

Abstract
Purpose of Review
Inflammation is a key component in cardiovascular disease. Controlling inflammatory events and their subsequent processes hold the potential for novel therapeutic options. Cytokines are the propagators of inflammation. In this review we will discuss important cytokines, including IL-6, TNF-alpha, MCP-1, M-CSF and GDF-15 and their effect on cardiac outcome.
Recent findings
IL-6 is a useful biomarker in patients with coronary artery disease.
Summary
Inflammation is part of the non specific immune response that occurs in reaction to any type of bodily injury. Cardinal signs of inflammation are increased blood flow, elevated cellular metabolism, vasodilatation, release of soluble mediators, extravasation of fluids and cellular influx. In some disorders the inflammatory process, which under normal conditions is self-limiting, becomes continuous and chronic, giving rise to inflammatory disease [1]. Over the last decade inflammatory immune responses have been implicated in several aspects of cardiovascular disease. Current concepts propose that the inflammation is aspectic and is not directly caused by infectious organisms. However, the precise relationship between inflammation, pathogens and cardiovascular disease remains incompletely understood. It is believed that depending on the specific disease different inflammatory pathways are of relevance. Manifestations of cardiovascular disease, such as hypertension and ischemic heart disease are now considered to be connected to inflammation. Equally important, inflammation is now firmly recognized to participate in the process of atherosclerosis. Clinical consequences of atherosclerosis, such as myocardial infarction (MI) caused by rupture of the atherosclerotic plaque, elicit a complex inflammatory response. Depending on the context and stage of the disease, different inflammatory events are in progress.
Cytokines and Cardiac Outcome
Increasing evidence suggests that inflammation plays an important role in heart failure and after MI. Inflammatory cascades are propagated by proximal mediators. More than 90 cytokines and cytokine receptors have been identified [2]. The early release of cytokines shapes the nature of inflammatory responses. These responses can be beneficial, such as driving protective pathways, or detrimental, such as the induction of immunopathology.
Interleukin-6
Interleukin-6 (IL-6) is considered to be one of the cytokines at the top of the inflammatory cascade. It is produced by various haematopoietic and non-haematopoietic cells and exerts proinflammatory effects including induction of acute phase proteins in the liver. Studies on the role of of IL-6 in coronary heart disease (CHD) have been relatively limited because of its short half-life and considerable intra-individual variability. However, several reports now describe IL-6 as a biomarker in CHD, highlighting a potential relevance for IL-6 mediated pathways. In a prospective study covering 2138 patients and 4267 controls Danesh et al showed that long term IL-6 levels are highly associated with CHD, with CHD risk increasing continuously with increasing levels of circulating IL-6 concentrations [3]. One of the concerns of the authors was the influence of fluctuating IL-6 levels over time. However, in a recent study IL-6 measurements showed good reproducibility and Karakas et al propose that in clinical practice misclassification should be minimal if at least 2 subsequent measurements are taken [4]. Several recent studies confirmed a risk association of IL-6 with CHD, including a possible role of IL-6 in mediating the associations of circulating inflammatory markers with risk of CHD in men [5]. In a heart failure cohort IL-6 was described as a stronger predictor of prognosis than CRP [6]. Interestingly, Empana et al demonstrated that IL-6 is also an independent predictor of sudden death in asymptomatic European men [7]. However, Fraser et al did not find strong evidence of an association between IL-6 and incident CHD in older British women after controlling for established CHD risk factors [8]. Further studies need to address if this could reflect a gender difference.
Pathways mediated by chronically elevated IL-6 plasma levels are associated with CHD risk, but the question is whether there is already a genetic component to increased IL-6 levels. Analysis of the 6kb upstream promoter region of the IL-6 transcription start site reveled variant −6331T/C to be associated with higher IL-6 levels in an acute inflammatory state due to an Oct1 transcription binding mechanism [9]. A prospective population based study described an association of the IL-6 polymorphism –174 G/C with increased arterial stiffness, a precursor of cardiovascular disease [10]. The –174 G/C polymorphism has been already associated with increased IL-6 plasma levels [11]. In rheumatoid arthritis patients, who are at higher risk to develop cardiovascular disease [12], the –174 G/C polymorphism was associated with even more increased cardiovascular disease risk probably via increased inflammation [13]. Thus, in patient groups at higher risk of developing cardiovascular disease, genetic testing for an IL-6 polymorphism might be warranted. Of note, the −174G/C polymorphism showed no significance for atherosclerosis risk factors in women [14], suggesting again a possible gender difference.
IL-6 is produced by various cell types, including cardiomyocytes, fibroblasts and inflammatory cells after MI or after an inflammatory stimulus. So far animal studies have described a mixed picture of the effect of IL-6 and its downstream signaling. IL-6 is a member of the gp130 receptor ligand family [15]. After binding to its receptor it activates 3 main signaling cascades, the JAK/STAT1/3 signaling pathway, Ras/ERK1/2 signaling pathway and the PI3K/AKT pathway [16] (Figure 2, [17-20]). In animal models, high IL-6 levels are associated with adverse post-MI remodeling, and activation of gp130 was found to be cardioprotective, in contrast to the adverse outcome effects of high IL-6 levels in human studies [21]. To shed light on the role of gp130 signaling Hilfiker-Kleiner et al. used cardiomyocyte specific mutant mice with a mutation of the gp130 signaling including a failure to abrogate STAT3 signaling [22]. Mutant mice displayed normal function and heart morphology after 3 months. However, after MI, sustained cardiac inflammation, increased left ventricular (LV) rupture rate, HF and higher mortality was observed. This was associated with a prolonged and enhanced activation of STAT3 and increased expression of IL-6. High levels of IL-6 might therefore increase adverse LV remodeling and heart failure after MI because of impaired downregulation of gp130 mediated STAT3 signaling. Therefore it seems to be of great importance to target IL-6 signaling at the right time point in disease progression. Notably, another gp130 ligand oncostatin M (OsM) was reported to have more positive effects on inducing a pro-healing environment6 Recent results showed an upregulation of stromal derived factor 1, a key factor in progenitor cell homing, in cardiac myocytes in vitro and in the heart in vivo after stimulation with OsM as well as upregulation of VEGF, a main factor in inducing angiogenesis [23].
Figure 2.
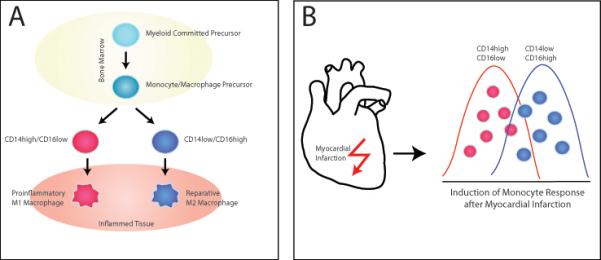
(A) Differentiation of Monocytes and Macrophages: Monocytes develop from the myeloid committed precursor in the bone marrow and enter the circulation as either CD14high/CD16low or CD14low/CD16high monocytes. At sites of injury and inflammation monocytes differentiate into a proinflammatory M1 macrophage subtype for CD14high/CD16low or reparative M2 macrophage subtype for CD14low/CD16high.
(B) Mobilization of Monocytes and Macrophages: Myocardial infarction induces the mobilization of different monocytic subsets with different kinetics. CD14high/CD16low proinflammatory monocytes respond first followed by the reparative CD14high/CD16low monocyte subset.
Currently, an antibody for IL-6 receptor inhibition, tocilizumab, is used to treat patients with RA. Tocilizumab in combination with methotrexat as a treatment for RA is associated with a small but significantly increased risk of adverse events, which is comparable with that of other biologics [24]. Unexpected, however, is the reported increase of total cholesterol, HDL cholesterol and LDL cholesterol for all measured time points [25]. How blockade of the IL-6 pathway interferes with the regulation of lipids is not understood, but obviously this is an important aspect when patients with advanced vascular disease are treated. Similarly, these findings will impact on considerations to utilize IL-6 receptor inhibition for the management of cardiovascular disease. Currently, lowering of cholesterol levels is considered a therapeutic cornerstone in the treatment of cardiovascular disease. If treatment-induced elevation of LDL cholesterol is associated with negative effects on cardiovascular outcomes remains to be investigated. Prospective studies will be necessary to collect relevant data sets.
Tumor necrosis factor-alpha
One of the first inflammatory pathways addressed in the setting of HF was tumor necrosis factor-alpha (TNF-alpha). Early studies identified the proinflammatory cytokine as a mediator of disease progression in the failing heart [26]. However, results of clinical trials using antibodies to block TNF-alpha gave disappointing results. In contrast to the expectations higher rates of mortality were observed in the treated individuals as compared to the placebo control [27]. A possible explanation might be that low physiologic levels of TNF-alpha might be required for proper tissue remodeling and repair. A recent report describes that TNF-alpha could protect cardiac mitochondria independent of its cell surface receptor [28]. Furthermore, the group of Mann showed that the cytoprotective effects of TNF-alpha might depend on tumor necrosis factor receptor-associated factor 2 in the heart [29]. According to the Translational Research Committee of the HF Association of the European Society of Cardiology future research should clarify the best type of anti-TNF-alpha therapy, the optimal dosage, and what subgroups of chronic HF to treat before any firm conclusions regarding efficacy in chronic HF can be drawn [26]. Special focus should be put on defined patient groups, e.g. inflammatory cardiomyopathies secondary to viral infection to finally determine a possible use of anti TNF-alpha therapy in heart disease.
Monocyte chemotactic protein-1, fractalkine and macrophage colony stimulating factor
Monocyte-derived cytokines have been shown to be of particular value in determining the risk of adverse events in patients with MI or HF. Monocyte chemotactic protein-1 (MCP-1) proved to be an independent prognostic marker in the acute and chronic phase after acute coronary syndrome (ACS) [30]. Furthermore MCP-1 is a predictor for all cause mortality in HF [31]. Interestingly, MCP-1 is a chemoattractant for a proinflammatory subset of monocytes characterized by CD14high CD16low. In contrast, the reparative subset of monocytes, characterized by CD14low CD16high, was described to be mainly attracted by fractalkine via the receptor CX3CR1 [32]. The two subgroups of monocytes give rise to two distinct sets of macrophages, a proinflammatory macrophage subset expressing TNF-alpha, IL-1beta, myeloperoxidase and matrix metalloproteinases, and a reparative macrophage subset expressing IL-10, TGF-beta and VEGF [32, 33]. Patients with MI show a biphasic monocyte response for the two subsets (Figure 2B). Proinflammatory monocytes peak after 3 days, whereas reparative monocytes have a peak at five days [34]. This biphasic monocyte pattern is in line with mouse experiments, showing a similar pattern [32]. Interfering with MCP-1 chemotactic signaling might be an interesting therapeutic target. A MCP-1 competitor, which was functionally inactive in CCR2 binding and calcium influx, but inhibited monocyte chemotaxis or transendothelial migration demonstrated reduced inflammatory monocyte recruitment, limited neointimal hyperplasia, and attenuated myocardial ischemia/reperfusion injury in mice [35]. Unfortunately, the effect of the MCP-1 competitor on the different subsets of monocytes was not evaluated. The two monocytic subsets might be of special interest in the setting of atherosclerosis. Atherosclerosis was reported to interfere with the infarct healing process due to an increase in proinflammatory monocytes [36].
The recruitment of the reparative monocytic subset was described as being mainly driven by fractalkine via the receptor CX3CR1 [32]. However, fractalkine was described to be involved in the development of numerous inflammatory pathologies [37]. Attention has been mainly given to the role of fractalkine in atherosclerotic development. Animals genetically defective for fractalkine and apoE show smaller atherosclerotic lesions. However, the role of fractalkine in respect to cardiac function and wound healing after MI needs further clarification, especially considering the data on reparative monocytes. Fractalkine was reported to be upregulated in human failing hearts and was increased in patients with chronic heart failure in accordance with disease severity. Furthermore, fractalkine itself induced the expression of markers of cardiac hypertrophy and protein phosphatases in neonatal cardiomyocytes [38]. The receptor CX3CR1 was described to be upregulated on monocytes from patients with heart failure [39]. Further clarification of the role of CXCR1 and reparative monocytes after MI and in HF seems warranted.
The role of macrophage modulating cytokines in the context of MI and heart failure is currently not well understood. Cytokines mediating monocyte chemotaxis are known risk factors after MI or in HF. However, macrophage colony stimulating factor (M-CSF), a key survival signal for monocytes and macrophages, was even stronger associated with all cause mortality and the composite endpoint, implying that survival of macrophages might be a more important contributor to adverse outcome than chemotaxis in late stage HF [31]. Currently it is unknown whether a specific cytokine environment in HF might favor the proinflammatory macrophage or reparative macrophage subset and if M-CSF might be part of this regulatory network. Furthermore, no data is available if reparative macrophages can be reeducated to proinflammatory macrophages in the context of HF.
Growth differentiation factor-15
Besides proteins modulating monocytes and macrophages also proteins expressed by macrophages might be useful in determining the risk and outcome in patients with ACS, CHD or HF. Recently, growth differentiation factor 15 (GDF-15), also known as macrophage inhibitory cytokine 1 has been implicated in cardiovascular pathophysiology. GDF-15 is mainly expressed by macrophages activated by inflammatory stimuli [40]. Furthermore cardiac myocytes under stress conditions such as stretch or inflammation have been shown to express GDF-15. The protein might control different developmental and physiological processes and tissue homeostasis and repair in adult life [41]. Although GDF-15 is not or only expressed at low abundance in the adult heart, it may be induced under certain pathologic conditions. In 2002, a link of GDF-15 with MI was proposed in healthy elderly women with no prior evidence of cardiovascular disease [42]. The study described a three times higher increased risk of future MI in women with the highest concentration of GDF-15. Future studies expanded the clinical importance of GDF-15 measurements by showing that high levels of GDF-15 predict adverse outcome in patients with HF [43] and is a prognostic marker in patients with acute MI [44]. Additionally, GDF-15 was described to predict the risk of a recurrent event in patients stabilized after acute coronary syndrome [45] as well as to be a long term risk prediction factor in patients after an episode of non-ST-segment-elevation acute coronary syndrome [46]. Furthermore, GDF-15 was proven to be an independent marker of cardiovascular dysfunction and disease in the elderly [47] as well as in patients with HF and in patients with HF and normal ejection fraction [48]. A multiple marker approach proved that GDF-15 has incremental predictive ability over established risk factors [49]. GDF-15 might already be important in unselected patients as it proved to be an early risk stratification marker in patients with acute chest pain [50]. Although there is a lot of clinical evidence for a role of GDF-15 in heart disease, the underlying molecular mechanisms still need to be clarified. GDF-15 inhibits the secretion of TNF-alpha from activated macrophages. Kempf et al suggested GDF-15 as a novel defense mechanism via an AKT dependent pathway [51]. But how GDF-15 might interact with the different macrophage subsets during MI and HF needs clarification. GDF-15 was recently shown to have a direct action on cardiac myocytes themselves by promoting hypertrophic growth and acting anti apoptotic in cultured cardiac myocytes [52]. It remains currently unknown whether the increase in GDF-15 levels seen in HF is a protective mechanism of the body in response to injury or if such high GDF-15 levels might be detrimental and cause disease progression.
Conclusion
Cytokines represent a major mediator of inflammation and therefore are potential drug targets. IL-6 is a central player in inflammation and antibodies against IL-6 are already used in certain disease settings. Whether application of these biologics is also warranted in heart disease needs further clarification, especially because IL-6 blockade is associated with increased lipid levels in clinical trials. Macrophage modulating cytokines are more downstream in the inflammatory cascade and might therefore allow amending disease with greater specificity.
Figure 1.
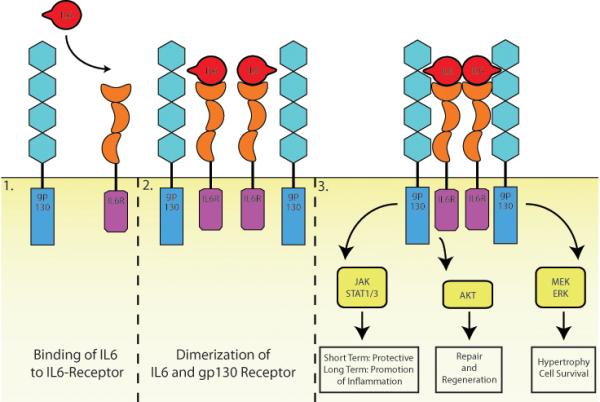
IL-6 Receptor and gp130 Receptor signaling: IL-6 binds to the IL-6 receptor (1). The complex of the cytokine with the receptor allows then the recruitment and dimerization of the signaling receptor unit gp130 (2). Downstream signaling of the gp130 receptor include activation of JAK/STAT, Akt and MEK/ERK. Activation of the JAK/STAT pathway was reported to have short term cardioprotective effects as well as inflammation-promoting long term effects [17]. AKT signaling in the heart includes activation of repair and regeneration pathways [18, 19]. MEK/ERK activation might lead to hypertrophy and cell survival [20].
References
- [1].Ferrero-Miliani L, Nielsen OH, Andersen PS, Girardin SE. Chronic inflammation: importance of NOD2 and NALP3 in interleukin-1beta generation. Clin Exp Immunol. 2007 Feb;147(2):227–35. doi: 10.1111/j.1365-2249.2006.03261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kopf M, Bachmann MF, Marsland BJ. Averting inflammation by targeting the cytokine environment. Nat Rev Drug Discov. Sep;9(9):703–18. doi: 10.1038/nrd2805. [DOI] [PubMed] [Google Scholar]
- [3].Danesh J, Kaptoge S, Mann AG, Sarwar N, Wood A, Angleman SB, et al. Long-term interleukin-6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PLoS Med. 2008 Apr 8;5(4):e78. doi: 10.1371/journal.pmed.0050078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Karakas M, Baumert J, Greven S, Ruckerl R, Peters A, Koenig W. Reproducibility in serial C-reactive protein and interleukin-6 measurements in post-myocardial infarction patients: results from the AIRGENE study. Clin Chem. May;56(5):861–4. doi: 10.1373/clinchem.2010.143719. [DOI] [PubMed] [Google Scholar]
- [5].Patterson CC, Smith AE, Yarnell JW, Rumley A, Ben-Shlomo Y, Lowe GD. The associations of interleukin-6 (IL-6) and downstream inflammatory markers with risk of cardiovascular disease: the Caerphilly Study. Atherosclerosis. Apr;209(2):551–7. doi: 10.1016/j.atherosclerosis.2009.09.030. [DOI] [PubMed] [Google Scholar]
- [6].Jug B, Salobir BG, Vene N, Sebestjen M, Sabovic M, Keber I. Interleukin-6 is a stronger prognostic predictor than high-sensitive C-reactive protein in patients with chronic stable heart failure. Heart Vessels. 2009 Jul;24(4):271–6. doi: 10.1007/s00380-008-1111-4. [DOI] [PubMed] [Google Scholar]
- [7].Empana JP, Jouven X, Canoui-Poitrine F, Luc G, Tafflet M, Haas B, et al. C-reactive protein, interleukin 6, fibrinogen and risk of sudden death in European middle-aged men: the PRIME study. Arterioscler Thromb Vasc Biol. Oct;30(10):2047–52. doi: 10.1161/ATVBAHA.110.208785. [DOI] [PubMed] [Google Scholar]
- [8].Fraser A, May M, Lowe G, Rumley A, Smith GD, Ebrahim S, et al. Interleukin-6 and incident coronary heart disease: results from the British Women's Heart and Health Study. Atherosclerosis. 2009 Feb;202(2):567–72. doi: 10.1016/j.atherosclerosis.2008.04.048. [DOI] [PubMed] [Google Scholar]
- [9].Smith AJ, D'Aiuto F, Palmen J, Cooper JA, Samuel J, Thompson S, et al. Association of serum interleukin-6 concentration with a functional IL6 -6331T>C polymorphism. Clin Chem. 2008 May;54(5):841–50. doi: 10.1373/clinchem.2007.098608. [DOI] [PubMed] [Google Scholar]
- [10].Sie MP, Mattace-Raso FU, Uitterlinden AG, Arp PP, Hofman A, Pols HA, et al. The interleukin-6-174 G/C promoter polymorphism and arterial stiffness; the Rotterdam Study. Vasc Health Risk Manag. 2008;4(4):863–9. doi: 10.2147/vhrm.s1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Smith AJ, Humphries SE. Cytokine and cytokine receptor gene polymorphisms and their functionality. Cytokine Growth Factor Rev. 2009 Feb;20(1):43–59. doi: 10.1016/j.cytogfr.2008.11.006. [DOI] [PubMed] [Google Scholar]
- [12].Weyand CM, Fujii H, Shao L, Goronzy JJ. Rejuvenating the immune system in rheumatoid arthritis. Nat Rev Rheumatol. 2009 Oct;5(10):583–8. doi: 10.1038/nrrheum.2009.180. [DOI] [PubMed] [Google Scholar]
- [13].Panoulas VF, Stavropoulos-Kalinoglou A, Metsios GS, Smith JP, Milionis HJ, Douglas KM, et al. Association of interleukin-6 (IL-6)-174G/C gene polymorphism with cardiovascular disease in patients with rheumatoid arthritis: the role of obesity and smoking. Atherosclerosis. 2009 May;204(1):178–83. doi: 10.1016/j.atherosclerosis.2008.08.036. [DOI] [PubMed] [Google Scholar]
- [14].Riikola A, Sipila K, Kahonen M, Jula A, Nieminen MS, Moilanen L, et al. Interleukin-6 promoter polymorphism and cardiovascular risk factors: the Health 2000 Survey. Atherosclerosis. 2009 Dec;207(2):466–70. doi: 10.1016/j.atherosclerosis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- [15].Rychli K, Kaun C, Hohensinner PJ, Rega G, Pfaffenberger S, Vyskocil E, et al. The inflammatory mediator oncostatin M induces angiopoietin 2 expression in endothelial cells in vitro and in vivo. J Thromb Haemost. Mar;8(3):596–604. doi: 10.1111/j.1538-7836.2010.03741.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fischer P, Hilfiker-Kleiner D. Role of gp130-mediated signalling pathways in the heart and its impact on potential therapeutic aspects. Br J Pharmacol. 2008 Mar;153(Suppl 1):S414–27. doi: 10.1038/bjp.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kurdi M, Booz GW. JAK redux: a second look at the regulation and role of JAKs in the heart. Am J Physiol Heart Circ Physiol. 2009 Nov;297(5):H1545–56. doi: 10.1152/ajpheart.00032.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Miyamoto S, Rubio M, Sussman MA. Nuclear and mitochondrial signalling Akts in cardiomyocytes. Cardiovasc Res. 2009 May 1;82(2):272–85. doi: 10.1093/cvr/cvp087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sussman M. "AKT"ing lessons for stem cells: regulation of cardiac myocyte and progenitor cell proliferation. Trends Cardiovasc Med. 2007 Oct;17(7):235–40. doi: 10.1016/j.tcm.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lorenz K, Schmitt JP, Vidal M, Lohse MJ. Cardiac hypertrophy: targeting Raf/MEK/ERK1/2-signaling. Int J Biochem Cell Biol. 2009 Dec;41(12):2351–5. doi: 10.1016/j.biocel.2009.08.002. [DOI] [PubMed] [Google Scholar]
- [21].Jugdutt BI. Preventing adverse remodeling and rupture during healing after myocardial infarction in mice and humans. Circulation. Jul 13;122(2):103–5. doi: 10.1161/CIRCULATIONAHA.110.969410. [DOI] [PubMed] [Google Scholar]
- [22].Hilfiker-Kleiner D, Shukla P, Klein G, Schaefer A, Stapel B, Hoch M, et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. Jul 13;122(2):145–55. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- [23].Hohensinner PJ, Kaun C, Rychli K, Niessner A, Pfaffenberger S, Rega G, et al. The inflammatory mediator oncostatin M induces stromal derived factor-1 in human adult cardiac cells. FASEB J. 2009 Mar;23(3):774–82. doi: 10.1096/fj.08-108035. [DOI] [PubMed] [Google Scholar]
- [24].Campbell L, Chen C, Bhagat SS, Parker RA, Ostor AJ. Risk of adverse events including serious infections in rheumatoid arthritis patients treated with tocilizumab: a systematic literature review and meta-analysis of randomized controlled trials. Rheumatology (Oxford) Nov 14; doi: 10.1093/rheumatology/keq343. [DOI] [PubMed] [Google Scholar]
- [25].Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008 Mar 22;371(9617):987–97. doi: 10.1016/S0140-6736(08)60453-5. [DOI] [PubMed] [Google Scholar]
- [26].Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand JL, Cohen-Tervaert JW, et al. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2009 Feb;11(2):119–29. doi: 10.1093/eurjhf/hfn043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002 Nov 29;91(11):988–98. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- [28].Lacerda L, McCarthy J, Mungly SF, Lynn EG, Sack MN, Opie LH, et al. TNFalpha protects cardiac mitochondria independently of its cell surface receptors. Basic Res Cardiol. Nov;105(6):751–62. doi: 10.1007/s00395-010-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Burchfield JS, Dong JW, Sakata Y, Gao F, Tzeng HP, Topkara VK, et al. The cytoprotective effects of tumor necrosis factor are conveyed through tumor necrosis factor receptor-associated factor 2 in the heart. Circ Heart Fail. Jan;3(1):157–64. doi: 10.1161/CIRCHEARTFAILURE.109.899732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].de Lemos JA, Morrow DA, Blazing MA, Jarolim P, Wiviott SD, Sabatine MS, et al. Serial measurement of monocyte chemoattractant protein-1 after acute coronary syndromes: results from the A to Z trial. J Am Coll Cardiol. 2007 Nov 27;50(22):2117–24. doi: 10.1016/j.jacc.2007.06.057. [DOI] [PubMed] [Google Scholar]
- [31].Hohensinner PJ, Rychli K, Zorn G, Hulsmann M, Berger R, Mortl D, et al. Macrophage-modulating cytokines predict adverse outcome in heart failure. Thromb Haemost. Feb;103(2):435–41. doi: 10.1160/TH09-06-0399. [DOI] [PubMed] [Google Scholar]
- [32].Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. Jun 8;121(22):2437–45. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. Feb 5;327(5966):656–61. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol. 2009 Jul 7;54(2):130–8. doi: 10.1016/j.jacc.2009.04.021. [DOI] [PubMed] [Google Scholar]
- [35].Liehn EA, Piccinini AM, Koenen RR, Soehnlein O, Adage T, Fatu R, et al. A new monocyte chemotactic protein-1/chemokine CC motif ligand-2 competitor limiting neointima formation and myocardial ischemia/reperfusion injury in mice. J Am Coll Cardiol. Nov 23;56(22):1847–57. doi: 10.1016/j.jacc.2010.04.066. [DOI] [PubMed] [Google Scholar]
- [36].Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol. Apr 13;55(15):1629–38. doi: 10.1016/j.jacc.2009.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].White GE, Tan TC, John AE, Whatling C, McPheat WL, Greaves DR. Fractalkine has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epidermal growth factor receptor signalling. Cardiovasc Res. Mar 1;85(4):825–35. doi: 10.1093/cvr/cvp341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Husberg C, Nygard S, Finsen AV, Damas JK, Frigessi A, Oie E, et al. Cytokine expression profiling of the myocardium reveals a role for CX3CL1 (fractalkine) in heart failure. J Mol Cell Cardiol. 2008 Aug;45(2):261–9. doi: 10.1016/j.yjmcc.2008.05.009. [DOI] [PubMed] [Google Scholar]
- [39].Cappuzzello C, Napolitano M, Arcelli D, Melillo G, Melchionna R, Di Vito L, et al. Gene expression profiles in peripheral blood mononuclear cells of chronic heart failure patients. Physiol Genomics. 2009 Aug 7;38(3):233–40. doi: 10.1152/physiolgenomics.90364.2008. [DOI] [PubMed] [Google Scholar]
- [40].Taddei S, Virdis A. Growth differentiation factor-15 and cardiovascular dysfunction and disease: malefactor or innocent bystander? Eur Heart J. May;31(10):1168–71. doi: 10.1093/eurheartj/ehq077. [DOI] [PubMed] [Google Scholar]
- [41].Mimeault M, Batra SK. Divergent molecular mechanisms underlying the pleiotropic functions of macrophage inhibitory cytokine-1 in cancer. J Cell Physiol. Sep;224(3):626–35. doi: 10.1002/jcp.22196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Brown DA, Breit SN, Buring J, Fairlie WD, Bauskin AR, Liu T, et al. Concentration in plasma of macrophage inhibitory cytokine-1 and risk of cardiovascular events in women: a nested case-control study. Lancet. 2002 Jun 22;359(9324):2159–63. doi: 10.1016/S0140-6736(02)09093-1. [DOI] [PubMed] [Google Scholar]
- [43].Anand IS, Kempf T, Rector TS, Tapken H, Allhoff T, Jantzen F, et al. Serial measurement of growth-differentiation factor-15 in heart failure: relation to disease severity and prognosis in the Valsartan Heart Failure Trial. Circulation. Oct 5;122(14):1387–95. doi: 10.1161/CIRCULATIONAHA.109.928846. [DOI] [PubMed] [Google Scholar]
- [44].Khan SQ, Ng K, Dhillon O, Kelly D, Quinn P, Squire IB, et al. Growth differentiation factor-15 as a prognostic marker in patients with acute myocardial infarction. Eur Heart J. 2009 May;30(9):1057–65. doi: 10.1093/eurheartj/ehn600. [DOI] [PubMed] [Google Scholar]
- [45].Bonaca MP, Morrow DA, Braunwald E, Cannon CP, Jiang S, Breher S, et al. Growth Differentiation Factor-15 and Risk of Recurrent Events in Patients Stabilized After Acute Coronary Syndrome: Observations From PROVE IT-TIMI 22. Arterioscler Thromb Vasc Biol. Jan;31(1):203–10. doi: 10.1161/ATVBAHA.110.213512. [DOI] [PubMed] [Google Scholar]
- [46].Eggers KM, Kempf T, Lagerqvist B, Lindahl B, Olofsson S, Jantzen F, et al. Growth-differentiation factor-15 for long-term risk prediction in patients stabilized after an episode of non-ST-segment-elevation acute coronary syndrome. Circ Cardiovasc Genet. Feb 1;3(1):88–96. doi: 10.1161/CIRCGENETICS.109.877456. [DOI] [PubMed] [Google Scholar]
- [47].Lind L, Wallentin L, Kempf T, Tapken H, Quint A, Lindahl B, et al. Growth-differentiation factor-15 is an independent marker of cardiovascular dysfunction and disease in the elderly: results from the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) Study. Eur Heart J. 2009 Oct;30(19):2346–53. doi: 10.1093/eurheartj/ehp261. [DOI] [PubMed] [Google Scholar]
- [48].Stahrenberg R, Edelmann F, Mende M, Kockskamper A, Dungen HD, Luers C, et al. The novel biomarker growth differentiation factor 15 in heart failure with normal ejection fraction. Eur J Heart Fail. Dec;12(12):1309–16. doi: 10.1093/eurjhf/hfq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Schnabel RB, Schulz A, Messow CM, Lubos E, Wild PS, Zeller T, et al. Multiple marker approach to risk stratification in patients with stable coronary artery disease. Eur Heart J. Dec;31(24):3024–31. doi: 10.1093/eurheartj/ehq322. [DOI] [PubMed] [Google Scholar]
- [50].Eggers KM, Kempf T, Allhoff T, Lindahl B, Wallentin L, Wollert KC. Growth-differentiation factor-15 for early risk stratification in patients with acute chest pain. Eur Heart J. 2008 Oct;29(19):2327–35. doi: 10.1093/eurheartj/ehn339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, Tongers J, et al. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res. 2006 Feb 17;98(3):351–60. doi: 10.1161/01.RES.0000202805.73038.48. [DOI] [PubMed] [Google Scholar]
- [52].Heger J, Schiegnitz E, von Waldthausen D, Anwar MM, Piper HM, Euler G. Growth differentiation factor 15 acts anti-apoptotic and pro-hypertrophic in adult cardiomyocytes. J Cell Physiol. Jul;224(1):120–6. doi: 10.1002/jcp.22102. [DOI] [PubMed] [Google Scholar]