

Abstract
Background
Collagen type IV alpha 1 (COL4A1) and alpha 2 (COL4A2) form heterotrimers critical for vascular basement membrane stability and function. Patients with COL4A1 or COL4A2 mutations suffer from diverse cerebrovascular diseases including cerebral microbleeds, porencephaly and fatal intracerebral hemorrhage (ICH). However, the pathogenic mechanisms remain unknown and there is a lack of effective treatment.
Methods and Results
Using Col4a1 and Col4a2 mutant mouse models, we investigated the genetic complexity and cellular mechanisms underlying the disease. We found that Col4a1 mutations cause abnormal vascular development, which triggers small vessel disease, recurrent hemorrhagic strokes and age-related macro-angiopathy. We showed that allelic heterogeneity, genetic context and environmental factors, such as acute exercise or anticoagulant medication, modulated disease severity and contributed to phenotypic heterogeneity. We found that intracellular accumulation of mutant collagen in vascular endothelial cells and pericytes was a key triggering factor of ICH. Finally, we showed that treatment of mutant mice with a FDA-approved chemical chaperone resulted in a decreased collagen intracellular accumulation and a significant reduction of ICH severity.
Conclusions
Our data are the first to show therapeutic prevention in vivo of ICH due to Col4a1 mutation, and imply that a mechanism-based therapy promoting protein folding might also prevent ICH in patients with COL4A1 and COL4A2 mutations.
Keywords: cerebrovascular disorders, collagen, genetics, hemorrhage, stroke
Introduction
Strokes cause a death every four minutes in the United States, representing the fourth leading cause of death and a major cause of long-term disability1. Intracerebral hemorrhages (ICHs) are the most fatal form of stroke2. Lack of effective treatment options and poor clinical outcomes for ICH patients suggest that prevention is paramount for reducing the tremendous personal and societal burden. Development of preventive strategies relies on understanding environmental and genetic factors that contribute to ICH risk. Mutations in the genes encoding collagen type IV alpha 1 (COL4A1) and alpha 2 (COL4A2) cause highly penetrant cerebrovascular disease with variable expressivity3. COL4A1 mutations are a significant cause of porencephaly 4 and pediatric ICH, which are associated with particularly poor outcomes including cerebral palsy, intellectual disabilities, developmental and behavioral disorders, and epilepsy. COL4A1 and COL4A2 mutations also cause spontaneous ICHs in adults5, 6. Thus, COL4A1 and COL4A2 mutations are important causes of highly penetrant perinatal ICH and may play a substantial role in age-related cerebrovascular diseases.
COL4A1 and COL4A2 are extracellular matrix molecules that form a network integral to basement membranes7, 8. They are co-translationally translocated into the endoplasmic reticulum (ER) where they assemble into heterotrimers composed of one COL4A2 and two COL4A1 molecules9. Each protein has a large triple-helical domain flanked by the 7S10 and non-collagenous (NC1) domains at the amino and carboxy terminus, respectively11. The globular NC1 domains are responsible for initiating heterotrimer assembly, which proceeds by the progressive inter-winding of the triple-helical domains12, 13. Triple-helical domains are characterized by repeated Gly-Xaa-Yaa motifs (Xaa and Yaa represent variable amino acids) and form greater than 80% of the proteins. Like in other collagens14, 15, glycine missense mutations are the most common type of mutation16 and, in vitro, the primary consequence appears to be impaired heterotrimer biosynthesis5, 17, 18. How this may contribute to ICH in vivo remains unknown. Pathogenesis could involve toxic intracellular heterotrimer accumulation and/or extracellular deficiency of normal collagen and/or extracellular presence of mutant collagen19. Our objectives were to identify the molecular and cellular events that occur in the neurovascular unit leading to ICH in Col4a1 mutant mice and to identify preventative therapeutics that target these events. We investigated the timing and location of pathogenesis and potential roles of intracellular and extracellular insults using multiple mouse models. Importantly, we identified modifiable ICH risk factors and a pharmacologic intervention that reduced ICH in vivo. These data provide proof-of-principle for mechanism-based interventions to reduce, delay or prevent ICH in patients with COL4A1 and COL4A2 mutations.
Materials and Methods
Animals
Procedures were in accordance with Institutional Animal Care and Use Committee guidelines. Col4a1 and Col4a2 mutant mice were described previously18, 20. Each strain was iteratively crossed to C57BL/6J (B6) mice for at least five generations. CAST/EiJ (CAST) and 129S6/SvEvTac (129) breeders were mated with B6 mice to produce CASTB6F1 and 129B6F1 mice respectively. The Col4a1Flex41 conditional mutant mouse was produced by InGenious Targeting Laboratory (Stony Brook, NY). Rosa26-CreER 21 were used for ubiquitous inducible CRE expression. Tie2-Cre22, Pdgfrb-Cre23 and Gfap-Cre24 were used for cell-type specific CRE expression. We used ROSA26tm14(CAG-tdTomato) reporter mice25 to validate CRE-mediated recombination. Unless specified, all mutant mice were heterozygous and both sexes were used.
In vivo procedures
CRE was activated with tamoxifen (10 mg.mL−1, Sigma-Aldrich, St Louis, MO). Pregnant females received one intraperitoneal tamoxifen injection (2 mg) mixed with progesterone (1 mg, Sigma-Aldrich, St Louis, MO). Pups received one intragastric tamoxifen injection (50 μg) for three consecutive days, and 3-week-old mice received one intraperitoneal tamoxifen injection (2 mg) for two consecutive days.
Virgin 3-month-old females were exercised on a horizontal treadmill (Exer 3/6, Columbus Instruments, Columbus, OH) for five sessions. Each session included a 2-minute acclimation period (speed: 0 m.min−1), 8-minute warm-up (3 min at 6 m.min−1, 3 min at 9 m.min−1, 2 min at 12 m.min−1) and five 1-min sprints (1 min at 15 m.min−1 followed by 1 min rest at 0 m.min−1). Sessions were performed 5 days apart and animals were sacrificed 12 days after the last session.
Mice received Warfarin (Warfarin Sodium Tablets, Amneal Pharmaceuticals, Bridgewater, NJ) via drinking water at the estimated dose of 0.4 mg.kg−1 per day (2.5 mg dissolved in 800mL water) for 4 days, water without Warfarin for 2 days, then water with Warfarin for 3 days. Because two mutant mice died, treatment was discontinued and remaining animals were sacrificed 11 days later.
Mice received sodium 4-phenylbutyrate (4PBA, Enzo Life Sciences Inc., Farmingdale, NY) diluted in Phosphate Buffered Saline (PBS) by injection. Pups received intragastric injection of 0.1 mg 4PBA at postnatal day 1 (P1), P3, P5 and P7, then intraperitoneal injection of 0.5 mg 4PBA at P10, P12, P14, P16, P18 and P20, and finally received intraperitoneal injection of 1.0 mg 4PBA at P23 and P26. Mice were sacrificed at 1 month of age.
Retinal fluorescein angiography was performed using a Micron III (Phoenix Research Labs Inc., Pleasanton, CA). One-month old mice were anesthetized with Ketamine (100 mg.kg−1) – Xylazine (10 mg.kg−1) and received an intraperitoneal 20 μL Fluorescein Sodium injection (25 mg.mL−1 in PBS, Altaire Pharmaceuticals Inc., Riverhead, NY).
Histology and immunofluorescence labeling
Anesthetized mice were transcardially perfused with saline followed by 4% paraformaldehyde (PFA) in PBS. Brains were post-fixed in 4% PFA for 16 hours, cryo-protected in 30% sucrose/PBS and embedded in OCT (Sakura Finetek, Torrance, CA). For ICH quantification, coronal cryo-sections (35 μm) regularly spaced along the raustro-codal axis were stained with Prussian blue/Fast red. Images were acquired using a SteREO Discovery.V8 microscope, an AxioCam ICc3 camera and AxioVision 4.6 software (Carl Zeiss Microscopy, LLC, Germany). On each section, the percentage of brain area with Prussian blue staining was calculated using ImageJ software (NIH). Hemorrhage severity was expressed as the average percentage of hemosiderin surface area on 27 sections for each brain. Trichrome staining was performed on 5 μm paraffin sections according to the manufacturer protocol (One step Trichrome Blue/Red Stain kit, American Mastertech Scientific Inc., Lodi, CA).
For the investigation of blood-brain-barrier integrity, 10 mL of biotin (EZ-link sulfo-NHS-Biotin, 0.5 mg.mL−1 in PBS, Thermo Scientific, Waltham, MA) was transcardially perfused, after washing with saline and before fixation with 4% PFA. Coronal cryo-sections were fixed 15 min in 4% PFA and labeled with Streptavidin Alexa Fluor® 488 (2 pg.μL−1, Invitrogen-Molecular Probes, Carlsbad, CA). Alternatively, a 2% Evans blue solution in saline (4 mL.kg−1) was injected intraperitoneally and allowed to circulate 24 hours.
Embryos were fixed 1 hour in cold methanol, cryo-protected in 30% sucrose/PBS and cryo-sectioned (20 μm). For embryonic angiogenesis, hindbrains were fixed 1 hour in 4% PFA before flat-mounting. For retinal analysis, enucleated eyes were fixed 16 hours in 4% PFA and dissected retinas were flat mounted. Cultured primary fibroblasts were fixed 15 min in 4% PFA and permeabilized in PBS/0.1% Triton X-100 before immunolabelling. Cryo-sections were post-fixed in 4% PFA for 15 min prior to immunolabelling. All specimens were blocked in PBS with 10% normal donkey serum, 1% bovine serum albumin and 0.3% Triton X-100. Primary antibodies: rat CD31 (1:200, BD Biosciences, San Jose, CA), goat type IV collagen (1:200, Southern Biotech, Birmingham, AL), rat COL4A1 (1:100, H11 clone, Shigei Medical Research Institute, Japan26), rabbit ZO-1 (1:100, Abcam, Cambridge, UK), rabbit Claudin5 (1:100, Invitrogen, Carlsbad, CA) and mouse HSP47 (1:500, M16.10A1 clone, Stressgen Biotechnologies, San Diego, CA) were incubated 3 hours at room temperature for cryo-sections or for 48 hours at 4°C for flatmounts. After 3 washes in PBS/0.1% Triton X-100, secondary antibodies Alexa Fluor® 488 or 594 (1:500 dilution, Invitrogen-Molecular Probes, Carlsbad, CA) were incubated 1 hour for sections or 24 hours for flatmounts. After 3 washes, coverslips were mounted using Mowiol with DAPI (2 μg.mL−1). A Zeiss AxioImager M.1 and a Zeiss LSM700 with plan-Apochromat objectives (63x/1.4 oil-immersion or 20x/0.8) were used for fluorescence microscopy, with an AxioCam MRm camera and AxioVision and ZEN softwares (Carl Zeiss Microscopy, LLC, Germany). Embryonic vascular plexus density quantifications were performed using ImageJ. Vein branch point quantifications were realized by averaging numbers of at least three primary veins per retina.
Western Blot analysis
Mouse embryonic fibroblasts were cultured in DMEM with 10% fetal bovine serum, 2 mM L-glutamine, 0.2 mM penicillin/streptomycin at 37°C in 5% CO2. At confluence, cells were serum-deprived and treated with 50 μg.mL−1 of ascorbic acid for 16 hours. Proteins were separated on 4–15% gradient SDS-PAGE gels (Bio-Rad Laboratories Inc., Hercules, CA). Membranes were blocked 16 hours in Tris Buffered Saline/0.1% Tween-20 (TBS-T) with 5% non-fat milk, then incubated 16 hours with primary antibodies: rat COL4A1 (1:200, H11 clone, Shigei Medical Research Institute, Japan26), tubulin (1:10000, T6557, Sigma-Aldrich, St Louis, MO), laminin (1+2) (1:2000, ab7463, Abcam, Cambridge, UK). After 3 washes in TBS-T, membranes were incubated 1 hour with horseradish peroxidase-conjugated secondary antibodies (1:10000, Jackson ImmunoResearch, West Grove, PA). After 3 washes, SuperSignal West Pico Chemiluminescent substrate (Thermo Scientific, Waltham, MA) was used according to manufacturer’s instructions.
Statistical Analysis
Normality was assessed using Kolmogorov-Smirnov and Shapiro-Wilk tests. Two-group comparisons were carried out using Student’s t-test. Multiple group comparisons were performed using ANOVA followed by Tukey post-test or Kruskal-Wallis test followed by Dunn’s post-test for normally and not normally distributed variables, respectively. p values < 0.05 were considered statistically significant. Data are presented as mean + or ± standard deviation.
Results
Pathogenesis occurs in distinct stages
Col4a1Δex41 is a splice–site mutation that skips exon 41 in the triple-helical domain of murine Col4a13, 17, 27. Col4a1Δex41/Δex41 mice die during embryogenesis and Col4a1+/Δex41 mice exhibit embryonic growth retardation and reduced postnatal viability (Supplemental Figure 1). Modeling patients with COL4A1 mutations, surviving Col4a1+/Δex41 mice have multi-system disorders, including fully penetrant cerebrovascular disease presenting as porencephaly, prenatal, perinatal and recurrent multi-focal ICHs17, 27. At embryonic day (E) 10.5 we observed irregularly shaped and enlarged blood vessels, and multifocal intraparenchymal and intraventricular hemorrhages (Figure 1A). Analysis of cerebral angiogenesis at E12.5 revealed increased vessel density and tortuosity in Col4a1+/Δex41 hindbrains (Figure 1B). Mature retinal vasculature showed similar defects including persistent hyaloid vessels and abnormal crossing of excessively branched, tortuous arteries and veins with irregular diameters (Figure 1C). We also detected tortuous cerebral arteries, veins and capillaries in adult Col4a1+/Δex41 mice (Figure 1D). Cerebral and retinal vessels were not permeable to biotin, Evans blue dye or sodium fluorescein and tight junction proteins were normally expressed during brain development indicating that the blood-brain and blood-retinal barriers are intact (Figure 1D and Supplemental Figure 2).
Figure 1.
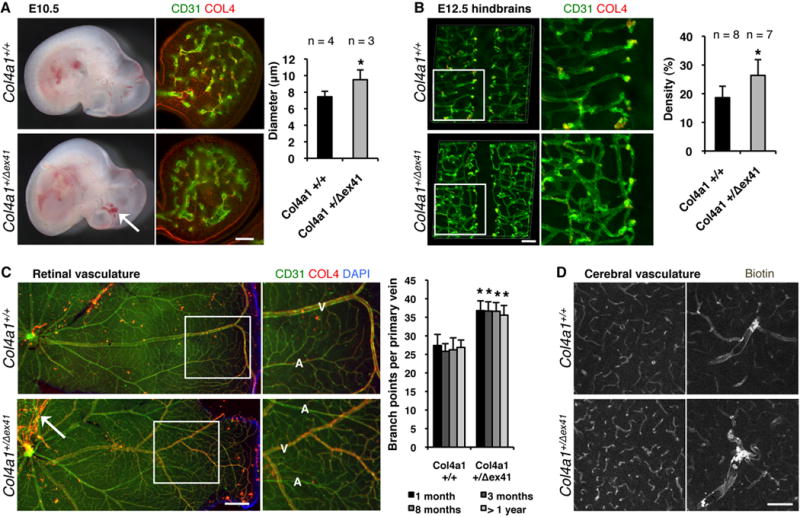
Col4a1 mutation causes abnormal cerebral and retinal vascular development. (A) At embryonic day (E) 10.5 Col4a1+/Δex41 mice have irregularly shaped, enlarged blood vessels (calculated in branchial arches using CD31 and Collagen type IV (COL4) labeling, scale bar: 100 μm) and intracerebral hemorrhages (arrow). (B) At E12.5 Col4a1+/Δex41 mice have abnormally tortuous vessels with increased density (calculated in hindbrain flatmounts using CD31 labeling, scale bar: 100 μm) (C) In mutant animals, retinal blood vessels are tortuous, arteries (A) and veins (V) cross each other and there is excess branching in the main veins (5≤n≤16, pair-wise comparisons per age, scale bar: 500 μm). (D) Intracardiac perfusion of biotin revealed that adult mutant animals have tortuous cerebral blood vessels without compromised blood-brain-barrier (scale bar: 100 μm). Data are reported as mean + standard deviation and * indicates p<0.05 compared to Col4a1+/+ by Student’s t-test.
At birth, Col4a1+/Δex41 pups had numerous macroscopic subcutaneous hematomas, extra-axial hemorrhages and frequent intraparenchymal hemorrhages (Supplemental Figure 1C). Consistent with highly penetrant intraventricular hemorrhages, 80% of Col4a1+/Δex41 mice had porencephaly (Figure 2A). ICH was completely penetrant, however, the location and character of the lesions changed with time suggesting different stages of pathology. At 1-month, Col4a1+/Δex41 mice had small vessel disease, with multiple, small hemosiderin deposits throughout the cortex, cerebral nuclei, brain stem and cerebellum. By 3-months, these were cleared but fewer and larger hemorrhages appeared in the basal ganglia. Reduced hemoglobin level and age-dependent hemosiderin accumulation indicated recurring ICHs (Figure 2A and Supplemental Figure 3). Importantly, cerebral macro-angiopathy with fibrotic vessel wall thickening and thrombus formation appeared with age (Figure 2B).
Figure 2.
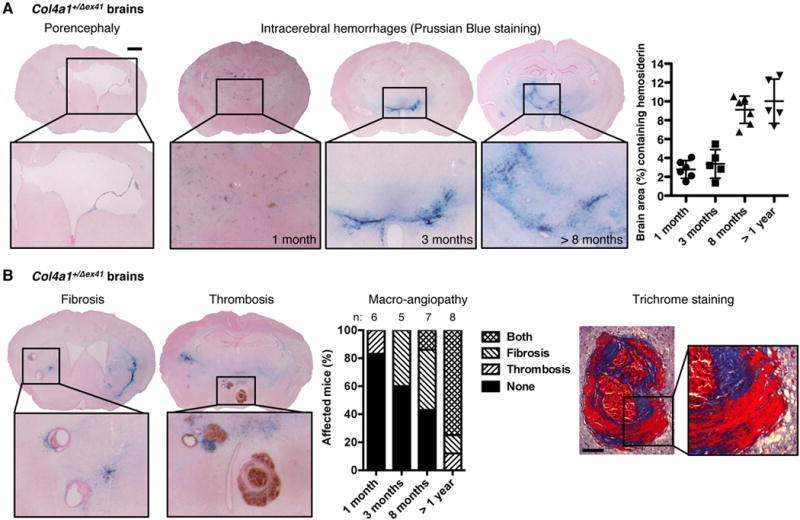
Col4a1 mutation causes small vessel disease, hemorrhagic stroke and macro-angiopathy. (A) Col4a1+/Δex41 mice have porencephaly (penetrance: 80%) and intracerebral hemorrhages (penetrance: 100%) quantified using Prussian blue staining of serial coronal brain sections (scale bar: 1 mm). Data are reported as mean ± standard deviation. (B) Macro-angiopathy with vessel wall thickening, fibrosis and thrombosis advanced with age. Trichrome staining shows collagen deposition (blue) and fibrin accumulation (red) in affected blood vessels (scale bar: 100 μm). No Col4a1+/+ mice had porencephaly, intracerebral hemorrhage or macro-angiopathy (n=32).
Age-related ICH and macro-angiopathy are consequences of developmental defects
To distinguish between potentially distinct roles for COL4A1 in cerebrovascular development and maintenance we engineered a conditional Col4a1 mutation by flanking exon 41 with LoxP sites (Col4a1Flex41). This allele recreates the Col4a1Δex41 mutation in a CRE-dependent manner (Supplemental Figure 4). We crossed Col4a1+/Flex41 mice to the inducible, ubiquitous CRE strain R26-CreER 21, injected tamoxifen at birth, 1 week or 3 weeks, validated CRE activation and quantified retinal branching and ICHs (Figure 3 and Supplemental Figure 5). In 8-month-old R26-CreER; Col4a1+/Flex41 mice, ICHs were more severe in mice that started to express mutant collagen at birth than at 1 week and were absent in mice that started to express mutant collagen at 3 weeks. Pathology in neither group was as severe as Col4a1+/Δex41 and none of the mice had porencephaly or macro-angiopathy. These data demonstrate that age-related, recurrent ICHs and macro-angiopathy do not occur in the absence of pre- and post-natal developmental defects. To refine the timing of pathogenesis we induced the Col4a1 mutation at E10.5 or E14.5 and quantified ICHs at 1 month. ICHs were more severe in mice induced at E10.5 than at E14.5. Again, neither group was as severe as Col4a1+/Δex41, however, controls revealed reduced recombination efficiency with embryonic induction of mutant Col4a1 (Supplemental Figure 5). Together, these data demonstrate that the effects of mutant COL4A1 require expression during embryogenesis and the early postnatal period in order to cause progressive, recurrent ICHs and age-related macro-angiopathy.
Figure 3.
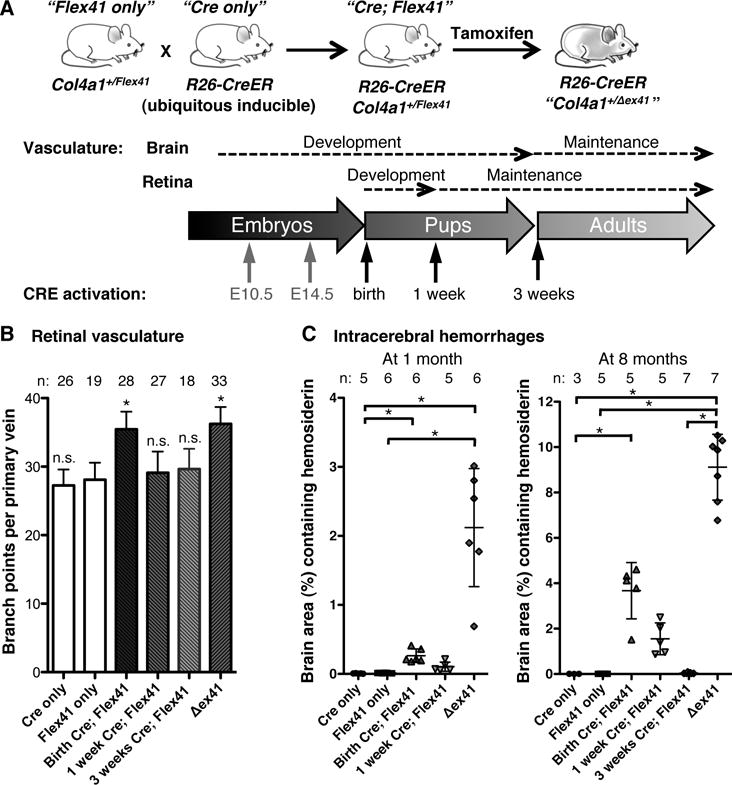
Mutant Col4a1 causes cerebrovascular disease when expressed during vascular development. (A) Col4a1+/Flex41 conditional mutant mice were crossed to R26-CreER ubiquitous inducible CRE mice and the CRE recombinase was activated at different time points during or after vascular development by tamoxifen injections. (B) Quantification of retinal vessels revealed excess branching in mice that started to express mutant COL4A1 at birth but not at 1- or 3-weeks. n.s.: p>0.05, *: p<0.05 compared to Flex41 only (Col4a1+/Flex41; R26-CreER−/-) mice by Student’s t-test. (C) ICH quantification at 1 or 8 months of age shows that ICH severity increases with earlier age of expression of the collagen mutation. *: p<0.05 by Kruskal-Wallis test followed by Dunn’s post-test. Data are reported as mean ± standard deviation.
ICH modulation by environmental and genetic factors
We sought ways to reduce the severity of progressive ICH. In the previous experiment, tamoxifen injection during pregnancy compromised natural birth and necessitated surgical delivery. A beneficial effect of surgical delivery was previously proposed but not quantified27. We found that tamoxifen-treated, surgically delivered Col4a1+/Δex41 mice had less ICHs than non-treated, naturally born Col4a1+/Δex41 mice (Figure 4A). Next, we quantified the impacts of other modifiable risk factors that have been anecdotally associated with ICH in patients with COL4A1 or COL4A2 mutations including physical exertion28–30, and anticoagulant administration5, 6, 27, 31. We found that exercise increased ICH severity (Figure 4B) and anticoagulants provoked fatal hemorrhages within just seven days of use (Figure 4C). Together, these data establish vaginal delivery, exercise and anticoagulants as modifiable risk factors that increase ICH severity in Col4a1+/Δex41 mice.
Figure 4.
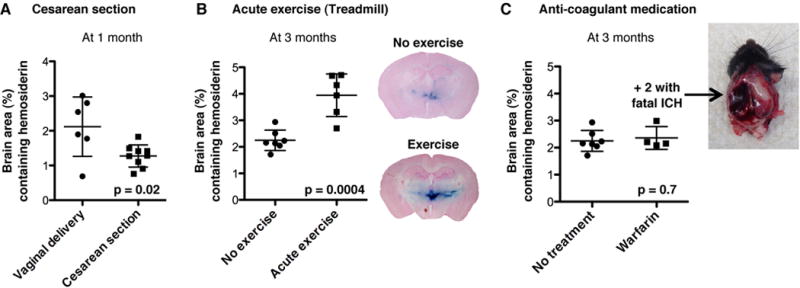
Environmental factors contribute to Col4a1 mutation expressivity. Intracerebral hemorrhage quantification in Col4a1+/Δex41 mice: (A) after vaginal delivery or Cesarean section, (B) without or with five sessions of five one-minute sprints on a treadmill, (C) without or with 7 days of Warfarin anticoagulant administration. Data are reported as mean ± standard deviation.
Genetic factors may also influence clinical heterogeneity among patients with COL4A1 and COL4A2 mutations32, 33 and can reveal pathogenic mechanisms and interventional approaches. We compared ICH severity in 1) mice with different mutations on the same genetic background and 2) mice with the same mutation on different genetic backgrounds. The allelic series comprised Col4a1Δex41, seven glycine mutations in the triple-helical domain (six in COL4A1, one in COL4A2) and one mutation in the COL4A1 NC1 domain (Figure 5A)18, 20. We backcrossed each strain to C57BL/6J (B6) mice, aged cohorts for 7.5–9.5 months (called 8-month cohort) and compared porencephaly, ICH and macro-angiopathy. We identified distinct classes of mutations and three potential genotype-phenotype correlations (Figure 5A and Supplemental Figure 6). First, there appears to be a ‘class effect’ whereby Col4a1Δex41 is more severe than missense mutations. Second is a domain effect whereby the NC1 domain mutation is milder than the triple-helical domain mutations. Third, for mutations within the triple-helical domain, there was a position effect whereby mutations nearer the carboxy terminus were more severe than mutations nearer the amino terminus. These data establish the powerful effects of allelic heterogeneity on ICH severity and suggest that there are class, domain and position effects of different alleles that resemble effects observed in other types of collagens19, 34–36. Next, we tested the effect of genetic context on ICH severity. We crossed B6 Col4a1+/Δex41 mice to CAST/EiJ (CAST) or 129S6/SvEvTac (129) mice to generate CASTB6F1 or 129B6F1 mice respectively. We compared ICH severity among Col4a1+/Δex41 mice and found that CASTB6F1 genetic context significantly suppressed ICH but 129B6F1 did not (Figure 5B). CASTB6F1 also reduced porencephaly penetrance, and delayed macro-angiopathy onset (Supplemental Figure 7). The effect was not allele- or gene-specific as CASTB6F1 also significantly suppressed ICH in Col4a1+/G1344D and Col4a2+/G646D mice (Figure 5C). Surprisingly, there were neither allelic nor genetic context effects on retinal or cerebral blood vessel defects (Figure 5D). The observations of allelic and genetic-context effects on ICH – but not on the vascular patterning defects – suggest that processes in addition to defective angiogenesis and vascular patterning may be required for ICH progression.
Figure 5.
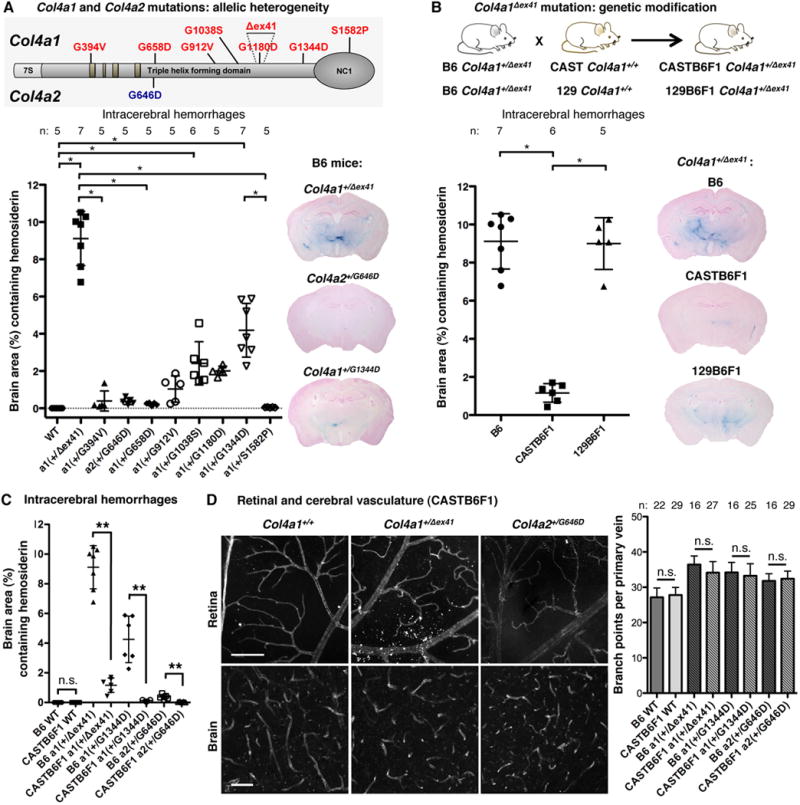
Allelic heterogeneity and genetic context contribute to the expressivity of Col4a1 and Col4a2 mutations. (A) Intracerebral hemorrhage (ICH) quantification in 8-month-old mice with different mutations but the same genetic background, C57BL/6J (B6), demonstrates that allelic heterogeneity influences ICH severity. *: p<0.05 by Kruskal-Wallis test followed by Dunn’s post-test. (B) ICH quantification in 8-month-old mice with the same Col4a1Δex41 mutation but different genetic contexts shows that genetic background influences ICH severity. A cross for one generation to CAST/EiJ (CAST), but not 129S6/SvEvTac (129) mice was sufficient to reduce ICH severity compared to Col4a1+/Δex41 mice on a B6 background. *: p<0.05 by ANOVA followed by Tukey post-test. (C) CAST suppression of ICH was consistent across different mutations, however, (D) did not extend to prevention of vascular tortuosity and excess branching (COL4 labeling in the retina, CD31 labeling in the brain, scale bars: 50 μm). In (C) and (D) n.s. indicates p>0.05 and ** indicates p<0.01 by Student’s t-test. Data are reported as mean ± (A, B and C) or + (D) standard deviation.
Pathogenic collagen accumulation in vascular endothelial cells and pericytes
Heterotrimers that incorporate mutant COL4A1 or COL4A2 tend to accumulate within cells at the expense of secretion. Because ICH severity correlates with intracellular accumulation in the allelic series18, we hypothesized that CASTB6F1 suppression of ICH might occur by reducing intracellular COL4A1. We compared intracellular and extracellular COL4A1 in primary fibroblasts and found that Col4a1+/Δex41 caused significantly increased intracellular COL4A1 in B6 but not CASTB6F1 cells (Figure 6A). Moreover, the CASTB6F1 background normalized intracellular without increasing extracellular COL4A1 levels – a distinction that was also striking in vivo. Blood vessels from B6 Col4a1+/Δex41 animals had punctuate intracellular COL4 labeling and less intense labeling of the vascular basement membrane than Col4a1+/+ mice. CASTB6F1 Col4a1+/Δex41 vessels do not show intracellular labeling yet still lacked intense labeling of the basement membrane (Figure 6B). Thus, mutant mice from both backgrounds have reduced extracellular COL4A1 yet only B6 mice have significant intracellular accumulation and severe ICH. Importantly, these data dissect the potential roles of intracellular accumulation and extracellular deficiency and suggest that chronic intracellular accumulation may cause ICH in the context of developmentally abnormal vessels.
Figure 6.
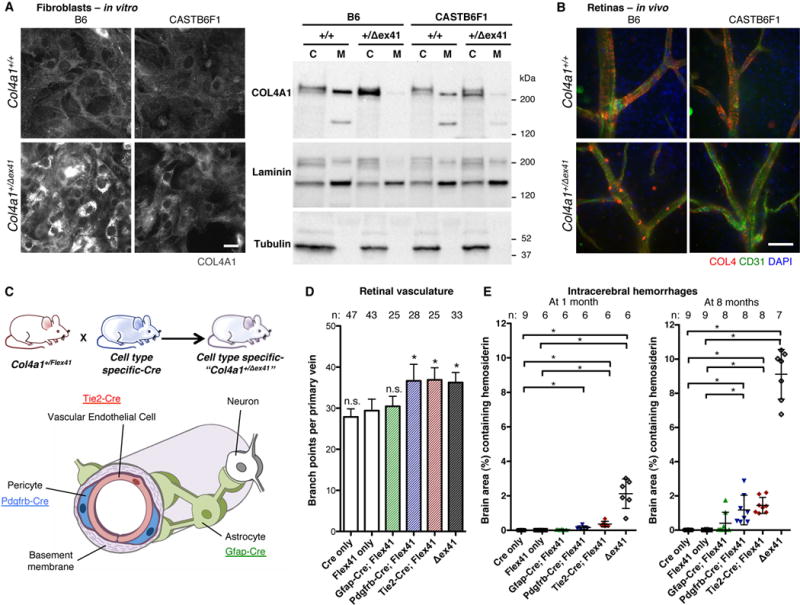
Intracerebral hemorrhages associate with intracellular COL4A1 accumulation in vascular endothelial cells and pericytes but not with extracellular deficiency. (A) Immunolabeling and Western blot show that intracellular COL4A1 in Col4a1+/Δex41 cells from B6 mice is greatly reduced in cells from CASTB6F1 mice but without an obvious increase in extracellular COL4A1 (C: cell lysate, M: conditioned medium, Laminin and Tubulin are controls for secreted and intracellular proteins, respectively). Results constitute representative examples from twelve independent biological replicates obtained from three independent experiments. (B) Immunolabeling of Col4a1+/Δex41 retinal vessels shows strong intracellular collagen type IV (COL4) in B6 but not CASTB6F1 mice. (C) Col4a1+/Flex41 mice were crossed to Tie2-Cre, Pdgfrb-Cre or Gfap-Cre strains for mutant Col4a1 conditional expression in vascular endothelial cells (VECs), pericytes or astrocytes respectively. (D) Retinal vessel quantification revealed excess branching with mutant expression in VECs and pericytes but not astrocytes. n.s.: p>0.05, *: p<0.05 compared to Flex41-only mice (Col4a1+/Flex41; Cre−/-) by Student’s t-test. (E) Intracerebral hemorrhage (ICH) quantification demonstrated that mutant Col4a1 expression in VECs or pericytes is sufficient to cause ICH but that neither cell-type alone phenocopies Col4a1+/Δex41. Conditional expression of mutant Col4a1 in astrocytes caused very mild ICHs in only three 8-month-old animals. *: p<0.05 by Kruskal-Wallis test followed by Dunn’s post-test. Scale bars: 50 μm. Data are reported as mean + (D) or ± (E) standard deviation.
To test the relative effects of the different cell types of the neurovascular unit, we conditionally expressed mutant Col4a1 in vascular endothelial cells (VECs), pericytes or astrocytes using Tie2-Cre22, Pdgfrb-Cre23 or Gfap-Cre24 transgenic mice respectively (Figure 6C). We validated these lines (Supplemental Figure 8A) and quantified ICH at 1 and 8 months. Specific expression of mutant Col4a1 only in astrocytes did not overall significantly affect retinal branching and gave very mild ICHs in only 3 out of 8 old animals (Figure 6D and E). Expression in VECs– or pericytes–only was sufficient to phenocopy excess retinal vascular branching of Col4a1+/Δex41 (Figure 6D), caused fully penetrant ICH (Figure 6E) and incompletely penetrant porencephaly and macro-angiopathy (Supplemental Figure 8B). However neither strain demonstrated ICH as severe as that in Col4a1+/Δex41 mice. Interestingly, when we tested E16.5 embryos, ICH was greater in Tie2-Cre; Col4a1+/Flex41 compared to Pdgfrb-Cre; Col4a1+/Flex41 mice and the postnatal viability of Tie2-Cre; Col4a1+/Flex41 mice was much lower (27% compared to 100% for Pdgfrb-Cre; Col4a1+/Flex41 mice) (Supplemental Figure 4D). Therefore, the most severely affected Tie2-Cre; Col4a1+/Flex41 mice may be underrepresented in postnatal analyses leading to an underestimate of the relative role of VECs in the pathogenesis. These data suggest that both VECs and pericytes contribute to vascular defects and that VECs may play a relatively larger role.
The results from CASTB6F1 mice suggest that reducing intracellular accumulation may suppress ICH, and results from the inducible mutation indicate that postnatal intervention may be effective. Sodium 4-phenylbutyrate (4PBA), an FDA-approved chemical chaperone, has been used in several models of disorders due to protein misfolding37 and we recently showed that its properties extend to COL4A1 in vitro18. To test our hypothesis in vivo, we treated pups with 4PBA from birth to weaning age and analyzed retinal vasculature and ICH at 1 month. Treatment had no effect on vessel branching, however, there was decreased intracellular accumulation and qualitatively more uniform labeling of vascular basement membranes. Importantly, compared to untreated Col4a1+/Δex41 mice, mutant animals treated with 4PBA had significantly less severe ICH (Figure 7). These data are the first to show therapeutic prevention of Col4a1-related ICH in vivo and support the hypothesis that promoting protein folding might also prevent ICH in patients with COL4A1 and COL4A2 mutations.
Figure 7.
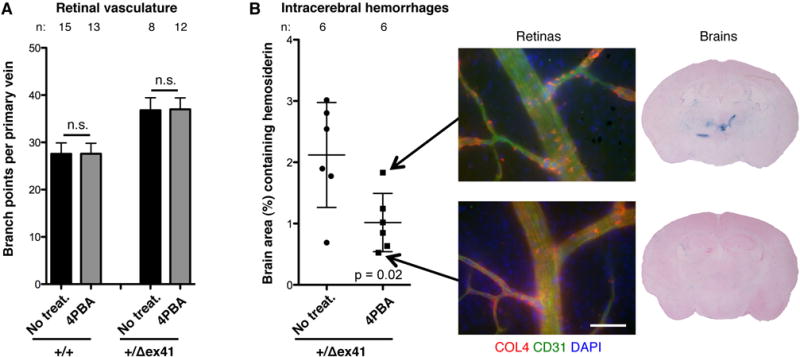
Treatment with chemical chaperone sodium 4-phenylbutyrate (4PBA) reduces intracerebral hemorrhage severity in vivo. Col4a1+/Δex41 mice treated from birth to 3 weeks with sodium 4-phenylbutyrate (4PBA) had similar levels of retinal vessel branching (A) but less severe intracerebral hemorrhages (ICHs) (B) compared to untreated Col4a1+/Δex41 mice. n.s.: p>0.05 by Student’s t-test. Immunolabeled vessels showed less intracellular and more extracellular collagen type IV (COL4) in the treated Col4a1+/Δex41 mouse with the least, compared to greatest, ICH severity. Scale bars: 50 μm. Data are reported as mean + (A) or ± (B) standard deviation.
Discussion
Col4a1 and Col4a2 mutant mice model important aspects of human disease. Embryonic germinal matrix hemorrhages cause porencephaly and early small vessel disease presents as microbleeds throughout the brain. Later, recurrent ICHs and age-related macro-angiopathy occur mainly in the basal ganglia. Importantly, our data bring new insights into the disease biology and suggest that two pathogenic mechanisms are necessary to cause ICH. First, we showed that Col4a1 and Col4a2 mutations cause abnormal angiogenesis. The fact that mice which start to express mutant collagen after the completion of vascular development do not suffer from ICH suggests that abnormal angiogenesis is required to trigger hemorrhage. Second, we showed that Col4a1 mutation causes increased intracellular and decreased extracellular collagen. The fact that ICH severity associates with the level of intracellular accumulation but not with extracellular deficiency (Figure 5B and 6A and B) suggests that the intracellular accumulation is a key molecular mechanism in the progression to vasculature rupture. A similar observation was reported in human cells cultured from an ICH patient with a COL4A2 mutation and his unaffected father who also carried the mutation38.The fact that the blood-brain-barrier of the abnormally developed vessels is not generally compromised also supports this hypothesis, suggesting that chronic intracellular accumulation could reach locally toxic thresholds and provoke focal ICH. Interestingly, we show that abnormal angiogenesis is independent from the level of intracellular collagen accumulation, suggesting that extracellular insults might underlie abnormal vascular development. To support this hypothesis, it would be interesting to investigate the COL4A1/COL4A2 network in the vascular basement membrane. Indeed, this network has been shown to interact directly with several signaling pathways involved in angiogenesis such as TGFβ/BMP signaling39, 40, Notch signaling41 and integrin signaling42 and Arresten, a proteolytic fragment of COL4A1 has anti-angiogenic properties43. Although further understanding the roles of COL4A1 and COL4A2 in angiogenesis is compelling, developmental defects remain challenging to target therapeutically. Thus, we showed that targeting intracellular collagen accumulation using 4PBA, even after birth, successfully decreases ICH severity in vivo. These data support that mechanism-based treatments, such as chemical chaperones, might also prevent ICH in patients with COL4A1 and COL4A2 mutations.
We investigated a clinically relevant series of Col4a1 and Col4a2 mutations and discovered important allelic effects that resemble those identified for other type of collagens19, 34–36. These data have implications for patient screening and for functionally validating mutations that are identified by genetic testing. We show that mutation class, domain and position all influence ICH severity and correlate with intracellular accumulation of heterotrimers18. Mutations that are near the amino terminus of the triple-helical domain may cause only mild intracellular collagen accumulation and therefore be overlooked by functional assays that measure relative levels of intracellular collagen resulting in false negatives (e.g. Col4a1+/G394V)18. Therefore position effects should be considered when functionally testing putative mutations and the incidence of COL4A1 and COL4A2 mutations may be higher than current estimates. Interestingly, despite differences in stoichiometric contributions to heterotrimer composition, positionally matched mutations in Col4a1 and Col4a2 caused similar ICH severity, which underscores the importance of analyzing both genes when genetically screening patients.
Importantly, our data suggests that the mechanism by which COL4A1 and COL4A2 mutations cause ICH might be specific to this phenotype, which may have important implications for patient prognosis and treatment. First, 129B6F1 genetic context modified ocular dysgenesis32 but not ICH. Second, in the allelic series, ICH severity correlates with levels of intracellular accumulation whereas myopathy does not18. This is illustrated by Col4a1G394V, which is among the mutations causing the most severe myopathy, but the least severe ICH. HANAC (hereditary angiopathy, nephropathy, aneurysms, and cramps) describes a sub-set of patients with myopathy, relatively mild cerebrovascular disease, and whose mutations cluster near integrin binding domains in the amino-terminal quarter of COL4A1. Our data (Figure 5A and ref.18) suggest a molecular explanation for HANAC whereby mutations in or near integrin binding domains lead to myopathy, and because these domains are near the amino terminus, the mutations cause only mild intracellular collagen accumulation and relatively mild ICH. Mechanistic heterogeneity has important implications for targeted therapies and patient treatment. These results suggest that pharmacologic interventions that prevent ICH might not be efficacious for other phenotypes and that comprehensive patient treatment might require distinct approaches for distinct pathologies.
We have identified vaginal delivery, acute exercise and anticoagulant use as modifiable environmental risk factors that exacerbate ICH severity in Col4a1+/Δex41 mice. Anticoagulation is a highly effective treatment for prevention of thromboembolic stroke. However, our results and reports of patients with COL4A1 or COL4A2 mutations suffering from hemorrhagic stroke while taking anticoagulant medication suggest that the risks of antithrombotic therapy may be judged to outweigh its benefits5, 6, 27, 31. These decisions will need to be measured against the increased risk for ICH. Similarly, Col4a1 mutations are not associated with hypertension27, 44, however, acute exercise in mice induced ICH and has been associated with hemorrhagic events in patients28–30. These data are important for patients with COL4A1 and COL4A2 mutations who are considering the beneficial or detrimental consequences of physical exercise. Although Cesarean delivery will not reduce or prevent embryonic ICH or porencephaly, our data demonstrate that Cesarean delivery of genetically at-risk individuals may reduce perinatal ICH. Because of the exacerbating effects of acute exercise, Cesarean delivery may also be warranted when the mother carries the mutation.
Altogether, we used an allelic series, genetic modification and a conditionally expressed Col4a1 mutation to distinguish the relative impacts of intracellular accumulation from extracellular COL4A1 deficiency, dissect cell-type specific contributions, and define a window for potential therapeutic intervention. Our data suggest that intracellular collagen accumulation is responsible for ICH progression. Thus, promoting either heterotrimer degradation or secretion may be viable approaches to reduce ICH severity. Importantly, we show that 4PBA, a readily available small molecule with chaperone properties, reduced ICH severity in vivo. Together this work demonstrates efficacy of controlling modifiable environmental risk factors and application of a mechanism-based therapy to prevent ICH caused by COL4A1 mutations.
Supplementary Material
Acknowledgments
We thank J. Rosand, R. Libby, R. Daneman, P. Yurchenco and members of the Gould lab for critical reviews of the manuscript.
Funding Sources: This study was funded by the American Heart Association (AHA), National Institutes of Heath (NS083830) and That Man May See (D.B.G.). Support was also provided by an AHA postdoctoral fellowship (M.J.) and by a core grant from the National Eye Institute (EY02162), a Research To Prevent Blindness Unrestricted Grant (UCSF Department of Ophthalmology), the Sandler Program in Basic Sciences (D.B.G.) and the Karl Kirchgessner Foundation (D.B.G.).
Footnotes
Journal Subject Code: [130] Animal models of human disease
Disclosures: None.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Spontaneous intracerebral hemorrhage. N Engl J Med. 2001;344:1450–60. doi: 10.1056/NEJM200105103441907. [DOI] [PubMed] [Google Scholar]
- 3.Kuo DS, Labelle-Dumais C, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet. 2012;21:R97–110. doi: 10.1093/hmg/dds346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoneda Y, Haginoya K, Arai H, Yamaoka S, Tsurusaki Y, Doi H, Miyake N, Yokochi K, Osaka H, Kato M, Matsumoto N, Saitsu H. De novo and inherited mutations in COL4A2, encoding the type IV collagen alpha2 chain cause porencephaly. Am J Hum Genet. 2012;90:86–90. doi: 10.1016/j.ajhg.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeanne M, Labelle-Dumais C, Jorgensen J, Kauffman WB, Mancini GM, Favor J, Valant V, Greenberg SM, Rosand J, Gould DB. COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke. Am J Hum Genet. 2012;90:91–101. doi: 10.1016/j.ajhg.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weng YC, Sonni A, Labelle-Dumais C, de Leau M, Kauffman WB, Jeanne M, Biffi A, Greenberg SM, Rosand J, Gould DB. COL4A1 mutations in patients with sporadic late-onset intracerebral hemorrhage. Ann Neurol. 2012;71:470–7. doi: 10.1002/ana.22682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yurchenco PD, Amenta PS, Patton BL. Basement membrane assembly, stability and activities observed through a developmental lens. Matrix Biol. 2004;22:521–38. doi: 10.1016/j.matbio.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Yurchenco PD, Ruben GC. Basement membrane structure in situ: evidence for lateral associations in the type IV collagen network. J Cell Biol. 1987;105:2559–68. doi: 10.1083/jcb.105.6.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khoshnoodi J, Sigmundsson K, Cartailler JP, Bondar O, Sundaramoorthy M, Hudson BG. Mechanism of chain selection in the assembly of collagen IV: a prominent role for the alpha2 chain. J Biol Chem. 2006;281:6058–69. doi: 10.1074/jbc.M506555200. [DOI] [PubMed] [Google Scholar]
- 10.Dixit SN, Stuart JM, Seyer JM, Risteli J, Timpl R, Kang AH. Type IV collagens’ isolation and characterization of 7S collagen from human kidney, liver and lung. Coll Relat Res. 1981;1:549–56. doi: 10.1016/s0174-173x(81)80036-2. [DOI] [PubMed] [Google Scholar]
- 11.Trueb B, Grobli B, Spiess M, Odermatt BF, Winterhalter KH. Basement membrane (type IV) collagen is a heteropolymer. J Biol Chem. 1982;257:5239–45. [PubMed] [Google Scholar]
- 12.Ries A, Engel J, Lustig A, Kuhn K. The function of the NC1 domains in type IV collagen. J Biol Chem. 1995;270:23790–4. doi: 10.1074/jbc.270.40.23790. [DOI] [PubMed] [Google Scholar]
- 13.Boutaud A, Borza DB, Bondar O, Gunwar S, Netzer KO, Singh N, Ninomiya Y, Sado Y, Noelken ME, Hudson BG. Type IV collagen of the glomerular basement membrane. Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J Biol Chem. 2000;275:30716–24. doi: 10.1074/jbc.M004569200. [DOI] [PubMed] [Google Scholar]
- 14.Kuivaniemi H, Tromp G, Prockop DJ. Mutations in collagen genes: causes of rare and some common diseases in humans. FASEB J. 1991;5:2052–60. doi: 10.1096/fasebj.5.7.2010058. [DOI] [PubMed] [Google Scholar]
- 15.Prockop DJ, Kivirikko KI. Heritable diseases of collagen. N Engl J Med. 1984;311:376–86. doi: 10.1056/NEJM198408093110606. [DOI] [PubMed] [Google Scholar]
- 16.Engel J, Prockop DJ. The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper. Annu Rev Biophys Biophys Chem. 1991;20:137–52. doi: 10.1146/annurev.bb.20.060191.001033. [DOI] [PubMed] [Google Scholar]
- 17.Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, Aguglia U, van der Knaap MS, Heutink P, John SW. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–71. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- 18.Kuo DS, Labelle-Dumais C, Mao M, Jeanne M, Kauffman WB, Allen J, Favor J, Gould DB. Allelic heterogeneity contributes to variability in ocular dysgenesis, myopathy and brain malformations caused by Col4a1 and Col4a2 mutations. Hum Mol Genet. 2014;23:1709–22. doi: 10.1093/hmg/ddt560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, Hyland JC, Korkko J, Prockop DJ, De Paepe A, Coucke P, Symoens S, Glorieux FH, Roughley PJ, Lund AM, Kuurila-Svahn K, Hartikka H, Cohn DH, Krakow D, Mottes M, Schwarze U, Chen D, Yang K, Kuslich C, Troendle J, Dalgleish R, Byers PH. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–21. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Favor J, Gloeckner CJ, Janik D, Klempt M, Neuhauser-Klaus A, Pretsch W, Schmahl W, Quintanilla-Fend L. Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics. 2007;175:725–36. doi: 10.1534/genetics.106.064733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Badea TC, Wang Y, Nathans J. A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J Neurosci. 2003;23:2314–22. doi: 10.1523/JNEUROSCI.23-06-02314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–42. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 23.Foo SS, Turner CJ, Adams S, Compagni A, Aubyn D, Kogata N, Lindblom P, Shani M, Zicha D, Adams RH. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell. 2006;124:161–73. doi: 10.1016/j.cell.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 24.Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 25.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–40. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sado Y, Kagawa M, Kishiro Y, Sugihara K, Naito I, Seyer JM, Sugimoto M, Oohashi T, Ninomiya Y. Establishment by the rat lymph node method of epitope-defined monoclonal antibodies recognizing the six different alpha chains of human type IV collagen. Histochem Cell Biol. 1995;104:267–75. doi: 10.1007/BF01464322. [DOI] [PubMed] [Google Scholar]
- 27.Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, Bousser MG, Heutink P, Miner JH, Tournier-Lasserve E, John SW. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354:1489–96. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
- 28.Vahedi K, Kubis N, Boukobza M, Arnoult M, Massin P, Tournier-Lasserve E, Bousser MG. COL4A1 mutation in a patient with sporadic, recurrent intracerebral hemorrhage. Stroke. 2007;38:1461–4. doi: 10.1161/STROKEAHA.106.475194. [DOI] [PubMed] [Google Scholar]
- 29.Gunda B, Mine M, Kovacs T, Hornyak C, Bereczki D, Varallyay G, Rudas G, Audrezet MP, Tournier-Lasserve E. COL4A2 mutation causing adult onset recurrent intracerebral hemorrhage and leukoencephalopathy. J Neurol. 2014;261:500–3. doi: 10.1007/s00415-013-7224-4. [DOI] [PubMed] [Google Scholar]
- 30.Corlobe A, Tournier-Lasserve E, Mine M, Menjot de Champfleur N, Carra Dalliere C, Ayrignac X, Labauge P, Arquizan C. COL4A1 mutation revealed by an isolated brain hemorrhage. Cerebrovasc Dis. 2013;35:593–4. doi: 10.1159/000351520. [DOI] [PubMed] [Google Scholar]
- 31.Alamowitch S, Plaisier E, Favrole P, Prost C, Chen Z, Van Agtmael T, Marro B, Ronco P. Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome. Neurology. 2009;73:1873–82. doi: 10.1212/WNL.0b013e3181c3fd12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gould DB, Marchant JK, Savinova OV, Smith RS, John SW. Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum Mol Genet. 2007;16:798–807. doi: 10.1093/hmg/ddm024. [DOI] [PubMed] [Google Scholar]
- 33.Labelle-Dumais C, Dilworth DJ, Harrington EP, de Leau M, Lyons D, Kabaeva Z, Manzini MC, Dobyns WB, Walsh CA, Michele DE, Gould DB. COL4A1 Mutations Cause Ocular Dysgenesis, Neuronal Localization Defects, and Myopathy in Mice and Walker-Warburg Syndrome in Humans. PLoS Genet. 2011;7:e1002062. doi: 10.1371/journal.pgen.1002062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Starman BJ, Eyre D, Charbonneau H, Harrylock M, Weis MA, Weiss L, Graham JM, Jr, Byers PH. Osteogenesis imperfecta. The position of substitution for glycine by cysteine in the triple helical domain of the pro alpha 1(I) chains of type I collagen determines the clinical phenotype. J Clin Invest. 1989;84:1206–14. doi: 10.1172/JCI114286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Byers PH, Wallis GA, Willing MC. Osteogenesis imperfecta: translation of mutation to phenotype. J Med Genet. 1991;28:433–42. doi: 10.1136/jmg.28.7.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willing MC, Deschenes SP, Scott DA, Byers PH, Slayton RL, Pitts SH, Arikat H, Roberts EJ. Osteogenesis imperfecta type I: molecular heterogeneity for COL1A1 null alleles of type I collagen. Am J Hum Genet. 1994;55:638–47. [PMC free article] [PubMed] [Google Scholar]
- 37.Iannitti T, Palmieri B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D. 2011;11:227–49. doi: 10.2165/11591280-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murray LS, Lu Y, Taggart A, Van Regemorter N, Vilain C, Abramowicz M, Kadler KE, Van Agtmael T. Chemical chaperone treatment reduces intracellular accumulation of mutant collagen IV and ameliorates the cellular phenotype of a COL4A2 mutation that causes haemorrhagic stroke. Hum Mol Genet. 2014;23:283–92. doi: 10.1093/hmg/ddt418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paralkar VM, Vukicevic S, Reddi AH. Transforming growth factor beta type 1 binds to collagen IV of basement membrane matrix: implications for development. Dev Biol. 1991;143:303–8. doi: 10.1016/0012-1606(91)90081-d. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Harris RE, Bayston LJ, Ashe HL. Type IV collagens regulate BMP signalling in Drosophila. Nature. 2008;455:72–7. doi: 10.1038/nature07214. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Meng H, Wang MM. Collagen represses canonical Notch signaling and binds to Notch ectodomain. Int J Biochem Cell Biol. 2013;45:1274–80. doi: 10.1016/j.biocel.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venstrom K, Reichardt L. Beta 8 integrins mediate interactions of chick sensory neurons with laminin-1, collagen IV, and fibronectin. Mol Biol Cell. 1995;6:419–31. doi: 10.1091/mbc.6.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colorado PC, Torre A, Kamphaus G, Maeshima Y, Hopfer H, Takahashi K, Volk R, Zamborsky ED, Herman S, Sarkar PK, Ericksen MB, Dhanabal M, Simons M, Post M, Kufe DW, Weichselbaum RR, Sukhatme VP, Kalluri R. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 2000;60:2520–6. [PubMed] [Google Scholar]
- 44.Van Agtmael T, Bailey MA, Schlotzer-Schrehardt U, Craigie E, Jackson IJ, Brownstein DG, Megson IL, Mullins JJ. Col4a1 mutation in mice causes defects in vascular function and low blood pressure associated with reduced red blood cell volume. Hum Mol Genet. 2010;19:1119–28. doi: 10.1093/hmg/ddp584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.