

Abstract
Diabetes associated metabolic syndrome has been shown to be an independent risk factor for the development of hepatocellular carcinoma (HCC). Cirrhosis, in fact, was not always a prerequisite of HCC development and this might particularly apply to the metabolic abnormality associated HCC. This study was to investigate diabetes associated HCC and the potential role of FGF21 during carcinogenetic transformation of HCC. Dimethylnitrosamine (DEN) was used to induce HCC in the diabetic OVE26 mice. Pronounced damage characterized by steatohepatitis was found in the liver of diabetic mice. Steatohepatitis accompanied by constant cell proliferation and tumor cell growth were also found in the hepatic tissues of diabetic OVE26 mice when DEN being administrated. FGF21 protein level increased in liver tissues at an early stage along with steatohepatitis in diabetic OVE26 mice, but decreased in liver tissues later when HCC was developed. In addition, decreased FGF21 protein level was associated with cancerous hyper-proliferation and aberrant p53 and TGF-β/Smad signaling during HCC development. Loss of FGF21 may play an important role in HCC carcinogenetic transformation during metabolic liver injury in diabetic animals. The present finding calls attention to the need to control metabolic disorders associated with diabetes and may further develop a protective strategy against HCC.
Keywords: Hepatocellular carcinoma, diabetes, OVE26 mice, dimethylnitrosamine, FGF21
Introduction
Metabolic disorders have been recognized as major risk factors for the development of certain types of human malignancies including hepatocellular carcinoma (HCC) [1]. Diabetes associated metabolic syndrome has been shown to be an independent risk factor for the development of HCC. This has been substantiated in large population-based cohort studies in which 4.5 and 1.86 fold increased risks of HCC were shown in male and female diabetic patients, respectively [2,3]. More than 70% of diabetic individuals suffer from a form of non-alcoholic fatty liver disease (NAFLD), which is known to be a hepatic manifestation of metabolic syndrome [4]. The estimated prevalence of NAFLD is 3-10 times higher than the prevalence of the hepatitis C virus (HCV) [5,6]. Among patients with NAFLD followed for a mean of 8 years, the occurrence of cirrhosis was 20% and the incidence of HCC was 1% [7]. However, cirrhosis is not always a prerequisite of HCC development and epidemiological data indicate this may be a metabolic abnormality associated with HCC [8,9]. A number of molecular mechanisms that may accelerate the development of HCC have been linked to metabolic disorders: adipose-derived inflammation [10], insulin resistance [11], and altered insulin-like growth factor (IGF) axis component [12]. The metabolic mechanism of the hepatocarcinogenesis during NAFLD progression remains to be determined.
Recent results suggest that fibroblast growth factor 21 (FGF21) is highly expressed in hepatocytes under metabolic stress caused by starvation, hepatosteatosis, obesity and diabetes [13,14]. Human FGF21 is mainly expressed in liver [15] and adipose tissue [16]. However, FGF21 synthesized by adipose tissue is not secreted and functions in an autocrine manner, while liver-derived FGF21 is secreted in order to exert systemic actions [13]. The metabolic effects of FGF21 on glucose and lipid metabolism involve a dual regulation by PPAR-α in liver [17] and by PPAR-γ in adipocytes [18]. Hepatic FGF21 elicits metabolic benefits by targeting adipocytes of the peripheral adipose tissue through the transmembrane FGFR1- co-factor βKlotho complex [19,20]. FGF21 has been considered as a potential agent to control metabolic disorders such as hyperglycemia and hyperlipidemia. Furthermore, administration of recombinant FGF21 lowered plasma glucose, increased insulin sensitivity, and reversed hepatic steatosis and obesity in a range of diabetes/obesity animal models [21,22]. Although predominantly expressed in liver and adipose tissue [23], FGF21 is also expressed in other tissues such as skeletal muscle and cardiac tissue [24]. Xiao et al. has demonstrated that serum FGF21 levels are increased in type 2 diabetes but decreased in type 1 diabetes [25]. The pathogenic implications of such distinct FGF21 expression in various tissues and conditions remain to be elucidated. Early studies have shown that FGF21 suppressed cerulein-induced pancreatitis in mice [26] and demonstrated anti-apoptotic effects in both islet β cells and endothelial cells [27,28]. Recent studies indicate that FGF21 functions as a hepatokine, adipokine, and myokine in metabolism, injury protection, and diseases [29], and may delay cancer progression [30,31]. In has even been reported that overexpression of FGF21 delays initiation of chemically induced hepatocarcinogenesis [32].
A recent study suggests that haploinsufficiency of p53 significantly affects FGF21 expression [33]. The transcription factor, p53, plays important roles in liver cancer, liver diseases and metabolic regulation. Additionally, loss of p53 function is known to contribute to tumorigenesis. Therefore, expression of the FGF21 gene might be regulated through p53 under tumorigenic conditions. However, the function of FGF21 with regard to HCC carcinogenetic transformation under the condition of a metabolic syndrome such as diabetes is largely unknown. In this study, we proposed to investigate diabetes associated HCC and the potential role of FGF21 related to cellular and molecular events during the carcinogenic transformation of HCC. Dimethylnitrosamine (DEN) was used to induce HCC in the OVE26 mice, which generally developed severe hyperglycemia between weeks 2 and 3 after birth and continued to develop metabolic abnormalities [34]. The pathological transformation of steatohepatitis as well as HCC was evaluated. Any alterations in the molecular events such as p53 signaling and transforming growth factor-β (TGF-β)/Smad signaling were determined.
Materials and methods
Animals and treatment
The OVE26 mice with FVB background as a type 1 diabetic model to study the diabetes associated HCC were generously granted by Dr. Paul Epstein [34]. FVB mice, obtained from Jackson Laboratory (Bar Harbor, Maine), were used as controls. A commonly-used type 1 diabetic mouse model is induction by streptozotocin, which is a limited by the potential toxic side effects of streptozotocin. Transgenic OVE26 mice normally develop severe hyperglycemia before 3 weeks of age, develop obvious albuminuria by 3 months, and mice can survive until 18 months without insulin supplements [34]. Therefore, OVE26 type 1 diabetic transgenic mice are an ideal model for the cancer research. Both FVB and OVE26 mice were housed four per cage, given commercial chow and tap water, and maintained at 22°C on a 12-hour light/dark cycle. Sexually matured male and female mice were set as mating pairs for breeding. At 15 days of age, male mice, yielding the F1 generation, 25 mg/kg body weight DEN (Sigma-Aldrich, St. Louis, MO) by intraperitoneal injection (i.p.). The mice were also injected with 100 mg/kg body DEN at weeks 6 and 10 of age. The dosage of DEN in this study was modified according to previous report [35]. We used only male mice because estrogen-mediated inhibition of interlukin-6 production reduces liver cancer risk by DEN in females [35]. The animals were assigned randomly to two groups and sacrificed at week 16 and week 20. Serum plasma and hepatic tissues were harvested for further analysis. Animal procedures were approved by the Institutional Animal Care and Use Committee of University of Louisville, which is certified by the American Association for Accreditation of Laboratory Animal Care.
Histopathological examination of hepatic tumors
Gross liver anatomy observation was performed to count foci nodules and masses in DEN challenged livers. The numbers of macroscopic foci nodules ≥ 1 mm was recorded for each animal. Liver weight and femur length were measured to determine the liver/femur ratio. Hepatic tissues with foci nodules and masses were fixed in 10% neutral phosphate buffered formalin. Tissues were embedded in paraffin and sectioned to a thickness of 5 µm for pathological examination. Hematoxylin-and-eosin (H & E) stained sections were evaluated microscopically for quantification of foci, and to confirm adenomas and carcinomas.
Biochemical analysis
Serum plasma alanine aminotransferase (ALT) was measured using an ALT infinity enzymatic assay kit (Thermo Fisher Scientific Inc., Waltham, MA). Serum glucose assay was performed using a Sigma assay kit (Sigma-Aldrich). Serum insulin was detected using an ultra sensitive mouse insulin ELISA kit (Crystal chemical incorporation, IL).
Immunohistochemical analysis in hepatic tissue
Immunohistochemical staining was performed on the paraffin embedded tissue sections. Endogenous peroxidase was blocked with 3% hydrogen peroxide, and then with 5% animal serum for 30 min to block non-specific reaction. These tissue sections were incubated with primary antibodies, alpha 1 fetoprotein (AFP) (Abcam, San Francisco, CA) and Proliferating Cell Nuclear Antigen (PCNA, Invitrogen, Camarillo, CA). Tissue sections were incubated with horseradish peroxidase-conjugated secondary antibody (1:300-400 dilutions with PBS) for 2 hr in room temperature, and then incubated with the peroxidase substrate DAB kit (Vector Laboratories, Inc., Burlingame, CA). Counterstaining was performed using hematoxylin. Computer image-analysis was performed under microscope at 20x magnification.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
TUNEL staining was performed using an ApopTag Peroxidase In Situ Apoptosis Detection Kit (Chemicon, Billerica, CA). Briefly, each slide was deparaffinized, rehydrated, and treated with proteinase K (20 mg/L) for 15 min. The slide was incubated with terminal deoxynucleotidyl transferase (TdT) and digoxigenin-11-dUTP for 1 hr at 37°C. Anti-digoxigenin antibody conjugated with horseradish peroxidase (HRP) along with the substrate (DAB-H2O2) was used for visualization. Apoptotic cell death was quantitatively analyzed by counting the TUNEL positive cells in ten fields for each section at 20X magnification. The apoptotic index was presented as TUNEL positive cells per 100 cells.
Western blot assay
Protein levels for the biomarkers were semi-quantified by Western blot analysis as described previously [36]. Electrophoresis was performed on 12% SDS-PAGE gel and the proteins were transferred to nitrocellulose membranes. Membranes were incubated with primary antibodies overnight at 4°C and with secondary antibody for 1 hr at room temperature. The antigen-antibody complexes were then visualized using ECL (Amersham, Piscataway, NJ). Primary antibodies used include those raised against Bcl2, Bax, caspase-8, P-P53, T-P53, TGF-β1, and P-smad3 (Cell Signaling Technology, Danvers, MA).
Cell lines and in vitro study
A mouse liver cell line, FL83B (ATCC CRL-2390), and a mouse hepatoma cell line, Hepa 1-6 (ATCC CRL-1830), were used for the in vitro study. FL83B cells was cultured in F12K medium (ATCC) supplemented with 10% fetal bovine serum. Hepa 1-6 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum. To study FGF21 expression, the two cell lines were seeded at 1 × 106 cells/well in 6-well plates and cultured for 24 hr. Then the hyperglycemic group was treated with 50 mM glucose while the osmotic control group was treated with 45 mM mannitol + 5 mM at 0, 1, 6, 12, 24 and 48 hr.
Real-time RT-PCR (qPCR)
Total RNA was extracted using the TRIzol reagent (Invitrogen). First-strand complimentary DNA (cDNA) was synthesized from total RNA according to the manufacturer’s protocol for the RNA PCR kit (Promega, Madison, WI, USA). Quantitative PCR was carried out using the ABI 7300 real-time PCR system (Applied Biosystems, Carlsbad, CA). FGF21 expression was quantified and β-actin was used as an endogenous reference. Results were expressed as fold change in gene expression.
Statistical analysis
Collected data from repeated experiments were presented as mean ± SD. One-way ANOVA was used to determine if differences existed. If so, a post hoc Tukey’s test was used for analysis of any differences between groups (Origin 8 laboratory data analysis and graphing software).
Results
DEN induced liver injury and HCC in OVE26/ FVB mice
Blood glucose levels were significantly increased while serum insulin levels were significantly decreased in OVE26 mice compared to the FVB control. This indicated type 1 diabetes occurred in the OVE26 mice [34]. A total of 36 mice (17 of FVB, 19 of OVE26) were randomly assigned to either DEN treated groups or saline injected controls. The OVE26 mice showed significantly increased blood glucose levels and decreased serum insulin levels when compared to FVB controls. DEN treatment did not affect the levels of glucose and insulin the OVE26 mice (Figure 1).
Figure 1.
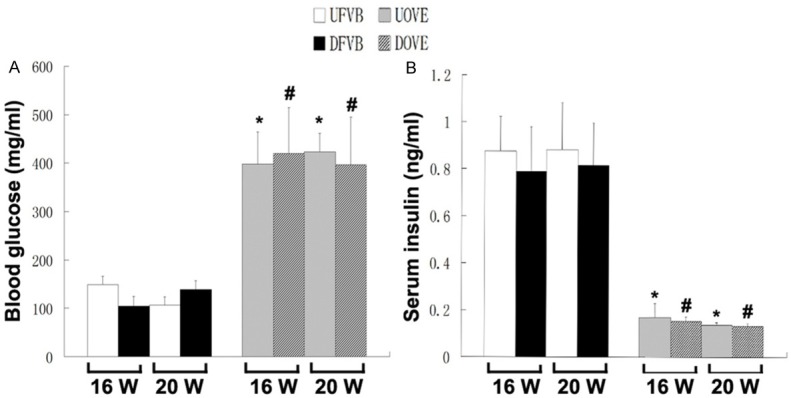
A. The blood glucose levels in the mice. B. The serum insulin levels in the mice. GLU: glucose; INS: insulin; UFVB: FVB untreated mice; DFVB: FVB DEN treated mice; UOVE: OVE untreated mice; DOVE: OVE DEN treated mice. *: vs UFVB, P < 0.05; #: vs DFVB, P < 0.05.
The survival rate in all groups was determined and shown in Figure 2A. A decreased trend of survival was observed in the OVE26 mice when treated with DEN (DEN-OVE26). The survival rate of FVB mice treated with DEN (DEN-FVB) also decreased, but much less so than the DEN-OVE26 mice. The reasons for animal death were most likely due to DEN related hepatic function failure at earlier stage and tumor related death at later stage. Surviving animals were sacrificed at 16 weeks and 20 week. The representative gross anatomy from one of the DEN-OVE26 mice showed HCC nodules in the liver (Figure 2B). All HCC foci were further confirmed by the histological examination of sections using H & E staining (Figure 2B). The numbers of HCC foci were counted in the H & E stained slides for all DEN treated groups. There were significantly greater numbers of HCC foci in the DEN-OVE26 mice compared to the DEN- FVB mice at both 16 weeks and 20 weeks. This implies that diabetes increases the susceptibility of liver cancer to DEN exposure (Figure 2C). Accordingly, hepatic AFP levels were significantly increased in all DEN treated mice compared to the saline treated controls at both 16 weeks and 20 weeks (Figure 2D). Serum ALT levels were also significantly increased in all DEN treated mice compared to the saline treated controls at both 16 weeks and 20 weeks (Figure 2E), indicating repetitive damage of the hepatocytes due to DEN treatment.
Figure 2.
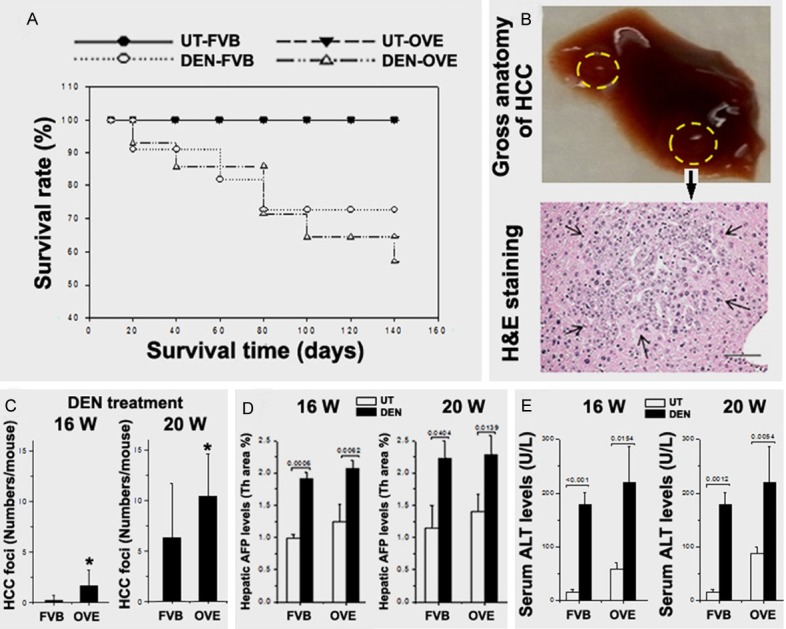
A. The animal survival rates in all 4 groups. B. Representative gross anatomy of HCC nodule on the liver lob and the histological confirmation for HCC. C. The numbers of HCC foci being counted in the tissue slides from DEN treated mice. D. The AFP levels in all 4 groups. E. The serum ALT levels in all 4 groups. *P < 0.05 vs DEN-FVB mice.
Metabolic abnormalities and FGF21 in OVE26 mice
Hepatic steatosis and steatohepatitis have been suggested as a predictor of liver tumorigenesis [37]. The current data provides further evidence that metabolic abnormalities are associated with development of HCC in the diabetic OVE26 mice. Fatty liver and steatohepatitis were detected in hepatic tissues adjacent to HCC foci, with the OVE26 mice aged at 20 weeks demonstrating the greatest degree of steatohepatitis (Figure 3A). Although HCC was detected in the DEN-FVB mice, few mice with steatohepatitis were observed (Figure 3A). Further analysis of metabolic syndrome factors indicated that triglycerides (TG) were significantly increased in OVE26 mice compared to FVB mice regardless of DEN treatment (Figure 3B). Although body weights were decreased, liver weights were significantly increased in OVE26 mice compared the FVB mice (Figure 3B). The increased hepatic TG levels and liver weights were also observed in the DEN-treated 20-week FVB mice and OVE26 mice (Figure 2B).
Figure 3.
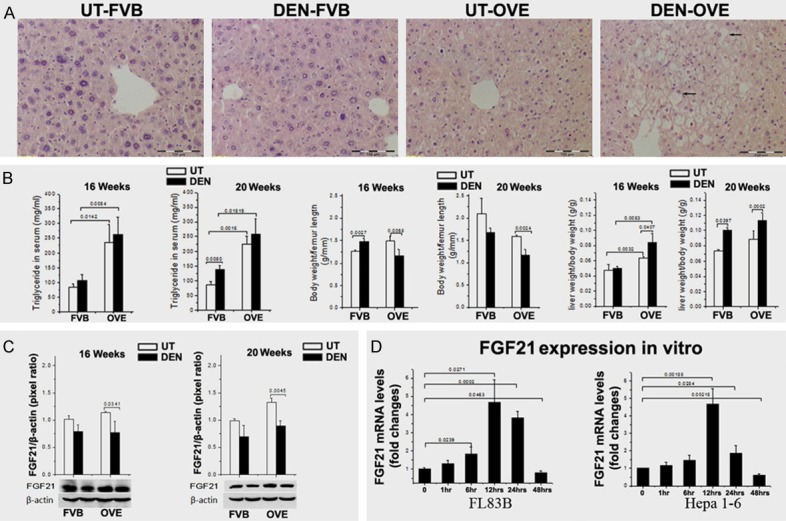
A. Steatohepatitis was detected in hepatic tissues from the DEN-OVE26 mice by H & E staining. The histology of steatohepatitis characterized by inflammation and hepatocellular ballooning. The arrows indicate Mallory- Denk bodies within ballooned hepatocytes. B. TG significantly increased in the OVE26 mice compared to FVB mice. The body weights were decreased in the OVE26 mice compared to FVB mice. Liver weights were significantly increased in the OVE26 mice compared to FVB mice. C. FGF21 protein levels were significantly decreased in the diabetic OVE26 mice treated with DEN, compared to the untreated OVE26 mice. D. High glucose induced FGF21 mRNA expression in both hepatocytes and hepatoma cells in vitro. However, FGF21 expression of hepatoma cells decreased after 12 hours and went back to the baseline.
These metabolic abnormalities concurrent with HCC in the DEN-OVE26 mice led to the hypothesis that FGF21 may be involved in the pathogenesis because of its known metabolic action in diabetes and cancer [37]. Therefore, FGF21 protein levels were evaluated in the hepatic samples. Results showed that FGF21 was increased in untreated, diabetic OVE26 mice and in the 20-week old mice in particular (P < 0.05) compared to FVB mice. Surprisingly, FGF-21 protein levels were significantly decreased in DEN-OVE26 mice compared to the untreated OVE26 controls (Figure 3C). Although FGF21 protein levels were also decreased in the FVB mice with DEN treatment, there was no statistical significance when compared to the untreated FVB controls (Figure 3C). A previous study indicated that FGF21 could be induced under diabetes and DEN treatment [33]. While our results did show an increase in diabetic mice, FGF21 levels decreased in the DEN treated diabetic mice.
To clarify whether the decreased FGF21 level could be associated with carcinogenetic transformation, an in vitro study was performed using a mouse liver cell line, FL83B, and a mouse hepatoma cell line, Hepa 1-6. Results showed that high glucose caused an increase in FGF21 mRNA expression in both hepatocytes and hepatoma cells which peaked at 12 hr post treatment. However, this mRNA decreased significantly after only 12 hr in the hepatoma cells when compared to hepatocytes (Figure 3D). This result implies that the carcinogenetic transformed cells may contribute to the loss of FGF21 in the DEN treated diabetic mice.
Proliferation and apoptosis in the OVE26 mice
The observation of decreased FGF21 levels in DEN treated, diabetic mice hinted that some metabolic events might be involve in the carcinogenetic process. To discover if FGF21 levels could affect the growth pattern during carcinogenetic transformation, cellular events were further evaluated. There were significant increases of proliferation rates in mice treated with DEN compared to the saline controls. The proliferation rate of DEN-OVE26 mice significantly increased at 20 weeks compared to the DEN-FVB mice (Figure 4A).
Figure 4.
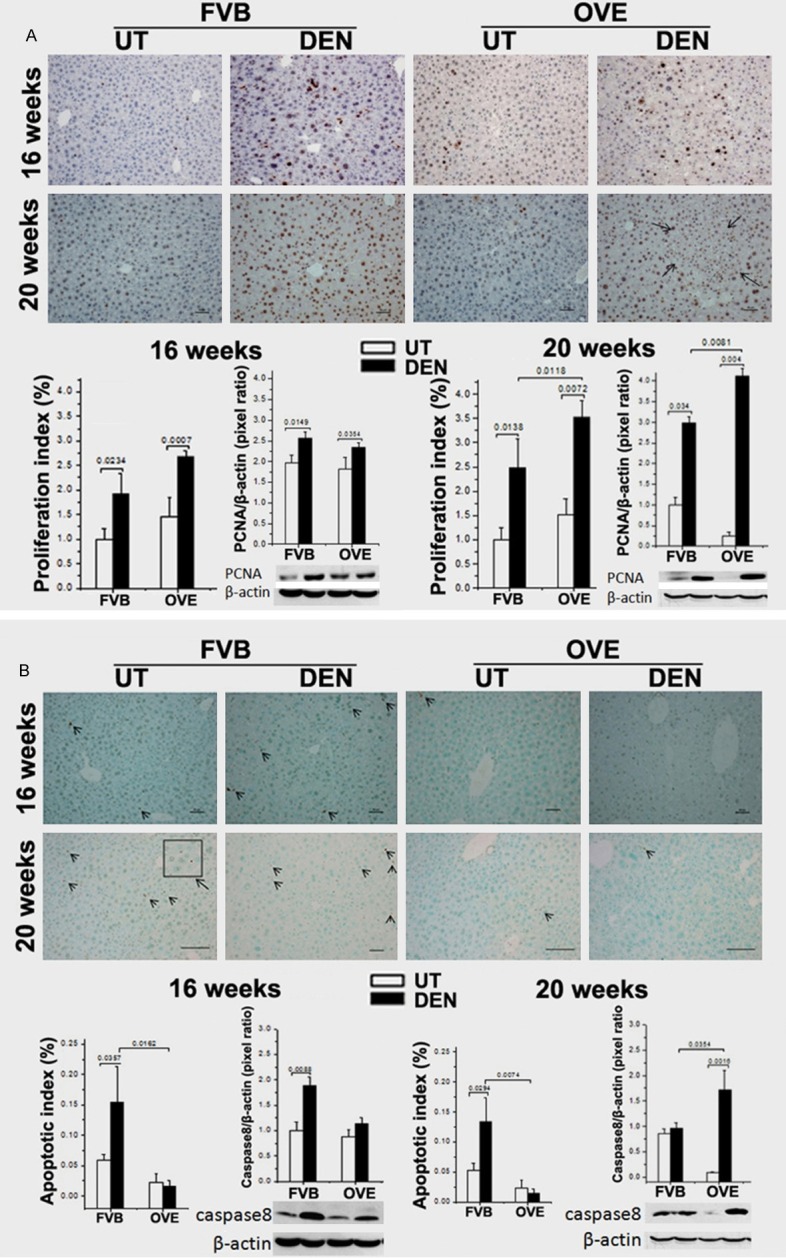
A. Upper, representative images of proliferation by PCNA staining in all 4 groups. Lower, the proliferation index by image-analysis of the positive PCNA cells and the PCNA protein levels in liver tissue by Western blot analysis. Arrow: HCC foci. B. Upper, representative images of apoptosis by TUNEL staining in all 4 groups. Lower, the apoptotic index by image-analysis of the positive apoptosis cells. The protein levels of caspase-8, in liver tissues by Western blot analysis. Arrow head: positive TUNEL staining cells.
Increased apoptotic rates were observed in DEN-FVB mice but not the DEN-OVE26 mice at both 16 and 20 weeks of age (Figure 4B). Consistent with apoptotic cell death, the cleavage level of caspase-8, a central executor of apoptosis in the Fas ligand/receptor pathway, was significantly increased in DEN-FVB mice. This increase was not observed in DEN-OVE26 mice 16-week old group nor in 20-week group (Figure 4B).
The signaling of p53 and TGF-β/Smad in the diabetes-HCC mice
The signaling of p53 and TGF-β/Smad is known to be a critical molecular event involved in HCC transformation. However, the interplay between these signaling events and FGF21 in diabetes-HCC was largely unknown. Consequently, the components in p53 and TGF-β/Smad signaling were further evaluated in the diabetic OVE26 mice as well as the FVB mice. The total and phosphorylated p53 protein levels T-p53 and P-p53, respectively) were determined by Western blot. Analysis of the ratio of P-p53/T-p53 showed that the levels of phosphorylated p53 were significantly down-regulated in both DEN-FVB and DEN-OVE26 mice at 16 weeks. Furthermore, this level was lowest in the DEN-OVE26 mice at 20 weeks of age (Figure 5A).
Figure 5.
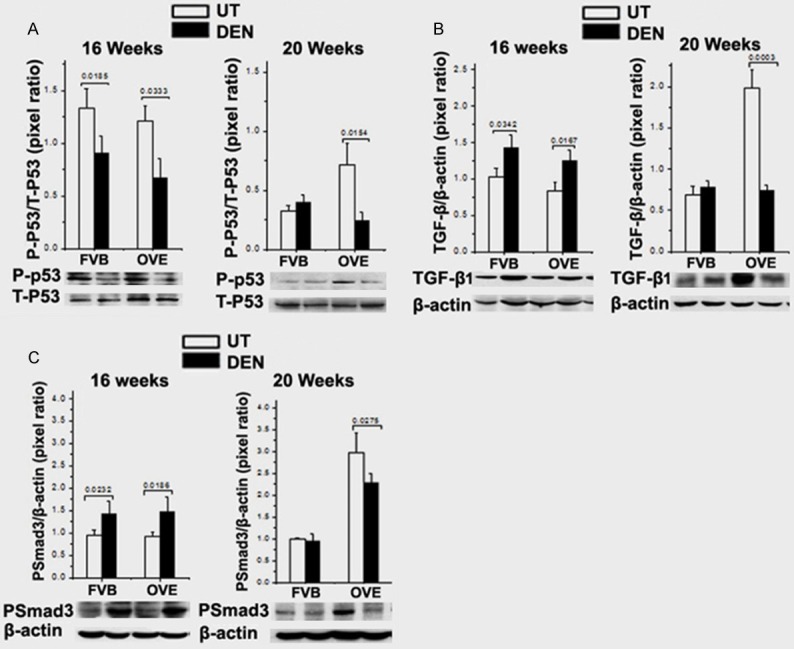
A. The P-P53 and T-P53 protein levels in the liver tissues by Western blot analysis. B. The TGF-β1 protein levels in liver tissues by Western blot analysis. C. The PSamd3 protein levels in liver tissue by Western blot analysis.
TGF-β1 levels were significantly up-regulated in the DEN-OVE26 and DEN-FVB mice at 16 weeks of age compared to the untreated mice. Interestingly, TGF-β1 was also significantly increased in the untreated OVE26 mice at 20 weeks but significantly decreased in the DEN-OVE26 mice compared to the untreated OVE26 mice (Figure 5B). Phosphorylated-Smad3 (P-Smad3) showed a similar trend of being significantly increased in the DEN-FVB and DEN-OVE26 mice at 16 weeks and did not change in FVB mice with or without DEN treatment at 20 weeks. P-Smad3 was significantly increased in OVE26 mice with a lesser increase in DEN-treated OVE26 mice (Figure 5C).
Discussion
Although cirrhosis is recognized as fertile ground for HCC with erratic tissue remodeling accompanying recurrent cycles of hepatocellular destruction and compensatory regeneration [39], evidence from epidemiologic investigations and clinical studies supported the hypothesis that metabolic diseases, such as diabetes and obesity, play an important role in HCC carcinogenesis [40]. However, the relevant experiments in animals were largely missing. In this study, we investigated, for the first time, the pathogenesis of metabolic liver injury in the OVE26 diabetic mouse model as well as diabetes-HCC in OVE26 mice treated with DEN. Results show pronounced liver damages characterized by steatohepatitis were found in the OVE26 diabetic mice. Steatohepatitis accompanied by constant cell proliferation and tumor cell growth were found in the hepatic tissues of OVE26 diabetic mice under DEN administration. Interestingly, increased FGF21 protein levels were found in the liver tissues with steatohepatitis in diabetic OVE26 mice. These levels decreased when HCC was induced in OVE26 mice. Additionally, decreased FGF21 protein levels associated with cancerous hyper-proliferation as well as aberrant p53 and TGF-β/Smad signaling during HCC development.
The FGF pathway promotes tumor growth and angiogenesis in many solid tumors including HCC [41]. Although many of the mechanisms discussed so far are the result of genetic dysregulation of the FGF/FGFR signaling axis, ligand-dependent signaling through either the hormone-like FGFs (FGF19, 21, and 23) or the canonical FGFs (FGF1-10, 16-18, and 20) is also likely to play a key role in carcinogenesis. For example, the relationship of FGF19 with cancer has been addressed [42]. Specifically FGF19 expression was found to correlate with tumor progression, recurrence, and poor prognosis [43,44]. In the experimental studies, HCC has been found to develop in transgenic mice overexpressing the hormonal FGF19 [45]. Contrary to FGF19, FGF21, as a recently-discovered liver safeguard, plays an important role in preventing liver damage caused by various pathogenic risks [46,47]. Liver FGF21 mRNA expression was significantly increased in the livers of mice at the early stage after DEN treatment [33] and also in the liver under several challenges; such as neoplastic and regenerating conditions after partial hepatectomy and CCl4 administration [32]. Here we found that FGF21 protein expression in OVE26 mice presenting diabetes-steatohepatitis is significantly increased at 16 weeks but at a low level when presenting diabetes-HCC. This may imply that the early increased expression of FGF21 is a protective or compensative mechanism and the late decreased expression of FGF21 may relate to chronic liver pathogenesis and including hepatocarcinogenesis. Our notion is consistent with a previous report, in which forced expression of FGF21 could delay DEN-initiated hepatocarcinogenesis [32].
There are several possible explanations for the loss of FGF21. First, FGF21 gene expression has been shown to be inversely related to hepatic triglyceride (TG) concentration [48]. Similar results were observed in our study, where the TG concentration reached its highest level in liver presenting diabetes-HCC while the hepatic FGF21 protein level was decreased. Second, there could be a dynamic, epigenetic modification during carcinogenetic transformation in order to control a robust increase of FGF21 followed by the promoter of FGF21 being turned off. This is supported by a recent study, in which epigenetic function of G9a, a histone methyltransferase, has been associated with the suppression of FGF21 even though the mechanism is unclear [49]. And third, physical hypoxia (1% oxygen) and chemical hypoxia inducers (such as cobalt chloride treatment) can decrease both FGF21 mRNA and secreted protein levels [50]. As we know, most solid tumors (including HCC) are in hypoxic microenvironment where the low levels of oxygen may contribute to the loss of FGF21 in the liver during HCC development. Nevertheless, the accurate mechanism regarding the loss of FGF21 in diabetes associated HCC is unclear and it will be of great interest to determine the suppression mechanism(s) of FGF21 in future studies.
The tumor suppressor p53 is a stress-responsive transcription factor that has been studied extensively. With regard to its metabolic role, it has been found that the protein abundance of p53 in the liver is elevated in rodents with fatty liver disease [51]. TGF-β/Smads is a classic pathway in NAFLD. Development of NAFLD activated canonical TGF-β signaling including TGF-β1 and PSmad3 in the TGF-β/Smads pathway [52]. Studies have discovered the active role of TGF-β/Smads signaling in both tumor-suppression and oncogenesis [53]. Hsu reported early on the association of the allotype of p53 gene in HCC with HBsAg and HBeAg seropositivities and diabetes, with the finding that loss of heterozygosity of p53 gene is a common event in HCC and correlates with diabetes [54]. A more recent study has revealed the importance of p53 in metabolic action beyond its canonical functions in processes such as cell-cycle arrest, apoptosis, and senescence [55]. However, the effect of diabetes on liver p53 expression with and without the administration of DEN has never been investigated. As reported by Yang [33], p53 negatively regulates FGF21 expression. They speculate that p53 may account for an additional or combinatory mechanism for regulating FGF21 expression in association with carcinogenesis beyond metabolic alterations. However, our data show the elevated p53 protein and FGF21 levels in the diabetes associated fatty liver, especially steatohepatitis, but decreased p53 protein and FGF21 levels in the DEN treated OVE26 mice with HCC. Likewise, TGF-β1 and PSmad3 are activated in diabetic OVE26 mice with steatohepatitis, but not in the diabetic OVE26 mice with HCC. Interestingly, all these factors, the metabolic regulator-FGF21, tumor suppressor-p53, tumor suppressor/initiator-TGF-β1 and PSmad3 show a similar expression pattern: simultaneously up-regulated in the untreated controls (benign tissues) and down-regulated in late stage DEN treated mice (malignant tissues). In this regard, there are two important issues remaining to be determined in future studies: 1) clarification of signaling cross talk between p53 and FGF21 regarding carcinogenetic transformation in the diabetes-HCC mice; 2) elucidation of any involvement of FGF21 with activin mediation of Smad3. As we know, activin phosphorylates Smads 2/3 while BMPs phosphorylate Smads 1/5/8 in the TGF-β signaling. It has been reported that FGF21 can enhance BMP-2-dependent transcription [56], however it is largely unknown about the interaction between FGF21 and activin. Therefore, it is valuable to know if FGF21 can interact with activin to mediate the Smad3 phosphorylation. The shifting of C-terminally phosphorylated Smad3 (pSmad3C) toward phosphorylation of the Smad3 linker region (pSmad3L) can promote the oncogenic activities which are necessary for HCC initiation [53].
In conclusion, loss of FGF21 may play an important role in HCC carcinogenetic transformation during metabolic liver injury in diabetic animals. The present findings call attention to preventing metabolic disorders in diabetes and may further develop a protective strategy against HCC.
Acknowledgements
This work was supported in part by American Diabetes Association Basic Science Award (1-13-BS-109) and Natural Science Foundation of Guizhou Educational Department (No. 2011037).
Disclosure of conflict of interest
None.
References
- 1.El Serag HB, Hampel H, Javadi F. The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol. 2006;4:369–80. doi: 10.1016/j.cgh.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Adami HO, Chow WH, Nyren O, Berne C, Linet MS, Ekbom A, Wolk A, McLaughlin JK, Fraumeni JF Jr. Excess risk of primary liver cancer in patients with diabetes mellitus. J Natl Cancer Inst. 1996;88:1472–7. doi: 10.1093/jnci/88.20.1472. [DOI] [PubMed] [Google Scholar]
- 3.Lagiou P, Kuper H, Stuver SO, Tzonou A, Trichopoulos D, Adami HO. Role of diabetes mellitus in the etiology of hepatocellular carcinoma. J Natl Cancer Inst. 2000;92:1096–9. doi: 10.1093/jnci/92.13.1096. [DOI] [PubMed] [Google Scholar]
- 4.Byrne CD, Olufadi R, Bruce KD, Cagampang FR, Ahmed MH. Metabolic disturbances in non-alcoholic fatty liver disease. Clin Sci (Lond) 2009;116:539–64. doi: 10.1042/CS20080253. [DOI] [PubMed] [Google Scholar]
- 5.Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol. 2003;98:960–7. doi: 10.1111/j.1572-0241.2003.07486.x. [DOI] [PubMed] [Google Scholar]
- 6.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–95. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 7.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–9. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 8.Torres DM, Harrison SA. Nonalcoholic steatohepatitis and noncirrhotic hepatocellular carcinoma: fertile soil. Semin Liver Dis. 2012;32:30–8. doi: 10.1055/s-0032-1306424. [DOI] [PubMed] [Google Scholar]
- 9.Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56:1384–91. doi: 10.1016/j.jhep.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 10.Monnier L, Mas E, Ginet C, Michel F, Villon L, Cristol JP, Colette C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA. 2006;295:1681–7. doi: 10.1001/jama.295.14.1681. [DOI] [PubMed] [Google Scholar]
- 11.Tsugane S, Inoue M. Insulin resistance and cancer: epidemiological evidence. Cancer Sci. 2010;101:1073–9. doi: 10.1111/j.1349-7006.2010.01521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scharf JG, Ramadori G, Dombrowski F. Analysis of the IGF axis in preneoplastic hepatic foci and hepatocellular neoplasms developing after low-number pancreatic islet transplantation into the livers of streptozotocin diabetic rats. Lab Invest. 2000;80:1399–411. doi: 10.1038/labinvest.3780147. [DOI] [PubMed] [Google Scholar]
- 13.Cuevas-Ramos D, Aguilar-Salinas CA, Gomez-Perez FJ. Metabolic actions of fibroblast growth factor 21. Curr Opin Pediatr. 2012;24:523–9. doi: 10.1097/MOP.0b013e3283557d22. [DOI] [PubMed] [Google Scholar]
- 14.Luo Y, McKeehan WL. Stressed Liver and Muscle Call on Adipocytes with FGF21. Front Endocrinol (Lausanne) 2013;4:194. doi: 10.3389/fendo.2013.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta. 2000;1492:203–6. doi: 10.1016/s0167-4781(00)00067-1. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Yeung DC, Karpisek M, Stejskal D, Zhou ZG, Liu F, Wong RL, Chow WS, Tso AW, Lam KS, Xu A. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes. 2008;57:1246–53. doi: 10.2337/db07-1476. [DOI] [PubMed] [Google Scholar]
- 17.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, Elmquist JK, Gerard RD, Burgess SC, Hammer RE, Mangelsdorf DJ, Kliewer SA. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–25. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Muise ES, Azzolina B, Kuo DW, El Sherbeini M, Tan Y, Yuan X, Mu J, Thompson JR, Berger JP, Wong KK. Adipose fibroblast growth factor 21 is up-regulated by peroxisome proliferator-activated receptor gamma and altered metabolic states. Mol Pharmacol. 2008;74:403–12. doi: 10.1124/mol.108.044826. [DOI] [PubMed] [Google Scholar]
- 19.Adams AC, Yang C, Coskun T, Cheng CC, Gimeno RE, Luo Y, Kharitonenkov A. The breadth of FGF21’s metabolic actions are governed by FGFR1 in adipose tissue. Mol Metab. 2012;2:31–7. doi: 10.1016/j.molmet.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen W, Hoo RL, Konishi M, Itoh N, Lee PC, Ye HY, Lam KS, Xu A. Growth hormone induces hepatic production of fibroblast growth factor 21 through a mechanism dependent on lipolysis in adipocytes. J Biol Chem. 2011;286:34559–66. doi: 10.1074/jbc.M111.285965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, Vonderfecht S, Hecht R, Li YS, Lindberg RA, Chen JL, Jung DY, Zhang Z, Ko HJ, Kim JK, Véniant MM. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–9. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen Y, Moller DE, Kharitonenkov A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–27. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 23.Mraz M, Bartlova M, Lacinova Z, Michalsky D, Kasalicky M, Haluzikova D, Matoulek M, Dostalova I, Humenanska V, Haluzik M. Serum concentrations and tissue expression of a novel endocrine regulator fibroblast growth factor-21 in patients with type 2 diabetes and obesity. Clin Endocrinol (Oxf) 2009;71:369–75. doi: 10.1111/j.1365-2265.2008.03502.x. [DOI] [PubMed] [Google Scholar]
- 24.Fon Tacer K, Bookout AL, Ding X, Kurosu H, John GB, Wang L, Goetz R, Mohammadi M, Kuro-o M, Mangelsdorf DJ, Kliewer SA. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol. 2010;24:2050–64. doi: 10.1210/me.2010-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao Y, Xu A, Law LS, Chen C, Li H, Li X, Yang L, Liu S, Zhou Z, Lam KS. Distinct changes in serum fibroblast growth factor 21 levels in different subtypes of diabetes. J Clin Endocrinol Metab. 2012;97:E54–8. doi: 10.1210/jc.2011-1930. [DOI] [PubMed] [Google Scholar]
- 26.Johnson CL, Weston JY, Chadi SA, Fazio EN, Huff MW, Kharitonenkov A, Köester A, Pin CL. Fibroblast growth factor 21 reduces the severity of cerulein-induced pancreatitis in mice. Gastroenterology. 2009;137:1795–804. doi: 10.1053/j.gastro.2009.07.064. [DOI] [PubMed] [Google Scholar]
- 27.Wente W, Efanov AM, Brenner M, Kharitonenkov A, Köster A, Sandusky GE, Sewing S, Treinies I, Zitzer H, Gromada J. Fibroblast growth factor-21 improves pancreatic beta-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Diabetes. 2006;55:2470–78. doi: 10.2337/db05-1435. [DOI] [PubMed] [Google Scholar]
- 28.Lu Y, Liu JH, Zhang LK, DU J, Zeng XJ, Hao G, Huang J, Zhao DH, Wang GZ, Zhang YC. Fibroblast growth factor 21 as a possible endogenous factor inhibits apoptosis in cardiac endothelial cells. Chin Med J (Engl) 2010;123:3417–21. [PubMed] [Google Scholar]
- 29.Itoh N. FGF21 as a Hepatokine, Adipokine, and Myokine in Metabolism and Diseases. Front Endocrinol (Lausanne) 2014;5:107. doi: 10.3389/fendo.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarty MF. GCN2 and FGF21 are likely mediators of the protection from cancer, autoimmunity, obesity, and diabetes afforded by vegan diets. Med Hypotheses. 2014;83:365–71. doi: 10.1016/j.mehy.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 31.Luo Y, Yang C, Ye M, Jin C, Abbruzzese JL, Lee MH, Yeung SC, McKeehan WL. Deficiency of metabolic regulator FGFR4 delays breast cancer progression through systemic and microenvironmental metabolic alterations. Cancer Metab. 2013;1:21. doi: 10.1186/2049-3002-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang X, Yu C, Jin C, Yang C, Xie R, Cao D, Wang F, McKeehan WL. Forced expression of hepatocyte-specific fibroblast growth factor 21 delays initiation of chemically induced hepatocarcinogenesis. Mol Carcinog. 2006;45:934–42. doi: 10.1002/mc.20241. [DOI] [PubMed] [Google Scholar]
- 33.Yang C, Lu W, Lin T, You P, Ye M, Huang Y, Jiang X, Wang C, Wang F, Lee MH, Yeung SC, Johnson RL, Wei C, Tsai RY, Frazier ML, McKeehan WL, Luo Y. Activation of Liver FGF21 in hepatocarcinogenesis and during hepatic stress. BMC Gastroenterol. 2013;13:67. doi: 10.1186/1471-230X-13-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Epstein PN, Overbeek PA, Means AR. Calmodulin-induced early-onset diabetes in transgenic mice. Cell. 1989;58:1067–73. doi: 10.1016/0092-8674(89)90505-9. [DOI] [PubMed] [Google Scholar]
- 35.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–4. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 36.Jiang X, Zhang C, Xin Y, Huang Z, Tan Y, Huang Y, Wang Y, Feng W, Li X, Li W, Qu Y, Cai L. Protective effect of FGF21 on type 1 diabetes-induced testicular apoptotic cell death probably via both mitochondrial- and endoplasmic reticulum stress-dependent pathways in the mouse model. Toxicol Lett. 2013;219:65–76. doi: 10.1016/j.toxlet.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura A, Tajima K, Zolzaya K, Sato K, Inoue R, Yoneda M, Fujita K, Nozaki Y, Kubota KC, Haga H, Kubota N, Nagashima Y, Nakajima A, Maeda S, Kadowaki T, Terauchi Y. Protection from non-alcoholic steatohepatitis and liver tumourigenesis in high fat-fed insulin receptor substrate-1-knockout mice despite insulin resistance. Diabetologia. 2012;55:3382–91. doi: 10.1007/s00125-012-2703-1. [DOI] [PubMed] [Google Scholar]
- 38.Cuevas-Ramos D, Aguilar-Salinas CA, Gomez-Perez FJ. Metabolic actions of fibroblast growth factor 21. Curr Opin Pediatr. 2012;24:523–9. doi: 10.1097/MOP.0b013e3283557d22. [DOI] [PubMed] [Google Scholar]
- 39.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 40.Tolman KG, Fonseca V, Dalpiaz A, Tan MH. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care. 2007;30:734–43. doi: 10.2337/dc06-1539. [DOI] [PubMed] [Google Scholar]
- 41.Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18:1855–62. doi: 10.1158/1078-0432.CCR-11-0699. [DOI] [PubMed] [Google Scholar]
- 42.Lin BC, Desnoyers LR. FGF19 and cancer. Adv Exp Med Biol. 2012;728:183–94. doi: 10.1007/978-1-4614-0887-1_12. [DOI] [PubMed] [Google Scholar]
- 43.Miura S, Mitsuhashi N, Shimizu H, Kimura F, Yoshidome H, Otsuka M, Kato A, Shida T, Okamura D, Miyazaki M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer. 2012;12:56–71. doi: 10.1186/1471-2407-12-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hyeon J, Ahn S, Lee JJ, Song DH, Park CK. Expression of fibroblast growth factor 19 is associated with recurrence and poor prognosis of hepatocellular carcinoma. Dig Dis Sci. 2013;58:1916–22. doi: 10.1007/s10620-013-2609-x. [DOI] [PubMed] [Google Scholar]
- 45.Nicholes K, Guillet S, Tomlinson E, Hillan K, Wright B, Frantz GD, Pham TA, Dillard-Telm L, Tsai SP, Stephan JP, Stinson J, Stewart T, French DM. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol. 2002;160:2295–307. doi: 10.1016/S0002-9440(10)61177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cariello M, Moschetta A. Fibroblast growth factor 21: A new liver safeguard. Hepatology. 2014;60:792–4. doi: 10.1002/hep.27147. [DOI] [PubMed] [Google Scholar]
- 47.Ye D, Wang Y, Li H, Jia W, Man K, Lo CM, Wang Y, Lam KS, Xu A. FGF21 protects against acetaminophen-induced hepatotoxicity by Potentiating PGC-1α-mediated antioxidant capacity in mice. Hepatology. 2014;60:977–89. doi: 10.1002/hep.27060. [DOI] [PubMed] [Google Scholar]
- 48.Uebanso T, Taketani Y, Fukaya M, Sato K, Takei Y, Sato T, Sawada N, Amo K, Harada N, Arai H, Yamamoto H, Takeda E. Hypocaloric high-protein diet improves fatty liver and hypertriglyceridemia in sucrose-fed obese rats via two pathways. Am J Physiol Endocrinol Metab. 2009;297:E76–84. doi: 10.1152/ajpendo.00014.2009. [DOI] [PubMed] [Google Scholar]
- 49.Tong X, Zhang D, Buelow K, Guha A, Arthurs B, Brady HJ, Yin L. Recruitment of histone methyltransferase G9a mediates transcriptional repression of Fgf21 gene by E4BP4 protein. J Biol Chem. 2013;288:5417–25. doi: 10.1074/jbc.M112.433482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Wang C, Wang Y, Ma Z, Xiao J, McClain C, Li X, Feng W. Cobalt chloride decreases fibroblast growth factor-21 expression dependent on oxidative stress but not hypoxia-inducible factor in Caco-2 cells. Toxicol Appl Pharmacol. 2012;264:212–21. doi: 10.1016/j.taap.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yahagi N, Shimano H, Matsuzaka T, Sekiya M, Najima Y, Okazaki S, Okazaki H, Tamura Y, Iizuka Y, Inoue N, Nakagawa Y, Takeuchi Y, Ohashi K, Harada K, Gotoda T, Nagai R, Kadowaki T, Ishibashi S, Osuga J, Yamada N. p53 involvement in the pathogenesis of fatty liver disease. J Biol Chem. 2004;279:20571–5. doi: 10.1074/jbc.M400884200. [DOI] [PubMed] [Google Scholar]
- 52.Xiao J, Ho CT, Liong EC, Nanji AA, Leung TM, Lau TY, Fung ML, Tipoe GL. Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3 K/Akt/FoxO1, and NF-kappa B pathways. Eur J Nutr. 2014;53:187–99. doi: 10.1007/s00394-013-0516-8. [DOI] [PubMed] [Google Scholar]
- 53.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 54.Hsu HC, Peng SY, Lai PL, Sheu JC, Chen DS, Lin LI, Slagle BL, Butel JS. Allelotype and loss of heterozygosity of p53 in primary and recurrent hepatocellular carcinomas. A study of 150 patients. Cancer. 1994;73:42–7. doi: 10.1002/1097-0142(19940101)73:1<42::aid-cncr2820730109>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 55.Zhang P, Tu B, Wang H, Cao Z, Tang M, Zhang C, Gu B, Li Z, Wang L, Yang Y, Zhao Y, Wang H, Luo J, Deng CX, Gao B, Roeder RG, Zhu WG. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc Natl Acad Sci U S A. 2014;111:10684–9. doi: 10.1073/pnas.1411026111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ishida K, Haudenschild DR. Interactions between FGF21 and BMP-2 in osteogenesis. Biochem Biophys Res Commun. 2013;432:677–82. doi: 10.1016/j.bbrc.2013.02.019. [DOI] [PubMed] [Google Scholar]