

Abstract
The cyclin-dependent kinase 2 (cdk2) is a serine/threonine protein kinase that plays a key role in the cell cycle control system of all eukaryotic organisms. It has been a much studied drug target for potential anticancer therapy. Most cdk2 inhibitors in clinical development target almost exclusively the catalytic ATP-binding pocket of cdk2. However, several five amino-acid peptide inhibitors that are directed towards a non-catalytic binding pocket of cdk2 are reported here. Upon binding to this new pocket located at the cdk2 and cyclin interface, these peptide inhibitors are found to disrupt the cdk2/cyclin E complex partially and diminish its kinase activity in vitro.
Keywords: cyclin dependent kinase, peptide inhibitor, binding pocket, cell cycle, computer docking simulations
Introduction
The orderly progression of cell division in higher eukaryotes is regulated by a series of complexes of cyclin and cyclin-dependent kinase (cdk)1. The catalytic kinase subunit of the complex is activated by the regulatory cyclin subunit. Upon binding to cdk, cyclin induces a large conformational change of the T-loop of cdk. This exposes the catalytic ATP-binding sites of cdk to which potential substrates will interact2. In mammalian cells, there are 13 known cdks. In particular, the cdk2/cyclin E complex starts to accumulate at the late G1 phase of the cell cycle and plays a critical role in the G1-S transition3. Cdk2 has thus become a popular drug target for potential anticancer therapies4,5. Indeed, quite a number of potent ATP-analogs such as roscovitine and flavopiridol have progressed into advanced stages of clinical development6. By binding directly to the ATP-binding pocket of cdks, these ATP-analogs have to overcome two potential hurdles of both competing with high cellular ATP concentrations and discriminating the rather conserved catalytic sites of the ATP-binding pocket among cdks for binding specificity7. Given the potential benefits of novel inhibitors of cdks with desired binding affinity and specificity, discovery of other new non-catalytic binding sites of cdk2 can be useful since non-catalytic sites tend to be less conserved among closely related cdks.
One possible strategy for developing inhibitors that target non-catalytic binding sites on the cdk2/cyclin complex is to block the substrate recruitment binding groove identified on the cyclin subunit8,9. Yet another strategy is to target the protein-protein interaction interface of cdk2 and, for instance, its regulatory cyclin partners. Indeed, a 22-mer peptide, which introduces a single mutation in the segment taken from amino acids 285 to 306 in the α-helix of cyclin A that interacts directly with cdk2, is found to inhibit cdk2 kinase activity with an IC50=1.8μM10 The peptide, however, does not compete with the cyclin for cdk2 binding since it does not disrupt the formation of cyclin A and cdk2 complex. Docking simulations appear to suggest that it instead binds to both cdk2 and cyclin A. This finding reflects the general challenge to design small molecule or short peptide inhibitors that directly target the protein-protein interaction interface because the interface typically spans a wide contact area without any obvious dominant contacts to target7. Nonetheless, there are remarkably successful instances of designing small molecule inhibitors that target protein-protein interfaces11.
Interestingly, in a rather different context of searching for peptide inhibitors that suppress the replication of human immunodeficiency virus 1 (HIV-1), we recently reported a short 5mer peptide TAALS that targets the cdk2/cyclin E complex and inhibits the phosphorylation of serine 5 of RNA polymerase II (RNAPII) by disrupting the complex formation12. To our knowledge, it is the only short peptide that succeeded in partially breaking the cdk2/cyclin E complex by binding to a new non-catalytic binding pocket of cdk2 that interferes with the interface formation of the complex. Here, we extend our previous work by characterizing this new binding pocket of cdk2 in greater details. Moreover, by zooming in on the local region of the pocket, we are able to computationally design several new peptide inhibitors of cdk2 and verify in vitro that these peptide inhibitors indeed break up the cdk2/cyclin E complex and inhibit the kinase activity of cdk2.
Materials and Methods
Peptide Docking
The peptide docking simulations were performed using AutoDock software package version 3.0513 where the protein receptor (cdk2) was held rigid. To model the flexibility of cdk2 within this framework, in particular, its flexible T-loop that can directly interfere with the targeted binding pocket at the interface, we selected two representative cdk2 conformations for docking. They were taken from PDB files 1E1X and 1FIN to represent inactive and active cdk2 conformations as in cdk2 alone and cdk2/cyclin complex, respectively, because, upon binding to cdk2, cyclin induces a large swing of the T-loop as part of activating cdk2. AutoDockTools was used to prepare the peptide and cdk2 molecules and Kollman charges were added. Mass-centered grid maps were generated with 0.36Å spacing for active state and 0.33Å spacing for inactive state by the AutoGrid program for the whole cdk2 with default parameters. Simulated annealing scheme was chosen for conformational searching and the annealing parameters were listed in Table I. First, we use TAALS to locate the binding pocket of cdk2. The initial temperature was set to 100kcal/mol and temperature was reduced with the ratio 0.995 per cycle. In each cycle, at least 100,000 acceptances or rejections were required for the flexible peptide to avoid being trapped in metastable states. The annealing procedure was terminated when the acceptance ratio in one cycle was less than 5 percent or the total number of the cycles. Each annealing run typically went through about 900 temperature cycles and took about 80 minutes of a Pentium4 2.4 GHz processor. Six different starting positions, which are the centers of the six faces of a box enclosing cdk2, are chosen for the initial position of the peptide. For each starting docking positions, 198 simulations were run, so a total of 1188 docked configurations were obtained for each target. Second, to screen for stronger binding peptides to cdk2, all possible 95 single mutants of the TAALS were examined. The docking simulations for these mutants, however, were only performed for the active cdk2 in the region around the newly discovered interface binding sites. A somewhat large local region of size 32Å × 32Å × 32Å was actually chosen so as to ensure the complete coverage of the binding pocket since the precise binding sites were unknown. The energy grid was generated with 0.32Å spacing. Simulated annealing scheme was chosen for conformational searching and the annealing parameters are also listed in Table I. For each mutant, 240 simulations were run. Finally, to evaluate the docking results, we used the following procedure. Cluster analysis was first applied to the docking results. Here the native clustering method in the AutoDock program was used and the root-mean-square distance (rmsd) threshold was set to 3Å. Top 10 binding modes were inspected visually. For the interesting binding modes, the energy breakdown analysis for kinase was performed and the contribution of each residue of cdk2 for the binding (including van der Walls, hydrogen bond, salvation, and electrostatic terms, using the same energy function in the AutoDock program) was obtained. To determine the key residues of cdk2 in these binding modes, the lowest binding free energy conformations were analyzed and the residues were ranked by the contribution for binding energy. The structures of the cdk2-peptide complex were visualized in the VMD program14 and the schematic diagrams of the binding mode were generated by LIGPLOT program15.
Table I.
Parameters used for the AutoDock simulated annealing runs
| Set 1 | Set 2 | |
|---|---|---|
| Initial translation step | 2Å | |
| Initial quaternion step | 5° | 50° |
| Initial torsion step | 5° | 50° |
| Initial annealing temperature | 100 kcal/mol | 10 kcal/mol |
| Reduction schedule | Geometric | |
| Temperature reduction factor per cycle | 0.995 | 0.99 |
| Translation reduction factor per cycle | 0.995 | 0.95 |
| Quaternion reduction factor per cycle | 0.995 | 0.95 |
| Torsion reduction factor per cycle | 0.995 | 0.95 |
| Maximum number of accepted steps per cycle | 100,000 | 40,000 |
| Maximum number of rejected steps per cycle | 100,000 | 40,000 |
| Starting state of new cycle | Minimum-energy state from previous cycle | |
| Energy if getting out of the grid | 1000 kcal/mol | |
Parameters in Set 1 were used for identifying the binding sites of cdk2 for peptide TAALS, and those in Set 2 were used when the docking simulations of 95 point mutants of TAALS were performed to screen for better binding free energy than that of TAALS to cdk2.
Cyclin E immunoprecipitation/Cdk2 Western
Size exclusion chromatography was performed on 1mL of C81 cell lysate (30 mg/mL) with a Superose 6 10/30 column. Samples were eluted and collected at 0.5mL for 50 fractions. Cdk2 containing fractions “28–32” were pooled together. The pooled C81 extracts (250μg each) were combined with 10μg/ul of each respective peptide. Cyclin E antibody (Santa Cruz, sc-198) was added to each reaction tube (10μL, 2μg) and allowed to incubate while rotating overnight at 4°C. The reaction mixture was brought up to 500μL with TNE50 + 0.1% NP-40. The following day, 30μL of 30% Protein A & G bead slurry was added to each reaction tube and allowed to incubate while rotating for 2 hours at 4°C. Samples were spun and washed 2X with TNE300 + 0.1% NP-40 and 1X with TNE50 + 0.1% NP-40. 2X Laemmli buffer was added to each sample and heated at 95°C for 3 minutes. Samples were loaded and run on a 4–20% Tris-Glycine gel. The gel was transferred overnight onto a nitrocellulose membrane, blocked for 2 hours with 3% BSA + 0.1% PBST, and incubated with cdk2 antibody (Santa Cruz, sc-163) at a 1:1000 dilution while rocking overnight at 4°C. The membrane was washed and incubated with anti-rabbit antibody for 2 hours at 4°C while rocking, and was developed with ECL reagent.
Phosphorylation of RNAPII CTD
Similar to the IP westerns, size exclusion chromatography was performed on 4mL of C81 cell lysate with a Superose 6 10/30 column. Samples were eluted and collected at 0.5mL for 50 fractions. Cdk2 containing fractions “28–32” were pooled together. CTD phosphorylation was performed using anti-cyclin E immunoprecipitates and fractions 28–32. The pooled fractions were combined with various amounts of peptides. Cyclin E antibody (Santa Cruz, sc-198) was added to each reaction tube (10μL, 1μg) and allowed to incubate while rotating overnight at 4°C. The reaction mixture was brought up to 500uL with TNE50 + 0.1% NP-40. The following day, 30uL of 30% Protein A & G bead slurry was added to each reaction tube and allowed to incubate while rotating for 2 hours at 4°C. Samples were spun and washed 2X with TNE300 + 0.1% NP-40 and 1X with TNE50 + 0.1% NP-40. CTD phosphorylation was performed using anti-cyclin E immunoprecipitates (1μg/reaction of antibody which brought down approximately 100 ng of an active cdk2/cyclin E complex). Four concentrations (0.01, 0.1, 1, and 10μM) of two Tat peptides were added to the reaction mixture overnight. The total kinase reaction volume was 20 μl for 1hr at 37C. 2X Laemmli buffer was added to each sample and heated at 95°C for 3 minutes. Samples were loaded and run on a 4–20% Tris-Glycine gel, dried and exposed to a Phosphor Imager cassette. 32P labeled bands were observed and counted the next day.
Results
Sites of the new binding pocket of cdk2
We previously reported that a short 5-mer peptide TAALS interacts directly with cdk2 and disrupts the cdk2/cyclin E complex. Furthermore, a number of point mutants at positions 126, 134, 150, 178, 180, and 234 of cdk2 were studied experimentally and two mutants, Y180A and K178A, in particular, showed a dramatic reduction in their binding affinity to the peptide, suggesting a binding pocket of cdk2 possibly around these two residues12.
To shed light on how the cdk2/cyclin interface might be targeted, we ran the PP_SITE program16 to rank the residues involved in the formation of cdk2/cyclin interface using structural information available for several cdk2/cyclin complexes in the Protein Data Bank (PDB). The results are tabulated in Table II. Unlike several other residues that are identified as the crucial residues across all cdk2/cyclin interfaces examined, K178 appears to play an important role only in the cdk2/cyclin E1 interface. As such, it would probably not have become a favorite residue to target if not for the existence of a suitable binding pocket near it, as reported in detail below. Again, this shows that targeting protein-protein interface is a difficult task in general.
Table II.
Key residues of cdk2 in the cdk2/cyclin interface are identified by PP_SITE
| 1FIN (CDK2/Cyclin A) | 1W98 (CDK2/Cyclin E1) | 2JGZ (CDK2/Cyclin B1) | 1E9H (CDK2/Cyclin A3) | 2CCH (CDK2/Cyclin A2) | ||||
|---|---|---|---|---|---|---|---|---|
| A/B | C/D | A/B | A/B | A/B | C/D | A/B | C/D | |
| L37 | + | + | + | + | ||||
| E40 | ++ | ++ | ++ | ++ | ||||
| T41 | + | |||||||
| E42 | ++ | ++ | ++ | ++ | ++ | ++ | ||
| V44 | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ |
| I49 | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ |
| R50 | + | + | ++ | ++ | ++ | ++ | ++ | ++ |
| I52 | + | ++ | ++ | + | ++ | ++ | ++ | ++ |
| S53 | + | |||||||
| K56 | + | + | + | + | + | + | ||
| E57 | + | + | ++ | + | + | + | + | + |
| V69 | + | ++ | + | + | + | + | + | + |
| H71 | ++ | + | + | ++ | ++ | + | ++ | ++ |
| E73 | + | + | + | + | ||||
| L76 | + | ++ | ++ | + | + | |||
| S120 | + | |||||||
| H121 | + | |||||||
| R122 | + | + | ++ | + | + | + | + | + |
| R150 | + | + | ++ | + | ++ | ++ | + | ++ |
| F152 | ++ | ++ | ++ | ++ | ++ | ++ | ++ | + |
| V154 | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ |
| P155 | + | + | ++ | + | ++ | ++ | ++ | ++ |
| V156 | ++ | ++ | ||||||
| R157 | + | ++ | + | + | + | + | + | + |
| Y159 | ++ | ++ | ||||||
| E162 | + | + | + | + | + | + | ||
| K178 | + | |||||||
| Y179 | + | ++ | + | |||||
| N272 | + | + | ||||||
| K278 | ++ | + | + | + | + | + | + | ++ |
Symbols of + and ++ denote the important residues in an increasing order of their contribution to the interaction energy of the interface. Residues shown in bold are significantly involved across all cdk2/cyclin interfaces that were analyzed using the structure of the cdk2/cyclin complex reported in PDB files: 1FIN, 1W98, 2JGZ, 1E9H, and 2CCH. Here A/B (C/D) indicates that the interface chosen for analysis is between chain A and chain B (chain C and chain D) in the corresponding PDB file.
Extensive docking simulations of peptide TAALS to the inactive and active states of cdk2 were performed and two binding conformations of comparable binding free energies of −5.11kcal/mol and −5.30kcal/mol were obtained13. The binding modes including the residues involved in the new binding pocket of cdk2 are shown in Figure 1(A) and Figure 1(B) for inactive and active states of cdk2 respectively. To find out exactly the sites on cdk2 that bind strongly with the peptide TAALS docked in this binding pocket, we computed the intermolecular energy between the peptide and each residue of cdk2 including van der Waals, hydrogen bond, salvation and electrostatic terms. The top ten residues of cdk2 that contribute most to the intermolecular interaction energy are tabulated in Table III. While both panels (A) and (B) in Fig. 1 as well as Table III indicate that residues K178 and Y180 are critical towards peptide binding, which is consistent with the result of alanine mutation experiments12, there is a distinction between the results obtained from the inactive and active states of cdk2. Only the result from the active state of cdk2 agrees with the alanine mutation experimental result suggesting that R126 does not participate significantly in the peptide binding to cdk212. Taking this as evidence that the peptide probably binds to the active form of cdk2, we focused ourselves to the active form of cdk2 in all subsequent simulations in search for possibly more potent peptide inhibitors.
Figure 1. Docking conformation of peptide TAALS to cdk2.

The final binding modes of peptide TAALS to the new binding pocket of cdk2 obtained from docking simulations were shown using LIGPLOT [13]. Results for inactive and active cdk2 are displayed in (A) and (B) respectively. Both panels indicate that residues of K178 and Y180 contribute significantly towards the binding of peptide TAALS to cdk2, which are consistent with the results of alanine mutation experiments [12].
Table III.
Key residues of the cdk2 for the docking of TAALS
| Top 10 Residues (inactive state) | Total (kcal/mol) | H-Bond (kcal/mol) | Vdw (kcal/mol) | Solvation (kcal/mol) | Electrostatic (kcal/mol) | ||
|---|---|---|---|---|---|---|---|
| 1 | 180 | TYR | −1.783 | −0.883 | −0.823 | −0.187 | 0.110 |
| 2 | 208 | GLU | −1.710 | −0.489 | −0.221 | −0.069 | −0.931 |
| 3 | 235 | ASP | −1.655 | −0.579 | −0.212 | −0.115 | −0.749 |
| 4 | 178 | LYS | −1.574 | −0.583 | −0.719 | −0.243 | −0.029 |
| 5 | 174 | LEU | −1.081 | −0.519 | −0.199 | −0.066 | −0.297 |
| 6 | 126 | ARG | −1.023 | −0.370 | −0.360 | −0.135 | −0.159 |
| 7 | 154 | VAL | −1.002 | −0.671 | −0.144 | −0.051 | −0.135 |
| 8 | 173 | ILE | −0.706 | −0.245 | −0.301 | −0.108 | −0.053 |
| 9 | 176 | GLY | −0.600 | −0.175 | −0.293 | −0.077 | −0.055 |
| 10 | 150 | ARG | −0.479 | −0.152 | −0.212 | −0.066 | −0.048 |
| Top 10 Residues (active state) | Total (kcal/mol) | H-Bond (kcal/mol) | Vdw (kcal/mol) | Solvation (kcal/mol) | Electrostatic (kcal/mol) | ||
|---|---|---|---|---|---|---|---|
| 1 | 179 | TYR | −2.080 | −0.706 | −1.083 | −0.275 | −0.015 |
| 2 | 227 | TRP | −1.240 | −0.951 | −0.373 | −0.124 | 0.209 |
| 3 | 228 | PRO | −1.041 | −0.596 | −0.100 | −0.033 | −0.313 |
| 4 | 155 | PRO | −0.896 | −0.270 | −0.346 | −0.082 | −0.198 |
| 5 | 178 | LYS | −0.882 | −0.408 | −0.295 | −0.103 | −0.076 |
| 6 | 180 | TYR | −0.840 | −0.312 | −0.402 | −0.115 | −0.011 |
| 7 | 156 | VAL | −0.815 | −0.490 | −0.169 | −0.049 | −0.107 |
| 8 | 271 | PRO | −0.752 | −0.104 | −0.515 | −0.145 | 0.012 |
| 9 | 233 | MET | −0.692 | −0.212 | −0.383 | −0.116 | 0.019 |
| 10 | 177 | CYS | −0.671 | −0.108 | −0.362 | −0.126 | −0.076 |
The energy breakdown analysis for TAALS and cdk2 complex was performed and the interface binding mode was chosen for analysis. The contribution of each residue in cdk2 for the binding (including van der Walls, hydrogen bond, salvation, and electrostatic terms, using the same energy function in the AutoDock program) is listed. The total intermolecular energy is the sum of four different types of interactions representing H-bond interaction, van der Waals interaction, salvation interaction and electrostatic interaction. Only the top 10 residues of cdk2 that contribute the most to total energy are included here.
Given the typical difficulty of assessing the accuracy of automated docking simulations as above, we sought alternative methods to evaluate the new binding pocket. We applied a new program named Cavity which was developed recently by Yuan et al17. The program not only detects possible cavites on target proteins but also evaluates the drugability of the cavities by assigning a score for each cavity detected. The score is essentially a weighted linear sum of several quantities including the geometric factors such as the pocket volume and depth that were previously considered in the LigandFit program of Venkatachalem et al18 as well as the chemical characteristics of the amino acids in the pocket. When compared with the experimentally available binding affinities of a set of small molecules to their known target protein binding pockets, a cutoff on the score for acceptance is identified that corresponds to high micromolar binding affinity. When the program was applied to the active state of cdk2, it found three binding pockets with accepted scores, as shown in Figure 2. The best binding pocket is the known ATP binding pocket of cdk2. The second best binding pocket, whose significance remains unknown to us, consists of amino acids at positions 97–101, 104, 194, 196–204, 214, 217–218, 246, 250–251, and 253–254. Finally, the third best binding pocket is our new binding pocket described above and it is composed of amino acids at positions 124, 152, 154–156, 172, 176–182, 184, 227–230, 232–234, and 270–272. This finding correlates well with the docking simulation results given in Table III as well as the experimental results of alanine scan12. For example, except for the mutant P234A, two mutants within this pocket, K178A and Y180A, are critical towards binding, while the other three outside the pocket, R126A, Leu134A, and R150A, are much less so.
Figure 2. Computed binding pockets of cdk2.

Three top-ranked binding pockets of cdk2 using the active conformation of cdk2 were detected by program Cavity [14]. The best binding pocket (in red) is the well-known ATP binding pocket. The significance of the second best (in blue) remains unknown at present. The third best (in green) is the new binding pocket examined in the present work. The amino acids in each pocket are provided in the text. Panel (B) is a simply a rotated view of panel (A). This picture was generated using PYMOL (DeLano, 2002, The PyMOL Molecular Graphics System, http://pymol.sourceforge.net/).
We also ran the program Cavity for three other cdks using theoretical models of cdk1 (PDBID: 1LC9) and cdk4 (PDBID: 1LD2), and experimentally determined structure of cdk6 (PDBID: 1BLX). Unlike the ATP binding pocket, which is detected and ranked the best for all three cdks, the new binding pocket at the interface is only detected in cdk1. This suggests that the new binding pocket may be less conserved and thus represents a more suitable target than the ATP-binding pocket to design inhibitors specific to cdk2.
New peptide inhibitors that disrupt cdk2/cyclin complex formation and inhibit its kinase activity
Aside from a solved structure of the cdk2-peptide complex, the next most stringent test on the validity of the new binding pocket of cdk2 is perhaps whether, using only local structural information around the pocket, more potent peptides can be designed to disrupt cdk2/cyclin complex and thereby inhibit its enzymatic activity. Moreover, if successful, these different peptide inhibitors may collectively provide a topographic map of the 3D pharmacophores, i.e., the structural regions of the peptide responsible for interacting with cdk2, which can in turn aid future design of small molecule inhibitors that specifically target this pocket19. To this end, we choose to dock all 95 possible single mutants of peptide TAALS to cdk2, but the docking simulations were performed only in the local region of the binding pocket13. Clearly, the choice of docking single mutants is rather biased towards TAALS, but the large combinatorial factor makes impractical the simulations of mutli-site mutatations.
We summarize our simulation results in Table IV. All mutants are ranked according to their lowest binding free energy to the pocket obtained from several hundred independent simulated annealing runs for each peptide. Fifteen mutants, or roughly 15% of all single mutants, have better binding energy than that of TAALS, which is −5.30kcal/mol. These binding free energies would suggest a disassociation constant of 1 ~ 100μM. Although the selection of single mutants can only provide a very limited picture of the critical residues of a potent peptide inhibitor, the top-ranked peptides examined so far nonetheless show that the two consecutive alanine residues in the middle are far more conserved than the residues at the ends of the peptides. To verify these computational results, we selected the top 5 peptides, i.e., TAALD, TAALE, LAALS, TAACS, and FAALS, for further experimental analysis on their ability of disrupting cdk2/cyclin E complex and inhibiting the enzymatic activity of the complex.
Table IV.
Docking results of all 95 single mutants of the TAALS
| Rank | Sequence | Binding free energy (kcal/mol) |
|---|---|---|
| 1 | TAALD | −8.04 |
| 2 | TAALE | −6.35 |
| 3 | LAALS | −5.85 |
| 3 | TAACS | −5.85 |
| 5 | FAALS | −5.83 |
| 6 | TAAVS | −5.59 |
| 7 | TAALA | −5.58 |
| 8 | DAALS | −5.52 |
| 9 | TGALS | −5.51 |
| 10 | TAHLS | −5.50 |
| 11 | TAALG | −5.44 |
| 12 | IAALS | −5.38 |
| 13 | TAAHS | −5.37 |
| 13 | TAALV | −5.37 |
| 15 | TAALF | −5.31 |
| 16 | AAALS | −5.29 |
| 17 | TAALM | −5.27 |
| 18 | TAASS | −5.24 |
| 19 | TAALL | −5.22 |
| 19 | TAANS | −5.22 |
| 21 | TAAIS | −5.17 |
| 22 | TAALW | −5.15 |
| 23 | VAALS | −5.13 |
| 24 | TAFLS | −5.11 |
| 25 | MAALS | −5.09 |
| 16 | TAALS | −5.30 |
Top 25 point mutants of peptide TAALS ranked by their lowest binding free energies to cdk2 is listed. Out of the 95 possible point mutants of TAALS examined, 15 of them resulted in better (lower) binding free energy than that of −5.30kcal/mol for TAALS (shown in bold), and the top 5 peptides, i.e., TAALD, TAALS, LAALS, TAACS, and FAALS, were selected for further experimental verification.
In order to visualize the dissociation of the cdk2/cyclin E complex in the presence of the peptides, immunoprecipitations against cyclin E were performed. Western blot results for cdk2 can be seen in Figure 3. In this experiment, the six peptides (Lanes 4–9) were incubated with cell extracts in the presence of the cdk2/cyclin E complex and consequently probed for the presence of cdk2, with the indication that the absence of cdk2 would assume dissociation of the complex. Lane 1 depicts the α-IgG pulldown as a negative control, as well as Lane 2, in which beads alone represents the amount of non-specific background binding expected from the experimental lanes. As can be seen from these results, there is a varying degree of dissociation amongst the peptides. Based on the average band intensities on the Western blot for cdk2, all five new peptides (Lane 4 – 8) have comparable or better levels of dissociation when compared to the positive control of TAALS. This is consistent with the computational results given in Table IV. Peptide LAALS in Lane 8 appears to have the highest level of dissociation followed by peptide TAACS in Lane 6. This strongly suggests that TAACS and, in particular, LAALS are more potent than TAALS in promoting the disassociation of cdk2/cyclin E complex.
Figure 3. Immunoprecipitation of cyclin E and Western blot for cdk2 in the presence of the peptides.

C81 fractionated cell extracts were incubated with α-cyclin E antibody in the presence of each of the six chosen peptides. Probing for cdk2 indicated a specificity of the peptides in targeting an increased dissociation of the cdk2/cyclin E complex. The average and standard deviation for the band intensities on the Western blot for cdk2 were obtained from three replicates. Peptides TAACS and LAALS were the most effective as compared to the control. The fully formed cdk2/cyclin E complex was included as a positive control.
Figure 4 illustrates the incorporation of peptides LAALS and TAALS in a dose-dependant manner for the purpose of inhibiting kinase activity. Here, the peptide LAALS is being compared to the previously effective peptide TAALS in order to pinpoint the concentration at which maximum inhibition occurs. It has been previously shown that cdk2 is part of a transcription complex that stimulates phosphorylation of serine residues in the C-terminal domain (CTD) of RNA polymerase II (RNAPII)20. Therefore, in vitro inhibition of CTD phosphorylation by cdk2/cyclin E is an accurate model of what would be happening in vivo. An immunoprecipitation of the cdk2/cyclin E complex was performed as previously described, followed by the kinase reaction with GST-CTD being added as the substrate. The levels of phosphorylation of the CTD are shown above the coomassie stained gel indicating the same amount of substrate was loaded. Figure 4 indicates that a smaller concentration of the peptide LAALS is necessary to induce inhibition of kinase activity as compared to TAALS. At an equal concentration of peptide in Lane 5 and Lane 9, it is LAALS that exhibits a loss of kinase activity, while TAALS still shows phosphorylation of the substrate. The measured IC50 for LAALS and TAALS are 4.26μM and 26.1μM, respectively. A lower IC50 for LAALS is advantageous when dealing with novel small peptide inhibitors as potential therapeutics.
Figure 4. Visualization of loss of kinase activity of cdk2/cyclin E in the presence of peptides TAALS and LAALS via (γ-32P) ATP labeling on the CTD of RNAPII.
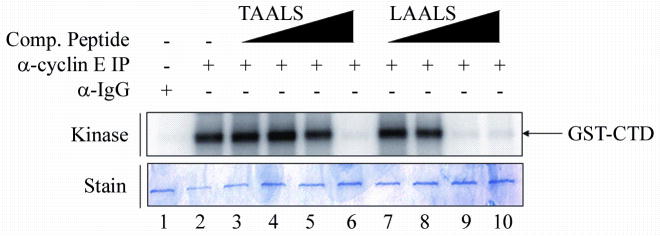
Immunoprecipitation of cyclin E in the presence of peptides was performed as in the Western blot analysis. Peptides TAALS and LAALS were added in increasing concentrations (0.01, 0.1, 1, and 10μM) to the kinase reaction. Kinase activity of cdk2/cyclin E was monitored via the addition of GST-CTD as a substrate. Dissociation of the cdk2/cyclin E complex is monitored by the decrease in phosphorylation of the CTD of RNAPII. Peptide LAALS required a lower concentration in order to exhibit a certain level of inhibition of kinase activity in vitro as compared to peptide TAALS. The lower panel shows the stained gel indicating equal loading of substrate among all lanes.
Discussion
Computational results and subsequent experimental verification were reported here to support the existence of a new non-catalytic binding pocket on cdk2 that several 5-mer peptides can target. The mechanism of inhibition on the enzymatic activity of cdk2/cyclin complex appears to be the suppression of cdk2/cyclin complex formation by these peptides. As such, these short peptides represent a distinct class of peptide inhibitors of cdk2 and may merit further investigation. To aid possible future research, several technical aspects of the computational as well as experimental results are discussed below.
Even though LAALS and TAACS represent novel peptide inhibitors of cdk2, the precise characterization of the binding sites of the new pocket at the interface of the cdk2/cyclin E complex is still difficult to pin down. One reason is that cdk2 is held rigid during the docking simulations, thus its flexibility cannot be accurately dealt with. Clearly cdk2 can possess an inactive (when disassociated from its cyclin partner) or active conformation (in complex with cyclin) or even perhaps anything in between. Simulation results indicated that the residue R126 played very different roles in the peptide binding, i.e., it is important in the inactive state of cdk2 but unimportant in the active state of cdk2. Based on the experimental finding that point mutant R126A does not affect the binding of peptide TAALS, we hypothesize that TAALS as well as its new variants LAALS and TAACS, or even this entire class of peptide inhibitors, recognize the active state of cdk2. However, there is a significant conformational change between the inactive and active state. It appears to suggest that these rather short peptides can induce similar conformational change of cdk2 as that of cyclin. It would be therefore highly desirable if the structure of cdk2-peptide complex can be solved experimentally.
We also clustered the final binding conformations out of several hundred independent runs for the three peptides TAALS, LAALS and TAACS with global docking being performed for the first peptide TAALS and local dockings for the last two peptides LAALS and TAACS. The results of clustering analysis are provided in Table V. For TAALS, the binding mode at the interface is only ranked third after the ATP binding pocket and another one according to the binding free energy. However, the interface binding mode has the largest cluster. In other words, this new binding pocket is most accessible. If now we rank the binding modes according to its cluster size, and draw the lowest energy conformation within the largest cluster, we observe results seen in Figure 5 which shows that all three peptides dock to the same binding pocket at the interface of cdk2. The consistency among these three peptides lends further support to the validity of this newly discovered binding pocket of cdk2.
Table V.
Cluster analysis for the docking results of TAALS, LAALS and TAACS
| Cluster Rank | Number in the Cluster | Lowest Docked Energy (kcal/mol) | Mean Docked Energy (kcal/mol) | Standard Deviation of Docked Energy(kcal/mol) | Lowest Binding Free Energy (kcal/mol) | |
|---|---|---|---|---|---|---|
| TAALS | 1 | 18 | −11.05 | −7.96 | 1.40 | −6.14 |
| 2 | 19 | −10.73 | −8.82 | 1.57 | −6.07 | |
| 3 | 57 | −10.41 | −7.01 | 1.13 | −5.30 | |
| 4 | 22 | −10.26 | −8.68 | 0.86 | −5.80 | |
| 5 | 8 | −10.04 | −6.87 | 1.48 | −4.65 | |
| LAALS | 1 | 61 | −11.94 | −8.29 | 1.09 | −5.85 |
| 2 | 24 | −9.45 | −7.32 | 1.06 | −3.40 | |
| 3 | 18 | −8.93 | −7.05 | 1.00 | −3.06 | |
| 4 | 7 | −8.78 | −7.36 | 1.03 | −3.44 | |
| 5 | 4 | −8.73 | −6.80 | 1.32 | −3.37 | |
| TAACS | 1 | 10 | −10.26 | −7.65 | 1.61 | −5.85 |
| 2 | 48 | −9.03 | −7.34 | 0.89 | −4.72 | |
| 3 | 24 | −8.70 | −6.79 | 0.96 | −4.43 | |
| 4 | 28 | −8.55 | −6.55 | 1.10 | −4.04 | |
| 5 | 3 | −8.32 | −6.79 | 1.61 | −4.31 |
The clustering analysis results for the global docking result of TAALS and local docking results of LAALS and TAACS are listed here. The data listed in bold are the biggest cluster and the lowest binding free energy conformation in this cluster was drawn in the Figure 5.
Figure 5. Binding modes of three peptides with cdk2.

To compare the binding mode of peptide inhibitor TAALS with those of peptide inhibitors of TAACS and LAALS that are found experimentally to be more potent in their ability of disrupting the formation of cdk2/cyclin E complex, we overlay the docked conformations of these three peptides in the binding pocket of cdk2. Red is for LAALS, green TAACS, and yellow TAALS.
Since the cell extract of cdk2/cyclin E complex was used in our experiments, we also simulated the docking of TAALS, TAACS, and LAALS to the local region of the new binding pocket of cdk2 obtained from PDB 1W98 (cdk2/cyclin E complex). The best binding free energies for TAALS, TAACS and LAALS were −4.44kcal/mol, −3.54kcal/mol and −4.75kcal/mol respectively. These values were much higher than those of using the cdk2 conformation obtained from 1FIN (cdk2/cyclin A complex). As shown in Figure 6, the superposition of the targeted binding pockets in 1W98 and 1FIN indicated that three key residues, C177, K178, and Y180 in 1W98 were pointing more inwardly to the pocket than those in 1FIN, and thus blocked the tight binding of those simulated peptide mutants to the pocket. This result didn’t contradict our experimental findings. From the viewpoint of thermodynamics, a peptide could break up the cdk2/cyclin E complex as long as the peptide could bind to a conformation that the cdk2 might adopt and the resulting cdk2-peptide complex would not bind with cyclin E anymore. This, however, also shows the limitation and drawback of a docking methodology that uses a rigid protein receptor (cdk2). Incorporating the flexibility of both the backbone and sidechains of a protein receptor in docking simulations remains a great challenge for the future.
Figure 6. Superposition of the binding pocket.

Panel (A) displays the superposition of cdk2 conformations obtained from PDB 1W98 (red, cdk2/cyclin E complex) and 1FIN (blue, cdk2/cyclin A complex). Panel (B) zooms in on the targeted binding pocket. In contrast to 1FIN, 1W98 has its three key residues, C177, K178, and Y180, point toward the pocket and thus block the tight binding of those simulated peptide mutants to the pocket. This picture was generated using PYMOL (DeLano, 2002, The PyMOL Molecular Graphics System, http://pymol.sourceforge.net/).
When comparing the simulated docking scenarios with experimental data, it is important to note that results shown in Figure 3 and 4 were obtained using in vitro immunoprecipitated purified cdk2/cyclin E complex. Different from an over-expressed, tagged-cyclin/cdk2 complex purified from a eukaryotic cell system, the immunoprecipitated complex used here most likely contains all the normal protein partners that would bind to cdk2/cyclin E and contribute to the kinase activity in vitro. While the implication of inhibition in this setting has been proven accurate enough as shown in Figure 3 where the immunoprecipitated complex dissociated in the presence of the peptide inhibitors, the dissociation of cdk2 and cyclin E will be only partial due to these other binding protein partners in the complex. This type of observation is expected when working with in vitro models.
Additionally, the immunoprecipitated complex has been exposed to various cellular modifications that can alter the otherwise idealized experimental setting. This, however, leads to a more accurate representation of what may be actually occurring in the cell. Figure 4 shows the phosphorylation events on the CTD (at either positions 2, 5, 7 at all or some of the 52 CTD repeats) in the presence and absence of the competing peptides. While these results demonstrate the inhibition of the enzymatic activity of cdk2/cyclin E complex, it also poses the question of what happens to the CTD of RNAPII in the cell because it is known that the cdk2/cyclin E complex plays an important role in in vivo phosphorylation of the CTD21. This lack of phosphorylation would presumably affect the activity of the entire preinitiation complex leading to either a reduced or completely silenced transcription at certain genes of interest. The phosphorylation of the RNAPII CTD is maintained from transcription initiation to termination and is also used as an indicator for RNA processing and chromatin remodeling22,23. Modifications of the RNAPII CTD have also been linked to the development and progression of certain cancers and viral infections24,25. The current study, however, does not address the significance of inhibition of CTD phosphorylation by these peptide inhibitors. Future studies using microarrays to screen for downstream transcriptional events in the presence of these peptide inhibitors would have important implications for in vivo analyses of which genes may be critically affected by these peptides.
Acknowledgments
We would like to acknowledge the NIH AIDS Research and Reference Reagent program for providing us with many of the reagents used in this study. This work was supported by grants from Conrad and the George Washington University REF and SE funds to FK, NIH grants AI065236 and AI043894 to FK, and NSF grant DMR0313129 to CZ. We also thank Xing Zheng for his participation at the early stage of the project and Yaxia Yuan, Jianfeng Pei, and Luhua Lai for sharing their program Cavity. NZ’s present address is National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health Bethesda, MD 20894, USA.
Contributor Information
Hao Chen, Email: hchen@gwu.edu.
Rachel Van Duyne, Email: rachel.vanduyne@gmail.com.
Naigong Zhang, Email: naigong@gmail.com.
Fatah Kashanchi, Email: bcmfxk@gwumc.edu.
Chen Zeng, Email: chenz@gwu.edu.
References
- 1.Morgan DO. The cell cycle : principles of control. London: New Science Press; 2007. p. 297. [Google Scholar]
- 2.Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376(6538):313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 3.Aleem E, Berthet C, Kaldis P. Cdk2 as a master of S phase entry: fact or fake? Cell Cycle. 2004;3(1):35–37. [PubMed] [Google Scholar]
- 4.Fischer PM. The use of CDK inhibitors in oncology: a pharmaceutical perspective. Cell Cycle. 2004;3(6):742–746. [PubMed] [Google Scholar]
- 5.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24(11):1770–1783. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 6.Fischer PM, Gianella-Borradori A. Recent progress in the discovery and development of cyclin-dependent kinase inhibitors. Expert Opin Investig Drugs. 2005;14(4):457–477. doi: 10.1517/13543784.14.4.457. [DOI] [PubMed] [Google Scholar]
- 7.Bogoyevitch MA, Barr RK, Ketterman AJ. Peptide inhibitors of protein kinases-discovery, characterisation and use. Biochim Biophys Acta. 2005;1754(1–2):79–99. doi: 10.1016/j.bbapap.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 8.McInnes C, Andrews MJ, Zheleva DI, Lane DP, Fischer PM. Peptidomimetic design of CDK inhibitors targeting the recruitment site of the cyclin subunit. Curr Med Chem Anticancer Agents. 2003;3(1):57–69. doi: 10.2174/1568011033353506. [DOI] [PubMed] [Google Scholar]
- 9.McInnes C, Fischer PM. Strategies for the design of potent and selective kinase inhibitors. Curr Pharm Des. 2005;11(14):1845–1863. doi: 10.2174/1381612053764850. [DOI] [PubMed] [Google Scholar]
- 10.Gondeau C, Gerbal-Chaloin S, Bello P, Aldrian-Herrada G, Morris MC, Divita G. Design of a novel class of peptide inhibitors of cyclin-dependent kinase/cyclin activation. J Biol Chem. 2005;280(14):13793–13800. doi: 10.1074/jbc.M413690200. [DOI] [PubMed] [Google Scholar]
- 11.Lu Y, Nikolovska-Coleska Z, Fang X, Gao W, Shangary S, Qiu S, Qin D, Wang S. Discovery of a nanomolar inhibitor of the human murine double minute 2 (MDM2)-p53 interaction through an integrated, virtual database screening strategy. J Med Chem. 2006;49(13):3759–3762. doi: 10.1021/jm060023+. [DOI] [PubMed] [Google Scholar]
- 12.Agbottah E, Zhang N, Dadgar S, Pumfery A, Wade JD, Zeng C, Kashanchi F. Inhibition of HIV-1 virus replication using small soluble Tat peptides. Virology. 2006;345(2):373–389. doi: 10.1016/j.virol.2005.09.062. [DOI] [PubMed] [Google Scholar]
- 13.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19(14):1639–1662. [Google Scholar]
- 14.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 15.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8(2):127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 16.Gao Y, Wang R, Lai L. Structure-based method for analyzing protein-protein interfaces. J Mol Model. 2004;10(1):44–54. doi: 10.1007/s00894-003-0168-3. [DOI] [PubMed] [Google Scholar]
- 17.Yuan Y, Pei J, Lai L. Cavity: a program for detecting and assessing binding sites on protein surfaces. To be submitted. [Google Scholar]
- 18.Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model. 2003;21(4):289–307. doi: 10.1016/s1093-3263(02)00164-x. [DOI] [PubMed] [Google Scholar]
- 19.Hruby VJ, Balse PM. Conformational and topographical considerations in designing agonist peptidomimetics from peptide leads. Curr Med Chem. 2000;7(9):945–970. doi: 10.2174/0929867003374499. [DOI] [PubMed] [Google Scholar]
- 20.Bres V, Kiernan RE, Linares LK, Chable-Bessia C, Plechakova O, Treand C, Emiliani S, Peloponese JM, Jeang KT, Coux O, Scheffner M, Benkirane M. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat Cell Biol. 2003;5(8):754–761. doi: 10.1038/ncb1023. [DOI] [PubMed] [Google Scholar]
- 21.Deng L, Ammosova T, Pumfery A, Kashanchi F, Nekhai S. HIV-1 Tat interaction with RNA polymerase II C-terminal domain (CTD) and a dynamic association with CDK2 induce CTD phosphorylation and transcription from HIV-1 promoter. J Biol Chem. 2002;277(37):33922–33929. doi: 10.1074/jbc.M111349200. [DOI] [PubMed] [Google Scholar]
- 22.Cho EJ. RNA polymerase II carboxy-terminal domain with multiple connections. Exp Mol Med. 2007;39(3):247–254. doi: 10.1038/emm.2007.28. [DOI] [PubMed] [Google Scholar]
- 23.Ammosova T, Berro R, Jerebtsova M, Jackson A, Charles S, Klase Z, Southerland W, Gordeuk VR, Kashanchi F, Nekhai S. Phosphorylation of HIV-1 Tat by CDK2 in HIV-1 transcription. Retrovirology. 2006;3:78. doi: 10.1186/1742-4690-3-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chun RF, Jeang KT. Requirements for RNA polymerase II carboxyl-terminal domain for activated transcription of human retroviruses human T-cell lymphotropic virus I and HIV-1. J Biol Chem. 1996;271(44):27888–27894. doi: 10.1074/jbc.271.44.27888. [DOI] [PubMed] [Google Scholar]
- 25.Zhou M, Halanski MA, Radonovich MF, Kashanchi F, Peng J, Price DH, Brady JN. Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Mol Cell Biol. 2000;20(14):5077–5086. doi: 10.1128/mcb.20.14.5077-5086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]