

Abstract
We have used Fourier transform infrared spectroscopy (FT-IR) to observe the photolysis and recombination of a novel EPR-silent CO-inhibited form of α-H195Q nitrogenase from Azotobacter vinelandii. Photolysis at 4 K yields a strong negative IR difference band at 1938 cm−1, along with a weaker negative feature at 1911 cm−1. These bands and the associated chemical species have both been assigned the label ‘Hi-3’. A positive band at 1921 cm−1 is assigned to the ‘Lo-3’ photoproduct. By using an isotopic mixture of 12C16O and 13C18O, we show that the Hi-3 bands arise from coupling of two similar CO oscillators with one uncoupled frequency at ~1917 cm−1. Although in previous studies Lo-3 was not observed to recombine, by extending the observation range to 200–240 K we found that recombination to Hi-3 does indeed occur, with an activation energy of ~6.5 kJ mol−1.
The frequencies of the Hi-3 bands suggest terminal CO ligation. We tested this hypothesis with DFT calculations on models with terminal CO ligands on Fe2 and Fe6 of the FeMo-cofactor. An S = 0 model with both CO ligands in exo positions predicts symmetric and asymmetric stretches at 1938 and 1909 cm−1 respectively, with relative band intensities of ~3.5:1, in good agreement with experiment. From the observed IR intensities, we find that Hi-3 is present at a concentration about equal to that of the EPR-active Hi-1 species. The relevance of Hi-3 to the nitrogenase catalytic mechanism and its recently discovered Fischer-Tropsch chemistry is discussed.
Keywords: nitrogenase, Fischer-Tropsch, photolysis, CO, DFT
Introduction
The enzymatic nitrogen fixation system known as nitrogenase (N2ase) catalyzes the reduction of dinitrogen to ammonia at ambient temperatures and atmospheric pressure.[1] For the Mo-dependent N2ase, binding of substrates and inhibitors involves a [Mo-7Fe-9S-X]-homocitrate-containing prosthetic group, called the FeMo-cofactor, of the larger MoFe protein (Av1). A recent crystal structure at 1.0 Å resolution[2] and an X-ray emission study[3] favor assigning C4− carbide as the much-debated atom X at the center of the cofactor. The mechanism by which this biological nanoparticle lowers the activation energy for reduction of the N≡N triple bond remains incompletely understood.
Carbon monoxide inhibits the reduction of N2 reversibly and non-competitively, and CO has been an important probe molecule for characterizing the active site.[4] Recently, Ribbe and coworkers have shown that CO can actually be a substrate for the V version of N2ase,[5] and with less efficiency, even for the wild-type Mo-dependent N2ase.[6] Seefeldt and coworkers have also demonstrated CO fixation, using the α-V70A and other variants of the MoFe protein.[7] Thus, a second front of interest in N2ase as a Fischer-Tropsch catalyst has emerged.[8]
Exposure of N2ase to CO during turnover elicits species with a variety of EPR signals, depending on the partial pressure of CO ([CO]), and each of these species and its characteristic EPR signal is described by the [CO] required for its formation.[9] The chemical species and its associated axial EPR signal (g-values near 2.17 and 2.06) are both called hi-CO and are formed under ~101 kPa [CO]. This hi-CO N2ase species is proposed to contain two CO molecules bound to the FeMo-cofactor.[9–10] A species with a rhombic EPR spectrum (g-values of 2.09, 1.98, and 1.93) called lo-CO forms under a much lower [CO][11] and is proposed to contain only one bound CO.[9–10] A third species and EPR signal, both called hi(5)-CO, have also been identified.[11c, 11d] Not all of these EPR signals are generated when variant Mo N2ases are turned over under CO,[12] and the integrated EPR intensities rarely exceed 40% of the active site species.[11d] Presumably, EPR-silent species with bound CO must co-exist with the EPR-active species.
Another spectroscopic tool that complements EPR for these studies has been FT-IR spectroscopy.[4c, 13] In stopped flow FT-IR experiments, under low [CO], a single ν(CO) band appeared at 1904 cm−1 and peaked in intensity within ~7s before decaying. Under high [CO], a new ν(CO) band was observed at 1936 cm−1 together with a pair of weaker, possibly coupled bands at 1958 and 1880 cm−1. All of these features were much longer lived than the 1904 cm−1 band. All three species were suggested to arise from terminally bound CO. After relatively long times (>10s), under low [CO] the 1904 cm−1 peak is succeeded by a new ν(CO) band at 1715 cm−1. Recently, the rates and relative intensities of these ν(CO) bands were shown to be sensitive to variation of the side-chain at the α-70 position.[13c] Other CO-related IR bands have been observed during spectro-electrochemical studies of FeMoco (the solvent-extracted version of the FeMo-cofactor),[14] where bands at 1808 and 1835 cm−1 were proposed to arise from bridging CO, whereas features at 1885 and 1920 cm−1 were assigned to terminally bound CO species.
In a recent study of CO-inhibited Azotobacter vinelandii Mo nitrogenase, we showed that FT-IR can be used to monitor CO photolysis by visible light at cryogenic temperatures.[15] Three distinct types of photolyzable CO complexes were found under hi-CO conditions. We labeled these stable inhibited forms ‘Hi-1’, ‘Hi-2’, and ‘Hi-3’. The photolyses of Hi-1 and Hi-2 were found to be reversible at around 80 K, with activation energies on the order of 3–4 kJ mol−1. However, the Hi-3 photoproduct, labeled ‘Lo-3’, was stable with respect to recombination up to 110 K. The Hi-3 species was most abundant in the photolysis spectra of N2ase with the variant α-H195Q MoFe protein. In the current work, we have used this variant and an extended temperature range (~200–250 K) to discover that Hi-3 photolysis is also reversible. We have used mixed CO isotopes to demonstrate vibrational coupling in the Hi-3 species, and wavelength-dependent photolysis to determine the spectral region with the greatest photochemical activity. We have also estimated the relative amounts of the Hi-1 and Hi-3 species in our samples. Finally, we have employed DFT calculations to evaluate plausible structures for Hi-3 and Lo-3 CO-bound FeMo-cofactor species.
Results
Photolysis
Figure 1 shows the time courses of changes in the IR spectra of CO-inhibited α-H195Q N2ase samples illuminated with visible light at ~4 K under either 12C16O or 13C18O. Photolysis spectra under 13C16O are included in Figure 2. At long times, the largest spectral changes come from the Hi-3 species,[15] with a strong negative band at 1938 (1894, 1849) cm−1 and a weaker negative band at 1911 (1867, 1824) cm−1. A positive product band from Lo-3 is seen in between these two bands at 1921 (1877, 1833) cm−1. (In these and later descriptions, the first value refers to results with 12C16O whereas the second and third values in parentheses, if present, refer to results with 13C16O and 13C18O, respectively.) As noted before[15], for α-H195Q N2ase the spectra are complicated by the presence of other photolyzable species, including bands for Hi-1 at 1969 (1925, 1879) cm−1 and for Hi-2 at 1932 (1888, 1844) cm−1.
Figure 1.
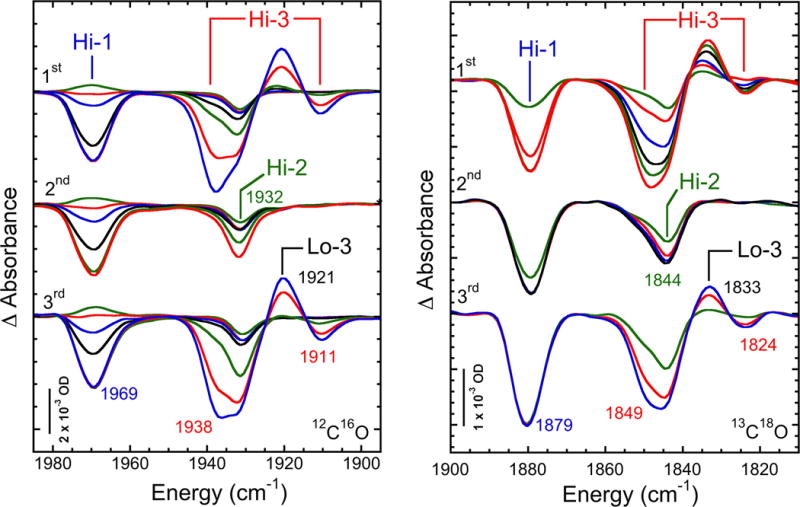
Time dependent absorbance changes upon photolysis at 4 K and thermal cycling of CO-inhibited α-H195Q N2ase in D2O-based 25 mM HEPES, pH 7.4 buffer with 40% ethylene glycol. Left: ‘hi-12C16O’ conditions. Top to bottom: 1st photolysis, time points shown are 0.5, 4, 9, 52s, 3, 30, 150 min; 2nd photolysis, after warming to 120 K for 20 min, time points shown are 0.5, 7, 13, 35s, 2, 30min; 3rd photolysis after poising at ~263 K for 16 h, time points shown are 0.5, 5, 13, 30s, 5, 70, 270 min. Right: ‘hi-13C18O’ conditions. Top to bottom: 1st photolysis, time points shown are: 0.5, 2, 10, 31, 60, and 122 min; 2nd photolysis, after warming sample to 120 K for 10 min, time points shown are: 2, 10, 30, and 60 min; 3rd photolysis, after warming sample to 233 K for 8 h, time points shown are: 3, 23, and 63 min.
Figure 2.
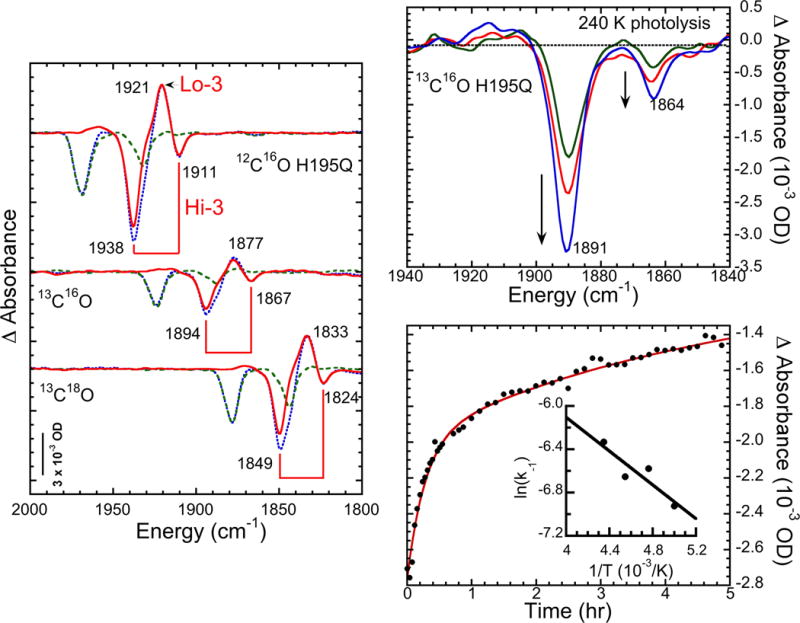
Left: extraction of Hi-3/Lo-3 spectra (—) by subtraction of 2nd photolysis spectra (- -) (photolyzed at 4K after recombination at 120 K) from 1st spectra (- -). Top right: photolysis at 240 K. Times are 40 (—), 60 (—), and 150 (—) min. Bottom right: recombination data for 210 K, fit with dual exponential. Inset: Arrhenius plot for fast phase of recombination, yielding activation energy of 6.5 kJ mol−1.
The spectra can be simplified by employing the fact that the photolysis product Lo-3 is relatively stable at temperatures where the photolysis products from Hi-1 and Hi-2 rapidly recombine.[15] Accordingly, we warmed the photolyzed samples to 120 K for 10–20 min and then photolyzed a second time. As shown in Figure 1, there was almost no trace of the Hi-3 or Lo-3 features in the second photolysis spectrum, consistent with the absence of Hi-3. By taking the difference between the two photolysis results, we obtained a relatively pure Hi-3→Lo-3 photolysis signal in the double difference spectrum (Figure 2). The extracted spectrum exhibited almost pure Hi-3 features at the same frequencies as noted in the mixed spectra. Note that in the third photolysis spectrum, which followed warming the sample from the second photolysis to above 230 K, the Hi-3→Lo-3 signals reappeared.
An alternative method of cleanly separating the Hi-3 features is to photolyze at higher temperatures, where the recombination of the other photolysis products is fast. As shown in Figure 2, photolysis of a 13C16O sample at 240 K yielded a spectrum with a pair of negative bands at 1891 and 1864 cm−1; nearly the same as the frequencies (1894 and 1867 cm−1) seen in the low-temperature photolysis. In fact, the 3 cm−1 downshifts are similar to those seen in myoglobin-CO upon raising the temperature from 4 K to room temperature.[16] This observation suggests that we should anticipate comparable shifts between our liquid helium-temperature photolysis results and the room-temperature stopped-flow FT-IR studies. In these higher temperature photolyses, there was less evidence of the Lo-3 photolysis product. We cannot exclude the possibility that additional processes involving CO migration and rearrangement and/or possible Lo-3 photolysis might be occurring at higher temperatures; these issues call for additional study.
Recombination
As seen in Figure 2, Hi-3 returns on the 1h time scale upon warming the sample to ~210 K. From monitoring this process at various temperatures, Arrhenius plot analysis using the initial rates yields an activation energy of ~6.5 kJ mol−1 (Figure 2), compared to the ~4 kJ mol−1 values previously seen for Hi-1 and Hi-2 recombination.[15] (In more recent studies we have found that with brief irradiation, the Hi-2 recombination can occur with a much smaller activation energy).[17] The Hi-3 value is similar to the barriers of 8–10 kJ mol−1 seen for Ni-SIa→NiSCO CO recombination in [NiFe] hydrogenases[18] and for photolyzed MbCO.[19]
Mixed Isotope Studies
The relative intensities of the negative Hi-3→Lo-3 features involve a higher 1938 (1894, 1849) cm−1 frequency band that is about four-times stronger than its lower 1911 (1867, 1824) cm−1 frequency partner. One explanation for the different intensities of the high and low frequency bands is that they represent the symmetric (with Isym intensity) and antisymmetric (Iasym) combinations of stretches for two distinct terminally bound CO molecules. Assuming that the symmetric combination has the higher frequency, the equation Isym/Iasym = cot2θ where θ is half the angle between the CO molecular axes, can be used to calculate a rough approximation of the relative orientations of the two molecules[20]. For an intensity ratio of ~4, the angle 2θ between CO axes is predicted to be ~50°. Such an angle is too small for two CO molecules bound at the same metal ion. Furthermore, coupling of vicinal 12C16O molecules usually produces splittings of about ~40–60 cm−1,[20] approximately twice as large as that observed for Hi-3. Both observations support the notion that there are two vibrationally coupled CO ligands, each of which is bound to a different FeMo-cofactor metal ion.
To confirm the assignment of the Hi-3 signals to coupled oscillators, we recorded photolysis spectra for α-H195Q N2ase inhibited by a mixture of ~25% 12C16O and ~75% 13C18O. In the absence of coupling, such a sample would produce a spectrum that is the 25:75 = 1:3 weighted average of the two pure 100% 12C16O and 100% 13C18O spectra, respectively. However, as seen in Figure 3, the mixed-CO spectrum has a new negative band at 1917 cm−1. This band does not appear in the 2nd photolysis spectrum after recombination at 120 K (see Figure 1 for the reference), so we can rule out assigning it to either Hi-1 or Hi-2. As expected for a Hi-3 assignment, the 1917 cm−1 band does reappear in the 3rd time photolysis, after recombination overnight at 233 K. We interpret this new band as resulting from photolysis of a Hi-3 species involving uncoupled CO oscillators, one with 12C16O and the other with 13C18O. Thus, at least one of the CO ligands is predicted to have an uncoupled frequency of 1917 cm−1.
Figure 3.
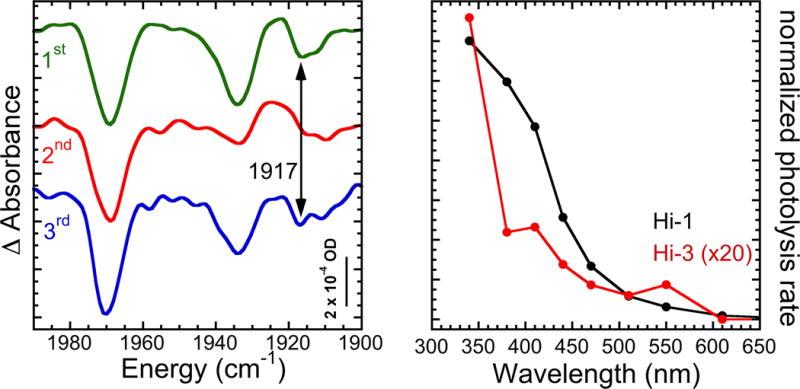
Left: Mixed isotope effects on CO-inhibited N2ase. Top to bottom: 1st photolysis for 122 min; 2nd photolysis for 60 min, after warming to 120 K for 10 min; 3rd photolysis for 63 min, after warming to 233 K for 8 h. Right: wavelength dependence of Hi-3 photolysis vs. Hi-1 photolysis.
Wavelength-Dependent Photolysis
So far, we have identified three forms of CO-inhibited N2ase that are produced under high [CO] conditions and are photolyzed by exposure to white light. To better distinguish these species, we investigated the wavelength dependence of the photolysis rates, and the results of these studies are shown in Figure 3. The Hi-3 photolysis rate rises from near zero at 600 nm to maximum efficiency at 340 nm, the shortest wavelength investigated. There appear to be minor features at ~550 and ~400 nm. By comparison, the Hi-1 species, which we associate with the hi-CO EPR species previously examined by Maskos and Hales, exhibits a structureless rise in photolysis efficiency towards shorter wavelengths. For Hi-1, we did not see the minor peak in photolysis yield at ~550 nm that was observed in the previous EPR experiments.[21]
DFT Calculations
To put speculation about the structure of the Hi-3 species on firmer ground, we used DFT calculations to investigate models that involve pairs of terminal CO ligands on different FeMo-cofactor metal sites. With 8 metal ions, there are 8(8−1)/2 = 28 possible pairs of binding sites on this prosthetic group. For the current study, we limited the calculations to binding at the most commonly proposed sites, the Fe2 and Fe6 ions on the face adjacent to the α-V70 residue.[13c, 22], see Figure 4. The atom and residue numbering here are as in the crystal structure.[23].As done in a recent study by Dance,[24] we considered exo (CO trans to the central X atom, now known to be carbon) and endo (CO trans to μ3 sulfurs, S1A/S1B when coordinated at Fe2/Fe6, respectively) terminal binding modes, which are subsequently referred as “Fe2/Fe6 exo/endo” geometries. A less popular bridging binding mode,[25] where the CO carbon occupies a position similar to that of μ2 S2B sulfur, was also considered. Below, we will discuss mostly those CO coordinations that were found relevant in context of the Hi-3 and Lo-3 photolytic species; a detailed description of the binding alternatives will be covered separately.
Figure 4.
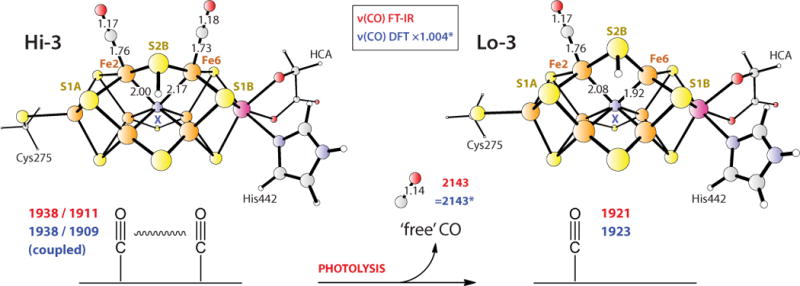
Favored DFT-optimized Hi-3 and Lo-3 structures. The observed FT-IR (red) and calculated DFT (blue) frequencies (cm−1) are given for the X = N3− models. Selected interatomic distances (Å) are shown. Similar results were obtained with the now-favored X = C4− assignment, provided S5A was also protonated (see text). The asterisk implies DFT frequency scaling as described in Methods. For the model coordinates and animated vibrational modes, see the Supporting Material section.
Our favored model for Hi-3 is shown in Figure 4. In the DFT calculation we assumed an EPR silent state for the transition metal ion core of the FeMo-cofactor, 1e− reduced from the S = 3/2 resting state. Other details on the metal oxidation states and spin coupling are given in the Discussion section. In line with earlier modelling by us and many others,[26] the central ligand X was initially assigned as N3−. Subsequently, as described below, we considered the X = C4− alternative which has recently received significant support.[2–3, 27] The μ2 S2B sulfur that bridges Fe2 and Fe6 and forms the hydrogen bond to α-H195 in the wild-type enzyme was chosen as the protonation site for all the models. Terminal CO ligands are at Fe2 and Fe6, both in the exo geometry, and the Fe-C≡O bond angles are essentially 180° (Figure 4). This model predicts two coupled 12C16O stretching modes, a symmetric stretch at 1938 cm−1 and an asymmetric stretch at 1909 cm−1, with relative band intensities of ~3.5:1, in good agreement with the experimental frequencies of 1938 and 1911 cm−1 and amplitude ratio of ~3.7:1. (Here and below, the broadened peak center heights are compared relative to the FT-IR, see Figure 5, in contrast to the raw ‘stick’ DFT mode intensities in Table S2 of the Supporting Information.) The optimized angle between the bound CO molecular axes is 37°, in line with the above ~50° rough estimate based on the relative FT-IR Hi-3 intensities. The basic FeMo-cofactor framework remained intact. However, we note a significant lengthening of the Fe6-X bond to 2.17 Å, compared to an average of 2.00 Å for the remaining optimized central Fe-X distances. The plasticity of the FeMo-cofactor core has been observed in other calculations; where even a complete loss of Fe coordination to X (Fe-X > 3.0 Å) upon ligand binding has been predicted.[24, 26c]
Figure 5.
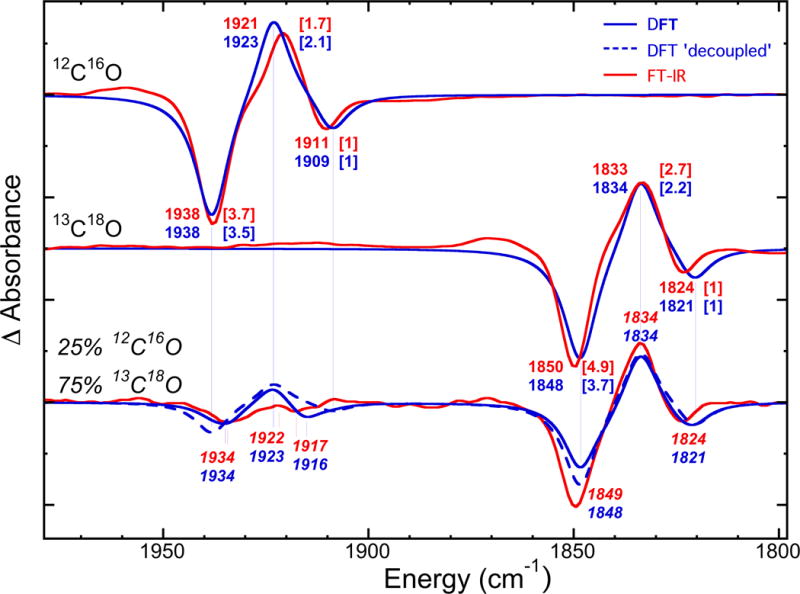
Overlay of the FT-IR (red) vs. DFT (blue) modelling (blue) of the Hi-3→Lo-3 photolysis difference spectra for pure 12C16O (top) and 13C18O (middle), and the mixed 25% 12C16O / 75% 13C18O (bottom) isotope experiments. The observed FT-IR and calculated DFT frequencies are in cm−1; the wave numbers for the mixed isotope case are in italic for clarity. For pure isotope spectra, the peak intensities are given in square brackets relatively to the lower frequency Hi-3 band. The simulation of the hypothetical ‘decoupled’ mixed isotope spectrum is presented as the broken line; the long vertical lines imply that this spectrum provides no peak shifts with respect to the pure isotope spectra.
For the Lo-3 photolysis product, the structure with the best match to the FT-IR Lo-3 band at 1921 cm−1 involved Fe2 exo binding (Figure 4), producing a 12C16O frequency at 1923 cm−1. The calculated band intensity of this ν(CO) stretch is ~2.1:1 relative to the asymmetric Hi-3 mode at 1909 cm−1, in reasonable agreement with the ~1.7:1 ratio seen in the experiment (Figure 2). For photolysis conducted using the pure 13C18O isotope, the DFT results are essentially of the same quality as those described above for 12C16O.
The calculations also shed light on our mixed isotope experiments. For models with 12C16O at Fe2 exo or Fe6 exo positions (and the other Fe site populated by 13C18O), they predict essentially ‘uncoupled’ pairs of ν(CO) frequencies of 1934/1825 or 1916/1842 cm−1 respectively, see the Supporting Information for the animated vibrational modes. We also modeled the actual Hi-3→Lo-3 photolysis IR spectra for 100% 12C16O, 100% 13C18O, and mixed isotope ~25% 12C16O / ~75% 13C18O experiments using our DFT frequency and intensity predictions, see Table S1 in the Supporting Material for the details. Figure 5 shows that in the low frequency 1810–1860 cm−1 region, the mixed isotope spectrum is dominated by the 13C18O/13C18O contributions, and the decoupled 13C18O bands are obscured by these features. However, in the higher 1900–1950 cm−1 frequency region, the mixed isotope species provides most of the intensity, and the DFT peaks at 1934 and 1916 cm−1 show noticeable shifts (of −4 and +7 cm−1) with respect to the pure 12C16O isotope Hi-3 features. The FT-IR bands seen at 1934 and 1917 cm−1 can now be assigned to essentially uncoupled stretching modes of the 12C16O at Fe2 and Fe6 respectively, when 13C18O binds at the other Fe site. The kinetic energy distribution (KED) factors in Table S2 of the Supporting Material indicate that less than 2% energy is accumulated in 13C18O stretch in these modes, and 12C16O stretch accounts for the rest.
As mentioned above, we also considered models with the same CO binding modes and with the now-favored X = C4− as the central ligand of the FeMo-cofactor. With the same μ2 S2B-H protonation scheme as for the X = N3− case, the calculations yielded bound ν(CO) frequencies >30 cm−1 red-shifted compared to the observed FT-IR values and optimized structures qualitatively identical to those presented in Figure 4. An extra proton addition at the μ2 S5A FeMo-cofactor sulfur places our X = C4− model at the same charge level (−3 units) as that of the X = N3− model, and it significantly improves the correspondence with the FT-IR experiment. For the calculated Hi-3 (12C16O and 13C18O) ν(CO) modes, the consistent negative deviation from the FT-IR peaks is 6–8 cm−1, while for the Lo-3 modes it is positive 2–3 cm−1, see Table S2 and S3 of the Supporting Material.
Discussion
Studies of the interaction of CO with N2ase have a long history dating back to at least 1941.[4a] The subject has taken on added significance with the discovery of N2ase Fischer-Tropsch-like CO reduction and condensation chemistry.[5–8] To date, nearly all the discussion about the structure and reactivity of CO intermediates has centered about two EPR-observable species, hi-CO and lo-CO.[9–10, 28] However, since integration of these EPR signals usually accounts for less than 50% of the enzyme, it is obvious that a good deal (50% or more) of N2ase-CO chemistry has not been accounted for.
To access these EPR/ENDOR-silent species, we have employed an FT-IR-monitored photolysis technique in much the same way as used in myoglobin-CO studies.[29] Our initial results[15] under high [CO] conditions indicated the presence of a previously unrecognized species that we labelled Hi-3, which was EPR-silent and so did not correlate with any of the known EPR-active species. In this current study, we have elucidated the spectroscopic properties, photochemistry, and likely structures of Hi-3 and its photolysis product Lo-3. As part of assessing the significance of these new EPR-silent species, an obvious question is: what fraction of our N2ase samples is present in these forms?
We can estimate Hi-3 abundance relative to Hi-1, by assuming (i) that Hi-1 contains a single terminal CO (as well as a second either bridging and/or more reduced CO species) bound at the FeMo-cofactor and (ii) that the sum of the oscillator strengths of the Hi-3 signals at 1938 and 1911 cm−1 (from two coupled CO oscillators) corresponds to twice the strength of the Hi-1 signal at 1969 cm−1 (from a single CO). From integrating the intensities of bands shown in Figure 1, the Hi-3 population is estimated as ~95% that of Hi-1. Although this is only an estimate, it is clear that the Hi-3 species is a major component of the reaction mixture. From the same assumptions, Hi-1 and Hi-3 signal concentrations are both significantly higher than that of Hi-2. Previous integrations of the EPR spectra of the α-H195Q MoFe protein under high [CO] conditions found 26% hi-CO, 6% hi(5)-CO, and 8% resting state enzyme.[11d] In some of our samples the hi-CO signal approaches 50% of the total MoFe protein. Thus, if we make the further assumption (iii) that the Hi-1 IR species is the same as the hi-CO EPR species,[15] then we conclude that at least a quarter and sometimes approaching half of our samples is present as Hi-3.
The combination of IR-detected photolysis experiments and DFT calculations has allowed us to propose a model for the Hi-3 species that involves two terminal exo CO ligands, one bound to each of the adjacent Fe2 and Fe6 sites of the FeMo-cofactor that are bridged by the protonated μ2 S2B sulfur (see Figure 4). Vibrational coupling between these two terminal CO molecules is proposed to contribute to the ~27 cm−1 frequency splitting and helps explain relative ~3.7:1 intensities of the Hi-3 bands at 1938 and 1911 cm−1. In the present study, such coupling was confirmed by the appearance of a new band at 1917 cm−1 in samples inhibited by a mixture of 12C16O and 13C18O isotopes. Our DFT model for Hi-3 lends support to this proposal. The model yields a 29 cm−1 splitting and relative intensities of ~3.5:1 for the coupled ν(CO) modes, symmetric at 1938 cm−1 vs. asymmetric at 1909 cm−1. The long-range vibrational coupling between CO molecules bound at the Fe sites ~2.7 Å apart can be rationalized by (i) approximate symmetry of the proposed Hi-3 structure with respect to the plane formed by the three μ2 S sulfurs (and the central X ligand), and (ii) electronic delocalization inherent to the iron-sulfur clusters such as FeMo-cofactor. Notably, this vibrational coupling may have implications to Hi-3 reactivity. Although our structural model is similar to one proposed by Hoffman and coworkers to account for properties of the hi-CO EPR signal,[10, 28b] we instead are using it to account for the properties of an EPR-silent species. Our prior results for the Hi-1 IR species, which we equate with the hi-CO EPR species,[15] indicate that this form instead has a single terminal CO with perhaps either a bridging CO or formyl ligand.
Dance has also conducted DFT calculations on CO-inhibited FeMo-cofactor with adjacent terminal CO ligands.[24] Two of his models (designated 10 and 11) with Fe2 exo and Fe6 exo CO ligands (for the resting state S = 1/2 cofactor, no protons added, and using X = N3−) most closely correspond to our current Hi-3 DFT proposal and carry the same total negative charge of 3e−. These models, which differ only by ‘coordinative allosterism’ in bonding with the central atom X, are predicted to have somewhat lower ν(CO) frequencies of 1914/1886 and 1904/1893 cm−1 for 10 and 11, respectively, than the 1938/1911 cm−1 pair observed in our experiments, and the intensity ratios were less than 2:1. From the present experience, these shifts could possibly be brought in line with our IR and DFT results via addition of one electron, and one proton at μ2 S sulfur. The importance of the electron and proton count for the bound ν(CO) frequencies and other FeMo-cofactor properties is also outlined below.
For the Lo-3 photolysis product, our favored DFT-based model places a single CO ligand in the Fe2 exo position (see Figure 4), implying that the net result of photolysis is displacement of the CO ligand from Fe6. We cannot exclude the possibility that photolysis of CO from the Fe2 position also occurs, with subsequent relaxation to the preferred structure. The DFT model yields a calculated ν(CO) band at 1923 cm−1, which compares well with the Lo-3 FT-IR signature at 1921 cm−1. The calculated intensity of this band is ~2.1:1 relative to the lower frequency Hi-3 mode, compared to the ~1.7:1 ratio seen in the FT-IR experiment. Dance also modeled FeMo-cofactor structures with only one CO ligand.[24] Most relevant to our proposed Lo-3 structure is his model 8, which places CO at Fe2 endo and yields a calculated ν(CO) frequency of 1904 cm−1. Other models with Fe2-H and/or Fe6-H hydrides in addition to a terminal CO (such as model 12), led to vibrational coupling of the C≡O and Fe-H stretches and introduced ν(CO) splittings that do not correspond to our Lo-3 experimental observations.
With the current DFT calculations, the FeMo-cofactor protonation scheme and the identity of the interstitial X (= C4−/N3−/O2−) ligand were both found to be important for reproducing the Hi-3 and Lo-3 FT-IR spectra. The interplay of these alternatives in defining the charge state of the FeMo-cofactor model was stressed earlier, for example in connection to Mössbauer isomer shifts[27, 30] and redox potential[27, 30b, 31] calculations. For our 1e− reduced Hi-3 and Lo-3 FeMo-cofactor states, we found single protonation of the cofactor at the μ2 S2B sulfur optimal for the X = N3− alternative, while double protonation at μ2 S2B and μ2 S5A gave the best results for X = C4−. The X = N3− option provides very good correspondence (≤2 cm−1 offset) to the Hi-3 and Lo-3 signatures and allows to interpret the minor peaks of the mixed isotope experiment (see Figure 5), while the X = C4− model results in somewhat larger frequency shifts of up to 8 cm−1 with respect to the FT-IR data (see Table S3 of the Supporting Material). However, since observed vs. computed frequency deviations of up to ~1% are inherent to the present DFT setup, our calculations cannot really discern whether X = N3− or C4−. We have no reason to doubt the X = C4− assignment that derives from other methods.[2–3]
Before discussing how or whether the Hi-3/Lo-3 couple has a role in the inhibition of N2 and other substrate reduction or perhaps in Fischer-Tropsch-like chemistry, we need to determine, in the nomenclature used by Hoffmann and coworkers,[28b, 32] its ‘electron inventory’. This refers to the number of electrons delivered from the Fe protein (called n) to form the Hi-3 species, starting from the resting state called E0. The equation n = s + m – p takes into account the number of electrons transferred to substrate (s), the number delivered to the FeMo-cofactor metals (m), and the number of electrons transferred from the P-cluster (p). The EPR silence of the Hi-3 species implies that n is odd, in order to produce an even-electron (most likely S = 0) Hi-3 species from the S = 3/2 EPR-active odd-electron resting (n = 0) state. The fact that Hi-3 is stable for days or longer argues against a highly reduced species with n ≥ 3, because such a species would presumably relax by H2 evolution. Given the preference of CO for reduced metal ions, chemical intuition also argues against a more oxidized form of the FeMo-cofactor (hence negative values for n). We are left with n = 1, a 1e− reduced FeMo-cofactor compared to resting MoFe protein. There is no evidence for P-cluster oxidation under these conditions, thus p = 0. Because the bound CO has not been reduced, s = 0. We thus get m = n = 1 for the FeMo-cofactor metal ion core electron count beyond the resting state.
A recent proposal suggests that the metal core of the FeMo-cofactor has only two accessible oxidation levels, the EPR-active resting state (MN) with m = 0 and the one-electron-reduced EPR-silent state (MR) with m = 1.[32] If so, then our Hi-3 species would retain the single electron on the FeMo-cofactor and be EPR-silent, whereas the lo-CO, hi-CO, and hi(5)-CO species, being more reduced than the resting state by an even number of electrons, would be EPR-active and these ‘extra’ electrons would be forced to reside on ‘substrates’. Thus, the lo-CO, hi-CO and hi(5)-CO EPR signals would come from forms of N2ase with m = 0 and s = 2 for a total of n = 2. The substrate-localized electrons could reside on either a formyl species or an as yet undetected hydride species. Partially reduced substrates are of course critical intermediates for C-C coupling reactions[33] as demonstrated previously by the formation of a similar range of hydrocarbons from N2ase-catalyzed isocyanide reduction.[34]
Is the Hi-3 (or Lo-3) species relevant to N2ase Fischer-Tropsch chemistry? Most mechanisms for Fe-catalyzed Fischer-Tropsch chemistry posit the presence of CO-derived reactants on adjacent Fe atoms,[35] and for N2ase, intermediates with adjacent CO or CHxO0,1 species[7, 33] have also been proposed. Evidence for multiple binding sites in N2ase dates back 40 years or more,[4b] but it has always been difficult to separate physically distinct binding sites on the same species from those produced by different redox levels and/or protonation states of the FeMo-cofactor. The current study is the first to demonstrate simultaneous terminal CO binding to distinct and adjacent Fe sites on the FeMo-cofactor. It thus demonstrates that two CO molecules can bind to Fe atoms only ~2.7 Å apart.
Yang et al. observed that the α-H195Q. α-V70A Av1 double mutant was much less efficient at hydrocarbon production than the α-V70A single mutant, and they suggested that α-H195 functions to deliver protons for the reduction of CO.[7, 36] Disruption of this proton transfer may also explain why we see a larger fraction of the Hi-3 species in the α-H195Q Av1 compared to wild-type enzyme.[15] Additional work is of course needed to determine whether Hi-3 represents a requisite intermediate or a side reaction resulting from a ‘proton bottleneck’.
Finally, the Hi-3 species is only 1-electron reduced from the resting state of the enzyme. Since the reduction of two CO molecules to C2H4 and 2 H2O requires 8 electrons and 8 protons, there is a good deal of chemistry required between Hi-3 and the major product observed during CO reduction. Efforts to identify and characterize those additional species are of course in progress.
Experimental Section
Sample Preparation
Purified α-H195Q MoFe protein and Fe protein had specific activities of 2000–2800 nmol of H2 (min/mg protein) −1 and 2000–2300 nmol of H2 (min/mg protein) −1 respectively. The purified component proteins were exchanged into D2O buffer containing 25 mM HEPES, pH 7.4, 10 mM MgCl2, 250 mM NaCl. For preparation of CO-inhibited samples, low electron-flux conditions were obtained using a 1:4 molar ratio of Fe protein:MoFe protein. The reaction-mixture components, which consisted of 2.5 mM ATP, 5 mM MgCl2, 30 mM creatine phosphate, 25 units/mL of creatine phosphokinase in 25 mM HEPES, pH 7.4 and 20 mM sodium dithionite were prepared in D2O. Turnover was accomplished under 101 kPa CO. The reaction was quenched by the addition of ethylene glycol to a final concentration of 40% after 15 minutes. The resulting turnover product was concentrated in an Amicon microfiltration pressure concentrator using a regenerated cellulose PLHK ultrafiltration membrane with a 100,000 molecular weight cut off under 202 kPa CO of the same composition used for the turnover reaction.
Photolysis and FT-IR spectroscopy
The sample photolysis was conducted in an Oxford liquid He flow cryostat using a Sutter Instruments 300 W Lambda LS xenon arc lamp. Spectra were recorded at 4 cm−1 resolution with a Bruker V-70v FT-IR spectrometer and a MCT detector. For photolysis, the lamp was shone through the side of the cryostat oriented 90° to the IR light path. The sample was held at 45° to both beams. This allowed the use of quartz windows for the visible light. For wavelength dependent photolysis, we used a set of VersaChrome® tunable bandpass filters (Semrock) with central wavelengths at 340, 380, 410, 440, 470, 510, 550, 610, 700 and 800 nm.
DFT
The DFT calculations were done using the PBE functional and the LACV3P** basis set, as implemented in JAGUAR 7.7 software. For the first- and second-row elements, LACV3P** implies a 6-311G** triple-zeta basis set including polarization function. For the Fe and Mo atoms, LACV3P** uses the Los Alamos effective core potential (ECP), and the valence part is essentially of triple-zeta quality. The geometries optimized at the PBE/LACV3P** level were used for the analytic Hessian calculations, resulting in the harmonic frequencies and IR intensities discussed in the text. The analysis of the computed vibrational normal modes has been facilitated using an in-house Q-SPECTOR Python tool, applied to model the FT-IR spectra and assess the FeMo-cofactor bound CO modes via kinetic energy distribution (KED) approach. Further important details are given in the Supporting Information sections.
Supplementary Material
Acknowledgments
This work was funded by NIH GM-65440 (SPC), NSF CHE-0745353 (SPC), the DOE Office of Biological and Environmental Research (SPC), the Alexander von Humboldt Foundation (VP), and UniCat Cluster of Excellence (VP).
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
Contributor Information
Dr. Vladimir Pelmenschikov, Email: pelmentschikov@mailbox.tu-berlin.de.
Prof. Stephen P. Cramer, Email: spjcramer@ucdavis.edu, spcramer@lbl.gov.
References
- 1.a) Peters JW, Szilagyi RK. Curr Opin Chem Biol. 2006;10:101–108. doi: 10.1016/j.cbpa.2006.02.019. [DOI] [PubMed] [Google Scholar]; b) Barney BM, Lee H-I, Santos PCD, Hoffman BM, Dean DR, Seefeldt LC. Dalton Trans. 2006:2277–2284. doi: 10.1039/b517633f. [DOI] [PubMed] [Google Scholar]; c) Dance I. Chem Asian J. 2007;2:936–946. doi: 10.1002/asia.200700131. [DOI] [PubMed] [Google Scholar]; d) Howard JB, Rees DC. Proc Natl Acad Sci USA. 2006;103:17088–17093. doi: 10.1073/pnas.0603978103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spatzal T, Aksoyoglu M, Zhang L, Andrade SLA, Schleicher E, Weber S, Rees DC, Einsle O. Science. 2011;334:940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lancaster KM, Roemelt M, Ettenhuber P, Hu YL, Ribbe MW, Neese F, Bergmann U, DeBeer S. Science. 2011;334:974–977. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Lind CJ, Wilson PW. J Am Chem Soc. 1941;63:3511–3514. [Google Scholar]; b) Hwang JC, Chen CH, Burris RH. Biochim Biophys Acta. 1973;292:256–270. doi: 10.1016/0005-2728(73)90270-3. [DOI] [PubMed] [Google Scholar]; c) Tolland JD, Thorneley RNF. Biochemistry. 2005;44:9520–9527. doi: 10.1021/bi050453m. [DOI] [PubMed] [Google Scholar]
- 5.a) Lee CC, Hu Y, Ribbe MW. Science. 2010;329:642. doi: 10.1126/science.1191455. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee CC, Hu YL, Ribbe MW. Angew Chem. 2011;50:5545–5547. doi: 10.1002/anie.201100869. [DOI] [PubMed] [Google Scholar]
- 6.Hu Y, Lee CC, Ribbe MW. Science. 2011;333:753–755. doi: 10.1126/science.1206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z-Y, Dean DR, Seefeldt LC. J Biol Chem. 2011;286:19417–19421. doi: 10.1074/jbc.M111.229344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerlach DL, Lehnert N. Ang Chem Int Ed. 2011;50:7984–7986. doi: 10.1002/anie.201102979. [DOI] [PubMed] [Google Scholar]
- 9.a) Pollock RC, Lee HI, Cameron LM, Derose VJ, Hales BJ, Orme-Johnson WH, Hoffman BM. J Am Chem Soc. 1995;117:8686–8687. [Google Scholar]; b) Christie PD, Lee HI, Cameron LM, Hales BJ, Orme-Johnson WH, Hoffman BM. J Am Chem Soc. 1996;118:8707–8709. [Google Scholar]
- 10.Lee H-I, Cameron LM, Hales BJ, Hoffman BM. J Am Chem Soc. 1997;119:10121–10126. [Google Scholar]
- 11.a) Lowe DJ, Eady RR, Thorneley RNF. Biochem J. 1978;173:277–290. doi: 10.1042/bj1730277. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Davis LC, Henzl MT, Burris RH, Orme-Johnson WH. Biochemistry. 1979;18:4860–4869. doi: 10.1021/bi00589a014. [DOI] [PubMed] [Google Scholar]; c) Cameron LM, Hales BJ. Biochemistry. 1998;37:9449–9456. doi: 10.1021/bi972667c. [DOI] [PubMed] [Google Scholar]; d) Sørlie M, Christiansen J, Lemon BJ, Peters JW, Dean DR, Hales BJ. Biochemistry. 2001;40:1540–1549. doi: 10.1021/bi0013997. [DOI] [PubMed] [Google Scholar]
- 12.a) Moore VG, Tittsworth RC, Hales BJ. J Am Chem Soc. 1994;116:12101–12102. [Google Scholar]; b) Maskos Z, Fisher K, Sørlie M, Newton WE, Hales BJ. J Biol Inorg Chem. 2005;10:394–406. doi: 10.1007/s00775-005-0648-2. [DOI] [PubMed] [Google Scholar]
- 13.a) George SJ, Ashby GA, Wharton CW, Thorneley RNF. J Am Chem Soc. 1997;119:6450–6451. [Google Scholar]; b) Thorneley RNF, Ashby GA, George SJ. In: Nitrogen Fixation: From Molecules to Crop Productivity. Pedrosa FO, Hungria M, Yates G, Newton WE, editors. Kluwer; The Netherlands: 2000. pp. 39–40. [Google Scholar]; c) Yang Z-Y, Seefeldt LC, Dean DR, Cramer SP, George SJ. Angew Chem. 2011;123:286–289. doi: 10.1002/anie.201005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pickett CJ, Vincent KA, Ibrahim SK, Gormal CA, Smith BE, Best SP. Chem Eur J. 2003;9:76–87. doi: 10.1002/chem.200390033. [DOI] [PubMed] [Google Scholar]
- 15.Yan L, Dapper CH, George SJ, Wang HX, Mitra D, Dong WB, Newton WE, Cramer SP. Eur J Inorg Chem. 2011:2064–2074. doi: 10.1002/ejic.201100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ansari A, Berendzen J, Braunstein D, Cowen BR, Frauenfelder H, Hong MK, Iben IET, Johnson JB, Ormos P, Sauke TB, Scholl R, Schulte A, Steinbach PJ, Vittitow J, Young RD. Biophys Chem. 1987;26:337–355. doi: 10.1016/0301-4622(87)80034-0. [DOI] [PubMed] [Google Scholar]
- 17.Myers WK, Dong W, Dapper CH, Yan L, Scott A, George SJ, Britt D, Newton WE, Cramer SP. in preparation 2012 [Google Scholar]
- 18.Bagley KA, Vangarderen CJ, Chen M, Duin EC, Albracht SPJ, Woodruff WH. Biochemistry. 1994;33:9229–9236. doi: 10.1021/bi00197a026. [DOI] [PubMed] [Google Scholar]
- 19.Austin RH, Beeson KW, Eisenstein L, Frauenfelder H, Gunsalus IC. Biochemistry. 1975;14:5355–5373. doi: 10.1021/bi00695a021. [DOI] [PubMed] [Google Scholar]
- 20.Darensbourg DJ. Inorg Chem. 1972;11:1606–1609. [Google Scholar]
- 21.Maskos Z, Hales BJ. J Inorg Biochem. 2003;93:11–17. doi: 10.1016/s0162-0134(02)00480-4. [DOI] [PubMed] [Google Scholar]
- 22.Sarma R, Barney BM, Keable S, Dean DR, Seefeldt LC, Peters JW. J Inorg Biochem. 2010;104:385–389. doi: 10.1016/j.jinorgbio.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 24.Dance I. Dalton Trans. 2011;40:6480–6489. doi: 10.1039/c1dt10505a. [DOI] [PubMed] [Google Scholar]
- 25.Durrant MC. Biochemistry. 2004;43:6030–6042. doi: 10.1021/bi036300l. [DOI] [PubMed] [Google Scholar]
- 26.a) Xiao Y, Fisher K, Smith MC, Newton W, Case DA, George SJ, Wang H, Sturhahn W, Alp EE, Zhao J, Yoda Y, Cramer SP. J Am Chem Soc. 2006;128:7608–7612. doi: 10.1021/ja0603655. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lukoyanov D, Pelmenschikov V, Maeser N, Laryukhin M, Yang TC, Noodleman L, Dean DR, Case DA, Seefeldt LC, Hoffman BM. Inorg Chem. 2007:46, 11437–11449. doi: 10.1021/ic7018814. [DOI] [PubMed] [Google Scholar]; c) Pelmenschikov V, Case DA, Noodleman L. Inorg Chem. 2008;47:6162–6172. doi: 10.1021/ic7022743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris TV, Szilagyi RK. Inorg Chem. 2011;50:4811–4824. doi: 10.1021/ic102446n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.a) Lee HI, Hales BJ, Hoffman BM. J Am Chem Soc. 1997;119:11395–11400. [Google Scholar]; b) Lee HI, Sørlie M, Christiansen J, Yang TC, Shao JL, Dean DR, Hales BJ, Hoffman BM. J Am Chem Soc. 2005;127:15880–15890. doi: 10.1021/ja054078x. [DOI] [PubMed] [Google Scholar]
- 29.Alben JO, Beece D, Bowne SF, Doster W, Eisenstein L, Frauenfelder H, Good D, McDonald JD, Marden MC, Moh PP, Reinisch L, Reynolds AH, Shyamsunder E, Yue KT. Proc Natl Acad Sci USA. 1982;79:3744–3748. doi: 10.1073/pnas.79.12.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Vrajmasu VV, Bominaar EL, Münck E. Inorg Chem. 2003;42:5974–5988. doi: 10.1021/ic0301371. [DOI] [PubMed] [Google Scholar]; b) Lovell T, Liu T, Case DA, Noodleman L. J Am Chem Soc. 2003:125, 8377–8383. doi: 10.1021/ja0301572. [DOI] [PubMed] [Google Scholar]
- 31.Dance I. Inorg Chem. 2006;45:5084–5091. doi: 10.1021/ic060438l. [DOI] [PubMed] [Google Scholar]
- 32.Doan PE, Telser J, Barney BM, Igarashi RY, Dean DR, Seefeldt LC, Hoffman BM. J Am Chem Soc. 2011;133:17329–17340. doi: 10.1021/ja205304t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dance I. Dalton Trans. 2011;40:5516–5527. doi: 10.1039/c1dt10240k. [DOI] [PubMed] [Google Scholar]
- 34.Kelly M. Biochem J. 1968;107:1–6. doi: 10.1042/bj1070001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Smit E, Weckhuysen BM. Chem Soc Rev. 2008;37:2758–2781. doi: 10.1039/b805427d. [DOI] [PubMed] [Google Scholar]
- 36.Fisher K, Dilworth MJ, Newton WE. Biochemistry. 2000;39:15570–15577. doi: 10.1021/bi0017834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.