

Abstract
AF4/AFF1 and AF5/AFF4 are the molecular backbone to assemble “super-elongation complexes” (SECs) that have two main functions: (1) control of transcriptional elongation by recruiting the positive transcription elongation factor b (P-TEFb = CyclinT1/CDK9) that is usually stored in inhibitory 7SK RNPs; (2) binding of different histone methyltransferases, like DOT1L, NSD1 and CARM1. This way, transcribed genes obtain specific histone signatures (e.g. H3K79me2/3, H3K36me2) to generate a transcriptional memory system. Here we addressed several questions: how is P-TEFb recruited into SEC, how is the AF4 interactome composed, and what is the function of the naturally occuring AF4N protein variant which exhibits only the first 360 amino acids of the AF4 full-length protein. Noteworthy, shorter protein variants are a specific feature of all AFF protein family members. Here, we demonstrate that full-length AF4 and AF4N are both catalyzing the transition of P-TEFb from 7SK RNP to their N-terminal domain. We have also mapped the protein-protein interaction network within both complexes. In addition, we have first evidence that the AF4N protein also recruits TFIIH and the tumor suppressor MEN1. This indicate that AF4N may have additional functions in transcriptional initiation and in MEN1-dependend transcriptional processes.
Keywords: AF4/AF4N, P-TEFb, elongation control, RNA polymerase II, 7SK RNP
Introduction
Gene transcription is a process that converts DNA-stored information into RNA molecules of which mRNA is the only species that is subsequently translated into protein. The enzymatic machinery transcribing protein-coding genes into mRNA is RNA polymerase II (RNAPII), which has been shown to be ‘elongation controlled’ for ~80-90% of all human genes [1]. The process of ‘transcriptional elongation’ is required because RNA synthesis of RNAPII becomes arrested after the production of the first ~50 nucleotides [2,3]. This is due to DSIF binding (DRB sensitive inducible factor) to the nascent mRNA emerging from the RNAPII, and binding of the NELF complex (negative elongation factor) in a DSIF-dependent manner [4]. Both are inhibitory proteins/protein complexes that block further transcription, but allow mRNA capping at this arrested state of RNAPII (POL A). The key regulator - capable to terminate this ‘promoter-proximal arrested state’ and allowing the transition into ‘elongating RNAPII’ (POL E) - is the positive transcription elongation factor b (P-TEFb). P-TEFb is a heterodimer composed of CDK9 and Cyclin T1 [5] which phosphorylates the RNAP II-associated negative elongation factors DSIF and NELF. This causes the proteasomal degradation of the NELF complex, but converts DSIF into an activator of transcriptional elongation [6]. Apart from these functions, P-TEFb also phosphorylates UBE2A that associates subsequently with two Ring finger proteins (RNF20 and RNF40) which in turn allows to execute histone mono-ubiquitinylation [7]. Finally, P-TEFb phosphorylates the C-terminal domain of RNAP II (CTD: 52 copies of YSPTSPS). The CTD of RNAPII is a target of multiple kinases [8] and displays distinct post-translational modifications at Ser-2, Thr-4, Ser-5 and Ser-7 residues [9,10]. These patterns are changing, starting from initiating RNAPII (P-Ser-5) [11], promoter-proximal arrested RNAPII (P-Ser-5; P-Ser-7 at nt +50) and elongating RNAP II (P-Ser-2; P-Thr-4 around nt +450). Each of these changes is associated with the assembly of additional proteins necessary for elongation (e.g. ELL), splicing or termination [12]. While TFIIH (Cyclin H and CDK7) is responsible for the Ser-5 phosphorylation step - necessary for transcriptional initiation - the P-TEFb kinase is responsible for the establishment of the P-Ser-2/P-Thr-4 signature [13,14], and thus, creates the environment for productive elongation of RNAPII.
Inactive P-TEFb is stored in 7SK RNPs [15,16]and displays a diffuse nuclear pattern [17]. Inside of the 7SK RNPs, P-TEFb interacts in a reversible manner with HEXIM1 (hexamethylene bis-acetamide inducible 1) and with the 332 nt-long 7SK RNA [15,16,18,19]. The 7SK RNA contains two distinct hairpin structures, bound by HEXIM1 and Cyclin T1, which are essential for the inhibitory effect of HEXIM1 towards P-TEFb [20,21]. Additional proteins in the 7SK RNP are MePCE (methylphosphate capping enzyme), LARP7 (La ribonucleoprotein domain family, member 7), several hnRNPs and RNA-Helicase A [22-24]. Since most P-TEFb is stored in the above mentioned complex, only a fraction of P-TEFb is functionally active due to its transient association with AF4- [25] or AF5-based ‘super elongation complexes’ (SECs) [26].
The precise molecular mechanism of releasing P-TEFb from the 7SK RNPs is yet unsolved. Studies with the HIV-1 Tat protein demonstrated its strong capability of binding P-TEFb (via Cyclin T1) and to actively recruit P-TEFb from the 7SK RNPs [27,28]. Tat competes directly with HEXIM1 for P-TEFb binding, thereby causing its dissociation by inducing conformational changes in 7SK RNA [29-32]. In addition, Tat is a direct competitor for BRD4 [33], and interacts with several SEC components [34,35].
SECs contain either the AFF family members AF4 or AF5, distinct combinations of ELL (ELL1-3), as well as AF9 or ENL, AF10, DOT1L, NSD1, BRD4 and P-TEFb [36-38]. Most of their coding genes were initially identified as translocation partner genes in MLL-rearranged leukemia (AF4, AF5, AF9, ENL, AF10), but their role as stimulators of transcriptional elongation has been shown in a more physiological context [26,39]. However, the precise mechanism of P-TEFb recruitment in the absence of viral or oncogenic proteins - like HIV-1 Tat or MLL-chimeras - remained elusive.
Here, we demonstrate that assembled AF4 complexes, and in particular the AF4N complex, mediates the release of P-TEFb from the 7SK SNPs. During the release process, P-TEFb dissociates from the 7SK RNP and translocates to the AF4 or AF4N protein complex.
Interestingly, the oncogenic AF4-MLL fusion protein - deriving from the leukemogenic t(4;11)(q21;q32) chromosomal translocation - retains the same AF4N portion and strongly activates transcriptional elongation, and moreover, exhibits in vitro and in vivo transformation capabilities [25,40,41]. Our findings contribute not only to understand elongation control in gene transcription, but also provides novel insights into a pathological mechanism, namely how the leukemogenic AF4-MLL fusion protein may contribute to a process that converts a normal hematopoietic cell into a pre-leukemic or leukemia-initiating cell.
Material and methods
Expression plasmids and vectors
The AF4 or AF4N cDNA were cloned into pEZP•CMV, pEZP•BGH, pTCZP. AF4N, HEXIM1 and HIV-1 Tat cDNA was additionally cloned into pGEX-5T. The pCDNA 3.0 vector (Life Technologies, Paisley, UK) was modified resulting into the pDF vector (new multiple cloning site, C-terminal FLAG-tag). Human CDK9 human cytomegalovirus IE1 were both cloned into pDF, resulting in pDF-CDK9 and pDF-IE1. The pDF-IE1 vector was further modified by cloning a C-terminal dTomato (pDTF-IE1). The lentiviral packaging vectors pCMV-dR8.91 and pMD2.G were a gift of Manuel Grez (GSH, Frankfurt, Germany), while the SB100X vector was kindly provided from Zoltán Ivics (PEI, Langen, Germany).
Cell culture and establishing stable cell lines
Adherent 293T and HeLa cells were maintained in DMEM with high glucose, supplemented with 10% FCS and antibiotics. Transfections were carried out with 2.5 x 107 cells and stable cell lines were generated after co-transfection of corresponding SB vectors with SB100X in a ratio of 20:1. Twenty-four hours post transfection cells were selected for 3 days by adding 1 µg/ml puromycin until a homogenous fluorescent population was established (normally achieved after 5 days). Cell proliferation and viability was measured with the Cell Couting Kit-8 (CCK-8; Dojindo Molecular Technologies, Kumamoto, Japan) according to manufacturer instructions.
Lentiviral knock-down of AF4
1 x 105 HeLa cells were grown to 80% confluency and transfected with pCMV-dR8.91, pMD2.G as well as the MISSION® shRNA Plasmid (Clone ID NM_005935.1-3282s1c1) for human AF4 (Sigma-Aldrich, Munich, Germany) to produce viral particles. Four h after transfection medium was replaced and 72 h after transfection the supernatant was removed and filtered through a 0.22 µm filter. Ten, 50 or 100 µl of viral supernatant (V10, V50, V100) were applied by centrifugation for 1.5 h at 32°C, 2,500 rpm to transduce protaminsulfate treated HeLa cells (spin transduction). Cells were selected as mentioned above by using 1 µg/ml puromycin.
Enzyme-linked immunosorbent assay (ELISA)
ELISAs were performed with the ELISA Kit for AF4/FMR2 Family, Member 1 (AFF1/AF4) (USCN Life Science, Houston, USA) according to manufacturer instructions. All measurements of the protein standard fulfilled a R2 > 0.99 criteria.
Cell lysis, nuclear extraction and salt fractionation
In general, cells were washed twice in cold 1 x PBS, counted and collected by centrifugation (5 min, 4,000 rpm, 4°C). Whole cell lysates were generated by resuspending cells in 300 µl IP buffer/1 x 107 cells (IP buffer: 150 mM NaCl, 20 mM HEPES, pH 7.5, 1% Triton X-100, 1 mM Na3VO4, 10 mM NaF, 1 mM PMSF, 1x Protease Inhibitor Cocktail (Calbiochem, Billercia, USA)) and incubated for 1 h at 4°C under rotation. Lysates were centrifuged (30 min, 4,000 rpm, 4°C) and supernatants were collected. Nuclear extracts were generated by resuspending cells in buffer C (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.4% NP-40, 1 mM DTT, 0.5 mM PMSF, protease inhibitor cocktail (Calbiochem)) and incubation for 10 min at room temperature. Lysates were centrifuged (3 min, 4,000 rpm, 4°C) and supernatants saved as cytosolic fractions. The nuclear pellet was resuspended in buffer D (20 mM HEPES pH 7.9, 0.4 M NaCl, 1 mM EDTA, 10% glycerol, 1 mM DTT, 0.5 mM PMSF, protease inhibitor cocktail (Calbiochem)) and incubated for 2 h at 4°C with constant vortexing. All samples were centrifuged and supernatants collected as nuclear extracts. Salt fractionation of cells were performed as recently described [42].
Expression and purification of recombinant proteins
Expression vectors for recombinant proteins (pGEX-5T) were transformed into E.coli One Shot BL21star (DE3, Life Technology, Germany) and selected. A single clone was chosen for inoculation of a 50 ml pre-culture and incubated for 16 h, 180 rpm at 37°C until stationary phase. 25 ml of pre-culture were used to inoculate a 1 l main culture and incubated to a OD600 of 0.6. Expression was induced with 1 mM IPTG for 3 h. Bacteria were harvested by centrifugation for 10 min, 5,000 rpm at 4°C, pellets resuspended in 3 ml/mg lysis buffer (50 mM NaH2PO4 pH 7.5, 300 mM NaCl, 10 mM imidazol, 1% (v/v) Triton X-100, 5% (v/v) glycerol, 10 mM β-mercaptoethanol) and incubated for 30 min on ice. Lysates were sonified for 6 x 10 sec and genomic DNA digested by addition of DNase I for 15 min at 4°C. Subsequently samples were centrifuged twice to remove cell debris (20 min, 10,000 rpm, 4°C and 1 h, 35,000 rpm, 4°C). Supernatants served as input for purification. The recombinant proteins were tandem purified batchwise by a Ni-NTA agarose resin (QIAGEN, Hilden, Germany) and subsequently by magnetic glutathione-sepharose beads (Thermo Scientific) according to manufacturer instructions. Purity of eluted recombinant proteins was controlled by 12% SDS-PAGE and coomassie/silver staining, protein concentrations were determined on a nanophotometer (P330, Implen, Munich, Germany).
Western-Blot experiments
Purified protein complexes, whole cell lysates or nuclear extracts were analyzed by western blot by using the following monoclonal antibodies: anti-AF4 (V-14), -CDK9 (C-20), -CDK7 (C-19), Cyclin T1 (H-245), -β-Actin (I-19) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, USA); anti-HEXIM1, -LARP7, -NFkB p65/p50 antibodies were obtained from Abcam (Cambridge, UK); anti-acetyl-lysine antibody was obtained from Cell Signaling Technology (Danvers, USA); anti-FLAG M2 antibody was obtained from Sigma Aldrich. Transferred proteins were visualized with the ECL detection system (GE Healthcare). Relative quantification of protein lanes was performed using the Image Lab 3.0 Software (Bio-Rad, Hercules, USA).
Affinity purification and immunoprecipitation experiments
Affinity purification of AF4 or AF4N protein complexes was performed as previously described [25] without adding MG132 and starting from only 5 x 106 293T cells. For immunoprecipitation, whole cell lysates or nuclear extracts were normalized according to the total protein concentration. All samples were pre-cleared by addition of 1 µg non-specific IgG and 20 µl Protein-G-Agarose (Santa Cruz Biotechnology) and incubated for 30 min at 4°C under rotation. Supernatants were collected and 0.5-2 µg of specific antibody was added and further incubated for 1 h. 15-30 µl of Protein-G-Agarose were added and incubated for additional 2 h. Supernatants were washed three times with lysis buffer, eluted by boiling for 5 min in Laemmli-buffer and subsequently analyzed.
Yeast-2-hybrid experiments
All yeast-2-hybrid screens of this study were conducted using the MATCHMAKER III Two-Hybrid System (Clontech) according to the manufacturers recommendations. The system provides the two vectors pGBKT7 (bait) and pGADT7 (prey). Bait and prey inserts are expressed as GAL4 fusions with c-Myc and hemagglutinin (HA) epitope tags, respectively. The haploid yeast strain AH109, which virtually eliminates false positives by using three reporters (ADE2, HIS3, MEL1 or lacZ) under the control of GAL4, was transformed with the bait vector expressing a fusion of Gal4 DNA-binding domain and the different AF4 protein fragments. The following cDNAs were clones into both plasmids in order to perform all experiments in both directions (bait and prey exchanged): AF4N (1-346), AF4•1 (1-58), AF4•2 (1-111), AF4•3 (1-150), AF4•4 (1-206), AF5•1 (1-505), AF5•2 (442-806), AF5•3 (795-1164), CCNT1 (1-727), CCNT1•1 (1-220), CCNT•2 (187-401), CDK9 (1-372), CDK9•1 (1-100), CDK9•2 (70-250), CDK9•3 (220-372), DOT1L•1 (1-422), DOT1L•2 (387-642), DOT1L•3 (612-972), DOT1L•4 (934-1275), NPM1 (1-295), NPM•1 (1-122), NPM•2 (91-201), NPM•3 (169-295), NSD1•1 (1-254), NSD1•2 (216-569), NSD1•3 (533-659), NSD1•4 (833-1175), NSD1•6 (1432-1870), NSD1•7 (1829-2240), NSD1•8 (2199-2695), RELA•1 (1-203) and RELA•2 (159-310).
P-TEFb release assay
Transiently transfected 293T cells (pDF-CDK9; 1 x 107) were incubated for 48 h and then harvested. Whole cell lysate (1 ml) was incubated with 20 µl equilibrated FLAG-M2 magnetic beads (Sigma-Aldrich) in the presence of RNasin. Samples were incubated under rotation for 2 h at room temperature and washed three times in 1xTBST without RNasin. Samples were split into aliquots and incubated with GST-HEXIM1 (N), 1 µg of RNase, GST-AF4N or cell lysates with overexpressed AF4 for 15 min at room temperature. Matrix bound P-TEFb and supernatant were magnetically separated and further analyzed by Western blots.
7SK RNA binding assay
Recombinant GST, GST-AF4N or GST-Tat (2 µg each) were bound to glutathione magnetic beads (Thermo Scientific, Waltham, USA) for 2 h at room temperature according to manufacturer instructions. Beads were subsequently washed three times and then incubated with 10 µg of total RNA from 293T cells in a total volume of 500 µl for another 2 h. Beads were washed three times and proteins were eluted in a total volume of 50 µl. An aliquot of these eluates (2 µl) was reverse transcribed using SuperScript II reverse transcriptase (Life Technology, Germany) and then amplified by standard PCR to analyze the amount of 7SK RNA (5’-AGGACCGGTCTTCGGTCAA-3’, 5’-TCATT-TGGATGTGTCTGCAGTCT-3’). The 133 bp amplicon was quantitatively analyzed by Q-PCR and compared to a defined 7SK RNA dilution series (108 to 101 copies), fulfilling the R2 > 0.9 criteria.
Glycerol gradients
A glycerol gradient was poured by adding 5, 10, 12.5, 15, 17.5, 20, 25, 30, 35 or 40% (v/v) glycerol to glycerol gradient buffer (20 mM HEPES pH 7.9, 300 mM KCl, 0.2 mM EDTA, 0.1% (v/v) NP-40). The gradient was sealed with parafilm and settled over 16 h at 4°C. Transiently transfected 293T cells (2 x 107) with pEZP-AF4ST, -AF4NST or pEZP (mock control) were harvested after 48 h and whole cell lysates of 1 ml prepared. Lysates were applied to glycerol gradients and ultracentrifuged (21 h, 38,000 rpm, 4°C). Ten fractions were carefully removed and analyzed in Western blot experiments.
Results
Establishment of a stable and inducible AF4 and AF4N expression system
Experimental studies of AF4 protein are hampered by the fact that AF4 is rapidly degraded via binding to the two E3 ligases SIAH1 and SIAH2 [40]. Consequently, proteasomal degradation must be blocked by addding MG132, but this treatment is rather toxic to cells and may influence experimental read-outs. Therefore, we cloned a C-terminally Strep-tagged AF4 into two constitutive sleeping beauty (SB) vector backbones (EZP•CMV-AF4ST; EZP•BGH-AF4ST) and a doxyxcycline-inducible SB vector backbone (TCZP-AF4ST; Figure 1A). All SB vectors were stably integrated in low copies (n = 2-6) into the genome of 293T cells. Q-PCR experiments demonstrated that AF4 mRNA levels increased up to 400-fold from the inducible vector backbone (Figure 1B). This allowed us to express the AF4 protein in an inducible manner without MG132 treatment, and AF4 protein could be readily detected 24h later from as little as 5 x 106 cells (Figure 1C, upper panel). Moreover, an AF4 expression kinetic demonstrated that a visible AF4 overexpression starts about 6h after induction, with high levels of AF4 remaining until 72 h (Figure 1C, lower panel). This indicates that the degradation pathway of the cells become outcompeted by the amount of recombinantly expressed AF4/AF4N protein, which allowed us to monitor their function, and more importantly, the purification of these complexes. AF4ST expressing cells displayed a more sustained proliferation and metabolic activity (CCK-8 activity) compared to regular 293T cells that display a rapid growth decline after having reached confluency (Figure 1D). This is in line with earlier observations where an overexpression of AF4 caused a loss-of-contact-inhibition (40). Based on all these data, we used the TCZP vector backbone for all further experiments. Finally, we tested the stability of expression of our integrated SB vector constructs by monitoring the expression of the ZsGreen reporter protein up to passage 37 without using selective media (Figure 1E).
Figure 1.
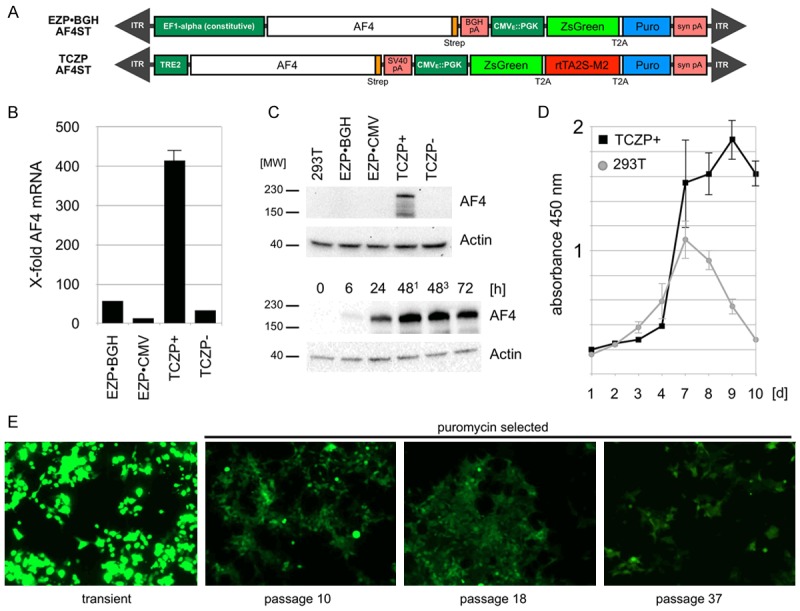
Characterization of the TCZP-AF4ST stable cell line. A. Stable integrated vectors used in this study. B. Quantitative analysis of relative amounts of AF4 mRNA by qPCR from prepared cDNA with (TCZP+) or without (TCZP-) application of doxycyclin compared to regular 293T cells. Amounts are normalized to Actin [n = 3, +S.E.M.]. C. Top: Representative Western-Blot experiments from TCZP-AF4ST whole cell lysates after 2 passages with or without doxycyclin compared to constitutively expressing constructs pEZP and pEZP-BGH. Faster migrating bands are degradation products of AF4. Bottom: Western-Blot experiments from TCZP-AF4ST whole cell lysates 0-72 h after induction of expression. Lanes 481 and 483 differ in the amount of applied doxycyclin (481 = 1 µg/ml, 483 = 3 µg/ml). D. CCK-8 proliferation assays of induced TCZP-AF4ST cells over a time course of 10 days, compared to non-treated 293T cells [n = 3, ±S.E.M.]. Decline of untransfected cells is due to reaching confluency. E. Representative in-vitro GFP fluorescence microscopy images of TCZP-AF4ST cells during the indicated passages (magnification 100x, 0.5 s. exposure).
AF4 knock-down reduces cell viability
Conversely, we also tried to use an shRNA-mediated knock-down of AF4 to investigate its loss-of-function effects. We have to mention that knockdown experiments have been attempted also in the past, where we observed very strong effects on cell viability when using siRNA mediated knock-down experiments (data not shown). Also the growth of murine Af4 knock-out cells (a gift of M. Djabali) displayed doubling times of ~14 days, indicating that AF4 is presumably a crucial protein for normal cell physiology (unpublished observations). Therefore, we used here only a short term observation period when we knocked-down endogenous AF4 by using 3 different amounts of viral supernatant in our transduction experiments (V10, V50 and V100). We were able to reduce the amount of endogenous AF4 mRNA only to about 40-50% when using the highest doses of virus (Figure 2A). This is perfectly in line with our unpublished observations that AF4 can only be knocked-down to a certain threshold. Similarly, the AF4 protein levels was reduced. Since the endogenous AF4 protein is even difficult to visualize under physiological conditions in Western-blots (see e.g. Figure 1C, lane 293T), a more sensitive AF4-specific ELISA was established here (Figure 2B). A maximum knock-down efficiency of ~60% of AF4 protein expression was achieved with V100, but even higher amounts of the shRNA-expressing lentiviral vector did not result in a higher knock-down efficiency (data not shown). However, the reduction of AF4 to about 50-60% was sufficient to affect the normal cell growth behavior. As shown in Figure 2C, the cell growth was affected and the AF4 V100 knock-down cells also displayeda morphology of stressed or resting cells (Figure 2D). In summary, the relative abundance of AF4 seems to be crucial for living cells, because AF4 overexpression seems to confer a growth advantage, while its knock-down resulted either in growth arrest or quiescence.
Figure 2.
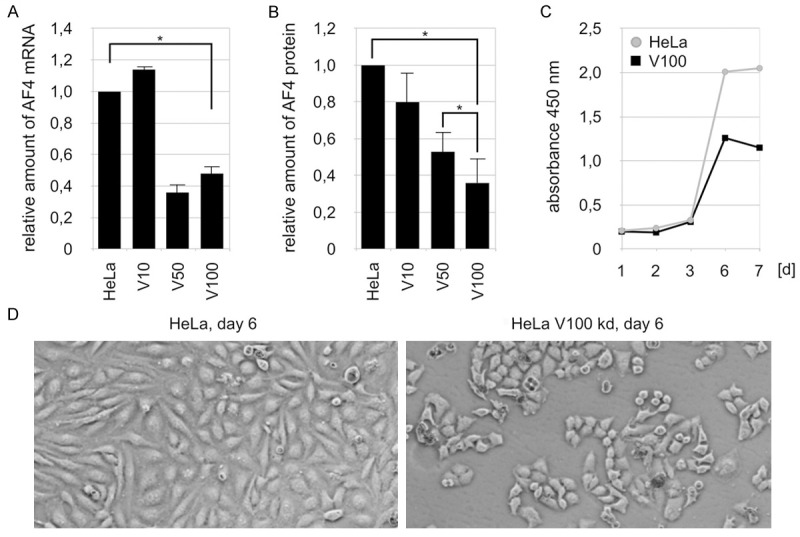
Characterization of the AF4kd V100 stable cell line. A. Results of the AF4-specific ELISA after transduction of HeLa cells with viral particles and establishment of resistance to puromycin. Relative amounts of AF4 protein in regular HeLa cells (100% = 1) and knock-down lines V10, V50 and V100 are depicted [n = 4, +S.E.M., *p < 0.05]. B. Quantitative analysis of relative amounts of AF4 mRNA by qPCR from prepared cDNA of the V10, V50 and V100 infected HeLa cells (100% = 1). Amounts are normalized to Actin [n = 3, +S.E.M., *p < 0.05]. C. CCK-8 proliferation assays of AF4kd V100 cells over a time course of 7 days, compared to non-treated HeLa cells [n = 3, ±S.E.M.]. D. Representative in-vitro microscopy images of AF4kd V100 cells during day 6 of the proliferation assays (magnification 100x, 10 ms. exposure) showing cowered morphology and non-commenced confluence.
Expression of SEC components after AF4 knock-down
Some of the already known constituents of the AF4 protein complex [25] were analyzed here in Western blot experiments as experimental read-out for potential changes. First, we tested the abundance of AF4 complex binding partners in the AF4 knock-down situation. Here, we tested for CDK9, CYCT1, NFkB1 (p65/p50), but also the functional important CDK9 T186-P modification which is a known marker for activated P-TEFb (Figure 3A). We used also three control proteins (CDK2, CDK7, MEN1). The only difference found in AF4 knock-down (V100) cells was a slight reduction of the T186-P modification (RQ = 0.42), supporting the notion that binding of P-TEFb to AF4 is crucial for its functional activation. We observed also a slight increase in MEN1 protein which was one of the control proteins. All other AF4 binding proteins were not affected in their steady-state abundance, suggesting that their steady-state expression is not influenced by the absence of AF4.
Figure 3.
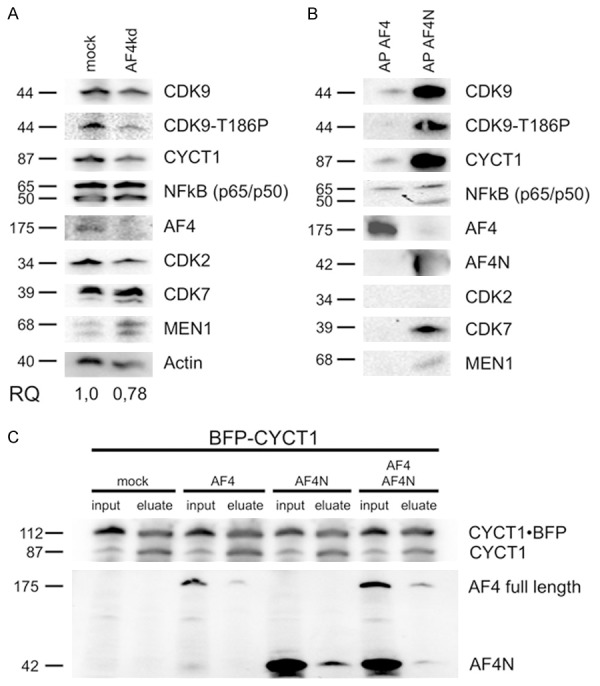
Interaction and dependency between AF4 and transcription-associated factors. A. Western-Blot experiment for CDK9, CDK9-T186A, CYCT1, NFkB, AF4, CDK2, CDK7 and MEN1 from whole cell lysates of non-transfected HeLa (left) compared to AF4kd V100 cells (right). Relative quantification was carried out by using the Image Lab 3.0 Software (Bio-Rad). Molecular weight of all proteins are indicated. B. Western-Blot experiments with affinity purified (AP) strep-tagged AF4 (left) and AF4N (right) probed with the same antibodies. C. Immunoprecipitation of BFP-CYCT1 from nuclear fractions of transiently transfected 293T cells. (mock, pETP-BFPCYCT1, pEZP-AF4ST or pEZP-AF4NST; input 0.2%, eluate 33%). BFP-CYCT1 (112 kDa) as well as endogenous CYCT1 (87 kDa) were precipitated. Lower panel: Western blot with antiserum against the AF4 N-terminus.
Composition of AF4 and AF4N SEC complexes
The AFF family proteins AF4 and AF5 are generally known to resemble a molecular platform for the assembly of high molecular weight protein complexes, called ‘super elongation complexes’ (SECs). We used our established Strep-tag affinity purification method [25] to isolate the nuclear complexes formed on strep-tagged full-length AF4 and the AF4N protein. AF4N is generated from an alternative AF4 transcript (FelC) that exhibits an open reading frame comprising the first 3 AF4 exons only and is terminated at a cryptic poly A site localized within intron 3. Noteworthy, the steady-state abundance of this FelC transcripts is strongly increased in leukemia cells that bear a t(4;11)(q21;q23) translocation [43]. After elution from the StrepTactin columns, bound proteins which interact with AF4/AF4N were detected by Western blot. As previously shown, we could detect CyclinT1, CDK9, NFkB1 (p65) and DOT1L (Figure 3B). The expression of AF4N and AF4 was strongly induced by doxycycline, but the shorter AF4N protein displayed a much higher abundance than the full-length AF4 protein. However, even concerning the difference in the abundance of both overexpressed proteins, the AF4N protein displayed a better binding to known interaction partners than the AF4 protein. Interestingly, the AF4N protein seems to make additional interactions with the p50 subunit of NFkB1, CDK7 as well as MEN1 (Figure 3B). Since CDK7 and Cyclin H1 form the transcription factor IIH (TFIIH) and AF4N seems to bind CDK7, it may indicate that AF4N is also involved in transcriptional initiation (TFIIH) and not only in transcriptional elongation (P-TEFb).
MEN1 has been described to counteract NFkB1-mediated activities (like the transcriptional activation of Cyclin D1) by binding directly to the p50 and p65 subunits of NFkB1. MEN1 is usually bound to MLL and was described to transcriptionally activate p18 and p27, leading to a growth arrest.
Since the smaller AF4N protein exhibited this prominent binding behavior, we further checked for a direct competition between full-length AF4 and AF4N by transfecting both expression constructs together with an BFP-tagged CYCT1 in 293T cells. Immunoprecipitation of BFP-CYCT1 resulted in the co-precipitation of AF4 as well as AF4N in single and double transfection experiments. This indicates that AF4 and AF4N are both able to bind to P-TEFb complexes even under competitive conditions (Figure 3C, lanes 4 vs 6 vs 8).
AF4 interactome
We also performed yeast-2-hybrid (Y-2-H) experiments to unravel the molecular interactions inside the SEC. We cloned AF4, AF5, NFkB1, CyclinT1, CDK9, BRD4, DOT1L, NSD1 and NPM1 as smaller, overlapping cDNA fragments in both Y-2-H vectors (bait and prey vectors), in order to perform all experiments in both directions and used the appropriate single transfections as control to exclude autoactivation. Despite the RELA protein fragments tested, no autoactivation was observed (data not shown). All protein interactions were classified upon the growth behavior of diploid yeast colonies on 4x drop-out media (4xDO) and classified as very weak (+) to very good (+++). Based on more than 1,000 tested interactions, a network emerged which is displayed in Figure 4A (right panel). A more detailed interaction map is displayed in Figure 4B, where we illustrate which portions of each protein is interacting with the other proteins. Based on these data, the N-terminal portion of AF4 or AF4N resembles a molecular hub for a multitude of protein interactions. All proteins make multiple interactions inside of these complexes, presumably allowing a very quick assembly of functional SECs from variable preformed smaller protein complexes.
Figure 4.

AF4/AF4N interactome mapping. A. The Yeast-2-hybrid Matchmaker III system was used to analyze the interactome of the core components of the protein assembly that occur within the AF4 N-terminal portion. B. More detailed interaction map displaying which portion of each protein which is binding to other proteins of the AF4/AF4N interactome. The experimentally observed strength of protein interactions is indicated by different sizes or dotted lines. Mapping of protein-protein interactions by others is indicated by colored lines.
AF4/AF4N protein complexes recruit P-TEFb from inhibitory 7SK RNPs
Next, we wanted to investigate the process that enables AF4 or AF4N to recruit P-TEFb from the inhibitory 7SK RNPs (Figure 5A). For this purpose we used purified 7SK RNPs which were obtained by precipitation of Flag-tagged CDK9 and bound to magnetic protein A/G beads. These loaded beads were used to establish a simple in vitro P-TEFb release assay. The bead-bound 7SK RNPs were incubated with cell lysates from untreated cells (N), or from cells that overexpressed CMV immediate early protein 1 (IE1; positive control), AF4 or the enzyme 5-lipoxygenase (5-LO; 2nd negative control). As shown in Figure 5B, we monitored the release of P-TEFb indirectly by measuring the loss of LARP7 from the bead fraction. LARP7 is an integral and most stable component of the 7SK RNPs. The release of LARP7 indicates the disintegration of the 7SK RNP complex, as observed and described for several viral proteins.
Figure 5.
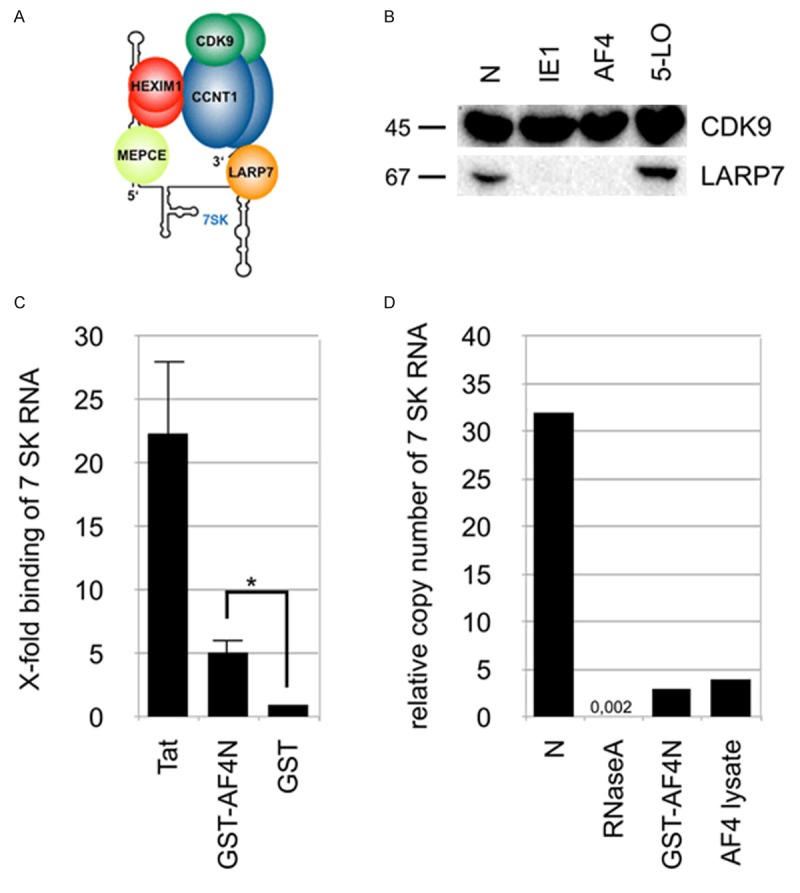
Recruitment of P-TEFb from storage complexes into AF4/AF4N. A. Schematic representation of the 7SK RNP. B. Western blots after CDK9 immunoprecipitation from whole cell lysates of transiently transfected 293T cells (mock, Strep-tagged AF4, HCMV IE1 or 5-LO). blots were probed with antibodies against CDK9 and LARP7. C. 7SK RNA binding assay. Results of specific quantitative PCR of 7SK RNA purified from elution fractions. Samples were subjected to quantitative PCR and quantified against a 7SK RNA dilution series. Shown is the x-fold binding of 7SK RNA compared to the GST negative control (set as 1) [n = 3, +S.E.M., *p < 0.05]. D. Results of quantitative PCR of 7SK RNA after incubation of whole cell lysates with RNase, recombinant GST-AF4N or a distinct whole cell lysate with overexpressed AF4ST. A non-treated lysate was used as negative control. Shown is the relative amount of 7SK RNA after incubation as the quotient of 7SK RNA/β-Actin copy number.
Potential mechanims of P-TEFb release
Next, we asked the question on how AF4, AF4N or viral proteins are able to perform the release of P-TEFb from 7SK RNPs. One idea came from a very early observation, namely that LAF4/AFF2 was able to bind directly to nucleic acids [44]. Therefore, we wanted to investigate whether AF4 or AF4N have similar properties. In addition, the use of both proteins would also allow to identify the responsible regions within the AF4 protein (N- or C-terminus). 7SK RNA was produced by in vitro transcription and incubated with recombinant TAT protein, GST-AF4N or GST as negative control. Bound 7SK RNA was then reverse transcribed, analyzed by QPCR and the x-fold enrichment relative to GST summarized in Figure 5C. This 7SK RNA binding assay revealed that Tat binds very effectively to 7SK RNA, AF4 does this to a lesser extent, but significantly better than the negative control. Therefore, we concluded that the N-terminal portion of AF4 is able to bind to 7SK RNA, presumably in a similar fashion as the TAT protein, which causes a conformational change of the 7SK RNA structure which is essential for P-TEFb release [45].
We quantified the amount of 7SK RNA by QPCR without any treatment (N) and after incubation with recombinant GST-AF4N, an AF4 overexpression whole cell lysate or with RNaseA as positive control. Notably, the presence of AF4 or GST-AF4N led to a comparable five- to tenfold loss of 7SK RNA (Figure 5D), which may indicate that full-length AF4 and AF4N are not only binding to 7SK RNA, but also cause their degradation, presumably as part of a potential mechanism to release P-TEFb kinase. However, RNaseA destroyed the 7SK RNA template about 1500 to 2000-fold more effectively.
AF4/AF4N protein complexes incorporate P-TEFb
Next, we investigated that AF4 or AF4N does not only cause a release of P-TEFb from 7SK RNPs, but also incorporates P-TEFb into the AF4 or AF4N protein complexes. A glycerol gradient experiment was performed to monitor the intracellular distribution of P-TEFb and LARP7 as 7SK RNP marker upon AF4N or AF4 overexpression. As shown in Figure 6, the distribution of CDK9 was mostly diffuse among all fractions of the gradient in mock-transfected cell lysates, displaying an overlap with LARP7 in high molecular weight (HMW) fractions. This indicates that under physiological conditions LARP7 and CDK9 are together part of high molecular weight complexes (7SK RNPs). This physiological distribution changes in the presence of overexpressed AF4N or AF4. In both cases, CDK9 co-fractionates together with AF4 or AF4N in high molecular weight complexes. Most importantly, the LARP7 protein disappears from the HMW fractions and shift towards fractions with lower molecular weight. This suggests that AF4N and AF4 are both able to release P-TEFb from the 7SK RNPs, thereby causing also a release of LARP7, and to recruit PTEFb into the assembled AF4N and AF4 complexes. A subsequently performed immunoprecipitation of AF4 from fractions 2-7 confirmed the association of AF4 with CDK9 in fractions 3-5.
Figure 6.
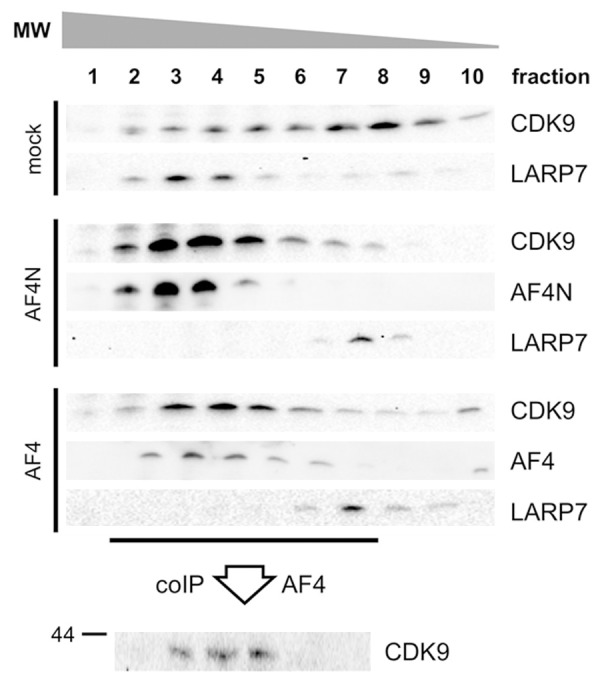
AF4N/AF4 transfers P-TEFb from 7SK RNPs to AF4N/AF4. 293T cells were transiently transfected with empty vector (mock), pEZP-AF4ST or pEZP-AF4NST. Whole cell lysates were prepared and separated on a 0-40% glycerol gradient by ultracentrifugation. Gradient were fractionated (n = 10) and analyzed for the presence of CDK9, LARP7 and AF4/AF4N. Dissociation of LARP7 from 7SK RNPs is equivalent to the disruption of existing 7SK RNPs. Bottom: AF4 immunoprecipitation from glycerol gradient fractions 2 to 7 was probed for the presence of CDK9 to demonstrate the P-TEFb transition from 7SK RNP to AF4N/AF4.
Discussion
The human AFF1/AF4 gene was identified as the major recombination partner gene in MLL-rearranged leukemia, associated with the development of proB acute lymphoblastic leukemia in infants, children and adults. For this reason, investigating the functions deriving from full-length AF4, tumor-overexpressed AF4N and the leukemogenic AF4-MLL fusion protein is of great value for our general understanding of cancer development. Here, we tried to answer different questions, namely the molecular requirements for the transitions of the P-TEFb kinase into AF4-mediated SECs, but also how the AF4 and the AF4N interactome is actually composed. From these studies we expected to learn about potential differences between full-length AF4 and the shorter protein variant AF4N. AF4N is a natural protein variant of AF4, exhibiting only the first 360 amino acids and is identical to the fused portion within the leukemogenic AF4-MLL fusion protein.
AFF family members (AF4, AF5) have been linked to transcriptional regulation and elongation [38,46,47]. This is such a fundamental process that overexpression of AF4 already confers a growth advantage with clear signs of malignant transformation [40]. These earlier findings could be confirmed here by using an inducible AF4 expression system, where cells induced to strongly overexpress AF4 displayed a growth advantage (indicated by CCK-8 activity) even on confluent petri dishes (Figure 1C). By contrast, an AF4 knock-down resulted in impaired cell growth and a morphology that reminded to severe cellular stress or quiescence (Figure 2D).
The AF4 protein complex contains several important constituents [25]. Here, we focussed specifically on CDK9, CYCT1 and a few other proteins (Figure 3B). The new inducible system allowed to affinity-purify and investigate AF4 or AF4N complexes from very few cells (5 x 106) in a reproducible manner. This is quite different to our earlier study [25] where we analyzed a priori the composition of these complexes always from 1 x 109 cells.
The question how P-TEFb is recruited into the AF4 protein complex has in part been solved: AF4 and AF4N are both able to bind directly to 7SK RNA (Figure 5D). This induces a release of LARP7 from the 7SK RNPs (Figures 5B, 6). Whether this is due to conformational changes in the 7SK RNA structure or to its degradation, is not completely clear, because the measured RNA degradation could be also an artifact of our applied method. Regardless whether this transition of P-TEFb is due to conformational changes within the 7SK RNA or by its degradation, AF4 and AF4N both caused the release of LARP7. LARP7 has been described as a robust complex partner within the 7SK RNP [22,24]. Thus, a release of LARP7 indicates a disintegration of the 7SK RNP complex. This was confirmed by the glycerol gradient experiments, where a release of P-TEFb and its incorporation into the AF4 or AF4N protein complexes was concomittantly accompanied by LARP7 release (Figure 6). In addition - and quite similar to the viral TAT protein - the N-terminal portion of AF4 displays a strong binding to P-TEFb and makes multiple interactions with other constituents of the SEC complex (e.g. NSD1, DOT1L, NFkB). To this end, the resulting SEC complex should be highly stable after P-TEFb has been included, and should become destabilized when the AF4 protein is degraded via the proteasomal pathway [40].
A completely new finding was that the AF4N protein binds to CDK7 (Figure 3B). CDK7 has an important function by phosphorylating the Ser-5 position within the CTD repeats of RNAPII, a process that is necessary for the recruitment of P-TEFb [48]. Since all members of the AFF protein family (AF4, AF5, LAF4 and FMR2) express such shorter protein variants [43], one might speculate that these shorter protein variants of the AFF protein family may have additional functions, e.g. to actively recruit TFIIH (CDK7/Cyclin H) which helps to initate transcription. CDK7 also functions as CDK-activating kinase (CAK) and phosphorylates e.g. CDK9. If so, then AF4N and AF4 complexes may even cooperate with each other, and may contribute to all initial events of the transcription process (initiation and elongation).
The interaction of AF4N with MEN1 is also a novel observation. MEN1 is known to transcriptionally activate the CDK inhibitors p18 and p27 via the interaction with the MLL complex [49]. Binding of MEN1 to overexpressed AF4N could be of importance in order to outcompete MEN1/MLL-derived functions, e.g. to upregulate cell cycle inhibitors. This would explain why AF4N is overexpressed in leukemia cells with a t(4;11) translocation, leading to a loss of cell cycle arrest. In addition, MEN1 is known to bind to p50 and p65 of NFkB1. This protein-protein interaction normally disables NFkB1 to mediate the transcription of downstream target genes like Cyclin D1. However, in the presence of AF4N, the specific MEN1-NFkB1 interaction may be compromised as well (due to their recruitment), and thus, AF4N would allow again the production of Cyclin D1. It would indicate that the AF4N protein – as well as the other shorter protein variants of the AFF protein family – have very specific functions, not only to serve as a hub controlling the process of transcription, but also to influence cell growth and cell cycle.
AFF family members represent versatile docking platforms because of their ‘flexible folding’ [50,51]. This allows to perform a series of actions necessary to recruit P-TEFb from 7SK RNPs, to activate P-TEFb and to convert promoter-proximal pausing RNAPII (POL A) into elongating RNAPII (POL E). In addition, several histone methyltransferases (DOT1L, NSD1, CARM1), all part of purified AF4 protein complexes, allow to imprint the transcribed region with H3K36me2, H3K79me2/3 and R17/R26me2 signatures. Pathological events such as the creation of the AF4-MLL fusion protein, which results in a strongly increased stability of AF4-MLL fusion protein, leads to a deregulation of these processes and finally to the malignant transformation of affected cells. Thus, interference with this tightly regulated process may be related to an oncogenic transition. Future work will help to identify critical targets and how they are related to cancer onset.
Acknowledgements
We thank Jennifer Merkens and Sylvia Bracharz for technical assistance. All FACS experiments were performed at the Georg-Speyer-Haus with the kind help of Tefik Merovci. This study was supported by research grants Ma1876/10-1, Ma1876/11-1 from the DFG to RM. KA is supported by the Else Kröner-Fresenius-Stiftung (Dr. Hans Kröner Graduiertenkolleg). DS and RM are PI’s within the CEF on Macromolecular Complexes funded by DFG grant EXC 115.
Disclosure of conflict of interest
The authorsdeclare no conflict of interest.
References
- 1.Cheng B, Li T, Rahl PB, Adamson TE, Loudas NB, Guo J, Varzavand K, Cooper JJ, Hu X, Gnatt A, Young RA, Price DH. Functional association of Gdown1 with RNA polymerase II poised on human genes. Mol Cell. 2012;45:38–50. doi: 10.1016/j.molcel.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiba K, Yamamoto J, Yamaguchi Y, Handa H. Promoter-proximal pausing and its release: molecular mechanisms and physiological functions. Exp Cell Res. 2010;316:2723–2730. doi: 10.1016/j.yexcr.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, Hasegawa J, Handa H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell. 1999;97:41–51. doi: 10.1016/s0092-8674(00)80713-8. [DOI] [PubMed] [Google Scholar]
- 4.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 5.Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;270:12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 6.Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol. 2000;20:2629–2634. doi: 10.1128/mcb.20.8.2629-2634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shchebet A, Karpiuk O, Kremmer E, Eick D, Johnsen SA. Phosphorylation by cyclin-dependent kinase-9 controls ubiquitin-conjugating enzyme-2A function. Cell Cycle. 2012;11:2122–2127. doi: 10.4161/cc.20548. [DOI] [PubMed] [Google Scholar]
- 8.Ramanathan Y, Rajpara SM, Reza SM, Lees E, Shuman S, Mathews MB, Pe’ery T. Three RNA polymerase II carboxyl-terminal domain kinases display distinct substrate preferences. J Biol Chem. 2001;276:10913–10920. doi: 10.1074/jbc.M010975200. [DOI] [PubMed] [Google Scholar]
- 9.Hintermair C, Heidemann M, Koch F, Descostes N, Gut M, Gut I, Fenouil R, Ferrier P, Flatley A, Kremmer E, Chapman RD, Andrau JC, Eick D. Threonine-4 of mammalian RNA polymerase II CTD is targeted by Polo-like kinase 3 and required for transcriptional elongation. EMBO J. 2012;31:2784–2797. doi: 10.1038/emboj.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu P, Kenney JM, Stiller JW, Greenleaf AL. Genetic organization, length conservation, and evolution of RNA polymerase II carboxyl-terminal domain. Mol Biol Evol. 2010;27:2628–2641. doi: 10.1093/molbev/msq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egloff S, Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet. 2008;24:280–288. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Corden JL, Patturajan M. A CTD function linking transcription to splicing. Trends Biochem Sci. 1997;22:413–416. doi: 10.1016/s0968-0004(97)01125-0. [DOI] [PubMed] [Google Scholar]
- 13.Hsin JP, Sheth A, Manley JL. RNAP II CTD phosphorylated on threonine-4 is required for histone mRNA 3’ end processing. Science. 2011;334:683–686. doi: 10.1126/science.1206034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshall NF, Peng J, Xie Z, Price DH. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem. 1996;271:27176–27183. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- 15.Michels AA, Nguyen VT, Fraldi A, Labas V, Edwards M, Bonnet F, Lania L, Bensaude O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol Cell Biol. 2003;23:4859–4869. doi: 10.1128/MCB.23.14.4859-4869.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- 17.Herrmann CH, Mancini MA. The Cdk9 and cyclin T subunits of TAK/P-TEFb localize to splicing factor-rich nuclear speckle regions. J Cell Sci. 2001;114:1491–1503. doi: 10.1242/jcs.114.8.1491. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z, Zhu Q, Luo K, Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- 19.Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK RNA. Mol Cell. 2003;12:971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- 20.Egloff S, Van Herreweghe E, Kiss T. Regulation of polymerase II transcription by 7SK RNA: two distinct RNA elements direct P-TEFb and HEXIM1 binding. Mol Cell Biol. 2006;26:630–642. doi: 10.1128/MCB.26.2.630-642.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, Sedore SC, Price JP, Price DH, Lania L, Bensaude O. Binding of the 7SK RNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 2004;23:2608–2619. doi: 10.1038/sj.emboj.7600275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He N, Jahchan NS, Hong E, Li Q, Bayfield MA, Maraia RJ, Luo K, Zhou Q. A La-related protein modulates 7SK RNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol Cell. 2008;29:588–599. doi: 10.1016/j.molcel.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michels AA, Bensaude O. RNA-driven cyclin-dependent kinase regulation: when CDK9/cyclin T subunits of P-TEFb meet their ribonucleoprotein partners. Biotechnol J. 2008;3:1022–1032. doi: 10.1002/biot.200800104. [DOI] [PubMed] [Google Scholar]
- 24.Xue Y, Yang Z, Chen R, Zhou Q. A capping-independent function of MePCE in stabilizing 7SK RNA and facilitating the assembly of 7SK RNP. Nucleic Acids Res. 2010;38:360–369. doi: 10.1093/nar/gkp977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benedikt A, Baltruschat S, Scholz B, Bursen A, Arrey TN, Meyer B, Varagnolo L, Müller AM, Karas M, Dingermann T, Marschalek R. The leukemogenic AF4-MLL fusion protein causes P-TEFb kinase activation and altered epigenetic signatures. Leukemia. 2011;25:135–144. doi: 10.1038/leu.2010.249. [DOI] [PubMed] [Google Scholar]
- 26.Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661–672. doi: 10.1101/gad.2015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, Price DH. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465:747–751. doi: 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 29.Barboric M, Yik JH, Czudnochowski N, Yang Z, Chen R, Contreras X, Geyer M, Matija Peterlin B, Zhou Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucl Acids Res. 2007;35:2003–2012. doi: 10.1093/nar/gkm063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krueger BJ, Varzavand K, Cooper JJ, Price DH. The mechanism of release of P-TEFb and HEXIM1 from the 7SK RNP by viral and cellular activators includes a conformational change in 7SK. PLoS One. 2010;5:e12335. doi: 10.1371/journal.pone.0012335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muniz L, Egloff S, Ughy B, Jady BE, Kiss T. Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog. 2010;6:e1001152. doi: 10.1371/journal.ppat.1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulte A, Czudnochowski N, Barboric M, Schönichen A, Blazek D, Peterlin BM, Geyer M. Identification of a cyclin T-binding domain in Hexim1 and biochemical analysis of its binding competition with HIV-1 Tat. J Biol Chem. 2005;280:24968–24977. doi: 10.1074/jbc.M501431200. [DOI] [PubMed] [Google Scholar]
- 33.Urano E, Kariya Y, Futahashi Y, Ichikawa R, Hamatake M, Fukazawa H, Morikawa Y, Yoshida T, Koyanagi Y, Yamamoto N, Komano J. Identification of the P-TEFb complex-interacting domain of Brd4 as an inhibitor of HIV-1 replication by functional cDNA library screening in MT-4 cells. FEBS Lett. 2008;582:4053–4058. doi: 10.1016/j.febslet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 34.He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell. 2010;38:428–438. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK RNP. Mol Cell. 2010;38:439–451. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo Z, Lin C, Guest E, Garrett AS, Mohaghegh N, Swanson S, Marshall S, Florens L, Washburn MP, Shilatifard A. The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol. 2012;32:2608–2617. doi: 10.1128/MCB.00182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeisig DT, Bittner CB, Zeisig BB, Garcia-Cuellar MP, Hess JL, Slany RK. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene. 2005;24:5525–5532. doi: 10.1038/sj.onc.1208699. [DOI] [PubMed] [Google Scholar]
- 39.Lin C, Garrett AS, De Kumar B, Smith ER, Gogol M, Seidel C, Krumlauf R, Shilatifard A. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC) Genes Dev. 2011;25:1486–1498. doi: 10.1101/gad.2059211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bursen A, Moritz S, Gaussmann A, Moritz S, Dingermann T, Marschalek R. Interaction of AF4 wild-type and AF4.MLL fusion protein with SIAH proteins: indication for t(4;11) pathobiology? Oncogene. 2004;23:6237–6249. doi: 10.1038/sj.onc.1207837. [DOI] [PubMed] [Google Scholar]
- 41.Bursen A, Schwabe K, Rüster B, Henschler R, Ruthardt M, Dingermann T, Marschalek R. The AF4. MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood. 2010;115:3570–3579. doi: 10.1182/blood-2009-06-229542. [DOI] [PubMed] [Google Scholar]
- 42.Fujinaga K, Barboric M, Li Q, Luo Z, Price DH, Peterlin BM. PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucl Acids Res. 2012;40:9160–9170. doi: 10.1093/nar/gks682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nilson I, Reichel M, Ennas MG, Greim R, Knörr C, Siegler G, Greil J, Fey GH, Marschalek R. Exon/intron structure of the human AF-4 gene, a member of the AF-4/LAF-4/FMR-2 gene family coding for a nuclear protein with structural alterations in acute leukaemia. Br J Haematol. 1997;98:157–169. doi: 10.1046/j.1365-2141.1997.1522966.x. [DOI] [PubMed] [Google Scholar]
- 44.Ma C, Staudt LM. LAF-4 encodes a lymphoid nuclear protein with transactivation potential that is homologous to AF-4, the gene fused to MLL in t(4;11) leukemias. Blood. 1996;87:734–745. [PubMed] [Google Scholar]
- 45.Lu J, Wong V, Zhang Y, Tran T, Zhao L, Xia A, Xia T, Qi X. Distinct conformational transition patterns of noncoding 7SK RNA and HIV TAR RNAs upon Tat binding. Biochem. 2014;53:675–681. doi: 10.1021/bi401131z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- 47.Estable MC, Naghavi MH, Kato H, Xiao H, Qin J, Vahlne A, Roeder RG. MCEF, the newest member of the AF4 family of transcription factors involved in leukemia, is a positive transcription elongation factor-b-associated protein. J Biomed Sci. 2002;9:234–245. doi: 10.1007/BF02256070. [DOI] [PubMed] [Google Scholar]
- 48.Garrett S, Barton WA, Knights R, Jin P, Morgan DO, Fisher RP. Reciprocal activation by cyclin-dependent kinases 2 and 7 is directed by substrate specificity determinants outside the T loop. Mol Cell Biol. 2001;21:88–99. doi: 10.1128/MCB.21.1.88-99.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 50.Chou S, Upton H, Bao K, Schulze-Gahmen U, Samelson AJ, He N, Nowak A, Lu H, Krogan NJ, Zhou Q, Alber T. HIV-1 Tat recruits transcription elongation factors dispersed along a flexible AFF4 scaffold. Proc Natl Acad Sci U S A. 2013;110:e123–131. doi: 10.1073/pnas.1216971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulze-Gahmen U, Upton H, Birnberg A, Bao K, Chou S, Krogan NJ, Zhou Q, Alber T. The AFF4 scaffold binds human P-TEFb adjacent to HIV Tat. Elife. 2013;2:e00327. doi: 10.7554/eLife.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]