

Abstract
Plasmodium falciparum , the causative agent of malaria, contributes to significant morbidity and mortality worldwide. Forward genetic analysis of the blood-stage asexual cycle identified the putative phosphatase from PF3D7_1305500 as an important element of intraerythrocytic development expressed throughout the life cycle. Our preliminary evaluation identified it as an atypical MAPK phosphatase. Additional bioinformatics analysis delineated a conserved signature motif and three residues with potential importance to functional activity of the atypical dual-specificity phosphatase (DUSP) domain. A homology model of the DUSP domain was developed for use in high-throughput in silico screening of the available library of antimalarial compounds from ChEMBL-NTD. Seven compounds from this set with predicted affinity to the active site were tested against in vitro cultures and three had reduced activity against a ΔPF3D7_1305500 parasite, suggesting PF3D7_1305500 is a potential target of the selected compounds. Identification of these compounds provides a novel starting point for a structure-based drug discovery strategy that moves us closer towards the discovery of new classes of clinical antimalarial drugs. These data suggest that MAPK phosphatases represent a potentially new class of P. falciparum drug target.
Introduction
Malaria is a terrible affliction of people in tropical and subtropical regions worldwide, putting the health of approximately 40% of the global population at risk with pregnant women and children most vulnerable (1). Currently, artemisinin combination therapies (ACTs) are the recommended first line therapy endorsed by the World Health Organization (WHO) (2–4), which have been highly successful in treating cases of uncomplicated malaria for several years. However, recent emergence of artemisinin treatment failures in Southeast Asia has intensified efforts for new chemotherapeutic agents with alternate modes of action to decrease the further development of multi-drug resistance (4–7).
In an effort to discover new antimalarial drug targets in P. falciparum we are utilizing a random transposon-mediated insertional mutagenesis strategy to identify metabolic processes and pathways that are important for asexual blood-stage growth (8, 9). A forward genetic screen in the laboratory-adapted clone of P. falciparum NF54 discovered that a disruption of the gene PF3D7_1305500 severely attenuated blood-stage P. falciparum growth (10). The primary phenotype of this mutant was due to a defect in cell cycle checkpoint with a significantly delayed progression out of pre-S phase (the trophozoite to schizont transition). The disruption was created by a single insertion of the transposon (piggyBac) into the ORF of PF3D7_1305500, creating a null mutant; normal growth could be completely restored by complementation with the intact gene (10). Bioinformatic analysis determined the PF3D7_1305500 protein product identified as homologous to the PTP superfamily and its structural characteristics similar to the Mitogen-Activated Protein Kinase (MAPK) phosphatase (MKP) subgroup. A key feature of this type of phosphatase is the tandem arrangement of a non-catalytic rhodanese domain followed by a dual-specificity phosphatase domain (11) and the gene is conserved in all Plasmodium species with a sequenced genome. MKPs in other eukaryotes can have similar functions and often are critical for intracellular signaling in response to numerous types of external stimuli (12–14).
The most detailed functional knowledge of signaling pathways in malaria research has revealed MAPK signaling cascades are critical components of sexual stage proliferation (11, 15–23). The MKP-type phosphatase is a likely regulator of these pathways. Interaction of the MKP with its phosphoprotein substrate depends on three conserved residues in the consensus DUSP domain binding pocket (11, 17, 24, 25). Analysis of the PF3D7_1305500 DUSP domain revealed that only two of the three conserved residues typically present in a DUSP signature motif are present. Instead the third residue, which is typically an arginine, aligns with an isoleucine that is conserved in each of the Plasmodium orthologs. Absence of this specific residue may reduce but may not totally abrogate phosphatase activity; therefore, PF3D7_1305500 is expected to have little or no phosphatase activity and possibly functioning as a pseudophosphatase (26–28). Conservation of the I to R substitution in all Plasmodium species is a unique characteristic indicative of an atypical MKP with an altered function within the malaria parasite lineage and requiring further study. This distinct active site and its potential involvement in regulating the MAPK pathway make this atypical MKP a promising candidate for antimalarial drug discovery. We have initiated identification of potential MKP inhibitory compounds through use of computational high-throughput screening (HTS) that allowed large sets of compounds to be investigated for possible biological activity (29). Using this method, suitable lead candidates can be identified from large drug-like data sets improving productivity and lowering costs to a level more favorable than in vitro screening methods, enhancing structure-based drug design (30, 31).
Materials and Methods
Identification of Conserved Domains and Evolutionary Lineage
The deduced amino acid sequence of PF3D7_1305500 (MKP) was retrieved from a public database (32) and physicochemical parameters were determined using ProtParam (33). Conserved domains were identified using the Conserved Domains Database (CDD) (34), Conserved Domain Architecture Retrieval Tool (CDART) (35), InterProScan (36, 37), Prosite (38, 39), Superfamily (40), and the Simple Modular Architecture Research Tool (SMART) (41, 42). The full deduced amino acid sequence and individual conserved domains were used with BLAST (BLASTP) to identify orthologs in NCBI protein. A multiple sequence alignment was constructed from the retrieved sequences with the lowest E-values to identify conserved regions. The phylogenetic tree was inferred using the Neighbor-joining method computing the evolutionary distance using the Poisson correlation with the Molecular Evolutionary Genetics Analysis software (MEGA5) (43–45).
Evaluation of Secondary Structure and Post-translational Modifications
The secondary structure of the PF3D7_1305500 product was evaluated using JPRED (46) and PSIPRED (47, 48). Phosphorylation sites were assessed using NetPhos 2.0 (49), which identifies serine, threonine and tyrosine phosphorylation sites, and NetPhosK 1.0 (50) to identify kinase binding sites. Prediction of a signal peptide and cleavage site was searched using Signal IP 3.0 (51). Mitochondrial and plastid targeting sequences were searched using the prediction servers Predotar (1) and PATS (52–54). N-terminal myristoylation was investigated using the Myristoylator from ExPASy (55).
Molecular Modeling and Structure Validation
The three-dimensional structure of PF3D7_1305500 has not been resolved so a homology model was built using the automated protein structure homology modeling server Swiss-Model. Suitable templates for modeling were identified using PSI-BLAST in the Swiss-Model repository (56–59). The crystal structure of MKP3 (NCBI Accession No. 1MKP_A) was the most suitable of the available templates with greatest sequence coverage and similarity (60). The remaining residues of PF3D7_1305500 not showing any significant similarity were not included for homology modeling. The model was assessed using the atomic empirical mean force potential with Atomic Non-Local Environment Assessment (ANOLEA), empirical force field with Gröningen Molecular Simulation program (GROMOS), and QMEAN6 (57, 61). The stereochemistry was assessed with a minimum resolution of 2.5 Å using PROCHECK (62). The final structure was also compared to the predicted secondary structure represented using DSSP and PROMOTIF (63). ERRAT plots were generated to check structure quality of the template and homology structure using a nine residue sliding window (64). This process was repeated in an iterative fashion until all the residues in the plot were not below 95% as done previously (65). The quality of the final structure was also verified using Verify 3D, Procheck and Ramachandran plots (62, 66, 67).
Identification of the Binding Pocket
The binding pocket was identified using Pocket Finder and Q-site finder that uses the Ligsite algorithm (68–70). The output of the predicted binding pocket was also compared to the Computed Atlas of Surface Topography of proteins (CASTp), which uses the alpha shape theory pocket algorithm (71, 72). The identified pocket was also validated through comparison to the structural alignment of the resolved homology model and template (MKP3).
Selection of the Compound Dataset and High-throughput in silico Docking
All docking and scoring calculations were performed using the 2012 Schrödinger Suite. The compound library was retrieved from ChEMBL-NTD (ftp://ftp.ebi.ac.uk/pub/databases/chembl/ChEMBLNTD/) and prepared using LigPrep (LigPrep v2.5, Schrödinger LLC). The homology model, made from sequence PF3D7_1305500, was minimized using the OPLS2005 (73) force-field algorithm and the grid files were generated in GLIDE (Glide v5.8, Schrödinger LLC) (74, 75). The modeled structure was used to identify small molecular inhibitors with affinity to the predicted active site through in silico docking experiments using extra precision (XP) mode (75) on a Dell Precision 490 workstation with an Intel Xeon dual quad-core processors. From the results obtained, the molecule structures with the highest predicted affinity; lowest docking scores within GLIDE’s error of 2 kcal/mol to the active site were selected. From this subset, the commercially available compounds were identified.
In vitro parasite culture conditions
P. falciparum NF54 and C9 were cultured according to standard methods at 37°C (5% O2 and 5% CO2, nitrogen balanced) in 5% hematocrit (O+ blood) and RPMI 1640 medium with 0.5% Albumax II, 0.25% sodium bicarbonate and 0.01 mg/ml gentamicin (76).
In vitro Drug Susceptibility Assay Using SYBR Green I
Synchronized cultures were seeded into 96-well plates at 0.5% parasitemia and cultured for 96 hours under the previously stated conditions. Plates were then frozen overnight at −80°C. Plates were thawed for 15 minutes then mixed by pipetting. Eighty microliters of each well were transferred to a new 96-well plate followed by 100 μL of SYBR Green I (Sigma Aldrich) in lysis buffer (0.2 μL of SYBR Green I/mL 2X lysis buffer). Plates were covered and incubated in the dark for 1 hour at room temperature. Fluorescence intensity was measured with a SpectraMax M2e microplate fluorescence reader (Molecular Devices) with excitation and emission wavelengths of 485 nm and 525 nm respectively. The sample values were expressed in relative fluorescence units (RFU). EC50 values were obtained by normalizing the values using control wells of samples cultured without drug and plotted using a one-phase exponential dose response curve using GraphPad Prism 6 (GraphPad Software Inc.).
Results
Evaluation of the Physical Properties of PF3D7_1305500
Initial evaluation of PF3D7_1305500 included an investigation of potential binding sites, phosphorylation sites, and post-translational modifications. These methods provided additional insight into potential interactions and functions of this putative phosphatase through the use of publicly available bioinformatics tools. This analysis found that there were not any significant phosphorylation sites or post-translational modifications. PF3D7_1305500 also did not show presence of a signal peptide. The predicted secondary structure was used to validate the homology model and supported the final structure.
Active Site Prediction
The predicted binding pocket for the PF3D7_1305500 protein product was identified and validated using Qsite-Finder, Pocket Finder and CASTp. A total of 10 binding pockets were found and compared to the active site of the template protein. The analysis revealed that the identified pocket in the region of the signature motif was highly conserved with the template active site as predicted through the multiple sequence alignment (Figure 1). Phylogenetic analysis showed the overall evolutionary distance between the Plasmodium sequences and the template (Figure 2). The residues within the binding pocket include the signature motif residues of previously characterized active phosphatases. For example, the residues C383, D345 and I398 of PF3D7_1305500 align with the conserved C293, D262 and R299 of MKP3. This comparison also suggests functional conservation can be inferred between this template and a homology model.
Figure 1.

Multiple sequence alignment of protozoan orthologs of PF2D7_1305500 with potential templates. Potential templates were identified from BLAST searches in Swiss Model. The obtained sequences were aligned using CLUSTALW. The conserved binding pocket residues are highlighted in black (
 ), whereas the main deviation in the binding pocket residues is the presence of an isoleucine in the Plasmodium sequences, which aligns with arginine present in the template sequences is highlighted in grey (
), whereas the main deviation in the binding pocket residues is the presence of an isoleucine in the Plasmodium sequences, which aligns with arginine present in the template sequences is highlighted in grey (
 ). Additionally there are additional residues in the region of the binding pocket following the cysteine. These two alterations are conserved in the Plasmodium sequences. Sequence orthologs in the alignment are from P. falciparum (PF3D7_1305500), P. knowlesi (PKH_140400), P. vivax (PVX_122110), P. berghei (PBANKA_140400) and Homo sapien (MKP3_1MKP_A, DUSP9_2HXP_A, MKP5_1ZZW_A).
). Additionally there are additional residues in the region of the binding pocket following the cysteine. These two alterations are conserved in the Plasmodium sequences. Sequence orthologs in the alignment are from P. falciparum (PF3D7_1305500), P. knowlesi (PKH_140400), P. vivax (PVX_122110), P. berghei (PBANKA_140400) and Homo sapien (MKP3_1MKP_A, DUSP9_2HXP_A, MKP5_1ZZW_A).
Figure 2.
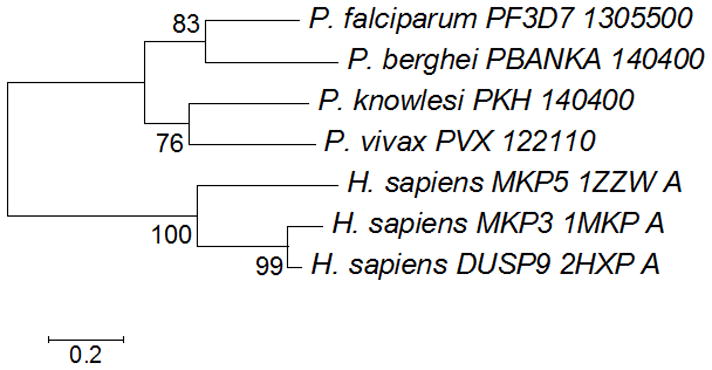
Phylogenetic analysis of PF3D7_1305500. The Neighbor-Joining method was used to construct the evolutionary history of the atypical phosphatase using the bootstrap test with 1000 replicates. The evolutionary distances were computed using the Poisson correction method.
Molecular Structure of PF3D7_1305500
A crystallographic structure of PF3D7_1305500 has not yet been resolved by experimental methods and neither is there a homologous protozoa protein that could be used for a template. The closest template available in the Swiss Model repository for homology modeling was the human phosphatase MKP3 with 21% similarity (Figure 3). The overall quality of the model predicted by ERRAT was 69.375 compared to 88.235 of the template, which was favorable considering the numerous INDELs in the primary sequence (Figure 4). Analysis and validation of the structure using the WHAT-IF web interface revealed that the structure was in agreement with standard structural conditions. Analysis of the Ramachandran plot gave a Z-score of −1.124 that was within the expected ranges for well-refined structures with 89.2% of the amino acid residues in favored regions (Figure 5). All bonds were in agreement with standard bond lengths with a RMS Z-score of 0.669 and RMS deviation of 0.015. Additionally, the RMSD score from DaliLite of Cα trace between 141 aligned residues of MKP3 and the homology model of the PF3D7 _1305500 DUSP was 0.5 Å with a Z-score of 26.7 and 21% sequence identity. Conservation of the predicted site in the homology model and the validated site in the template suggests that the selected pocket was the most favorable for HTS. The combined results from these various analyses suggest that the homology model of PF3D7_1305500 DUSP is reasonable and reliable.
Figure 3.
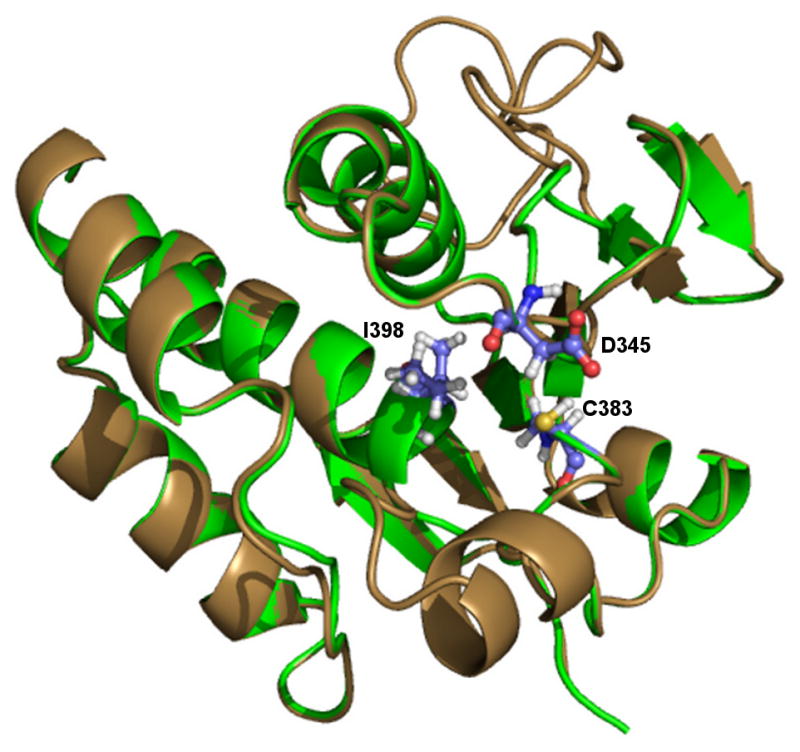
Homology model of PF3D7_1305500. The structure of PF3D7_1305500 (brown) was developed using the crystal structure of MKP3 as a template (PDB 1MKP). Alignment of the model with MKP3 (green) revealed that the final structure showed the catalytic residues aligned to the proposed positions in the active site. The presence of the signature motif insertion does not affect the shape of the active site and forms an alpha-helix adjacent to the binding pocket without obstructing the site.
Figure 4.
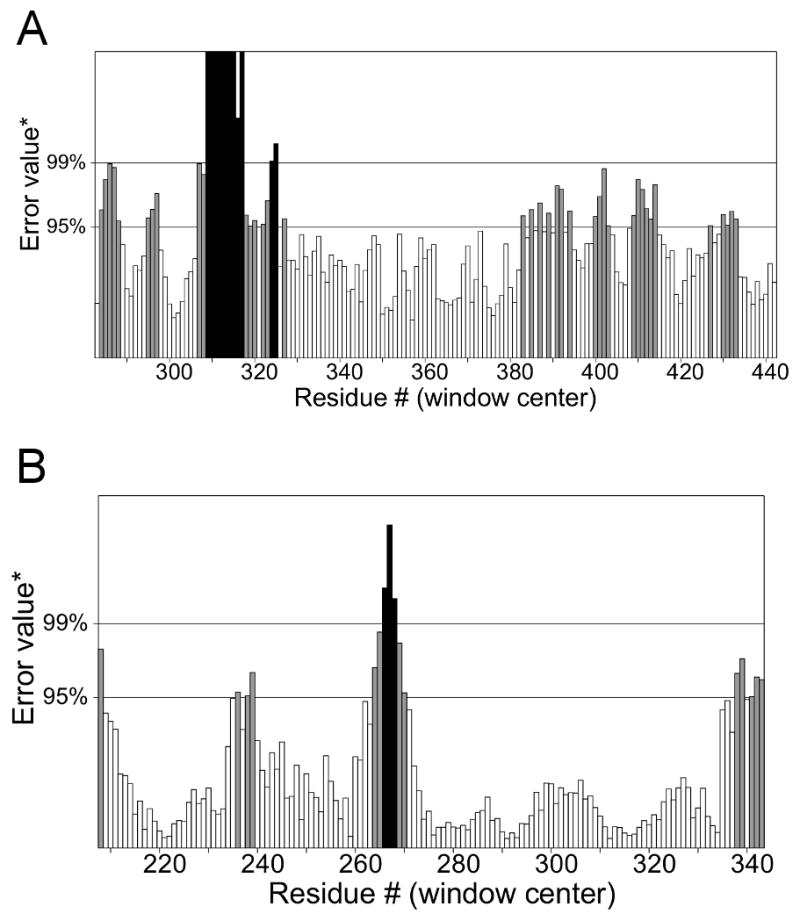
Overall quality assessment of the model evaluated using ERRAT. The figures compare the template crystal structure and homology model for (A) PF3D7_1305500 and (B) MKP3.
Figure 5.

Ramachandran plot. The quality of the homology model was validated and found to have 89.2% of the residues in favorable positions.
Ligand Selection and Drug Susceptibility Assay
The European Bioinformatics Institute (ChEMBL-NTD) has published open access phenotypic screening datasets that focus on neglected tropical diseases. Inhibitory compounds were previously screened against P. falciparum 3D7 and have a minimum inhibitory potential of 80% validated using LDH activity assays as an index of growth (GSK TCAMS Dataset) or erythrocyte-based proliferation assays (Novartis-GNF Malaria Box Dataset) (29, 77). The bioactive drug-like small molecules in the database adhere to the Lipinski rule-of-five and some provide abstracted bioactivities (78). Therefore, this collection of validated bioactive compounds represents the best, most diverse sets of compounds currently available for anti-malarial drug discovery research.
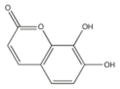
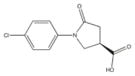
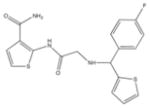
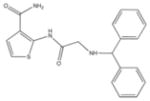
From the ChEMBL-NTD datasets, compounds were docked and ranked according to the GLIDE score. The 5% of the highest-ranking compounds were evaluated and selected on the basis of their quality of interaction represented by the GLIDE calculation. Poses of each compound were resolved to show the theoretical interaction of each molecule with the active site residues (Figure 6). Of the most promising compounds, seven were readily available by commercial sources (390097; 7,8-Dihydroxy-2H-chromen-2-one: 524725; 1-(4-Chlorophenyl)-5-oxo-3-pyrrolidinecarboxylic acid: 533073; 2-((N-[(4-Fluorophenyl)(2-thienyl)methyl]glycyl)amino)-3-thiophenecarboxamide: 533730; 2-([N-(Diphenylmethyl)glycyl]amino)-3-thiophenecarboxamide: 525841; 3-[(E)-(1H-Benzimidazol-2-ylhydrazono)methyl]-2-chloro-7-methoxyquinoline: 579624; 2-[(2E)-2-(1,3-Benzodioxol-5-ylmethylene)hydrazino]-1H-benzimidazole: 585222; 2-[(2E)-2-(3,4-Dimethoxybenzylidene)hydrazino]-1H-benzimidazole) (Table 1) and therefore selected for the validation.
Figure 6.

DUSP domain virtual screen. The homology model structure of the binding site of the PF3D7_1305500 DUSP domain was used for an in silico virtual screen of compound structures in ChEMBL database. (A) The binding pocket is surrounded by three conserved residues (D345, C383, I398) thought to be involved in substrate binding. Additional residues adjacent to the binding pocket with predicted polar interaction of the ligands were also included. (B–H) This pocket was targeted for virtual screening and 7 of the compounds with greatest affinity to this site were identified.
Table 1.
Selected ChEMBL-NTD compounds used for in vitro screening.
| Structure | ChEMBL ID | Name | MW | LogP | HBA | HBD | Score |
|---|---|---|---|---|---|---|---|
|
390097 | 7,8-Dihydroxy-2H-chromen-2-one | 178.1 | 1.42 | 4 | 2 | −8.35 |
|
524725 | 1-(4-Chlorophenyl)-5-oxo-3-pyrrolidinecarboxylic acid | 239.6 | 1.40 | 3 | 1 | −8.41 |
|
533073 | 2-((N-[(4-Fluorophenyl)(2-thienyl)methyl]glycyl)amino)-3- thiophenecarboxamide | 389.5 | 2.46 | 3 | 3 | −8.29 |
|
533730 | 2-([N-(Diphenylmethyl)glycyl]amino)-3-thiophenecarboxamide | 365.5 | 2.40 | 3 | 3 | −8.42 |
|
|
525841 | 3-[(E)-(1H-Benzimidazol-2-ylhydrazono)methyl]-2-chloro-7-methoxyquinoline | 351.8 | 3.91 | 5 | 2 | −8.36 |
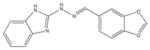
|
579624 | 2-[(2E)-2-(1,3-Benzodioxol-5-ylmethylene)hydrazino]-1H-benzimidazole | 280.3 | 3.27 | 6 | 2 | −7.23 |
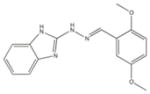
|
585222 | 2-[(2E)-2-(3,4-Dimethoxybenzylidene)hydrazino]-1H-benzimidazole | 298.3 | 3.47 | 6 | 2 | −7.44 |
The bioactivity of the selected compounds was contrasted between the wild-type parasite strain (NF54) and an attenuated line unable to express PF3D7_1305500 (C9) (Figure 7). These in vitro assays revealed that C9 had reduced susceptibility to 533073, 533730 and 579624 when compared with NF54 (Table 2). The mechanisms of action for each of these antimalarials is not known; however, considering the absence of the proposed target in the C9 parasite line it would be expected that there would be an altered response in these parasites when compared to the wild-type. Understanding the difference in susceptibility allows us to develop further studies to delineate the mechanisms of action for each of the selected molecular inhibitors.
Figure 7.

SYBR green I assay of selected compounds. Mean EC50 values were calculated and plotted in GraphPad Prism 6. Three compounds 533073, 533730 and 579624 shows significantly lower activity against C9 parasites.
Table 2.
Comparison of NF54 and C9 susceptibility to the selected compounds. EC50 values obtained from treating the wild type (NF54) and ΔPF3D7_1305500 (C9) parasites were compared using the Mann-Whitney test (α= 0.05). Compounds 5333073, 533730 and 579624 show NF54 to have significantly greater susceptibility (p< 0.05). This suggests that C9 parasites may have a slight advantage when not expressing the predicted target.
| Compound ID | EC50 NF54 (nM) | EC50C9 (nM) | p |
|---|---|---|---|
| 390097 | 340.2±53.6 | 248.9±33.7 | 0.1124 |
| 524725 | 503.1±36.5 | 248.4±62 | 0.0240 |
| 533073 | 471.9±37.4 | 1227.3±86.3 | 0.0013 |
| 533730 | 196.3±113.8 | 510.9±51.6 | 0.0073 |
| 525841 | 186.1±33.8 | 224.7±48.1 | 0.5467 |
| 579624 | 136±33.1 | 584±39.86 | 0.0010 |
| 585222 | 378.8±97.7 | 339.3±28 | 0.7070 |
Discussion
Phosphorylation cascades are important regulatory process that exert their influence on cellular development through signal transduction and as a result have been investigated extensively to elucidate targets of chemotherapeutic intervention (12, 25, 79–82). These pathways are not yet fully characterized in Plasmodium and consequently there have not been any classes of drugs developed to target phosphorylation-dependent signal transduction cascades. Based on our preliminary understanding of PF3D7_1305500, pathways utilizing this atypical phosphatase are involved in important cell cycle regulatory processes during intraerythrocytic asexual development. Through homology modeling of the atypical MKP DUSP domain and in silico high-throughput screening of its binding pocket, we investigated the possibility of PF3D7_1305500 as a drug target. Previously, these methods were successful in identifying potential inhibitory compounds using other components of the developmental cycle (83).
In P. falciparum, PF3D7_1305500 is a unique atypical phosphatase without any other identifiable paralogs in the genome. It is conserved among Plasmodium species as a single copy ortholog present in P. berghei (PBANKA_140400), P. c. chabaudi (PCHAS_140590), P. knowlesi (PKH_140400), P. vivax (PVX_12110), P. cynomolgi (PCYB_141500), and P. y. yoelii (PYYM_1407600). Conservation among the Plasmodium parasites, especially P. falciparum and P. vivax is favorable, since the ability to target both of these parasites with the same drug would present a clear advantage (84, 85). The conserved binding pocket in the DUSP domain homology model maintained the necessary features for activity and demonstrated an ability to accommodate an inhibitory substrate. Considering these structural characteristics, it is likely that PF3D7_1305500 interacts with the MAPK signaling pathway that is indispensable to P. falciparum development similar to its critical function in other eukaryote organisms (11, 17, 86–91). In addition, low homology with the closest mammalian orthologs is often an indicator of favorable drug targets. Since unique and conserved genes are typically under negative selection making their products essential, low homology limits adverse off-target effects in the mammalian host (83, 92).
The ChEMBL-NTD contains thousands of compounds with validated antimalarial activity profiles (29, 93, 94). In light of their inhibitory actions, the targets and mechanisms of action for most of them in P. falciparum are yet to be determined. Hypothetical malarial modes of action were developed for a few compounds to help facilitate their application against Plasmodium through historical GSK data regarding biochemical activity with human and microbial targets (29). In the previous studies, possible targets were inferred, but none of the hypothetical targets were associated with the compounds identified through the in silico screen in this current study.
To investigate the potential targeting of this P. falciparum atypical MKP by the selected inhibitory compounds, the susceptibility of NF54 wild-type parasite line was compared to the mutant P. falciparum clone C9, which is a genetically attenuated parasite line not expressing PF3D7_1305500. In vitro growth inhibition assays revealed that compounds 524725, 533073 and 533730 have lower efficacy against mutant parasites compared to the wild-type NF54. Although further definitive studies are required this finding indicated that the MKP DUSP is a target of these compounds, enabling us to postulate it as a therapeutic target. Investigation of potential off-target activity was not feasible in this study.
Conclusions and Future Directions
One of the main challenges to post genomic biology is translating a pathogen’s genome to new drug targets to take advantage of combinatorial methods. In this study we demonstrated the ability to utilize in silico HTS to identify from a chemical database suitable lead candidates for novel target validated in a forward genetic screen. Greater utilization of this approach offers a great opportunity to assist drug discovery efforts and accelerating structure-based design. In practice, experimentally determined structures are preferred for in silico studies; however, the number of pharmaceutical targets of interest has far outpaced the ability to experimentally develop protein structures (30). As a result, homology modeling has become the popular method of investigation for the growing number of interesting pharmaceutical targets. Comparisons of docking results from both homology models and experimentally validated structures have produced comparable results (95, 96). Exploitation of multiple strategies is necessary in order to advance the base of knowledge in this field.
Our findings show a proof-of concept using in silico screening of available compound libraries with a Plasmodium protein to identify inhibitors and potentially elucidate mechanisms of action. The approach used employs both experimental and computational methods to identify drug compounds, which would be vital to the search for new antimalarial drugs. This method expedites the screening of large sets of compounds, allowing the identification of more manageable compounds sets in a reduced timeframe.
Acknowledgments
This study was supported by funds from US National Institutes of Health (R01AI033656, R01AI094973, F31AI083053) and USF BITT Seed grant. The authors thank Robert Deschenes, Dennis Kyle, and Andreas Seyfang for their input on these studies.
Footnotes
Conflicts of Interest
All authors confirm we have no financial or commercial conflicts of interest associated with this study.
The authors confirm we have no conflicts of interest associated with this study.
References
- 1.Small I. Predotar v. 1.03 A prediction service for identifying putative N-terminal targeting sequences. Genoplante; 2003. Predotar v. 1.03 A prediction service for identifying putative N-terminal targeting sequences. [Google Scholar]
- 2.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–67. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude RJ, Pontavornpinyo W, Saralamba S, Aguas R, Yeung S, Dondorp AM, et al. The last man standing is the most resistant: eliminating artemisinin-resistant malaria in Cambodia. Malar J. 2009;8:31. doi: 10.1186/1475-2875-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dondorp AM, Yeung S, White L, Nguon C, Day NP, Socheat D, et al. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol. 2010;8:272–80. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- 5.WHO. World Malaria Report 2011. World Health Organization; 2011. World Malaria Report 2011. [Google Scholar]
- 6.Wells TN, Alonso PL, Gutteridge WE. New medicines to improve control and contribute to the eradication of malaria. Nat Rev Drug Discov. 2009;8:879–91. doi: 10.1038/nrd2972. [DOI] [PubMed] [Google Scholar]
- 7.Takala-Harrison S, Clark TG, Jacob CG, Cummings MP, Miotto O, Dondorp AM, et al. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:240–5. doi: 10.1073/pnas.1211205110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balu B, Chauhan C, Maher SP, Shoue DA, Kissinger JC, Fraser MJ, Jr, et al. piggyBac is an effective tool for functional analysis of the Plasmodium falciparum genome. BMC Microbiol. 2009;9:83. doi: 10.1186/1471-2180-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balu B, Singh N, Maher SP, Adams JH. A genetic screen for attenuated growth identifies genes crucial for intraerythrocytic development of Plasmodium falciparum. PLoS One. 2010;5:e13282. doi: 10.1371/journal.pone.0013282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balu B, Campbell C, Sedillo J, Maher S, Singh N, Thomas P, et al. Atypical mitogen-activated protein kinase phosphatase implicated in regulating transition from pre-S-Phase asexual intraerythrocytic development of Plasmodium falciparum. Eukaryotic cell. 2013;12:1171–8. doi: 10.1128/EC.00028-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farooq A, Zhou M-M. Structure and regulation of MAPK phosphatases. Cellular signalling. 2004;16:769–79. doi: 10.1016/j.cellsig.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 12.Barr AJ, Knapp S. MAPK-specific tyrosine phosphatases: new targets for drug discovery? Trends in pharmacological sciences. 2006;27:525–30. doi: 10.1016/j.tips.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Cohen PT, Philp A, Vazquez-Martin C. Protein phosphatase 4--from obscurity to vital functions. FEBS Lett. 2005;579:3278–86. doi: 10.1016/j.febslet.2005.04.070. [DOI] [PubMed] [Google Scholar]
- 14.Toth K, Djeha H, Ying B, Tollefson AE, Kuppuswamy M, Doronin K, et al. An oncolytic adenovirus vector combining enhanced cell-to-cell spreading, mediated by the ADP cytolytic protein, with selective replication in cancer cells with deregulated wnt signaling. Cancer Res. 2004;64:3638–44. doi: 10.1158/0008-5472.CAN-03-3882. [DOI] [PubMed] [Google Scholar]
- 15.Camps M, Nichols a, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2000;14:6–16. [PubMed] [Google Scholar]
- 16.Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Current opinion in cell biology. 2000;12:186–92. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 17.Farooq a, Chaturvedi G, Mujtaba S, Plotnikova O, Zeng L, Dhalluin C, et al. Solution structure of ERK2 binding domain of MAPK phosphatase MKP-3: structural insights into MKP-3 activation by ERK2. Molecular cell. 2001;7:387–99. doi: 10.1016/s1097-2765(01)00186-1. [DOI] [PubMed] [Google Scholar]
- 18.Noordman YE, Jansen PAM, Hendriks W. Tyrosine-specific MAPK Phosphatases and the control of ERK Signalling in PC12 Cells. Journal of Molecular Signaling. 2006;1:4. doi: 10.1186/1750-2187-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim Y, Gentry MS, Harris TE, Wiley SE, Lawrence JC, Dixon JE. A conserved phosphatase cascade that regulates nuclear membrane biogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6596–601. doi: 10.1073/pnas.0702099104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondoh K, Nishida E. Regulation of MAP kinases by MAP kinase phosphatases. Biochimica et biophysica acta. 2007;1773:1227–37. doi: 10.1016/j.bbamcr.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Rudolph J. Cdc25 phosphatases: structure, specificity, and mechanism. Biochemistry. 2007;46:3595–604. doi: 10.1021/bi700026j. [DOI] [PubMed] [Google Scholar]
- 22.Andreeva AV, Kutuzov MA. Protozoan protein tyrosine phosphatases. Int J Parasitol. 2008;38:1279–95. doi: 10.1016/j.ijpara.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 23.Szöor B, Ruberto I, Burchmore R, Matthews KR. A novel phosphatase cascade regulates differentiation in Trypanosoma brucei via a glycosomal signaling pathway. Genes & development. 2010;24:1306–16. doi: 10.1101/gad.570310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barford D, Das aK, Egloff MP. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annual review of biophysics and biomolecular structure. 1998;27:133–64. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 25.Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. 2008;60:261–310. doi: 10.1124/pr.107.00106. [DOI] [PubMed] [Google Scholar]
- 26.Denu JM, Dixon JE. Protein tyrosine phosphatases: mechanisms of catalysis and regulation. Current opinion in chemical biology. 1998;2:633–41. doi: 10.1016/s1367-5931(98)80095-1. [DOI] [PubMed] [Google Scholar]
- 27.Hinton SD, Myers MP, Roggero VR, Allison LA, Tonks NK. The pseudophosphatase MK-STYX interacts with G3BP and decreases stress granule formation. Biochem J. 2010;427:349–57. doi: 10.1042/BJ20091383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wishart MJ, Dixon JE. Gathering STYX: phosphatase-like form predicts functions for unique protein-interaction domains. Trends in biochemical sciences. 1998;23:301–6. doi: 10.1016/s0968-0004(98)01241-9. [DOI] [PubMed] [Google Scholar]
- 29.Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL, et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–10. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 30.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–49. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 31.McInnes C. Virtual screening strategies in drug discovery. Curr Opin Chem Biol. 2007;11:494–502. doi: 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 32.Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, et al. Plasmo DB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37:D539–43. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein Identification and Analysis Tool on the ExPASy Server. Humana Press; 2005. Protein Identification and Analysis Tool on the ExPASy Server. [Google Scholar]
- 34.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–9. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geer LY, Domrachev M, Lipman DJ, Bryant SH. CDART: protein homology by domain architecture. Genome Res. 2002;12:1619–23. doi: 10.1101/gr.278202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–5. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–20. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sigrist CJ, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, et al. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 2010;38:D161–6. doi: 10.1093/nar/gkp885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, et al. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34:W362–5. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001;313:903–19. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- 41.Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5857–64. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Letunic I, Doerks T, Bork P. SMART 6: recent updates and new developments. Nucleic Acids Res. 2009;37:D229–32. doi: 10.1093/nar/gkn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuckerkandl E, Pauling L. Molecules as documents of evolutionary history. Journal of theoretical biology. 1965;8:357–66. doi: 10.1016/0022-5193(65)90083-4. [DOI] [PubMed] [Google Scholar]
- 44.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular biology and evolution. 1987;4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 45.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nucleic Acids Res. 2005;33:W36–8. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 49.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–62. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 50.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–49. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 51.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 52.Zuegge J, Ralph S, Schmuker M, McFadden GI, Schneider G. Deciphering apicoplast targeting signals--feature extraction from nuclear-encoded precursors of Plasmodium falciparum apicoplast proteins. Gene. 2001;280:19–26. doi: 10.1016/s0378-1119(01)00776-4. [DOI] [PubMed] [Google Scholar]
- 53.Waller RF, Keeling PJ, Donald RG, Striepen B, Handman E, Lang-Unnasch N, et al. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12352–7. doi: 10.1073/pnas.95.21.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waller RF, Reed MB, Cowman AF, McFadden GI. Protein trafficking to the plastid of Plasmodium falciparum is via the secretory pathway. Embo J. 2000;19:1794–802. doi: 10.1093/emboj/19.8.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bologna G, Yvon C, Duvaud S, Veuthey AL. N-Terminal myristoylation predictions by ensembles of neural networks. Proteomics. 2004;4:1626–32. doi: 10.1002/pmic.200300783. [DOI] [PubMed] [Google Scholar]
- 56.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 57.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–92. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 60.Stewart AE, Dowd S, Keyse SM, Mcdonald NQ. Crystal structure of the MAPK phosphatase Pyst1 catalytic domain and implications for regulated activation. America. 1999;6:174–82. doi: 10.1038/5861. [DOI] [PubMed] [Google Scholar]
- 61.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 62.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 63.Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–6. 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 64.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein science: a publication of the Protein Society. 1993;2:1511–9. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trivedi V, Nag S. In silico characterization of atypical kinase PFD0975w from Plasmodium kinome: a suitable target for drug discovery. Chemical biology & drug design. 2012;79:600–9. doi: 10.1111/j.1747-0285.2012.01321.x. [DOI] [PubMed] [Google Scholar]
- 66.Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–70. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 67.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–5. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 68.Laurie AT, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–16. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 69.Burgoyne NJ, Jackson RM. Predicting protein interaction sites: binding hot-spots in protein-protein and protein-ligand interfaces. Bioinformatics. 2006;22:1335–42. doi: 10.1093/bioinformatics/btl079. [DOI] [PubMed] [Google Scholar]
- 70.Hendlich M, Rippmann F, Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. Journal of molecular graphics & modelling. 1997;15:359–63. 89. doi: 10.1016/s1093-3263(98)00002-3. [DOI] [PubMed] [Google Scholar]
- 71.Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein science: a publication of the Protein Society. 1998;7:1884–97. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liang J, Edelsbrunner H, Fu P, Sudhakar PV, Subramaniam S. Analytical shape computation of macromolecules: II. Inaccessible cavities in proteins. Proteins. 1998;33:18–29. [PubMed] [Google Scholar]
- 73.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. Journal of Physical Chemistry; 2001:6474–87. [Google Scholar]
- 74.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of medicinal chemistry. 2004;47:1739–49. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 75.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. Journal of medicinal chemistry. 2006;49:6177–96. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 76.MR4. Methods in Malaria Research. Paris, France: Malaria Research and Reference Reagent Resourse Center; 2008. [Google Scholar]
- 77.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9059–64. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lipinski C. Lead- and drug-like compounds: the rule-of-five revolution. Drug discovery today. 2004;1:337–41. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 79.Ham S. Studies on Menadione as an Inhibitor of the cdc25 Phosphatase. Bioorganic Chemistry. 1997;25:33–6. [Google Scholar]
- 80.Brohm D, Philippe N, Metzger S, Bhargava A, Müller O, Lieb F, et al. Solid-phase synthesis of dysidiolide-derived protein phosphatase inhibitors. Journal of the American Chemical Society. 2002;124:13171–8. doi: 10.1021/ja027609f. [DOI] [PubMed] [Google Scholar]
- 81.Dobson S, May T, Berriman M, Del Vecchio C, Fairlamb aH, Chakrabarti D, et al. Characterization of protein Ser/Thr phosphatases of the malaria parasite, Plasmodium falciparum: inhibition of the parasitic calcineurin by cyclophilin-cyclosporin complex. Molecular and biochemical parasitology. 1999;99:167–81. doi: 10.1016/s0166-6851(99)00010-9. [DOI] [PubMed] [Google Scholar]
- 82.Contour-Galcera MO, Sidhu A, Prevost G, Bigg D, Ducommun B. What’s new on CDC25 phosphatase inhibitors. Pharmacology & therapeutics. 2007;115:1–12. doi: 10.1016/j.pharmthera.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 83.Ludin P, Woodcroft B, Ralph S, Maser P. In silico prediction of antimalarial drug target candidates. Int J Parasitol. 2012;2:191–9. doi: 10.1016/j.ijpddr.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Delves M, Plouffe D, Scheurer C, Meister S, Wittlin S, Winzeler EA, et al. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS Med. 2012;9:e1001169. doi: 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Enayati A, Hemingway J. Malaria management: past, present, and future. Annual review of entomology. 2010;55:569–91. doi: 10.1146/annurev-ento-112408-085423. [DOI] [PubMed] [Google Scholar]
- 86.Gustin MC, Albertyn J, Alexander M, Davenport K. MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1998;62:1264–300. doi: 10.1128/mmbr.62.4.1264-1300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rangarajan R, Bei AK, Jethwaney D, Maldonado P, Dorin D, Sultan AA, et al. A Mitogen-activated Protein Kinase Regulates Male Gametogenesis and Transmission of the Malaria Parasite Plasmodium berghei. EMBO reports. 2005;6:464–9. doi: 10.1038/sj.embor.7400404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dorin-Semblat D, Quashie N, Halbert J, Sicard A, Doerig C, Peat E, et al. Functional characterization of both MAP kinases of the human malaria parasite Plasmodium falciparum by reverse genetics. Molecular microbiology. 2007;65:1170–80. doi: 10.1111/j.1365-2958.2007.05859.x. [DOI] [PubMed] [Google Scholar]
- 89.Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26:3203–13. doi: 10.1038/sj.onc.1210412. [DOI] [PubMed] [Google Scholar]
- 90.Low H, Chua CS, Sim T-S. Regulation of Plasmodium falciparum Pfnek3 relies on phosphorylation at its activation loop and at threonine 82. Cellular and molecular life sciences: CMLS. 2009;66:3081–90. doi: 10.1007/s00018-009-0101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Surachetpong W, Singh N, Cheung KW, Luckhart S. MAPK ERK signaling regulates the TGF-beta1-dependent mosquito response to Plasmodium falciparum. PLoS pathogens. 2009;5:e1000366. doi: 10.1371/journal.ppat.1000366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gagaring K, Borboa R, Francek C, Chen Z, Buenviaje J, Plouffe D, et al. Novartis, editor. Novartis-GNF Malaria Box Dataset. Genomics Institute of the Novartis Research Foundation (GNF); 2010. Novartis-GNF Malaria Box Dataset. [Google Scholar]
- 94.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, et al. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–5. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lye YM, Chan M, Sim TS. Pfnek3: an atypical activator of a MAP kinase in Plasmodium falciparum. FEBS Lett. 2006;580:6083–92. doi: 10.1016/j.febslet.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 96.McGovern SL, Shoichet BK. Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. Journal of medicinal chemistry. 2003;46:2895–907. doi: 10.1021/jm0300330. [DOI] [PubMed] [Google Scholar]