

Abstract
Insufficient oxygen leads to the cessation of growth in favor of cellular survival. Our unique model of high-altitude human pregnancy indicates that hypoxia-induced reductions in fetal growth occur at higher levels of oxygen than previously described. Fetal PO2 is surprisingly high and fetal oxygen consumption unaffected by high altitude, whereas fetal glucose delivery and consumption decrease. Placental delivery of energy-generating substrates to the fetus is thus altered by mild hypoxia, resulting in maintained fetal oxygenation but a relative fetal hypoglycemia. Our data point to this altered mix of substrates as a potential initiating factor in reduced fetal growth, since oxygen delivery is adequate. These data support the existence, in the placenta, of metabolic reprogramming mechanisms, previously documented in tumor cells, whereby HIF-1 stimulates reductions in mitochondrial oxygen consumption at the cost of increased glucose consumption. Decreased oxygen consumption is not due to substrate (oxygen) limitation but rather results from active inhibition of mitochondrial oxygen utilization. We suggest that under hypoxic conditions, metabolic reprogramming in the placenta decreases mitochondrial oxygen consumption and increases anerobic glucose consumption, altering the mix of energy-generating substrates available for transfer to the fetus. Increased oxygen is available to support the fetus, but at the cost of less glucose availability, leading to a hypoglycemia-mediated decrease in fetal growth. Our data suggest that metabolic reprogramming may be an initiating step in the progression to more severe forms of fetal growth restriction and points to the placenta as the pivotal source of fetal programming in response to an adverse intrauterine environment.
Keywords: hypoxia, glucose, trophoblast, mitochondria, HIF-1
Studies of fetal growth restriction
A majority of the studies examining the fetal growth restriction have chosen to investigate the differences between normally grown fetuses and those whose birth weight is below a particular cutoff, e.g. the 10th, 5th or 3rd centile on the growth curve. The more detailed studies have also required additional criteria to distinguish small-for-gestational-age fetuses (SGA) from those fetuses failing to reach their growth potential, defined as intrauterine growth retardation (IUGR). These criteria include parameters such as abnormal fetal heart rate, fetal Doppler resistance indices and fetal distress. In these latter studies, the fetuses are usually at the extreme of abnormal growth. This presents both advantages and disadvantages for study and interpretation. As a growth extreme, the group is clearly defined and well separated from the normal growth controls. Moreover abnormalities in many growth parameters such as concentrations of metabolites or growth factors are maximized by comparison with the normal growth group. The disadvantage of investigating this extreme of fetal growth is the likely presence of multiple abnormalities in metabolism, gene expression and morphology, some of which may be causal while others are the consequence of fetal growth reduction. In human studies especially, where only a single sampling point is available, distinguishing between cause and effect is frequently problematic.
Current studies of chronic (altitude-induced) hypoxia and fetal growth
In contrast to the investigations described above, we will describe in this article our studies on the effects of chronic (altitude-induced) hypoxia on human pregnancy, placental function and the resultant decrease in fetal growth. In these studies the initiating agent is clear and well defined. The difference is that while there are significant reductions in fetal growth as a result of chronic hypoxia, these pregnancies are normal by all other standards and the degree of growth reduction does not generally extend into the SGA/IUGR range. Instead the entire birth weight distribution is right shifted such that mean birth weights are reduced, and ~ 10% of infants fall into the SGA category vs. 2.5% at sea level (Yip, 1987; for a more recent review see Moore, 2003), using appropriate sea level standards (Williams et al., 1982). We are looking therefore at a “gray” area between normal fetal growth and extreme growth restriction where we believe it is possible to dissect out the initiating mechanisms in growth reduction without the overlying complications of the multiple alterations observed in clinically significant IUGR. As a result of detailed ultrasound measurements enabling calculation of uterine and umbilical volumetric blood flows, we have been able, for the first time, to generate data on the supply to and the consumption by the fetus of the energy-generating substrates, oxygen and glucose. The results of these studies led us to develop a model of growth reduction consistent not only with our data but also with the other, more extreme forms of growth restriction. As part of this model we also introduce into perinatal biology the concept of placental metabolic reprogramming, a mechanism which results in the delivery of an altered mix of metabolic substrates to the fetus in hypoxic conditions. Metabolic reprogramming has only recently emerged within the hypoxia literature and has thus far been confined to fields such as tumorbiology (Koumenis and Wouters, 2006; Pouyssegur et al., 2006) and muscle molecular physiology (Aragones et al., 2008; Mason et al., 2007).
Although the mechanisms we describe are likely to be applicable to animal as well as human systems, we have in large part focused on the human data supporting changes in fetal growth as a result of metabolic reprogramming rather than to write an extensive review of the broader array of experimental animal models. We have, referenced the experimental animal literature where data on the human is unavailable or minimal.
Glucose and fetal growth
Studies on the association between fetal glycemic status and fetal growth have clearly shown that a reduced fetal blood glucose concentration is associated with fetal growth restriction (Bagga et al., 1990; Delmis et al., 1992; Economides and Nicolaides, 1989; Hubinont et al., 1991; Marconi et al., 1996; Nicolini et al., 1989; Setia et al., 2006; Soothill et al., 1987). In particular, the studies by Marconi et al., indicate that over gestation there is an increase in the maternal arterial-fetal venous glucose gradient and that this gradient is increased in IUGR as a consequence of lower fetal umbilical venous glucose concentration (Marconi et al., 1996). An important corollary to these observations is the confirmation that there is minimal gluconeogenesis in the fetus during conditions of normal growth and that this is unchanged in IUGR (Marconi et al., 1993). Hence fetoplacental glucose is derived entirely from maternal supply, and the difference in umbilical venous to arterial concentrations represents fetal glucose consumption.
Fetal growth decreases as a result of hypoglycemia. In animal models, hypoglycemia due to maternal hyperinsulinemia (Gruppuso et al., 1981; Lueder et al., 1992) or EGF deficiency (Kamei et al., 1999) leads to fetal growth restriction. These conditions also lead to increasing protein breakdown as oxidation of amino acid carbon substitutes for loss of glucose. Similarly, ovine growth restriction models show fetal hypoxia, hypoglycemia, hypoinsulinemia and protein breakdown associated with the reductions in fetal growth (Carver et al., 1997; Hay, 2006; van Veen et al., 1987; Wallace et al., 2004), but whether hypoglycemia precedes a fall in the growth trajectory has not been established. There is less directly applicable data available for the human. Rosenn and Miodovnik showed that maternal hypoglycemia in diabetics induced by (over)administration of insulin results in fetal hypoglycemia and reductions in fetal growth (Rosenn and Miodovnik, 2000). Other reports link maternal hypoglycemia to reductions in fetal growth (Khouzami et al., 1981; Langer et al., 1986; Sokol et al., 1982; Van Assche and De Prins, 1983), although some studies focus on SGA rather than IUGR fetuses. In aggregate these data support that fetal hypoglycemia is strongly associated with fetal growth restriction. It is thus widely acknowledged that relative hypoglycemia is present in the IUGR fetus, but a clinically significant hypoglycemia in utero has not been defined. There have been no large-scale studies of fetal growth in relation to glucose concentrations at birth, again because underlying pregnancy complications make it difficult to attribute reductions in fetal growth specifically to hypoglycemia.
Oxygen, hypoxia and fetal growth
Oxygen is a primary substrate for energy generation and therefore crucial for fetal and placental growth. An insufficient O2 supply leads to the cessation of growth in favor of cellular survival. In pregnancy this is exemplified by fetal metabolic and circulatory adaptations during hypoxia to ensure adequate O2 supplies to brain and heart, at the cost of restricted fetal growth. The association of lowered fetal PO2 with growth restriction is well documented. The >19,000 births reported by the Richardson group in Canada showed that fetal PO2 declined in a stepwise manner by ~ 2 mmHg per decrement with increasing severity of fetal growth restriction (<8th centile, <2nd centile) (Lackman et al., 2001). The specific mechanisms by which O2 deficiency affects growth have not been elucidated. The general assumption has been that when supply of a major energy-generating substrate such as O2 is limited, fetal growth is reduced to enable cell/tissue survival. It would be predicted therefore that pathophysiologies which alter fetal O2 availability would also alter fetal growth. Thus decreased pre-placental levels of oxygen caused by high-altitude residence, cyanotic heart disease or anemia have all been shown to produce decreases in fetal growth (Kingdom and Kaufmann, 1997; Moore et al., 1998; Shime et al., 1987). At the level of the placenta, reduced uteroplacental perfusion leading to fetal hypoxia contributes to preeclampsia and/or fetal growth restriction (Aardema et al., 2001; Bower et al., 1993; Campbell et al., 1986; Konje et al., 2003; Lunell et al., 1984; Lunell et al., 1979; Zamudio et al., 1995), frequently associated with shallow or inadequate placentation (Brosens et al., 1972; Labarrere and Faulk, 1994; Meekins et al., 1994). Similarly, altered placental structure, diffusional distance or reduced fetoplacental perfusion have been associated with decreased oxygen transfer and reduced fetal growth (Aardema et al., 2001; Ferrazzi et al., 2000; Giles et al., 1986; Kreczy et al., 1995; Mayhew et al., 2004; Meekins et al., 1994; Todros et al., 1999). Although the relationship between hypoxia and decreased fetal growth is clear, the specific mechanism by which growth reduction takes place has not been described.
The high altitude model
We have carried out a program of investigation comparing fetal growth at low altitude (LA) and high altitude (HA), a natural experiment comparing maternal and neonatal parameters from pregnancies exposed to normoxia or hypobaric hypoxia over the entire gestational course. The demographic and clinical characteristics of the mothers and the neonates from LA and HA are summarized in Table 1. In general, maternal characteristics were very similar between altitudes; the only differences were that the women in the study at HA were slightly older, slightly shorter and showed an increased mean arterial blood pressure, the latter consistent with prior studies in North America (Palmer et al., 1999). The infant characteristics showed a clear reduction in birth weight at HA, amounting to 304 ± 43 g (mean ± SEM), after adjustment for maternal age and parity, gestational age and fetal sex. Despite this substantial reduction in birth weight, there was no change in placental weight between altitudes, and thus there was a decreased birth/placental weight ratio. The HA infants had a lower birth length than the LA infants and tended to have decreased abdominal circumference, indicative of reduced third trimester growth.
TABLE 1.
MATERNAL AND NEONATAL CHARACTERISTICS (MEAN±SEM)
| Maternal characteristics | 400 m n=90 | 3600 m n=80 | p values |
|---|---|---|---|
| Gestational age at ultrasound (weeks) | 38.0 ± 0.2 | 37.9 ± 0.2 | p=0.70 |
| Age (years) | 28 ± 1 | 32 ± 1 | p<0.0001 |
| Gravidity | 2.7 ± 0.2 | 2.7 ± .3 | p=0.98 |
| Parity | 1.3 ± 0.2 | 1.2 ± −/2 | p=0.64 |
| Height (cm) | 159 ± 1 | 157 ± 1 | p=0.12 |
| Non-pregnant weight (kg) | 60 ± 1 | 60 ± 1 | p=0.65 |
| Non-pregnant Body Mass Index (kg/m2) | 24 ± 1 | 24 ± 1 | p=0.60 |
| Weight gain with pregnancy (kg) | 12 ± 1 | 13 ± 2 | p=0.05 |
| Mean arterial blood pressure (mm Hg) | 93 ± 1 | 97 ± 1 | P<0.0005 |
|
Infant characteristics
| |||
| Birth weight (grams, (unadjusted values) | 3480 ± 39 | 3167 ± 44 | p <0.0001 |
| Birth weight (grams, adjusted values) | 3485 ± 20 | 3168 ± 30 | p <0.0001 |
| Placental weight (g) | 470 ± 10 | 489 ± 13 | p = 0.25 |
| Birth/placental weight ratio | 7.6 ± 0.2 | 6.7 ± 0.2 | p < 0.0001 |
| Clinically assessed gestational age at birth (weeks) | 38.7± 0.1 | 38.5 ± 0.2 | p = 0.29 |
| Birth length (cm) | 50.5 ± 0.2 | 48.6 ± 0.2 | p < 0.0001 |
| Head circumference (cm) | 34.8 ± 0.2 | 34.5 ± 0.2 | p = 0.21 |
| Abdominal circumference (cm) | 34.3 ± 0.2 | 33.8 ± 0.2 | p =0.04 |
| Sex ratio M/F | 46/44 | 32/48 | p = 0.19 |
Altitude, blood flow and oxygenation
Prior to delivery (4±1 day, range, 0–10), maternal uterine artery diameters and Doppler blood flows were assessed as described (Palmer et al., 1992; Zamudio et al., 2007b). The results showed that there was a reduction in uterine diameter and a decrease in the mean blood flow velocity at HA. This data was used to calculate the total uterine volumetric blood flow, which decreased by 25%, from 690 ± 35 ml/min/kg uterine contents (fetal and placental weight) at LA to 514 ± 32 ml/min/kg at HA (p < 0.01). In a similar manner, calculations of umbilical venous volumetric blood flows were made using ultrasound measurements of the umbilical vein diameter and blood flows. Umbilical blood flow decreased from 101 ± 3 ml/min/ kg fetal weight at LA to 82 ± 4 ml/min/kg at HA (p<0.001).
At the same time as the ultrasound measurements were made, arterialized maternal blood was drawn for blood gas measurements and for analysis of hematocrit and hemoglobin. Umbilical arterial and venous blood samples were obtained from the doubly-clamped cord at elective cesarean section delivery for assessment of the same parameters. The calculated values for uterine oxygen delivery and fetal oxygen consumption are summarized in Fig. 1. What is apparent is that despite the differences in uterine blood flow and arterial PO2, oxygen delivery to the placenta does not differ between altitudes. Perhaps even more importantly, O2 delivery to delivery to the fetus (10.1 ml/min/kg at 400 m, 9.7 ml/min/kg at 3600 m, p=0.46) and O2 consumption by the fetus do not differ between the altitudes, despite differences in umbilical blood flow and umbilical venous PO2. This is due in large part to the increased hemoglobin observed in the maternal and fetal circulations at HA. Given that maternal arterial PO2 is reduced by >30 mmHg, our finding that umbilical venous PO2 is reduced by only 3 mmHg at 3600 m was very surprising. Moreover the fetal PO2 we report is well within the range of values reported at sea level (Postigo et al., 2009; Zamudio et al., 2007b).
Fig. 1. Oxygen delivery and consumption at low and high altitude.

Summary of the data calculated from blood flow and blood gas measurements describing maternal and fetal oxygenation, maternal oxygen delivery and fetal oxygen consumption at 400 m (low) and 3600 m (high) altitude.
Blood glucose and fetal glucose consumption
Blood samples obtained at the time of ultrasound measurements (maternal) or at delivery (fetal) were assayed for glucose and the results are shown in Fig. 2 with the calculated values for the maternal delivery and fetal uptake values (Zamudio et al., in press). It is apparent that maternal arterial glucose concentrations are similar between altitudes, but the venous concentrations are reduced at HA compared to LA (Fig. 2A). Therefore maternal extraction of glucose is increased in the HA pregnancies. Uteroplacental delivery of glucose is not different between altitudes (Fig. 2B, p=0.11). By contrast, both fetal umbilical venous and arterial glucose concentrations are lower at HA (Fig. 2C), although the venous-arterial difference is the same. Most importantly, the fetal consumption of glucose at HA is decreased by 35% under conditions of chronic hypoxia (Fig. 2D).
Fig. 2. Glucose delivery and consumption at low and high altitude.

(A) Maternal arterial and venous glucose concentrations at 400 m (low; red) and 3600 m (high; blue) altitude. (B) Uteroplacental delivery of glucose at 400 m and 3600 m, normalized per kg of uterine contents. (C) Umbilical venous and arterial blood glucose concentrations at 400 m and 3600 m. (D) Fetal uptake of glucose at 400 m and 3600 m, normalized per kg fetus.
Modeling growth restriction in chronic (altitude-induced) hypoxia
The primary results obtained from these investigations show that although birth weight is significantly reduced at HA, there is no alteration in fetal oxygen consumption under conditions of chronic hypoxia. Despite the sizeable decrease in maternal arterial PO2 and the probable reduction in intervillous PO2, the fall in umbilical venous PO2 is proportionately much smaller than might be predicted. In contrast, there is a highly significant reduction in umbilical venous and arterial glucose concentrations (p<0.0001) and a decrease in the rate of glucose consumption at HA. These results raise two primary questions.
Given that fetal oxygen delivery and oxygen consumption are unaltered in conditions of chronic hypoxia, why do HA fetuses still show growth reduction?
What is the mechanism behind these reductions?
We expected that hypoxic conditions would lead to a reduction in oxygen consumption and a compensatory increase in glucose consumption as the rate of anerobic glycolysis is increased to make up for decreased oxidative metabolism. This reciprocal relationship was not apparent however, since there was no change in fetal oxygen consumption. Instead the obvious candidate for reduced fetal growth is the decrease in fetal blood glucose levels and the decrease in fetal glucose consumption. As noted above, fetal hypoglycemia is associated with hypoxia in a variety of conditions and fetal hypoglycemia, in the absence of hypoxia, can produce fetal growth restriction, so such a scenario is plausible. By extension, therefore, the problem becomes a question of how fetal glucose delivery and consumption are decreased at HA despite the fact that maternal glucose delivery to the placenta does not differ between altitudes. Clearly there must be increased placental glucose consumption at HA, but why is it increased when there is apparently adequate oxygen to support oxidative metabolism? Our challenge was to develop a model which can account for the metabolic changes between the normoxic and chronically hypoxic placenta, consistent with reductions in fetal, but not placental growth.
Hypoxia responses and metabolic reprogramming
We believe that the answer to this challenge lies in the concept of metabolic reprogramming. Traditionally, hypoxia triggers a number of responses, common to most tissues and cells, responses designed either to increase tissue and cellular O2 supply or to enhance cellular survival under hypoxic conditions (Fig. 3). The first response consists of increased angiogenesis and erythropoiesis which, in the placenta, results in increased villous capillary density (Zamudio, 2003), and elevated secretion of angiogenic or hematopoietic factors such as VEGF and erythropoietin (Zamudio et al., 2006; Zamudio et al., 2007c). The second type of response, which enhances cell survival, is the metabolic adaptation to augment the energy supply via non-oxidative (i.e. anaerobic) pathways. In the placenta this process has been demonstrated most clearly by an increase in glucose consumption and concomitant lactate production during hypoxia (Cannell et al., 1988; Illsley et al., 1984; Kay et al., 2007) as well as upregulation of glucose transporters (Baumann et al., 2007; Esterman et al., 1997) and other key elements of the glycolytic pathway (Fukasawa et al., 2004). The third response is broadly defined as “demand reduction” and consists of the attenuation of energy-requiring processes which are not essential to cellular survival, including gross energy demands such as fetal movement (Hooper, 1995). At the tissue level, however, the primary means of demand reduction is the inhibition of transcription, translation and related processes which rely on protein synthesis, such as cellular growth and proliferation (Hochachka and Lutz, 2001). Although the mechanisms regulating demand reduction have not yet been examined in the placenta, their operation in pathological conditions can be inferred from the decreased placental growth often observed in conditions associated with reduced O2 delivery, such as fetal growth restriction and preeclampsia (Aardema et al., 2001; Han, 1993; Pardi et al., 2002). That is why the altitude model is of particular importance: placental growth is maintained, in the face of chronically reduced fetal growth that does not reach the level seen in pathological IUGR, or even clinical SGA in most cases. This implies the absence of placental demand reduction, despite chronic hypoxia. This is consistent with the concept that the high-altitude pregnancies fall into a group intermediate to the more frequently studied paradigm of normal versus severe IUGR pregnancies, a group where early reductions in fetal growth occur as a result of altered substrate delivery by the placenta.
Fig. 3. Responses to hypoxia.

Cellular responses to hypoxia, including the recently described “metabolic reprogramming”.
To these traditional responses can be added another response to decreased O2, “metabolic reprogramming”, described first in the tumor biology literature (Brahimi-Horn and Pouyssegur, 2006; Koumenis and Wouters, 2006; Pouyssegur et al., 2006) and more recently in muscle biochemistry (Aragones et al., 2008; Mason et al., 2007). Although comprised of several different and separate mechanisms (see below), collectively metabolic reprogramming produces a hypometabolic state characterized by a reduction in oxygen consumption that increases cellular oxygen availability. It is a reversible metabolic switch from carbohydrate oxidation to anaerobic glycolysis, driven by hypoxia. Each of the specific mechanisms, by different means and with different temporal responses, decreases oxidative metabolism. It is not a forced increase in anaerobic glycoysis due to irreversible losses in mitochondrial oxidative metabolism as characterized by the Warburg effect. Neither does this decrease in oxygen consumption arise from substrate (O2) limitation. Rather it is a direct result of processes which actively limit cellular O2 consumption.
Metabolic reprogramming mechanisms
Metabolic reprogramming consists of mechanisms which cause (1) the direct inhibition of mitochondrial oxygen consumption, (2) the diversion of carbohydrates away from oxidative pathways and (3) a reduction in mitochondrial mass, described briefly below. Each of the relevant mechanisms discussed below are associated with increased levels of Hypoxia Inducible Factor 1α (HIF-1α). Of critical importance to the model we propose here is that HIF-1α levels are doubled in the high altitude placenta, with concomitant increases in multiple target genes (Ietta et al., 2006; Nevo et al., 2006; Soleymanlou et al., 2005; Zamudio et al., 2007c).
Direct inhibition of mitochondrial oxygen consumption
In the first category of metabolic reprogramming systems are two specific mechanisms which directly inhibit mitochondrial oxygen consumption (Fig. 4). The first of these is the reversible inhibition of complex IV of the mitochondrial electron transport chain, cytochrome c oxidase, by nitric oxide (NO) which acts as a competitive inhibitor for O2 binding to cytochrome c (for a review, see Erusalimsky and Moncada, 2007). The IC50 for NO decreases substantially as O2 concentrations decrease, such that a drop in cellular O2 levels from 30 μM to 10 μM decreases the IC50 for NO from 60 nM to 20 nM, well within the range of measured endogenous, cellular NO levels (Malinski and Taha, 1992; Shibuki, 1990). As a result, with decreasing O2 concentration, endogenous NO will increasingly compete for binding to cytochrome c oxidase, decreasing oxygen consumption and leading to increased cellular oxygen availability (Hagen et al., 2003). In many cell types, NOS isoforms (eNOS, iNOS) are reported to be upregulated by hypoxia via a HIF-1-mediated mechanism (Coulet et al., 2003; Gao et al., 2005; Hellwig-Burgel et al., 2005; Land and Rae, 2005; Lu et al., 2006; Matrone et al., 2004; Melillo et al., 1997; Min et al., 2006; Semenza et al., 1997), thus increasing the production of NO at the same time that oxygen concentration is falling. Thus in vivo, NO and HIF-1 appear to act together to diminish the effects of hypoxia.
Fig. 4. Hypoxia inducible factor1 (HIF-1)-mediated inhibition of mitochondrial electron transport.
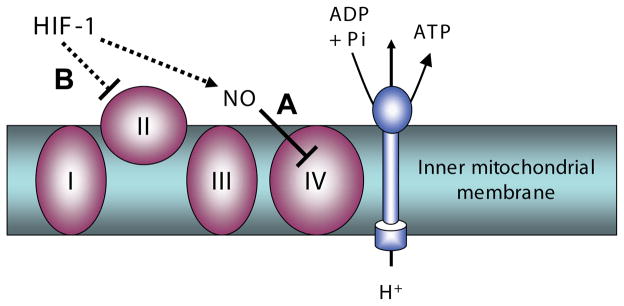
Mechanisms which reduce mitochondrial oxygen consumption. (A) Nitric oxide (NO) inhibition of mitochondrial electron transport chain complex IV, cytochrome c oxidase, stimulated by HIF-1. (B) Inhibition of mitochondrial electron transport chain complex II (succinate dehydrogenase, SDH) mediated by HIF-1 suppression of SDH subunit B expression.
The second mechanism involves the enzyme succinate dehydrogenase (SDH; Fig. 4). SDH is a component protein of the tricarboxylic acid (TCA) cycle, not only catalyzing the conversion of succinate to fumarate but also functioning as complex II in the mitochondrial electron transport chain, enabling transfer of electrons from FADH to ubiquinone. Under hypoxic conditions, where HIF-1 levels are elevated, expression of the SDH B subunit (SDHB) is suppressed, resulting in the inhibition of complex II of the electron transport chain (Dahia, 2006; Dahia et al., 2005), decreasing oxygen consumption and leading to increased oxygen availability. In addition, the inhibition of complex II increases levels of succinate (Guzy et al., 2008; King et al., 2006), which acts as a competitive inhibitor of the enzyme regulating HIF-1α degradations, the prolyl hydroxlase domain-containing protein (PHD)(Lu et al., 2005; Stolze et al., 2006). Thus, under hypoxic conditions, increased HIF-1 results in the suppression of SDHB expression, leading to increased succinate concentrations which, in turn, stabilize the increased level of HIF-1 (MacKenzie et al., 2007; Selak et al., 2005). This mechanism represents not only an oxygen sparing mechanism but also a means by which HIF-1 can be stabilized in the chronically hypoxic conditions documented in the HA placenta (Zamudio et al., 2007c).
Diversion of metabolic substrate from oxidative to anaerobic metabolism
The second category of metabolic reprogramming systems consists of two mechanisms which alter the flux of glucose carbon away from oxidative metabolism (Fig. 5). Pyruvate dehydrogenase (PDH) is a multisubunit enzyme which forms the gateway to the TCA cycle, converting pyruvate, the terminal metabolite of the glycolytic pathway, to acetyl CoA, through an irreversible reaction which directs carbon into the TCA cycle. The balance between aerobic and anaerobic metabolism is controlled in large part by PDH and thus it is under tight regulation. Pyruvate dehydrogenase kinase (PDK) inhibits PDH by phosphorylation of the PDH E1 regulatory subunit (Sugden and Holness, 2003), blocking carbon entry into the TCA cycle and forcing carbon flux from glucose toward lactate rather than providing reducing equivalents for oxidative metabolism. PDK consists of four isoforms which show differential activity towards the three phosphorylation sites on PDH (Kolobova et al., 2001); all the isoforms are capable of inactivating PDH by phosphorylation of site 1, however only PDK1 also phosphorylates both sites 2 and 3, which appear to regulate PDH phosphatase (reactivation) activity. Most importantly, the PDK1 isoform is significantly induced by hypoxia (Kim et al., 2006; Papandreou et al., 2006) so that under hypoxic conditions the increased expression of PDK1 leads to PDH inhibition. This decreases the flux through the TCA cycle, reducing the substrate availability for oxidative metabolism, thus decreasing oxygen consumption and leading to increased oxygen availability (Papandreou et al., 2006). This is a HIF-mediated process which, in addition to increasing oxygen availability, attenuates ROS production and increases cell survival (Kim et al., 2006; Papandreou et al., 2006).
Fig. 5. Diversion of glucose carbon flux away from oxidative metabolism.

(A) HIF-1 stimulation of the inhibition of pyruvate dehydrogenase (PDH) through pyruvate dehydrogenase kinase (PDK)-mediated phosphorylation. (B) HIF-1 stimulation of the expression of the lactate dehydrogenase A subunit.
The second of the diversion mechanisms is complementary to the alterations in PDH activity regulated by PDK and involves the enzyme lactate dehydrogenase (LDH). LDH catalyses the reversible interconversion of pyruvate and lactate at the end of the glycolytic pathway (Fig. 5). Most tissues express both the A (or M/muscle) and B (or H/heart) isoforms. The primary difference between the A and B isoforms is the binding of pyruvate. The A isoform preferentially catalyzes conversion of pyruvate to lactate, whereas the B isoform preferentially catalyses the reverse reaction (Markert, 1984; Read et al., 2001). The gene encoding LDHA has HIF-1-binding hypoxia response elements in its promoter sequence and responds to hypoxia by HIF-1-mediated upregulation of LDHA expression (Firth et al., 1995; Semenza et al., 1996). The upregulation of LDHA serves to channel pyruvate, the product of the glycolytic pathway, towards generation of lactate rather than providing substrates for the TCA cycle via conversion to acetyl CoA, thus decreasing oxygen consumption and leading to increased oxygen availability. The combination of HIF-upregulated LDHA and PDK will diminish substantially the supply of reducing equivalents to the mitochondrial electron transport chain.
Changes in mitochondrial mass
The third category of metabolic reprogramming systems encompasses mechanisms which alter cellular mitochondrial mass. Changes in mitochondrial mass, while a much slower response than those outlined above, have been observed under hypoxic conditions. These changes will be the sum of mitochondrial biogenesis and mitochondrial autophagy, both of which appear be regulated by hypoxia. Recent reports have shown that when HIF-1 is elevated mitochondrial mass is decreased whilst when HIF-1 is suppressed, mitochondrial DNA is increased relative to nuclear DNA (Zhang et al., 2007). In the former case mitochondrial biogenesis is decreased by HIF-1 mediated inhibition of expression and activity of the C-MYC transcription factor, leading to reduced expression of the PPAR-γ coactivator 1β (PGC-1β) and decreased biogenesis (Fig. 6)(Zhang et al., 2007). Mitochondrial O2 consumption and ROS production are decreased simultaneously. This report is consistent with the model of hypoxia in which HIF-1 upregulation stimulates a hypometabolic state, including decreased cellular respiration (Semenza, 2007; Zhang et al., 2007). The other factor contributing to mitochondrial mass is autophagy, a protein and organelle degradation process which has been observed in response to both hypoxia and nutrient deprivation (Hamacher-Brady et al., 2007; Yan et al., 2005; Zhu et al., 2007). Mitochondrial autophagy in response to hypoxia was recently reported as a HIF-1 dependent adaptive metabolic response, which decreases O2 consumption and ROS generation and as a result, prevents cell death (Zhang et al., 2008). The mechanism by which hypoxia induces mitochondrial autophagy appears to involve the apoptosis-related protein, BNIP3 (Bcl-2/adenovirus E1B 19kDa interacting protein 3) (Mellor and Harris, 2007; Tracy and Macleod, 2007) as well as components of the autophagic machinery, including Bcl-2 and the Bcl-2 interacting protein, Beclin-1 (Fig. 6). In combination, HIF-1-mediated changes in biogenesis and autophagy will favor a reduction in cellular mitochondrial mass, decreasing oxygen consumption and leading to increased oxygen availability.
Fig. 6. Pathways of hypoxia inducible factor1 (HIF-1)-mediated alterations in mitochondrial biogenesis and autophagy.

The partial signaling pathways by which hypoxia, via HIF-1, alters the rates of (A) mitochondrial biogenesis and (B) mitochondrial autophagy (from Zhang et al., 2008; Zhang et al., 2007).
Hypoxia and HIF-1 mediation of metabolic reprogramming
Cellular responses to hypoxia, are governed largely via the activation of gene transcription through the binding of the hypoxia-inducible factor-1 (HIF-1) to the hypoxia response elements on numerous genes (Semenza, 1998). Under normoxic conditions, the prolyl hydroxylase domain-containing proteins or PHDs utilize molecular O2 and the TCA cycle intermediate, 2-oxoglutarate, as substrates in reactions which hydroxylate two proline residues on HIF-1α while converting 2-oxoglutarate to succinate and CO2 (D’Angelo et al., 2003). The proline hydroxylation provides sites for the ubiquitination of HIF-1α by the von Hippel Lindau (VHL)/E3 ubiquitin ligase complex; ubiquitination of HIF-1α in turn targets the protein for rapid proteasomal degradation. Under hypoxic conditions the substrate limitation imposed by a decreased oxygen concentration reduces PHD activity, decreasing the rate of HIF-1α degradation and increasing HIF-1 levels. Although O2 concentration is the primary effector, activity of the PHDs is also modulated by metabolic intermediates (succinate, oxaloacetate, pyruvate) and by agents such as nitric oxide (NO) and reactive oxygen species (ROS) which alter the redox status of the non-heme iron in the catalytic core (Cash et al., 2007; Lu et al., 2005; Pan et al., 2007).
The elevation of HIF-1 by hypoxia appears to be the central regulatory element for metabolic reprogramming. HIF-1 increases the expression of endothelial nitric oxide synthase (eNOS) and the extent of eNOS phosphorylation (Chen and Meyrick, 2004; Dedkova et al., 2004; Shi et al., 2002), HIF-1 suppresses SDHB (Dahia et al., 2005), stimulates expression of PDK and LDHA (Firth et al., 1995; Kim et al., 2006; Papandreou et al., 2006; Semenza et al., 1996) and is involved in the regulation of mitochondrial mass via inhibition of biogenesis (Zhang et al., 2007) and the stimulation of mitochondrial autophagy (Zhang et al., 2008).
Metabolic reprogramming and placental function
We postulate that within the range of PO2 we observe in our high-altitude fetuses, metabolic reprogramming mechanisms are activated in the placental syncytiotrophoblast. As described above, activation of these mechanisms leads to decreased oxygen consumption and increased cellular availability of oxygen. We predict that that in the placenta, hypoxia-stimulated metabolic reprogramming will lead to (1) decreased O2 consumption and to (2) increased glucose consumption. The altered syncytial consumption of O2- and glucose produced by metabolic reprogramming will lead to an increased intrasyncytial level of O2 and a decreased intrasyncytial concentration of glucose. As the placental transfer of these substrates takes place by transsyncytial diffusional mechanisms, the altered syncytial levels alter placental-to-fetal gradients, increasing oxygen transfer and decreasing glucose transfer to the fetal circulation. This change in the availability of energy substrates, while resulting in the maintenance of fetal oxygenation, will also result in a relative fetal hypoglycemia that, in turn, appears to drive the reduced fetal growth observed under conditions of mild-moderate hypoxia.
Evidence supporting placental metabolic reprogramming
The data obtained from studies of high altitude pregnancy is consistent with the operation of metabolic reprogramming in conditions of chronic hypoxia. Umbilical venous PO2 was reduced by only 10% under hypoxic conditions, despite a 33% decrease in uterine arterial PO2, and fetal oxygen delivery and consumption were unchanged, consistent with the maintenance of oxygen transfer predicted by the operation of metabolic reprogramming mechanisms. By contrast, umbilical venous glucose concentration was decreased by 18% under conditions of chronic hypoxia, while fetal glucose consumption was reduced by 35%, consistent with increased placental glucose consumption in the face of oxygen sparing caused by metabolic reprogramming. As a result, birth weight was reduced by 9% while placental weight was unchanged.
In addition to this data, there is further evidence to support the possibility of placental metabolic reprogramming. Recent data shows that there is a clear increase in HIF-1α protein in syncytiotrophoblast cells in vivo under conditions of chronic hypoxia, as demonstrated both by Western blotting and by immunohistochemistry (Zamudio et al., 2007c). Other markers of hypoxia are also elevated in samples from HA pregnancies, including maternal erythropoietin and VEGF (Zamudio et al., 2007c), maternal and placental sFlt1 (Nevo et al., 2006) and increased syncytial erythropoietin and transferrin receptor (Zamudio et al., 2006). Global patterns of placental gene expression are markedly similar in the high altitude placentas, placental explants incubated in 3% oxygen and in preeclampsia, a disease in which placental HIF and HIF gene targets such as TGFβ3) levels are elevated (Caniggia et al., 2000; Soleymanlou et al., 2005). In addition, nitric oxide generation appears also to be upregulated, as demonstrated indirectly by the increase in the staining for nitrotyrosine observed in placental sections obtained from HA pregnancies (Zamudio et al., 2007a).
Implication for studies of fetal growth restriction
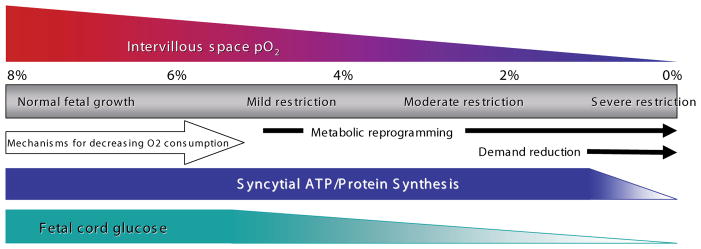
The placenta is positioned at the beginning of the fetoplacental supply line, thus placental O2 consumption and transfer are crucial components of the fetal response to hypoxia. Not only is the placenta the conduit for transfer of O2 to the fetus, but it is also an active metabolic organ that consumes up to 40% of total uterine O2 uptake (Carter, 2000). Changes in substrates used in placental energy generation will thus have significant effects on the extent and mix of the primary energy substrates (glucose and O2) delivered to the fetus. Our novel extension to the placenta of the metabolic reprogramming model developed in solid tumors argues that these processes are beneficial in normal physiology, supporting increased placental O2 transfer and decreased glucose transport, which in turn promote fetal survival at the cost of limiting fetal growth. The diagram in Fig. 7 illustrates our conceptualization of these processes, although not an exact delimitation of the initiation points. At the normal intervillous PO2 level of 6–8% (Jauniaux et al., 2001; Soothill et al., 1986) both placental and fetal growth are in the normal range as reflected by normal rates of protein synthesis, stable syncytial ATP levels and normal fetal blood glucose. As intervilllous PO2 drops below 5% there is an increase in syncytial HIF-1 which stimulates metabolic reprogramming, including oxygen sparing, which in turn maintains fetal oxygenation at the cost of increased placental glucose consumption. Excess placental glucose consumption maintains syncytial ATP levels and energy-intensive processes such as protein synthesis and placental growth, but leads to decreased transfer to the fetus and a drop in fetal blood glucose levels. This is associated with mild growth restriction as the reduced glucose supply (and consumption) leads to a reduced growth stimulus. As intervillous PO2 drops further, there is continued oxygen sparing and a further increase in placental glucose consumption leading to a greater drop in fetal blood glucose and moderate growth restriction. Finally as intervillous PO2 reaches low levels (<2%), the supply of energy substrates affects placental function, leading to decreased syncytial ATP levels, inhibition of protein synthesis and compromised function, including the transfer functions supplying the fetus. At this point, the restrictions in oxygen and glucose supply to the fetus are exacerbated not only by placental metabolic abnormalities and decreased transport function but also, in the case of chronic hypoxia, by structural abnormalities as placental growth is not only reduced but modified by the hypoxic environment.
Fig. 7. Model of growth restriction processes stimulated by hypoxia.
Conclusions
The data generated from the high altitude model and the metabolic programming model gives us the opportunity to look at the mid-section of the model presented in Fig. 7, an area that has hitherto not received detailed cellular, molecular or integrative physiological attention. It is critical to define the earliest changes that occur in fetal growth restriction, so that we may develop biomarkers that would refine our ability to engage in nutritional or other interventions, to assess longer term fetal risk and the timing of delivery, and to avoid unnecessary pre-term deliveries where fetal growth is compromised, but placental function is adequate. Perhaps as interesting is the impetus this concept gives to placental programming of adult health. The alteration in the mix of energy-generating substrates envisioned in this model suggests that the placenta plays a pivotal role in regulating the fetal growth response to an abnormal intrauterine environment. If further research validates this model, the idea that the placenta is a major component in fetal programming raises interesting diagnostic and therapeutic possibilities. If lowered fetal glucose concentrations are the earliest detectable step in decreased fetal growth, the rapid advances in metabolomics and imaging techniques such as magnetic resonance imaging may make non-invasive detection of cord blood glucose concentrations very useful diagnostically. This model also raises the possibility of a simpler therapeutic modality for early fetal growth restriction, given the greater accessibility of placental metabolic systems.
Acknowledgments
The authors would like to gratefully acknowledgement the support provided by the National Institutes of Health (NIH HD042737, HD046982, TW007444) the Canadian Institutes of Health Research (MT14096) and the National Science Foundation (BCS-0309142).
Abbreviations used in this paper
- HIF
hypoxia inducible factor
- IUGR
intrauterinegrowthretardation
- NO
nitricoxide
- NOS
nitricoxidesynthase
- PHD
prolyl hydroxylase domain
- SGA
small for gestational age
- TCA
tricarboxylic acid
References
- AARDEMA MW, OOSTERHOF H, TIMMER A, VAN ROOY I, AARNOUDSE JG. Uterine artery doppler flow and uteroplacental vascular pathology in normal pregnancies and pregnancies complicated by preeclampsia and small for gestational age fetuses. Placenta. 2001;22:405–411. doi: 10.1053/plac.2001.0676. [DOI] [PubMed] [Google Scholar]
- ARAGONES J, SCHNEIDER M, VAN GEYTE K, FRAISL P, DRESSELAERS T, MAZZONE M, DIRKX R, ZACCHIGNA S, LEMIEUX H, JEOUNG NH, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40:170–80. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- BAGGA R, VASISHTA K, MAJUMDAR S, GARG SK. Correlation between human placental lactogen levels and glucose metabolism in pregnant women with intrauterine growth retardation. Aust N Z J Obstet Gynaecol. 1990;30:310–3. doi: 10.1111/j.1479-828x.1990.tb02017.x. [DOI] [PubMed] [Google Scholar]
- BAUMANN MU, ZAMUDIO S, ILLSLEY NP. Hypoxic upregulation of glucose transporters in BeWo choriocarcinoma cells is mediated by hypoxia-inducible factor-1. Am J Physiol Cell Physiol. 2007;293:C477–85. doi: 10.1152/ajpcell.00075.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOWER S, BEWLEY S, CAMPBELL S. Improved prediction of preeclampsia by two-stage screening of uterine arteries using the early diastolic notch and color Doppler imaging. Obstet Gynecol. 1993;82:78–83. [PubMed] [Google Scholar]
- BRAHIMI-HORN C, POUYSSEGUR J. The role of the hypoxia-inducible factor in tumor metabolism growth and invasion. Bull Cancer. 2006;93:E73–80. [PubMed] [Google Scholar]
- BROSENS IA, ROBERTSON WB, DIXON HG. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet Gynecol Annu. 1972;1:177–91. [PubMed] [Google Scholar]
- CAMPBELL S, PEARCE JM, HACKETT G, COHEN-OVERBEEK T, HERNANDEZ C. Qualitative assessment of uteroplacental blood flow: early screening test for high-risk pregnancies. Obstet Gynecol. 1986;68:649–53. [PubMed] [Google Scholar]
- CANIGGIA I, MOSTACHFI H, WINTER J, GASSMANN M, LYE SJ, KULISZEWSKI M, POST M. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3) J Clin Invest. 2000;105:577–87. doi: 10.1172/JCI8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANNELL GR, KLUCK RM, HAMILTON SE, MORTIMER RH, HOOPER WD, DICKINSON RG. Markers of physical integrity and metabolic viability of the perfused human placental lobule. Clin Exp Pharmacol Physiol. 1988;15:837–44. doi: 10.1111/j.1440-1681.1988.tb01026.x. [DOI] [PubMed] [Google Scholar]
- CARTER A. Placental oxygen consumption. Part I: in vivo studies-a review. Placenta. 2000;21(Suppl A):S31–S37. doi: 10.1053/plac.1999.0513. [DOI] [PubMed] [Google Scholar]
- CARVER TD, QUICK AA, TENG CC, PIKE AW, FENNESSEY PV, HAY WW., JR Leucine metabolism in chronically hypoglycemic hypoinsulinemic growth-restricted fetal sheep. Am J Physiol. 1997;272:E107–17. doi: 10.1152/ajpendo.1997.272.1.E107. [DOI] [PubMed] [Google Scholar]
- CASH TP, PAN Y, SIMON MC. Reactive oxygen species and cellular oxygen sensing. Free Radic Biol Med. 2007;43:1219–25. doi: 10.1016/j.freeradbiomed.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN JX, MEYRICK B. Hypoxia increases Hsp90 binding to eNOS via PI3K-Akt in porcine coronary artery endothelium. Lab Invest. 2004;84:182–90. doi: 10.1038/labinvest.3700027. [DOI] [PubMed] [Google Scholar]
- COULET F, NADAUD S, AGRAPART M, SOUBRIER F. Identification of hypoxia-response element in the human endothelial nitric-oxide synthase gene promoter. J Biol Chem. 2003;278:46230–40. doi: 10.1074/jbc.M305420200. [DOI] [PubMed] [Google Scholar]
- D’ANGELO G, DUPLAN E, BOYER N, VIGNE P, FRELIN C. Hypoxia up-regulates prolyl hydroxylase activity: a feedback mechanism that limits HIF-1 responses during reoxygenation. J Biol Chem. 2003;278:38183–7. doi: 10.1074/jbc.M302244200. [DOI] [PubMed] [Google Scholar]
- DAHIA PL. Transcription association of VHL and SDH mutations link hypoxia and oxidoreductase signals in pheochromocytomas. Ann N Y Acad Sci. 2006;1073:208–20. doi: 10.1196/annals.1353.023. [DOI] [PubMed] [Google Scholar]
- DAHIA PL, ROSS KN, WRIGHT ME, HAYASHIDA CY, SANTAGATA S, BARONTINI M, KUNG AL, SANSO G, POWERS JF, TISCHLER AS, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005;1:72–80. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEDKOVA EN, JI X, LIPSIUS SL, BLATTER LA. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am J Physiol Cell Physiol. 2004;286:C406–15. doi: 10.1152/ajpcell.00155.2003. [DOI] [PubMed] [Google Scholar]
- DELMIS J, DRAZANCIC A, IV, ANISEVIC M, SUCHANEK E. Glucose, insulin, HGH and IGF-I levels in maternal serum, amniotic fluid and umbilical venous serum: a comparison between late normal pregnancy and pregnancies complicated with diabetes and fetal growth retardation. J Perinat Med. 1992;20:47–56. doi: 10.1515/jpme.1992.20.1.47. [DOI] [PubMed] [Google Scholar]
- ECONOMIDES DL, NICOLAIDES KH. Blood glucose and oxygen tension levels in small-for-gestational-age fetuses [see comment] Am J Obstet Gynaecol. 1989;160:385–9. doi: 10.1016/0002-9378(89)90453-5. [DOI] [PubMed] [Google Scholar]
- ERUSALIMSKY JD, MONCADA S. Nitric Oxide and Mitochondrial Signaling. From Physiology to Pathophysiology. Arterioscler Thromb Vasc Biol. 2007 doi: 10.1161/ATVBAHA.107.151167. [DOI] [PubMed] [Google Scholar]
- ESTERMAN A, GRECO MA, MITANI Y, FINLAY TH, ISMAIL-BEIGI F, DANCIS J. The effect of hypoxia on human trophoblast in culture: morphology, glucose transport and metabolism. Placenta. 1997;18:129–36. doi: 10.1016/s0143-4004(97)90084-9. [DOI] [PubMed] [Google Scholar]
- FERRAZZI E, RIGANO S, BOZZO M, BELLOTTI M, GIOVANNINI N, GALAN H, BATTAGLIA FC. Umbilical vein blood flow in growth-restricted fetuses. Ultrasound Obstet Gynecol. 2000;16:432–8. doi: 10.1046/j.1469-0705.2000.00208.x. [DOI] [PubMed] [Google Scholar]
- FIRTH JD, EBERT BL, RATCLIFFE PJ. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. J Biol Chem. 1995;270:21021–7. doi: 10.1074/jbc.270.36.21021. [DOI] [PubMed] [Google Scholar]
- FUKASAWA M, TSUCHIYA T, TAKAYAMA E, SHINOMIYA N, UYEDA K, SAKAKIBARA R, SEKI S. Identification and characterization of the hypoxia-responsive element of the human placental 6-phosphofructo-2-kinase/ fructose-2,6-bisphosphatase gene. J Biochem (Tokyo) 2004;136:273–7. doi: 10.1093/jb/mvh137. [DOI] [PubMed] [Google Scholar]
- GAO W, GAO Y, ZHANG G, SONG L, SUN B, SHI J. Hypoxia-induced expression of HIF-1alpha and its target genes in umbilical venous endothelial cells of Tibetans and immigrant Han. Comp Biochem Physiol Toxicol Pharmacol. 2005;141:93–100. doi: 10.1016/j.cca.2005.05.006. [DOI] [PubMed] [Google Scholar]
- GILES WB, LINGMAN G, MARSAL K, TRUDINGER BJ. Fetal volume blood flow and umbilical artery flow velocity waveform analysis: a comparison. Br J Obstet Gynaecol. 1986:93. [PubMed] [Google Scholar]
- GRUPPUSO PA, MIGLIORI R, SUSA JB, SCHWARTZ R. Chronic maternal hyperinsulinemia and hypoglycemia. A model for experimental intrauterine growth retardation. Biol Neonate. 1981;40:113–20. doi: 10.1159/000241479. [DOI] [PubMed] [Google Scholar]
- GUZY RD, SHARMA B, BELL E, CHANDEL NS, SCHUMACKER PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–31. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAGEN T, TAYLOR CT, LAM F, MONCADA S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–8. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- HAMACHER-BRADY A, BRADY NR, LOGUE SE, SAYEN MR, JINNO M, KIRSHENBAUM LA, GOTTLIEB RA, GUSTAFSSON AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–57. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- HAN VK. Pathophysiology, cellular and molecular mechanisms of foetal growth retardation. Equine Vet J Suppl. 1993:12–6. doi: 10.1111/j.2042-3306.1993.tb04802.x. [DOI] [PubMed] [Google Scholar]
- HAY WW., JR Recent observations on the regulation of fetal metabolism by glucose. J Physiol. 2006;572:17–24. doi: 10.1113/jphysiol.2006.105072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HELLWIG-BURGEL T, STIEHL DP, WAGNER AE, METZEN E, JELKMANN W. Review: hypoxia-inducible factor-1 (HIF-1): a novel transcription factor in immune reactions. J Interferon Cytokine Res. 2005;25:297–310. doi: 10.1089/jir.2005.25.297. [DOI] [PubMed] [Google Scholar]
- HOCHACHKA PW, LUTZ PL. Mechanism, origin, and evolution of anoxia tolerance in animals. Comp Biochem Physiol B Biochem Mol Biol. 2001;130:435–59. doi: 10.1016/s1096-4959(01)00408-0. [DOI] [PubMed] [Google Scholar]
- HOOPER SB. Fetal metabolic responses to hypoxia. Reprod Fert Dev. 1995;7:527–38. doi: 10.1071/rd9950527. [DOI] [PubMed] [Google Scholar]
- HUBINONT C, NICOLINI U, FISK NM, TANNIRANDORN Y, RODECK CH. Endocrine pancreatic function in growth-retarded fetuses. Obstet Gynecol. 1991;77:541–4. [PubMed] [Google Scholar]
- IETTA F, WU Y, WINTER J, XU J, WANG J, POST M, CANIGGIA I. Dynamic HIF1A regulation during human placental development. Biol Reprod. 2006;75:112–21. doi: 10.1095/biolreprod.106.051557. [DOI] [PubMed] [Google Scholar]
- ILLSLEY NP, AARNOUDSE JG, PENFOLD P, BARDSLEY SE, COADE SB, STACEY TE, HYTTEN FE. Mechanical and metabolic viability of a placental perfusion system in vitro under oxygenated and anoxic conditions. Placenta. 1984;5:213–25. doi: 10.1016/s0143-4004(84)80031-4. [DOI] [PubMed] [Google Scholar]
- JAUNIAUX E, WATSON AL, BURTON G. Evaluation of respiratory gases and acid-base gradients in fetal tissue and uteroplacental tissue between 7–16 weeks. Am J Obstet Gynaecol. 2001;184:998–1003. doi: 10.1067/mob.2001.111935. [DOI] [PubMed] [Google Scholar]
- KAMEI Y, TSUTSUMI O, YAMAKAWA A, OKA Y, TAKETANI Y, IMAKI J. Maternal epidermal growth factor deficiency causes fetal hypoglycemia and intrauterine growth retardation in mice: possible involvement of placental glucose transporter GLUT3 expression. Endocrinology. 1999;140:4236–43. doi: 10.1210/endo.140.9.6993. [DOI] [PubMed] [Google Scholar]
- KAY HH, ZHU S, TSOI S. Hypoxia and lactate production in trophoblast cells. Placenta. 2007;28:854–60. doi: 10.1016/j.placenta.2006.11.011. [DOI] [PubMed] [Google Scholar]
- KHOUZAMI VA, GINSBURG DS, DAIKOKU NH, JOHNSON JW. The glucose tolerance test as a means of identifying intrauterine growth retardation. Am J Obstet Gynaecol. 1981;139:423–6. doi: 10.1016/0002-9378(81)90319-7. [DOI] [PubMed] [Google Scholar]
- KIM JW, TCHERNYSHYOV I, SEMENZA GL, DANG CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- KING A, SELAK MA, GOTTLIEB E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25:4675–82. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- KINGDOM JC, KAUFMANN P. Oxygen and placental villous development: origins of fetal hypoxia. Placenta. 1997;18:613–21. doi: 10.1016/s0143-4004(97)90000-x. discussion 623–6. [DOI] [PubMed] [Google Scholar]
- KOLOBOVA E, TUGANOVA A, BOULATNIKOV I, POPOV KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J. 2001;358:69–77. doi: 10.1042/0264-6021:3580069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONJE JC, HOWARTH ES, KAUFMANN P, TAYLOR DJ. Longitudinal quantification of uterine artery blood volume flow changes during gestation in pregnancies complicated by intrauterine growth restriction. Bjog. 2003;110:301–5. [PubMed] [Google Scholar]
- KOUMENIS C, WOUTERS BG. «Translating» tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol Cancer Res. 2006;4:423–36. doi: 10.1158/1541-7786.MCR-06-0150. [DOI] [PubMed] [Google Scholar]
- KRECZY A, FUSI L, WIGGLESWORTH JS. Correlation between umbilical arterial flow and placental morphology. Int J Gynecol Pathol. 1995;14:306–9. doi: 10.1097/00004347-199510000-00004. [DOI] [PubMed] [Google Scholar]
- LABARRERE CA, FAULK WP. Antigenic identification of cells in spiral artery trophoblastic invasion: validation of histologic studies by triple-antibody immunocytochemistry. Am J Obstet Gynaecol. 1994;171:165–71. doi: 10.1016/0002-9378(94)90464-2. [DOI] [PubMed] [Google Scholar]
- LACKMAN F, CAPEWELL V, GAGNON R, RICHARDSON B. Fetal umbilical cord oxygen values and birth to placental weight ratio in relation to size at birth. Am J Obstet Gynaecol. 2001;185:674–82. doi: 10.1067/mob.2001.116686. [DOI] [PubMed] [Google Scholar]
- LAND SC, RAE C. iNOS initiates and sustains metabolic arrest in hypoxic lung adenocarcinoma cells: mechanism of cell survival in solid tumor core. American J Physiol - Cell Physiology. 2005;289:C918–33. doi: 10.1152/ajpcell.00476.2004. [DOI] [PubMed] [Google Scholar]
- LANGER O, DAMUS K, MAIMAN M, DIVON M, LEVY J, BAUMAN W. A link between relative hypoglycemia-hypoinsulinemia during oral glucose tolerance tests and intrauterine growth retardation. Am J Obstet Gynaecol. 1986;155:711–6. doi: 10.1016/s0002-9378(86)80004-7. [DOI] [PubMed] [Google Scholar]
- LU DY, LIOU HC, TANG CH, FU WM. Hypoxia-induced iNOS expression in microglia is regulated by the PI3-kinase/Akt/mTOR signaling pathway and activation of hypoxia inducible factor-1alpha. Biochemical Pharmacology. 2006;72:992–1000. doi: 10.1016/j.bcp.2006.06.038. [DOI] [PubMed] [Google Scholar]
- LU H, DALGARD CL, MOHYELDIN A, MCFATE T, TAIT AS, VERMA A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem. 2005;280:41928–39. doi: 10.1074/jbc.M508718200. [DOI] [PubMed] [Google Scholar]
- LUEDER FL, BUROKER CA, KIM SB, FLOZAK AS, OGATA ES. Differential effects of short and long durations of insulin-induced maternal hypoglycemia upon fetal rat tissue growth and glucose utilization. Pediatr Res. 1992;32:436–40. doi: 10.1203/00006450-199210000-00013. [DOI] [PubMed] [Google Scholar]
- LUNELL NO, LEWANDER R, MAMOUN I, NYLUND L, SARBY S, THORNSTROM S. Uteroplacental blood flow in pregnancy induced hypertension. Scand J Clin Lab Invest Suppl. 1984;169:28–35. doi: 10.3109/00365518409085374. [DOI] [PubMed] [Google Scholar]
- LUNELL NO, SARBY B, LEWANDER R, NYLUND L. Comparison of uteroplacental blood flow in normal and in intrauterine growth-retarded pregnancy. Measurements with Indium-113m and a computer-linked gammacamera. Gynecol Obstet Invest. 1979;10:106–18. doi: 10.1159/000299924. [DOI] [PubMed] [Google Scholar]
- MACKENZIE ED, SELAK MA, TENNANT DA, PAYNE LJ, CROSBY S, FREDERIKSEN CM, WATSON DG, GOTTLIEB E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol. 2007;27:3282–9. doi: 10.1128/MCB.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALINSKI T, TAHA Z. Nitric oxide release from a single cell measured in situ by a porphyrinic-based microsensor. Nature. 1992;358:676–8. doi: 10.1038/358676a0. [DOI] [PubMed] [Google Scholar]
- MARCONI AM, CETIN I, DAVOLI E, BAGGIANI AM, FANELLI R, FENNESSEY PV, BATTAGLIA FC, PARDI G. An evaluation of fetal glucogenesis in intrauterine growth-retarded pregnancies. Metabolism. 1993;42:860–4. doi: 10.1016/0026-0495(93)90060-2. [DOI] [PubMed] [Google Scholar]
- MARCONI AM, PAOLINI C, BUSCAGLIA M, ZERBE G, BATTAGLIA FC, PARDI G. The impact of gestational age and fetal growth on the maternal-fetal glucose concentration difference. Obstet Gynecol. 1996;87:937–42. doi: 10.1016/0029-7844(96)00048-8. [DOI] [PubMed] [Google Scholar]
- MARKERT CL. Lactate dehydrogenase. Biochemistry and function of lactate dehydrogenase. Cell Biochem Funct. 1984;2:131–4. doi: 10.1002/cbf.290020302. [DOI] [PubMed] [Google Scholar]
- MASON SD, RUNDQVIST H, PAPANDREOU I, DUH R, MCNULTY WJ, HOWLETT RA, OLFERT IM, SUNDBERG CJ, DENKO NC, POELLINGER L, et al. HIF-1alpha in endurance training: suppression of oxidative metabolism. American J Physiol - Reg Integ Comp Physiol. 2007;293:R2059–69. doi: 10.1152/ajpregu.00335.2007. [DOI] [PubMed] [Google Scholar]
- MATRONE C, PIGNATARO G, MOLINARO P, IRACE C, SCORZIELLO A, DI RENZO GF, ANNUNZIATO L. HIF-1alpha reveals a binding activity to the promoter of iNOS gene after permanent middle cerebral artery occlusion. J Neurochem. 2004;90:368–78. doi: 10.1111/j.1471-4159.2004.02483.x. [DOI] [PubMed] [Google Scholar]
- MAYHEW TM, CHARNOCK-JONES DS, KAUFMANN P. Aspects of human fetoplacental vasculogenesis and angiogenesis. III Changes in complicated pregnancies. Placenta. 2004;25:127–39. doi: 10.1016/j.placenta.2003.10.010. [DOI] [PubMed] [Google Scholar]
- MEEKINS JW, PIJNENBORG R, HANSSENS M, MCFADYEN IR, VAN ASSHE A. A study of placental bed spiral arteries and trophoblast invasion in normal and severe pre-eclamptic pregnancies. Brit J Obstet Gynaecol. 1994;101:669–74. doi: 10.1111/j.1471-0528.1994.tb13182.x. [DOI] [PubMed] [Google Scholar]
- MELILLO G, TAYLOR LS, BROOKS A, MUSSO T, COX GW, VARESIO L. Functional requirement of the hypoxia-responsive element in the activation of the inducible nitric oxide synthase promoter by the iron chelator desferrioxamine. J Biol Chem. 1997;272:12236–43. doi: 10.1074/jbc.272.18.12236. [DOI] [PubMed] [Google Scholar]
- MELLOR HR, HARRIS AL. The role of the hypoxia-inducible BH3-only proteins BNIP3 and BNIP3L in cancer. Cancer Metastat Rev. 2007;26:553–66. doi: 10.1007/s10555-007-9080-0. [DOI] [PubMed] [Google Scholar]
- MIN J, JIN YM, MOON JS, SUNG MS, JO SA, JO I. Hypoxia-induced endothelial NO synthase gene transcriptional activation is mediated through the tax-responsive element in endothelial cells. Hypertension. 2006;47:1189–96. doi: 10.1161/01.HYP.0000222892.37375.4d. [DOI] [PubMed] [Google Scholar]
- MOORE LG. Fetal growth restriction and maternal oxygen transport during high altitude pregnancy. High Alt Med Biol. 2003;4:141–56. doi: 10.1089/152702903322022767. [DOI] [PubMed] [Google Scholar]
- MOORE LG, NIERMEYER S, ZAMUDIO S. Human adaptation to high altitude: regional and life-cycle perspectives. Am J Phys Anthropol. 1998;(Suppl):25–64. doi: 10.1002/(sici)1096-8644(1998)107:27+<25::aid-ajpa3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- NEVO O, SOLEYMANLOU N, WU Y, XU J, KINGDOM J, MANY A, ZAMUDIO S, CANIGGIA I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1085–93. doi: 10.1152/ajpregu.00794.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICOLINI U, HUBINONT C, SANTOLAYA J, FISK NM, COE AM, RODECK CH. Maternal-fetal glucose gradient in normal pregnancies and in pregnancies complicated by alloimmunization and fetal growth retardation. Am J Obstet Gynaecol. 1989;161:924–7. doi: 10.1016/0002-9378(89)90753-9. [DOI] [PubMed] [Google Scholar]
- PALMER SK, MOORE LG, YOUNG D, CREGGER B, BERMAN JC, ZAMUDIO S. Altered blood pressure course during normal pregnancy and increased preeclampsia at high altitude (3100 meters) in Colorado. Am J Obstet Gynaecol. 1999;180:1161–8. doi: 10.1016/s0002-9378(99)70611-3. [DOI] [PubMed] [Google Scholar]
- PALMER SK, ZAMUDIO S, COFFIN C, PARKER S, STAMM E, MOORE LG. Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet Gynecol. 1992;80:1000–6. [PubMed] [Google Scholar]
- PAN Y, MANSFIELD KD, BERTOZZI CC, RUDENKO V, CHAN DA, GIACCIA AJ, SIMON MC. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol. 2007;27:912–25. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAPANDREOU I, CAIRNS RA, FONTANA L, LIM AL, DENKO NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–97. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- PARDI G, MARCONI AM, CETIN I. Placental-fetal interrelationship in IUGR fetuses—a review. Placenta. 2002;23(Suppl A):S136–41. doi: 10.1053/plac.2002.0802. [DOI] [PubMed] [Google Scholar]
- POSTIGO L, HEREDIA G, ILLSLEY N, TORRICOS T, ECHELAR L, TELLEZ W, RODRIGUEZ C, MALDONADO I, BRIMACOMBE M, BALANZA E, et al. Where the O2 goes to: Preservation of human fetal oxygen deivery and consumptio at high altitude. J Physiol. 2009;587:693–708. doi: 10.1113/jphysiol.2008.163634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POUYSSEGUR J, DAYAN F, MAZURE NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–43. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- READ JA, WINTER VJ, ESZES CM, SESSIONS RB, BRADY RL. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins. 2001;43:175–85. doi: 10.1002/1097-0134(20010501)43:2<175::aid-prot1029>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- ROSENN BM, MIODOVNIK M. Glycemic control in the diabetic pregnancy: is tighter always better? J Matern Fetal Med. 2000;9:29–34. doi: 10.1002/(SICI)1520-6661(200001/02)9:1<29::AID-MFM7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- SELAK MA, ARMOUR SM, MACKENZIE ED, BOULAHBEL H, WATSON DG, MANSFIELD KD, PAN Y, SIMON MC, THOMPSON CB, GOTTLIEB E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- SEMENZA GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8:588–94. doi: 10.1016/s0959-437x(98)80016-6. [DOI] [PubMed] [Google Scholar]
- SEMENZA GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- SEMENZA GL, AGANI F, BOOTH G, FORSYTHE J, IYER N, JIANG BH, LEUNG S, ROE R, WIENER C, YU A. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 1997;51:553–5. doi: 10.1038/ki.1997.77. [DOI] [PubMed] [Google Scholar]
- SEMENZA GL, JIANG BH, LEUNG SW, PASSANTINO R, CONCORDET JP, MAIRE P, GIALLONGO A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–37. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- SETIA S, SRIDHAR MG, BHAT V, CHATURVEDULA L, VINAYAGAMOORTI R, JOHN M. Insulin sensitivity and insulin secretion at birth in intrauterine growth retarded infants. Pathology. 2006;38:236–8. doi: 10.1080/00313020600696256. [DOI] [PubMed] [Google Scholar]
- SHI Y, BAKER JE, ZHANG C, TWEDDELL JS, SU J, PRITCHARD KA., JR Chronic hypoxia increases endothelial nitric oxide synthase generation of nitric oxide by increasing heat shock protein 90 association and serine phosphorylation. Circulation Res. 2002;91:300–6. doi: 10.1161/01.res.0000031799.12850.1e. [DOI] [PubMed] [Google Scholar]
- SHIBUKI K. An electrochemical microprobe for detecting nitric oxide release in brain tissue. Neurosci Res. 1990;9:69–76. doi: 10.1016/0168-0102(90)90048-j. [DOI] [PubMed] [Google Scholar]
- SHIME J, MOCARSKI EJ, HASTINGS D, WEBB GD, MCLAUGHLIN PR. Congenital heart disease in pregnancy: short- and long-term implications. Am J Obstet Gynaecol. 1987;156:313–22. doi: 10.1016/0002-9378(87)90275-4. [DOI] [PubMed] [Google Scholar]
- SOKOL RJ, KAZZI GM, KALHAN SC, PILLAY SK. Identifying the pregnancy at risk for intrauterine growth retardation: possible usefulness of the intravenous glucose tolerance test. Am J Obstet Gynaecol. 1982;143:220–3. doi: 10.1016/0002-9378(82)90658-5. [DOI] [PubMed] [Google Scholar]
- SOLEYMANLOU N, JURISICA I, NEVO O, IETTA F, ZHANG X, ZAMUDIO S, POST M, CANIGGIA I. Molecular evidence of placental hypoxia in preeclampsia. J Clin Endocrinol Metab. 2005;90:4299–308. doi: 10.1210/jc.2005-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOOTHILL PW, NICOLAIDES KH, CAMPBELL S. Prenatal asphyxia, hyperlacticaemia, hypoglycaemia, and erythroblastosis in growth retarded fetuses. Brit Med J Clin Res. 1987;294:1051–3. doi: 10.1136/bmj.294.6579.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOOTHILL PW, NICOLAIDES KH, RODECK CH, CAMPBELL S. Effect of gestational age on fetal and intervillous blood gas and acid-base values in human pregnancy. Fetal Ther. 1986;1:168–75. doi: 10.1159/000262264. [DOI] [PubMed] [Google Scholar]
- STOLZE IP, MOLE DR, RATCLIFFE PJ. Regulation of HIF: prolyl hydroxylases. Novartis Found Symp. 2006;272:15–25. discussion 25–36. [PubMed] [Google Scholar]
- SUGDEN MC, HOLNESS MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003;284:E855–62. doi: 10.1152/ajpendo.00526.2002. [DOI] [PubMed] [Google Scholar]
- TODROS T, SCIARRONE A, PICCOLI E, GUIOT C, KAUFMANN P, KINGDOM J. Umbilical Doppler waveforms and placental villous angiogenesis in pregnancies complicated by fetal growth restriction. Obstet Gynecol. 1999;93:499–503. doi: 10.1016/s0029-7844(98)00440-2. [DOI] [PubMed] [Google Scholar]
- TRACY K, MACLEOD KF. Regulation of mitochondrial integrity, autophagy and cell survival by BNIP3. Autophagy. 2007;3:616–9. doi: 10.4161/auto.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN ASSCHE FA, DE PRINS FA. Maternal hypoglycemia and intrauterine growth retardation. Am J Obstet Gynaecol. 1983;146:349–50. doi: 10.1016/0002-9378(83)90775-5. [DOI] [PubMed] [Google Scholar]
- VAN VEEN LC, TENG C, HAY WW, JR, MESCHIA G, BATTAGLIA FC. Leucine disposal and oxidation rates in the fetal lamb. Metabolism. 1987;36:48–53. doi: 10.1016/0026-0495(87)90062-x. [DOI] [PubMed] [Google Scholar]
- WALLACE JM, AITKEN RP, MILNE JS, HAY WW., JR Nutritionally mediated placental growth restriction in the growing adolescent: consequences for the fetus. Biol Reprod. 2004;71:1055–62. doi: 10.1095/biolreprod.104.030965. [DOI] [PubMed] [Google Scholar]
- WILLIAMS RL, CREASY RK, CUNNINGHAM GC, HAWES WE, NORRIS FD, TASHIRO M. Fetal growth and perinatal viability in California. Obstet Gynecol. 1982;59:624–32. [PubMed] [Google Scholar]
- YAN L, VATNER DE, KIM SJ, GE H, MASUREKAR M, MASSOVER WH, YANG G, MATSUI Y, SADOSHIMA J, VATNER SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci USA. 2005;102:13807–12. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YIP R. Altitude and birth weight. J Pediatr. 1987;111:869–76. doi: 10.1016/s0022-3476(87)80209-3. [DOI] [PubMed] [Google Scholar]
- ZAMUDIO S. The placenta at high altitude. High Alt Med Biol. 2003;4:171–91. doi: 10.1089/152702903322022785. [DOI] [PubMed] [Google Scholar]
- ZAMUDIO S, BAUMANN MU, ILLSLEY NP. Effects of chronic hypoxia in vivo on the expression of human placental glucose transporters. Placenta. 2006;27:49–55. doi: 10.1016/j.placenta.2004.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAMUDIO S, KOVALENKO O, VANDERLELIE J, ILLSLEY NP, HELLER D, BELLIAPPA S, PERKINS AV. Chronic hypoxia in vivo reduces placental oxidative stress. Placenta. 2007a;28:846–53. doi: 10.1016/j.placenta.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAMUDIO S, PALMER S, DROMA T, STAMM E, COFFIN C, MOORE L. Effect of altitude on uterine artery blood flow during normal pregnancy. J Appl Physiol. 1995;79:7–14. doi: 10.1152/jappl.1995.79.1.7. [DOI] [PubMed] [Google Scholar]
- ZAMUDIO S, POSTIGO L, ILLSLEY NP, RODRIGUEZ C, HEREDIA G, BRIMACOMBE M, ECHALAR L, TORRICOS T, TELLEZ W, MALDONADO I, et al. Maternal oxygen delivery is not related to altitude- and ancestry-associated differences in human fetal growth. J Physiol. 2007b;582:883–95. doi: 10.1113/jphysiol.2007.130708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAMUDIO S, TORRICOS T, FIK-RYMARKIEWICA E, OYALA M, ECHELAR L, PULLOCKARAN J, TUTINO E, BELLIAPA S, BALANZA E, ILLSLEY N. Hypoglycemia and the origin of hypoxia-induced reduction in human fetal growth. PLoS ONE. doi: 10.1371/journal.pone.0008551. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAMUDIO S, WU Y, IETTA F, ROLFO A, CROSS A, WHEELER T, POST M, ILLSLEY NP, CANIGGIA I. Human placental hypoxia-inducible factor-1alpha expression correlates with clinical outcomes in chronic hypoxia in vivo. Am J Pathol. 2007c;170:2171–9. doi: 10.2353/ajpath.2007.061185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG H, BOSCH-MARCE M, SHIMODA LA, TAN YS, BAEK JH, WESLEY JB, GONZALEZ FJ, SEMENZA GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- ZHANG H, GAO P, FUKUDA R, KUMAR G, KRISHNAMACHARY B, ZELLER KI, DANG CV, SEMENZA GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–20. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- ZHU H, TANNOUS P, JOHNSTONE JL, KONG Y, SHELTON JM, RICHARDSON JA, LE V, LEVINE B, ROTHERMEL BA, HILL JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–93. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]