central area. Floating cells and debris were removed, and the medium was changed to serum-free with or without TNF-α (10 ng/ml). The cells were incubated for 24 h to allow cells to grow and close the wound. Photographs were taken at the same position of the wound at the indicated time points.
Flow cytometry
For flow cytometric cell-cycle analysis, the cells were synchronized with serum-free medium for 18 h, released and then cultured using the medium with or without TNF-α (10 ng/ml) for 48 h. Then cells were detached from the culture plate with trypsin, washed PBS, and then resuspended in 75% alcohol. The prepared cells were stained with 100 mg/ml of propidium iodide (BD Pharmingen, San Jose, CA, USA) prior to analysis using flow cytometry with a BD FACS Calibur (BD Biosciences) and Cell Quest Pro software (BD Biosciences).
Immunofluorescence
After serum starvation for 18 h, OSCC cells were incubated with or without TNF-α (10 ng/ml) for 72 h. Cells were washed three times with PBS, fixed with 4% PFA for 20 min at room temperature, permeabilized with 1% Triton X-100 for 15 min. After blocked with goat serum albumin for 30 min at 37°C, the cells were incubated with specific antibodies anti-E-cadherin (1:100, Cell Signaling Technology) and anti-vimentin (1:100, Santa Cruz Biotechnology), or anti-Snail (1:100, Abcam) and anti-Id2 (1:100, Santa Cruz Biotechnology) at 4°C overnight. The appropriate secondary antibodies (diluted 1:50) were used, and then nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI, 1:1000, Invitrogen) for 2 min. Immunofluorescence was visualized using a Zeiss LSM-710 laser-scanning con-focal microscopy.
Co-immunoprecipitation (co-IP) assays
After serum starvation for 18 h, OSCC cells were treated with or without TNF-α (10 ng/ml) for 72 h. In brief, cells were lysed in RIPA buffer on ice for 20 min, then 300 μg total cellular protein were incubated at 4°C for 2 h with 1.0 μg of the appropriate control lgG (Santa Cruz Biotechnology) or 2.0 μg anti-Snail (Abcam), after which 20 μl of resuspended volume of Protein A/G Plus Agarose (Santa Cruz Biotechnology) were added, and the mixture was incubated on a rocker at 4°C overnight. The protein A/G Plus Agarose bound immunocomplexes were washed 4 times with ice-cold RIPA buffer to eliminate non-specific interactions, isolated by centrifugation and then analyzed by Western blotting protocols as introduced above.
Statistical analysis
The assays were repeated three or more independent experiments as the mean ± s.d. Statistical significance was assessed using independent-samples t test. P-value<0.05 was considered significant.
Results
TNF-α induces Snail stabilization through NF-κB activation
Snail as one of the transcription factors plays a fundamental role in EMT [18]. Our published data demonstrated that overexpression of Snail repressed expression of E-cadherin and induced EMT in SCC9 cells [21]. Recent research revealed that TNF-α was the major inflammatory cytokine to induce Snail stabilization in tumor cells [23]. In the current study, Snail was detected to increase significantly after TNF-α stimulation in SCC4 and SCC9 cells. The results of real-time RT-PCR and western blot revealed that the level of Snail was obviously activated after 0.5 h of TNF-α treatment and remained stabilization at all time points in the OSCC cells (Figure 1A and 1B). Western blotting analysis was performed to assess the activation of the components of the NF-κB pathway. The results showed that nuclear translocation of p65 was detected in SCC9 and SCC25 cells dealing by TNF-α treatment (Figure 1C). This result indicated that TNF-α induced Snail stabilization was correlated with the activation of NF-κB pathway in oral cancer cells.
Figure 1.

TNF-α induces Snail stabilization through NF-κB activation. OSCC cells were treated with TNF-α (10 ng/ml) for the indicated times. A. The mRNA level of Snail was analyzed by real-time RT-PCR. GAPDH was used as a control. Each bar represents the mean ± s.d. *P<0.05, **P<0.01, ***P<0.001. B. The western blot analysis was performed to assess the expression of Snail on the protein level. β-actin was employed as a loading control. C. SCC9 and SCC25 cells were treated with TNF-α (10 ng/ml) for 48 h. Western blotting was performed to assess the expression of NF-κB pathway-related proteins in cytoplasmic and nuclear extracts in SCC9 and SCC25 cells with or without dealing by TNF-α. β-actin and Histone 3 (H3) were employed as the positive controls for cytoplasmic and nuclear proteins, respectively.
TNF-α promotes migration and invasion of OSCC cells
The wound healing assay showed that OSCC cells treated with TNF-α could close the scratch wound at 24 h while untreated groups could not, especially in SCC9 and SCC25 cells (Figure 2A). Flow cytometric cell-cycle analysis showed that there was no significant difference of proliferation between TNF-α treatment and nontreatment groups (Figure 2B). All these indicated that TNF-α could promote migratory ability of OSCC cells. The photographs of transwell invasion assay demonstrated that the cells treated with TNF-α were more invasive than untreated group (Figure 3A). The quantitative analysis confirmed that the invasive abilities were significantly increased in SCC4 (1.591-fold), SCC9 (1.812-fold) and SCC25 (1.772-fold) after TNF-α treatment (Figure 3B). Thus, we conclude that TNF-α can promote the migratory and invasive ability of OSCC cells.
Figure 2.

TNF-α could enhance migration behavior of OSCC cells. A. Photographs were taken at the same position of the wound at the indicated time points (×40 magnification). Bar, 100 μm. B. OSCC cells were incubated in the absence or presence of TNF-α (10 ng/ml) for 48 h, flow cytometric cell-cycle analysis showed that there was no significant difference of proliferation ability between TNF-α treatment group and nontreatment group.
Figure 3.
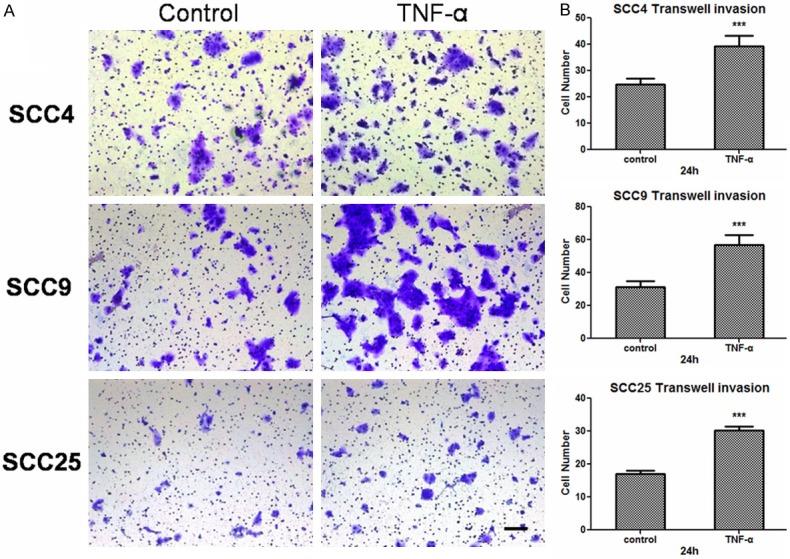
TNF-α promotes the invasive ability of OSCC cells. A. Pictures presenting the cells penetrating the Matrigel basement membrane matrix after 24 h. Bar, 100 μm. B. Quantitative analysis of cell invasion in ten different random fields. The data were calculated as the mean ± s.d. ***P<0.001.
TNF-α induces EMT in OSCC
After treated with 10 ng/ml TNF-α for 0 h, 0.5 h, 1 h, 2 h, 4 h, 8 h, 24 h, 48 h and 72 h, three OSCC cell lines gradually changed from typical cobblestone appearance to a spindle-like fibroblastic appearance. The cell-cell junction presented to become loose. The morphological changes of cells were captured at the time point of dealing after 72 h (Figure 4A). The results of real-time RT-PCR revealed that TNF-α treatment down-regulated E-cadherin expression and up-regulated vimentin expression. However, the down-regulation of E-cadherin expression only occurred at the beginning of the stimulus, whereas the maximal level of vimentin were seen at 48 h of SCC4, 24 h of SCC9, 72 h of SCC25, respectively (Figure 4B). Western blotting analysis showed that the down-regulation of E-cadherin and the up-regulation of vimentin on the protein level, and the decreased trends of E-cadherin expression were more obvious than the mRNA level (Figure 4C). Additionally, immunofluorescence analysis further confirmed that the three OSCC cells underwent EMT by TNF-α treatment. The morphological changes presented with a decreased expression of E-cadherin and an increased expression of vimentin (Figure 5). Collectively, these results demonstrated that three OSCC cells underwent an EMT after treating with TNF-α.
Figure 4.

TNF-α induces EMT in OSCC. A. OSCC cells were treated with TNF-α (10 ng/ml) for 0 h, 24 h, 48 h, 72 h. Cell morphological changes associated with EMT are shown in the phase contrast image. Photographs were taken under inverted microscope (Olympus). Bar, 100 μm. B. OSCC cells were treated with TNF-α (10 ng/ml) for the indicated times. The mRNA levels of E-cadherin and vimentin were analyzed by real-time RT-PCR. GAPDH was used as a control. Each bar represents the mean ± s.d. *P<0.05, **P<0.01. C. OSCC cells were incubated in the presence of TNF-α (10 ng/ml) for the indicated times, and the protein levels of E-cadherin and vimentin were analyzed by western blotting. β-actin was employed as a loading control.
Figure 5.

Immunofluorescence analysis ofE-cadherin and vimentin in the three OSCC cell lines. Double immunofluorescence staining of E-cadherin (E-cad: red) and vimentin (Vim: green) in the three OSCC cells with or without dealing by TNF-α treatment. The nuclei in image sets were stained with DAPI (blue). Images were taken at ×200 magnification. Bar, 100 μm.
TNF-α-mediated Snail expression interacts with Id2
The data shown in Figure 4B, E-cadherin expression was not repressed so much while Snail was extensively activated in three OSCC cells. These results illuminated that there may exist some factors which constrained the repressive function of Snail. Of the many factors that associated with tumor invasion and metastasis, we found that the expression of Id2 was up-regulated after treating with TNF-α in the three OSCCcells (Figure 6A and 6B). As shown in Figure 6C, TNF-α significantly activated the nuclear expression of Snail compared with the control group. Simultaneously, immunofluorescence pictures showed that the expression of Id2 also increased in the three OSCC cells (Figure 6C). All these results suggested that TNF-α induces Snail stabilization meanwhile up-regulates Id2 expression.
Figure 6.

TNF-α up-regulates Id2 while inducing Snail stabilization. OSCC cells were incubated in the presence of TNF-α (10 ng/ml) for the indicated times. A. Quantitative determination of Id2 mRNA expression in three OSCC cells. Each bar represents the mean ± s.d. *P<0.05, **P<0.01. B. The western blot analysis was performed to assess the expression of Id2 on the protein level. β-actin was employed as a loading control. C.Immunofluorescence analysis of Snail and Id2 in three OSCC cells. Double immunofluorescence staining of Snail (green) and Id2 (red) in the three OSCC cells culturing in the absence or presence of TNF-α. The nuclei in image sets were stained with DAPI (blue). Images were taken at ×400 magnification. Bar, 100 μm.
Although Id2 can interact with bHLH protein [25,28], little is known about its interaction with zinc finger proteins. The immunofluorescence images showed that Id2 was localized in both the nucleus and cytoplasm of the three cell lines, while Snail was detected activation in nucleus (Figure 6C). Thus co-immunoprecipitation assay was performed to test the possibility that Snail partners with Id2. After pulling down the Snail protein, Id2 was also detected by western blot in SCC4, SCC9 and SCC25 (Figure 7A). To further investigate the interaction of Id2 and Snail in TNF-α-induced EMT, siRNA was constructed to interfere the expression of Id2. When Id2 was partially depleted by siRNA, Snail shown to be downregulated and E-cadherin was performed to increase in SCC9 cells compared to control siRNA (Figure 7B and 7C). These data indicate that Snail protein interacts with Id2, which may contribute to regulate the function of Snail in repressing E-cadherin in OSCC.
Figure 7.

Snail interacts with Id2. A. OSCC cells were treated with or without TNF-α (10 ng/ml) for 72 h, then cells were immunoprecipitated with anti-Snail antibodies and control IgG and analyzed by Western blotting with the indicated anti-Snail and anti-Id2 antibodies. B and C. Id2 knockdown prevent Snail expression in SCC9 cells after incubation with TNF-α. SCC9 cells after dealing with TNF-α were transfected with either control siRNA (siNC) or Id2 siRNA (siId2). Cells were analyzed for Id2, Snail and epithelial and mesenchymal markers by real-time RT-PCR and western blot. GAPDH was used as a control. Each bar represents the mean ± s.d. *P<0.05. β-actin was employed as a loading control.
Discussion
Invasion and metastasis induced by chronic inflammation is a main challenge in cancer therapy [1]. Some inflammatory mediators have been demonstrated to as key initiators of EMT in various cancers [8,36,37]. In this study, we showed that TNF-α-induced Snail stabilization played a critical role in oral cancer cell invasion and metastasis. Our research provides insights to the regulation of inflammatory cytokine and tumor invasion in OSCC. First, our study indicates that TNF-α induces Snail stabilization, dependent on the activation of NF-κB pathway and promotes the invasive and migratory activities in oral cancer cells. Second, TNF-α induces an EMT and up-regulates the expression of Id2. In addition, Id2 complexes with Snail and may constrain the repressive function of E-cadherin mediated by Snail.
Snail, as one of the pivotal EMT regulators, controls repression of E-cadherin by binding to three independent E-boxes in the E-cadherin promoter [15,18,38]. Our previous study demonstrated that overexpression of Snail played an important role in inducing EMT in SCC9 cells [21]. Recent study indicated that TNF-α induced EMT through mediating Snail stabilization in breast cancer cells [23]. In this study, we detected the expression of transcriptional factors in three OSCC cells after TNF-α treatment, there was no significant difference in the mRNA levels of Slug, Twist, ZEB1 and ZEB2 (data not shown). However, Snail was detected to present a rapid activation and stable expression in three OSCC cells after TNF-α stimulation. Based on that Snail acting as a labile protein, had a half-life of about 25 min, our study indicated that TNF-α could regulate the nuclear stabilization of Snail in OSCC. This Snail stabilization mediated by TNF-α was dependent on the activation of NF-κB pathway. Recent study reported that activation of NF-κB pathway was required for the stabilization of Snail [23]. Our research was consistent with this viewpoint. It has become increasing clear that inflammation is correlated with tumor progression [1,39]. Our study verified that TNF-α promoted cancer cell invasion and migration in OSCC. TNF-α treatment resulted in a change in the biological properties of the three OSCC cells. The cells acquired highly invasive and migratory capacities after interacting with TNF-α.
Studies have showed that TNF-α can induce EMT with the down-regulation of E-cadherin and promotes invasion and metastasis in cancer cells [9,23]. In our study, TNF-α induced morphological changes in three OSCC cells that are consistent with the acquisition of EMT phenotype, presenting the up-regulation of mesenchymal marker vimentin. However, after stimulating by TNF-α, the E-cadherin expression was not repressed so much while Snail exhibited high activation. It is known that Snail has a preeminent role in the repression of E-cadherin. Thus we suppose that there may exist some factor impedes the ability of Snail to repress E-cadherin. We proceeded to detect key factors involved in EMT process and Id2 was observed to display highly expression in the three OSCC cells by TNF-α treatment. It has been reported that Id2 is repressed dramatically in TGF-β-induced EMT of epithelial cells [34]. Based on our observation, it is possible that there may be interaction between Id2 and Snail.
Id2 belongs to the HLH subfamily protein, which do not contain a basic region preventing to bind to DNA [40]. It has been proposed that Id2 can interact with bHLH proteins, and recent study revealed that Id2 complexes with SNAG domain of Snail on the β4 promoter and constrains the repressive function of Snail [41]. In our study, Snail and Id2 was observed to colocalize within the nuclei of OSCC cells. The results of co-immunoprecipitation assay and siRNA interference of Id2 further indicated that Id2 interacted with Snail protein, and thus we infer that this interaction may inhibit the function of Snail to regulate E-cadherin. However, further investigation is required, exploring the mechanistic relationship between Id2 and Snail and the Id2-mediated regulation of E-cadherin expression.
In conclusion, our study demonstrated that TNF-α induces Snail stabilization is dependent on the activation of NF-κB pathway and results in increasing cell invasion and migration in OSCC cells. Furthermore, Id2 may contribute to regulate the function of Snail during TNF-α-mediated EMT in OSCC. Collectively, our data may have significant implications for inflammation-induced tumor promotion in OSCC.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81272968) and from Provincial Natural Science Research Project of Anhui Colleges (KJ2014A269) and from A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD, 2014-37) and from the Science and Technology Development Fund of Nanjing Medical University (2013NJMU065).
Disclosure of conflict of interest
The authors have no conflict of interest.
References
- 1.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 3.Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park) 2002;16:217–226. 229. discussion 230-212. [PubMed] [Google Scholar]
- 4.van Kempen LC, Ruiter DJ, van Muijen GN, Coussens LM. The tumor microenvironment: a critical determinant of neoplastic evolution. Eur J Cell Biol. 2003;82:539–548. doi: 10.1078/0171-9335-00346. [DOI] [PubMed] [Google Scholar]
- 5.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361–371. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- 7.Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006;25:409–416. doi: 10.1007/s10555-006-9005-3. [DOI] [PubMed] [Google Scholar]
- 8.Ho MY, Tang SJ, Chuang MJ, Cha TL, Li JY, Sun GH, Sun KH. TNF-alpha induces epithelial-mesenchymal transition of renal cell carcinoma cells via a GSK3beta-dependent mechanism. Mol Cancer Res. 2012;10:1109–1119. doi: 10.1158/1541-7786.MCR-12-0160. [DOI] [PubMed] [Google Scholar]
- 9.Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, Wang Y, Lai YJ, LaBaff AM, Wu TJ, Lin BR, Yang MH, Hortobagyi GN, Hung MC. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72:1290–1300. doi: 10.1158/0008-5472.CAN-11-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Fang R, Wang XF, Zhang F, Chen DY, Zhou B, Wang HS, Cai SH, Du J. Stabilization of Snail through AKT/GSK-3beta signaling pathway is required for TNF-alpha-induced epithelial-mesenchymal transition in prostate cancer PC3 cells. Eur J Pharmacol. 2013;714:48–55. doi: 10.1016/j.ejphar.2013.05.046. [DOI] [PubMed] [Google Scholar]
- 11.Vairaktaris E, Yapijakis C, Serefoglou Z, Avgoustidis D, Critselis E, Spyridonidou S, Vylliotis A, Derka S, Vassiliou S, Nkenke E, Patsouris E. Gene expression polymorphisms of interleukins-1 beta, -4, -6, -8, -10, and tumor necrosis factors-alpha, -beta: regression analysis of their effect upon oral squamous cell carcinoma. J Cancer Res Clin Oncol. 2008;134:821–832. doi: 10.1007/s00432-008-0360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 14.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 15.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 17.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 18.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 19.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 20.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 21.Zhu LF, Hu Y, Yang CC, Xu XH, Ning TY, Wang ZL, Ye JH, Liu LK. Snail overexpression induces an epithelial to mesenchymal transition and cancer stem cell-like properties in SCC9 cells. Lab Invest. 2012;92:744–752. doi: 10.1038/labinvest.2012.8. [DOI] [PubMed] [Google Scholar]
- 22.Wu Y, Zhou BP. Snail: More than EMT. Cell Adh Migr. 2010;4:199–203. doi: 10.4161/cam.4.2.10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sikder HA, Devlin MK, Dunlap S, Ryu B, Alani RM. Id proteins in cell growth and tumorigenesis. Cancer Cell. 2003;3:525–530. doi: 10.1016/s1535-6108(03)00141-7. [DOI] [PubMed] [Google Scholar]
- 25.Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci. 2000;113:3897–3905. doi: 10.1242/jcs.113.22.3897. [DOI] [PubMed] [Google Scholar]
- 26.Coppe JP, Smith AP, Desprez PY. Id proteins in epithelial cells. Exp Cell Res. 2003;285:131–145. doi: 10.1016/s0014-4827(03)00014-4. [DOI] [PubMed] [Google Scholar]
- 27.Sun XH, Copeland NG, Jenkins NA, Baltimore D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol Cell Biol. 1991;11:5603–5611. doi: 10.1128/mcb.11.11.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- 29.Kleeff J, Ishiwata T, Friess H, Buchler MW, Israel MA, Korc M. The helix-loop-helix protein Id2 is overexpressed in human pancreatic cancer. Cancer Res. 1998;58:3769–3772. [PubMed] [Google Scholar]
- 30.Rollin J, Blechet C, Regina S, Tenenhaus A, Guyetant S, Gidrol X. The intracellular localization of ID2 expression has a predictive value in non small cell lung cancer. PLoS One. 2009;4:e4158. doi: 10.1371/journal.pone.0004158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lasorella A, Iavarone A. The protein ENH is a cytoplasmic sequestration factor for Id2 in normal and tumor cells from the nervous system. Proc Natl Acad Sci U S A. 2006;103:4976–4981. doi: 10.1073/pnas.0600168103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kowanetz M, Valcourt U, Bergstrom R, Heldin CH, Moustakas A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol Cell Biol. 2004;24:4241–4254. doi: 10.1128/MCB.24.10.4241-4254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J, Hong SJ, Park JY, Park JH, Yu YS, Park SY, Lim EK, Choi KY, Lee EK, Paik SS, Lee KG, Wang HJ, Do IG, Joh JW, Kim DS. Epithelial-mesenchymal transition gene signature to predict clinical outcome of hepatocellular carcinoma. Cancer Sci. 2010;101:1521–1528. doi: 10.1111/j.1349-7006.2010.01536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondo M, Cubillo E, Tobiume K, Shirakihara T, Fukuda N, Suzuki H, Shimizu K, Takehara K, Cano A, Saitoh M, Miyazono K. A role for Id in the regulation of TGF-beta-induced epithelial-mesenchymal transdifferentiation. Cell Death Differ. 2004;11:1092–1101. doi: 10.1038/sj.cdd.4401467. [DOI] [PubMed] [Google Scholar]
- 35.Meng Y, Gu C, Wu Z, Zhao Y, Si Y, Fu X, Han W. Id2 promotes the invasive growth of MCF-7 and SKOV-3 cells by a novel mechanism independent of dimerization to basic helix-loop-helix factors. BMC Cancer. 2009;9:75. doi: 10.1186/1471-2407-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopez-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. 2009;1:303–314. doi: 10.1002/emmm.200900043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dudas J, Fullar A, Bitsche M, Schartinger V, Kovalszky I, Sprinzl GM, Riechelmann H. Tumor-produced, active interleukin-1beta regulates gene expression in carcinoma-associated fibroblasts. Exp Cell Res. 2011;317:2222–2229. doi: 10.1016/j.yexcr.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 40.Lasorella A, Benezra R, Iavarone A. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat Rev Cancer. 2014;14:77–91. doi: 10.1038/nrc3638. [DOI] [PubMed] [Google Scholar]
- 41.Chang C, Yang X, Pursell B, Mercurio AM. Id2 complexes with the SNAG domain of Snai1 inhibiting Snai1-mediated repression of integrin beta4. Mol Cell Biol. 2013;33:3795–3804. doi: 10.1128/MCB.00434-13. [DOI] [PMC free article] [PubMed] [Google Scholar]