

Abstract
Phosphatidylinositol 3-kinase (PI3K) promotes cell survival and communication by activating its downstream effector Akt kinase. Here we show that PS1, a protein involved in familial Alzheimer's disease (FAD), promotes cell survival by activating the PI3K/Akt cell survival signaling. This function of PS1 is unaffected by γ-secretase inhibitors. Pharmacological and genetic evidence indicates that PS1 acts upstream of Akt, at or before PI3K kinase. PS1 forms complexes with the p85 subunit of PI3K and promotes cadherin/PI3K association. Furthermore, conditions that inhibit this association prevent the PS1-induced PI3K/Akt activation, indicating that PS1 stimulates PI3K/Akt signaling by promoting cadherin/PI3K association. By activating PI3K/Akt signaling, PS1 promotes phosphorylation/inactivation of glycogen synthase kinase-3 (GSK-3), suppresses GSK-3-dependent phosphorylation of tau at residues overphosphorylated in AD and prevents apoptosis of confluent cells. PS1 FAD mutations inhibit the PS1-dependent PI3K/Akt activation, thus promoting GSK-3 activity and tau overphosphorylation at AD-related residues. Our data raise the possibility that PS1 may prevent development of AD pathology by activating the PI3K/Akt signaling pathway. In contrast, FAD mutations may promote AD pathology by inhibiting this pathway.
Keywords: Alzheimer's disease, cadherin, PI3K, Presenilin, tau
Introduction
Increased neuronal cell death, tau overphosphorylation and accumulation of neurofibrillary tangles (NFTs) and amyloid plaques are the main pathological hallmarks of Alzheimer's disease (AD) brains. The phosphatidylinositol 3-kinase (PI3K) signaling pathway plays crucial roles in the transmission of survival signals in a wide range of cell types including neurons (for reviews, see Chan et al, 1999; Brunet et al, 2001). PI3K activates its downstream effector Akt/protein kinase B (Akt) by promoting its phosphorylation at residues serine 473 (Ser473) and threonine 308 (Thr308). Activated Akt, in turn, phosphorylates a wide range of substrates activating anti-apoptotic (survival) factors and inactivating pro-apoptotic factors (Brunet et al, 2001). The PI3K/Akt pathway is activated following recruitment of PI3K to the plasma membrane in response to a number of extracellular stimuli including growth factors (Brunet et al, 2001) and cadherin homophilic cell–cell adhesions, which result in the recruitment of PI3K to adhesion complexes (Pece et al, 1999; Kovacs et al, 2002; Tran et al, 2002; Yap and Kovacs, 2003). Akt downregulates the activities of glycogen synthase kinases 3α (GSK-3α) and 3β (GSK-3β) by phosphorylating the former at residue serine 21 (Ser21) and the latter at residue serine 9 (Ser9) (Cross et al, 1995; Kaytor and Orr, 2002). Increased GSK-3β activity has been implicated in neuronal cell death (Pap and Cooper, 1998; Hetman et al, 2000; Cross et al, 2001; Lucas et al, 2001) and tau overphosphorylation (Hanger et al, 1992; Hong et al, 1997; Pei et al, 1999; Lucas et al, 2001), while GSK-3α was recently implicated in the production of Aβ peptide, the principal protein component of amyloid plaques (Phiel et al, 2003).
Presenilin1 (PS1), a multipass transmembrane protein important in development (Shen et al, 1997), is tightly linked to many cases of early-onset familial Alzheimer's disease (FAD) indicating a causal relationship between these mutants and AD pathology. As a result, the biological function of PS1 and the mechanism(s) involved in the pathogenic activities of PS1 mutants are under intense investigation. PS1 is cleaved in vivo to yield an N-terminal (PS1/NTF) fragment and a C-terminal (PS1/CTF) fragment that associate to form a functional heterodimer (Thinakaran et al, 1996). PS1 forms complexes with and promotes the γ-secretase-like processing of APP and several other type I transmembrane proteins (for review, see Fortini, 2002). Recently, we reported that PS1 binds cadherins and promotes cadherin–cadherin cell–cell adhesion interactions (Georgakopoulos et al, 1999; Baki et al, 2001). Ca2+ influx however, including activation of NMDA receptor, stimulates a PS1/γ-secretase-dependent processing of cadherins at the ɛ-cleavage site resulting in the production of gene expression signals (Marambaud et al, 2002, 2003). PS1 is expressed in neurons and plays crucial roles in the development of the CNS (Elder et al, 1996; Shen et al, 1997). In vitro experiments showed that overexpression of PS1 FAD mutants promotes apoptosis (Weihl et al, 1999; Leroy et al, 2003) and may decrease Akt activity (Weihl et al, 1999; Vestling et al, 2001). However, it remains unclear how PS1 promotes development (Shen et al, 1997) and cell survival (Hong et al, 1999) and whether these functions of PS1 are related to the mechanisms by which PS1 FAD mutants promote tau overphosphorylation and neuronal cell death (Irving and Miller, 1997; Takashima et al, 1998; Pigino et al, 2001). Here we show that PS1 promotes survival of confluent cell cultures by increasing cadherin/PI3K association, thus stimulating the PI3K/Akt cell survival signaling. This function of PS1 is independent of its γ-secretase activity. We also show that by promoting the PI3K/Akt signaling, PS1 downregulates GSK-3 activity and reduces overphosphorylation of tau at AD-related residues. In contrast, PS1 FAD mutations inhibit this signaling, thus compromising the cell survival function of PS1 and its ability to suppress GSK-3 and tau overphosphorylation.
Results
Absence of PS1 triggers density-dependent apoptosis
Postconfluent E-cadherin-expressing PS1−/− cell cultures displayed increased cell death compared to control (PS1+/+) cells (Figure 1A). DNA fragmentation assays showed a clear laddering pattern in DNA from postconfluent PS1−/− cell cultures but not in DNA from postconfluent PS1+/+ cultures, indicating apoptotic cell death in the former cultures (Figure 1B). Accordingly, prominent TUNEL staining and progressive accumulation of pro-apoptotic cleaved (activated), caspase-3 were observed in postconfluent PS1−/− but not in PS1+/+ cells (Figures 1C and D, respectively). To exclude clonal effects, we established additional E-cadherin-transfected embryonic fibroblast clones derived from independent matings. Prominent TUNEL staining was observed in all postconfluent PS1−/− clones but not in PS1+/+ clones (data not shown). Apoptotic changes, revealed by the progressive activation of apoptotic caspase-3, are also observed in non-E-cadherin-transfected postconfluent PS1−/− cells (Figure 1E, lanes 1–6); E-cadherin, however, accelerates these changes as indicated by the presence of activated caspase-3 in 2 days postconfluent E-cadherin-transfected but not in nontransfected PS1−/− cell cultures (Figure 1E, compare lane 2 to lane 8). Thus, in the absence of PS1, cells undergo apoptosis following confluence. Since E-cadherin expression accelerates apoptosis, all following cell culture experiments were performed using E-cadherin-expressing cells.
Figure 1.
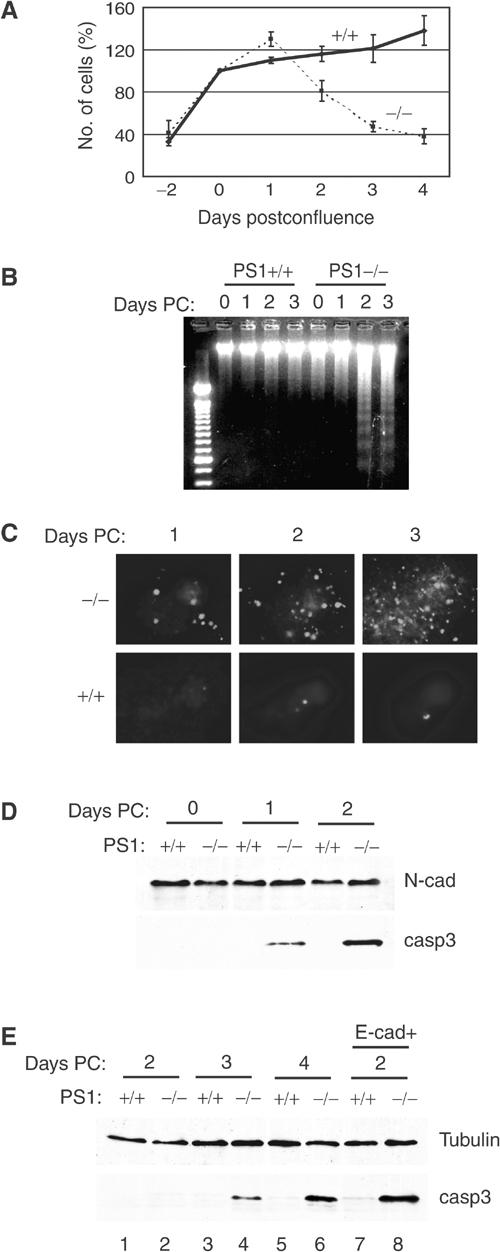
Absence of PS1 triggers density-dependent apoptosis. (A) Survival analysis of E-cadherin-transfected PS1+/+ or PS1−/− cells (continuous and dotted lines, respectively) cultured up to 4 days postconfluence. Mean values, ±s.e.m., from two independent experiments (each in triplicate) are presented. (B) Kinetics of the appearance of apoptotic (fragmented) DNA in nuclei of E-cadherin-transfected PS1+/+ and PS1−/− cells, cultured for up to 3 days postconfluence (Days PC). (C) Kinetics of the appearance of TUNEL-positive (apoptotic) nuclei in E-cadherin-transfected PS1+/+ and PS1−/− cells, cultured for up to 3 days postconfluence. (D) Lysates from 0-, 1- and 2-day postconfluent E-cadherin-transfected PS1+/+ and PS1−/− cells were analyzed with anti-N-cadherin (loading control) and anti-activated (cleaved) caspase-3 antibodies. N-cad: N-cadherin; casp3: activated caspase-3. (E) Lysates from 2- to 4-day postconfluent nontransfected PS1+/+ and PS1−/− cells (lanes 1–6) and from 2-day postconfluent E-cadherin-transfected PS1+/+ and PS1−/− cells (lanes 7 and 8) were analyzed with anti-b-tubulin (control) and anti-activated (cleaved) caspase-3 antibodies.
PS1 inhibits apoptosis by activating PI3K/Akt signaling
Inadequate activation of the PI3K/Akt signaling pathway may result in impaired transmission of survival signals leading to apoptosis (Brunet et al, 2001). PI3K kinase promotes cell survival by activating its downstream effector Akt, an event indicated by phosphorylation of Akt residue Ser473 (for review, see Chan and Tsichlis, 2001). Figure 2A shows that Akt Ser473 is underphosphorylated in PS1−/− cells suggesting that PS1 is needed for full activation of the PI3K/Akt cell survival pathway. Indeed, expression of PS1 in PS1−/− cells stimulated phosphorylation of Akt at Ser473 (Figure 2B, lanes 1 and 2). Pharmacological inhibition of PI3K activity strongly suppressed phosphorylation of Akt in PS1+/+ cells (Figure 2A, lane 3) and prevented the PS1-induced Akt phosphorylation in PS1−/− cells (Figure 2B, lane 3), suggesting that PI3K mediates the PS1-induced Akt phosphorylation. We next asked whether PS1-induced activation of the PI3K/Akt survival pathway is sufficient to rescue PS1−/− cells from apoptosis. Indeed, re-introduction of PS1 into PS1−/− cells decreased the number of annexin-positive (apoptotic) PS1−/− cells from 68 to 22% (Figure 2C) and strongly suppressed activation of apoptotic caspase-3 (Figure 2B, panel c, lanes 1 and 2). Pharmacological inhibition of PI3K activity prevented the PS1-induced inhibition of caspase-3 cleavage (Figure 2B, panel c) indicating that the anti-apoptotic function of PS1 depends on its ability to activate the PI3k/Akt signaling cascade.
Figure 2.
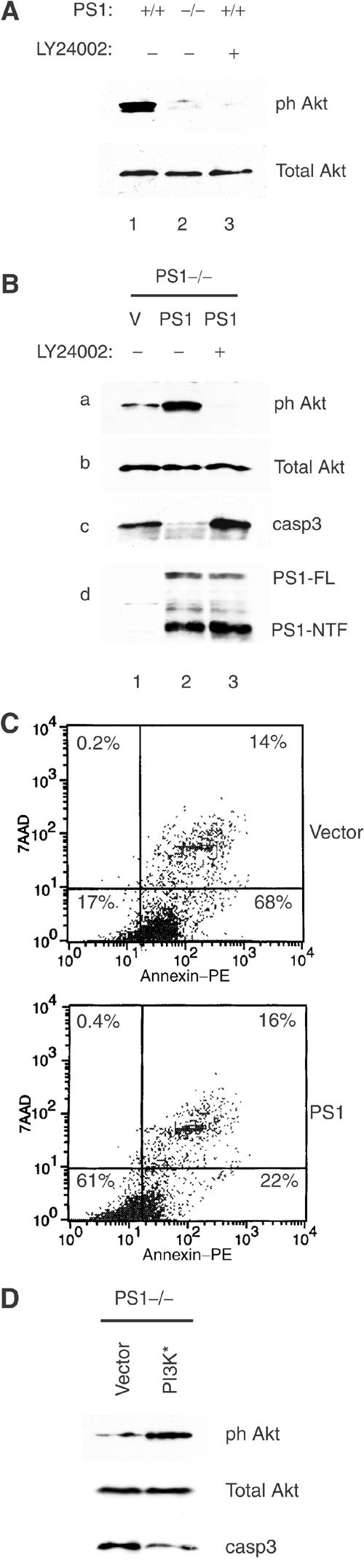
PS1 inhibits apoptosis by activating PI3K/Akt signaling. (A) Lysates from confluent E-cadherin-transfected PS1+/+ and PS1−/− fibroblasts, cultured in the absence or presence of the PI3K inhibitor LY24002, were analyzed for phosphorylated Akt at Ser473 (ph Akt) or total Akt. (B) PS1−/− cells infected with HSV vector (V) or HSV PS1 (PS1) were cultured in the presence or absence of PI3K inhibitor (LY24002). Lysates were analyzed as shown. ph Akt: phosphorylated Akt at Ser473; casp3: activated caspase-3; PS1-FL: full-length PS1; PS1-NTF: NTF fragment of PS1. (C) PS1−/− cells, transiently transfected either with EGFP vector (V) or with EGFP-PS1 (PS1), were subjected to annexin/7AAD staining and analyzed by flow cytometry. Only the EGFP-positive (transfected) cells are shown. Percentages refer to total EGFP-positive cells. Nonapoptotic cells are shown in the lower left square. A total of 10 000 EGFP-positive cells were analyzed. (D) PS1−/− cells were transfected with mutant (active) PI3K (PI3K*) or empty vector and lysates were analyzed as shown.
The fact that inhibition of PI3K activity prevents the PS1-dependent Akt phosphorylation suggests that PS1 does not act directly on Akt but rather at an upstream step of the PI3K/Akt signaling pathway. PTEN phosphates negatively regulates the PI3K pathway by blocking signal transmission from PI3K to Akt. Examination of PTEN phosphorylation (Stambolic et al, 1998), however, showed that PTEN activity is not involved in the reduced Akt phosphorylation of PS1−/− cells (data not shown). Furthermore, a PI3K mutant with high constitutive activity (Rodriguez-Viciana et al, 1996) is able to stimulate Akt phosphorylation and to suppress activation of caspase-3 even in the absence of PS1 (Figure 2D). Together, these data indicate that PS1 promotes activation of the PI3K/Akt survival pathway by affecting a step at or before PI3K.
PS1 regulates GSK-3 activity via the PI3K/Akt pathway
To further explore the downstream physiological consequences of the PS1-dependent PI3K/Akt activation, we examined the phosphorylation of GSK-3α and GSK-3β kinases at the Akt-specific epitopes Ser21 and Ser9, respectively. Figure 3A shows that compared to wild-type (WT) cells, phosphorylation of both GSK-3 kinases is decreased in PS1−/− cells, suggesting that PS1 promotes the Akt-dependent phosphorylation/inactivation of both kinases (Cross et al, 1995; Kaytor and Orr, 2002). Indeed, re-introduction of PS1 in these cells increased phosphorylation of both kinases (Figure 3B, lanes 1 and 2) and this increase was prevented by pharmacological inhibition of either PI3K or Akt activities (Figure 3B, lanes 3 and 4). As expected, PS1 suppressed apoptotic caspace-3 and this suppression was abolished in the presence of either PI3K or Akt inhibitors (panel e). Together, these results show that PS1 increases GSK-3 phosphorylation via the PI3K/Akt signaling. Since this phosphorylation suppresses GSK-3 activity (Cross et al, 1995; Kaytor and Orr, 2002), our data indicate that PS1 downregulates the activity of both GSK-3 isoforms by stimulating the PI3K/Akt pathway.
Figure 3.
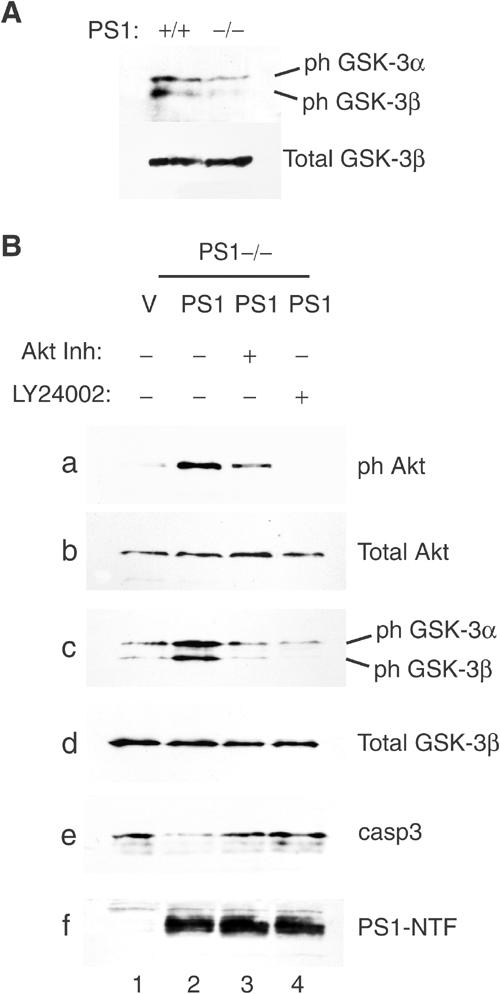
PS1 regulates GSK-3 activity via the PI3K/Akt pathway. (A) Lysates from confluent PS1+/+ or PS1−/− fibroblasts were analyzed for phosphorylation of GSK-3α (ph GSK-3α) and GSK-3β (ph GSK-3β) at Ser21 and Ser9, respectively. (B) PS1−/− cells infected with HSV vector (V) or HSV PS1 (PS1) were cultured in the presence or absence of either Akt inhibitor (Akt Inh) or PI3K inhibitor (LY24002). Lysates were analyzed as shown. PS1-NTF: NTF fragment of PS1.
PS1-mediated activation of the PI3K–Akt pathway is independent of γ-secretase activity
PS1 is important for the γ-secretase cleavage of many proteins including E- and N-cadherin. Inhibition of this cleavage by γ-secretase inhibitors or absence of PS1 results in the accumulation of cadherin fragments N-Cad/CTF1 and E-Cad/CTF1, which are substrates for the PS1/γ-secretase system (Marambaud et al, 2002, 2003). To explore any potential role of this system in the activation of PI3K/Akt signaling, we used two distinct γ-secretase inhibitors, L-685,458 and Compound E (see Materials and methods), to ask whether inhibition of γ-secretase affects Akt phosphorylation and apoptosis. Figure 4A shows that, in contrast to PS1−/− cells, PS1+/+ cells contain no detectable levels of the γ-secretase substrate N-Cad/CTF1 (lanes 1 and 2). As expected, treatment of PS1+/+ cells with either inhibitor resulted in the accumulation of N-Cad/CTF1 indicating inhibition of γ-secretase activity (Marambaud et al, 2003). These treatments, however, did not affect phosphorylation of Akt nor did they promote caspase-3 activation (Figure 4A, lanes 2–5).
Figure 4.
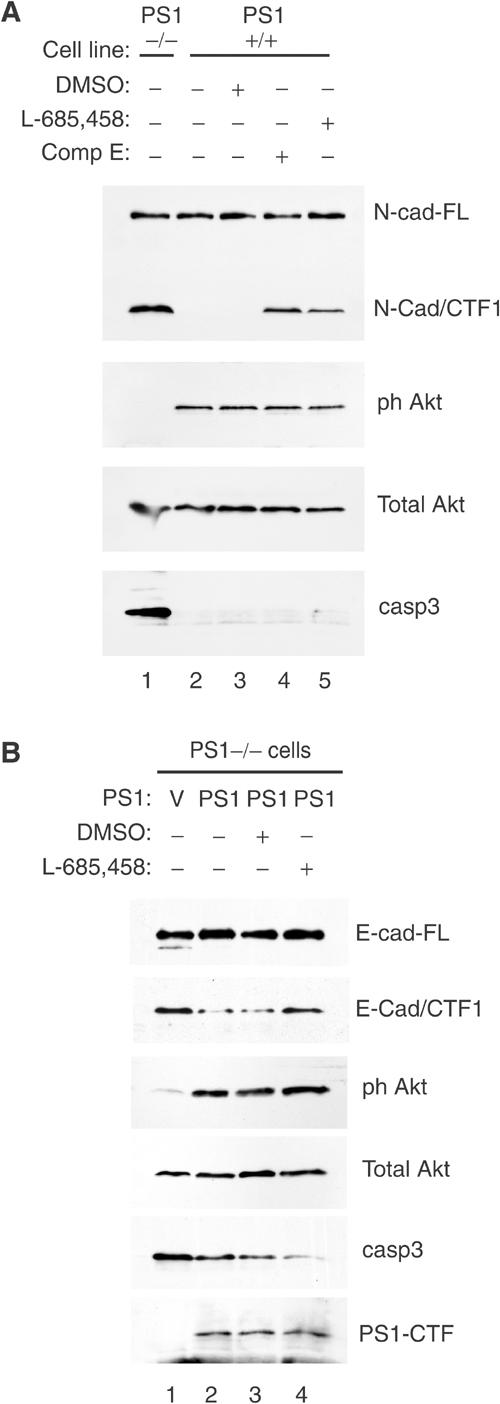
PS1-mediated activation of the PI3K–Akt pathway is independent of γ-secretase activity. (A) Confluent PS1−/− (lane 1) or PS1+/+ cells (lanes 2–5) were cultured overnight in the presence or absence of γ-secretase inhibitor XVIII (Compound E, lane 4), γ-secretase inhibitor L-685,458 (lane 5) or vehicle (DMSO, lane 3). Lysates were analyzed as shown. N-cad-FL: full-length N-cadherin; N-cad-CTF1: CTF1 fragment of N-cadherin (Marambaud et al, 2003). (B) PS1−/− cells were infected with HSV vector (V) or HSV PS1 (PS1) and cultured in the presence or absence of γ-secretase inhibitor. Lysates were analyzed as shown. E-cad-FL: full-length E-cadherin; E-cad-CTF1: CTF1 fragment of E-cadherin (Marambaud et al, 2002); PS1-CTF: CTF fragment of PS1.
In a different approach we asked whether inhibition of γ-secretase activity prevents the PS1-induced activation of the PI3K pathway. Figure 4B (lanes 1 and 2) shows that re-introduction of PS1 in PS1−/− cells reduced E-cadherin fragment CTF1, increased Akt activation and reduced apoptotic caspase-3. However, although inhibitor L-685,458 almost completely prevented the γ-secretase activity-dependent metabolism of E-Cad/CTF1 (Marambaud et al, 2002), it affected neither the PS1-induced Akt phosphorylation nor the PS1-dependent inhibition of caspase-3 (Figure 4B, lanes 3 and 4). Together, the above data show that the PS1-dependent activation of the PI3K/Akt cell survival pathway is a novel function of PS1, unrelated to PS1-mediated γ-secretase activity.
PS1 activates PI3K/Akt signaling by promoting association of the p85 subunit of PI3K with E- and N-cadherin
The increased apoptosis manifested by confluent E-cadherin-transfected PS1−/− cells suggested that cadherins, proteins that associate and functionally interact with both PS1 and PI3K (Pece et al, 1999; Baki et al, 2001; Laprise et al, 2002; Tran et al, 2002), might be involved. Furthermore, PS1 promotes cadherin–cadherin adhesive interactions (Baki et al, 2001), a key prerequisite for cadherin-mediated activation of PI3K (Pece et al, 1999; Kovacs et al, 2002; Tran et al, 2002). To explore the possibility that cadherins are the mediators of the PS1 effect on PI3K/Akt signaling, we asked whether inhibition of adhesion function of either E- or N-cadherin would prevent the PS1-induced phosphorylation of Akt. Function-blocking antibodies against E- or N-cadherin inhibit cadherin–cadherin homophilic interactions and prevent cadherin-mediated activation of PI3K (Pece et al, 1999; Laprise et al, 2002; Tran et al, 2002). Figure 5A shows that either one of these antibodies suppressed phosphorylation of both Akt and its substrate GSK-3β in WT cells, but had no significant effect on the already low levels of phosphorylated Akt or GSK-3β of PS1−/− cells. Furthermore, a combination of the above antibodies prevented the PS1-induced phosphorylation of Akt (Figure 5B), showing that functional cadherins mediate the PS1-induced stimulation of PI3K/Akt signaling.
Figure 5.
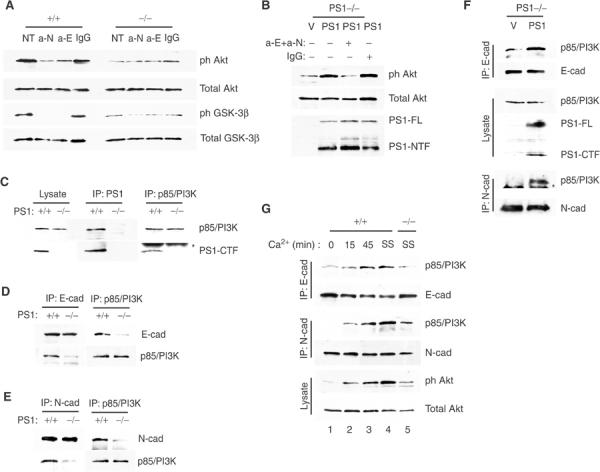
PS1 activates the PI3K/Akt pathway by promoting association of the p85 subunit of PI3K with E- and N-cadherin. (A) Postconfluent PS1+/+ or PS1−/− cells were cultured in the presence of anti-N-cadherin and anti-E-cadherin antibodies (a-N and a-E, respectively) or isotypic IgG (IgG). Lysates were analyzed as shown. NT: no treatment. (B) PS1−/− cells were infected with HSV vector (V) or HSV PS1 (PS1) and cultured in the presence of both anti-E- and anti-N-cadherin antibodies (anti-E+anti-N) or isotypic IgG (IgG). Lysates were analyzed as shown. (C) Lysates from postconfluent PS1+/+ or PS1−/− cells were immunoprecipitated (IPed) with anti-PS1 (IP: PS1) or anti-p85/PI3K (IP: p85/PI3K) antibodies and obtained immunoprecipitates (IPs) were analyzed with anti-p85/PI3K or anti-PS1-CTF antibodies as shown. Asterisk denotes IgG. (D) Lysates from postconfluent PS1+/+ or PS1−/− cells were IPed with anti-E-cadherin or anti-p85/PI3K antibodies and obtained IPs were analyzed for E-cadherin or p85/PI3K. (E) Lysates as in (D) were IPed with anti-N-cadherin or anti-p85/PI3K antibodies and obtained IPs were analyzed as shown. (F) PS1−/− cells were infected with HSV vector (V) or HSV PS1 (PS1). Lysates were IPed with anti-E- or anti-N-cadherin antibodies and analyzed as shown. Asterisk denotes IgG. (G) Confluent PS1+/+ cells were preincubated for 45 min in the presence of 4 mM EGTA to break cadherin/PI3K complexes (Pece et al, 1999; Tran et al, 2002) and then cultures were switched to Ca2+-containing medium at time zero and followed for the times shown (lanes 1–3). Lysates were IPed with anti-E- or anti-N-cadherin antibodies and IPs were analyzed with anti-p85/PI3K, or anti-E- and anti-N-cadherin antibodies as shown. Steady-state (SS) levels of cadherin/PI3K complexes in the absence of EGTA were monitored in PS1+/+ and PS1−/− cultures (lanes 4 and 5, respectively). All cultures were in serum-free media.
Cadherin-mediated activation of PI3K involves association of the p85 regulatory subunit of PI3K with either E- or N-cadherin (Pece et al, 1999; Baki et al, 2001; Laprise et al, 2002; Tran et al, 2002). Co-immunoprecipitation experiments reveal that PS1 forms a complex with the p85 subunit of PI3K (Figure 5C), raising the possibility that PS1 plays a role in the association of cadherins and p85. Indeed, association of p85 subunit of PI3K with either E- or N-cadherin is severely impaired in PS1−/− cells (Figures 5D and E, respectively). Moreover, re-introduction of PS1 into PS1−/− cells stimulates association of PI3K with both cadherins (Figure 5F). Thus, PS1 promotes association of the p85 subunit of PI3K with E- and N-cadherin.
To further examine the involvement of the cadherin/PI3K complexes in the PS1-stimulated activation of the PI3K/Akt pathway, we asked whether disruption of these complexes suppresses Akt phosphorylation even in the presence of PS1 and whether de novo formation of the complexes would re-activate Akt. To this aim, we used a calcium switch approach to disrupt and re-form cadherin/PI3K complexes (Pece et al, 1999; Tran et al, 2002). Figure 5G shows that calcium deprivation of PS1+/+ cultures dissociates PI3K from both cadherins and strongly suppresses phosphorylation of Akt (compare lanes 1 and 4). Restoration of calcium results in a progressive re-formation of cadherin/PI3K complexes and restores Akt phosphorylation to steady-state levels (Figure 5G, lanes 1–4). In contrast, in the absence of PS1, both cadherin/PI3K complexes and Akt phosphorylation are significantly decreased despite the continuous presence of calcium (Figure 5G, compare lanes 4 and 5). Together, the above data show that PS1 promotes association of the p85 regulatory subunit of PI3K with E- and N-cadherin and that this association is necessary for the PS1-induced activation of the PI3K/Akt pathway.
PS1 knockout embryos show reduced cadherin–PI3K complexes, impaired Akt activity and increased GSK-3-dependent phosphorylation of tau
The physiological significance of PS1 for the cadherin–PI3K association and PI3K/Akt activation was examined in vivo using PS1 null mice. Figure 6A shows that, compared to WT embryos, PS1−/− embryos contain significantly lower amounts of the p85/E-cadherin complexes, whereas an even more dramatic reduction is observed in the levels of the N-cadherin/p85 complexes. Phosphorylation of both Akt and its substrate GSK-3β is also reduced in PS1−/− embryonic brains compared to WT littermates, indicating reduced activation of the PI3K/Akt pathway and increased GSK-3 activity in the absence of PS1 (Figure 6B).
Figure 6.
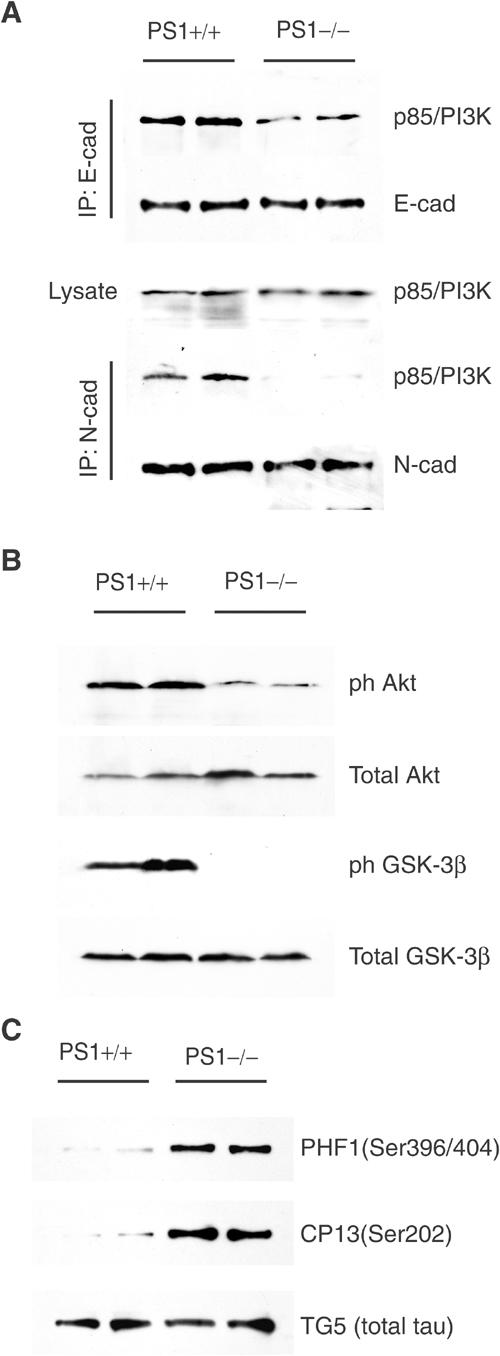
PS1 knockout embryos show reduced cadherin/PI3K complexes, decreased phosphorylation of Akt and GSK-3 and increased GSK-3-dependent phosphorylation of tau. (A) Total embryo homogenates prepared from PS1+/+ or PS1−/− mouse embryo littermates were immunoprecipitated with anti-E-cadherin (IP: E-cad) or anti-N-cadherin (IP: N-cad) antibodies and analyzed as shown. (B) Lysates were prepared from PS1+/+ or PS1−/− embryonic brains and analyzed for phosphorylated Akt and GSK-3β as shown. (C) Lysates were prepared from PS1+/− and PS1−/− mouse embryonic brains. The heat-stable fraction of lysates was analyzed with phosphorylation-dependent (PHF1, CP13) and phosphorylation-independent (TG5) anti-tau antibodies. Duplicate samples each from a littermate embryo are shown.
GSK-3β (also called tau kinase 1) phosphorylates tau at several serine and threonine residues found hyperphosphorylated in AD brains (Hanger et al, 1992; Pei et al, 1999). The reduced phosphorylation of GSK-3β at Akt-dependent residues observed in brains from PS1−/− mice suggested an increased GSK-3 activity and prompted us to examine phosphorylation of tau at residues targeted by GSK-3β. Figure 6C shows that, consistent with the low levels of GSK-3β phosphorylation, brain tau of PS1 null mice is hyperphosphorylated at GSK-3-dependent epitopes compared to tau of WT (PS1+/+) brains. Thus, the absence of PS1 results in reduced cadherin/PI3K association, impaired PI3K/Akt signaling and in increased GSK-3 activity and tau phosphorylation in vivo.
PS1 controls tau phosphorylation via the PI3K/Akt/GSK-3β pathway
To further explore the role of PS1 in GSK-3β-dependent phosphorylation of tau, we transfected PS1+/+ and PS1−/− fibroblasts with the longest human tau isoform and then examined phosphorylation of tau residues Ser396/404 and Ser202 that are targets of GSK-3β and are overphosphorylated in AD brains (Sperber et al, 1995). Figure 7A shows that, in agreement with the low levels of phosphorylated GSK-3β in PS1−/− cells (Figure 7A, lanes 1 and 4, panels f and g), exogenous tau is hyperphosphorylated in these cells (Figure 7A, lanes 1 and 4, panels a–c) suggesting that absence of PS1 activity results in tau hyperphosphorylation. Since cells were transfected with one tau isoform, the multiple bands detected by anti-phosphorylated tau antibodies indicate multiple phosphorylation sites. LiCl, an inhibitor of GSK-3 activity, reduced tau phosphorylation in PS1−/− cells (Figure 7A, lanes 4 and 6, panels a–c), indicating involvement of GSK-3β activity in the observed tau overphosphorylation. Thus, absence of PS1 activity results in increased GSK-3-dependent tau phosphorylation. Inhibition of PI3K activity by LY24002 in PS1+/+ cells reduced GSK-3β phosphorylation and increased tau phosphorylation (Figure 7A, lanes 1 and 2, panels f–g and a–c, respectively), indicating that in the presence of PS1 maintenance of low levels of tau phosphorylation requires PI3K activity. In contrast, in the absence of PS1, LY24002 had no detectable effect on the already high levels of tau phosphorylation (Figure 7A, lanes 4 and 5, panels a–c).
Figure 7.
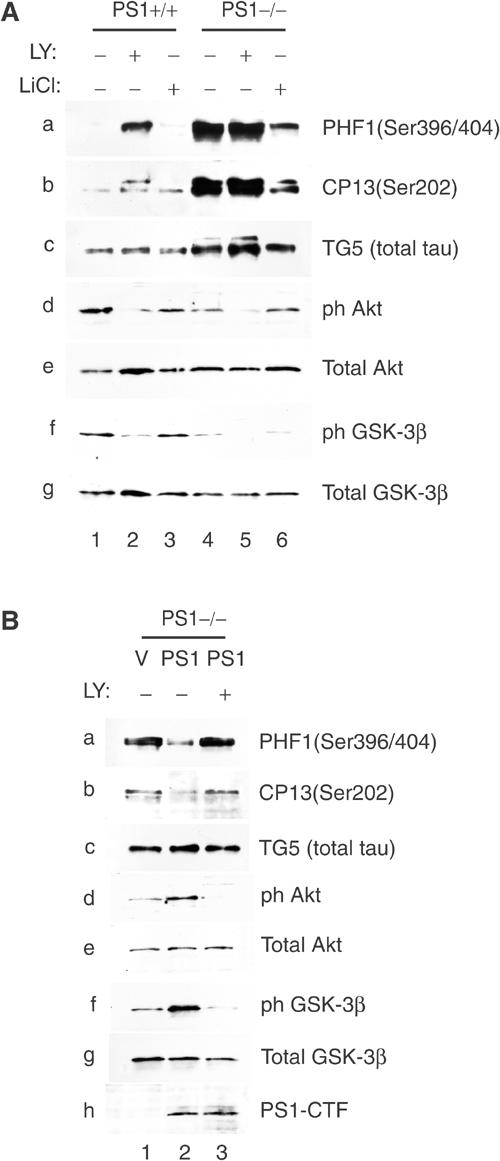
PS1 controls tau phosphorylation via the PI3K/Akt/GSK-3β pathway. (A) PS1+/+ or PS1−/− fibroblasts transiently expressing the longest human tau isoform were cultured in the presence or absence of PI3K inhibitor LY24002 (LY) or GSK-3 inhibitor LiCl. Lysates were analyzed for phosphorylation of tau (panels a–c), phosphorylation of Akt (panels d and e) and phosphorylation of GSK-3β (panels f and g). Transfected tau is detected by anti-tau antibody TG5 (panel c, total tau). (B) Human tau-expressing PS1−/− fibroblasts (see A) were infected with HSV vector (V) or HSV PS1 (PS1) and cultured in the presence or absence of PI3K inhibitor LY24002. Lysates were analyzed on Western blots as shown.
Re-introduction of PS1 in tau-expressing PS1−/− cells resulted in increased phosphorylation of both Akt and GSK-3β and in reduced phosphorylation of tau (Figure 7B, compare lane 1 to lane 2). Furthermore, these PS1 effects require PI3K activity as they were inhibited by LY24002 (Figure 5B, compare lane 2 to lane 3). Together, these data show that PS1 suppresses tau phosphorylation through the PI3K/Akt/GSK-3β pathway.
PS1 FAD mutants are defective in their ability to activate the PI3K–Akt pathway and to suppress tau phosphorylation
That PS1 activates the cadherin/PI3K/Akt pathway suggests that this protein may control numerous cellular events regulated by PI3K signaling, raising the possibility that interference of FAD mutations with this PS1 function may disrupt key cellular events including tau phosphorylation. To examine this possibility, PS1−/− cells were infected with recombinant viruses carrying either WT PS1 or one of the following PS1 FAD mutants: M146L, A246E, E280A and deletion of exon 9 (ΔE9). In contrast to missense mutants that are cleaved similar to WT PS1 (see Introduction), deletion FAD mutant ΔE9 is not processed by the cell (Thinakaran et al, 1996). Figure 8A shows that, compared to WT PS1, all four mutants were significantly impaired in their ability to promote Akt phosphorylation (panels a and b). Consistent with the inability of the FAD mutants to activate Akt, phosphorylation of GSK-3β at the Akt-specific epitope Ser9 was impaired in cells expressing any one of the four mutants (panels c and d). Furthermore, in contrast to WT PS1, none of the FAD mutants was able to suppress activation of apoptotic caspase-3 (panel e).
Figure 8.
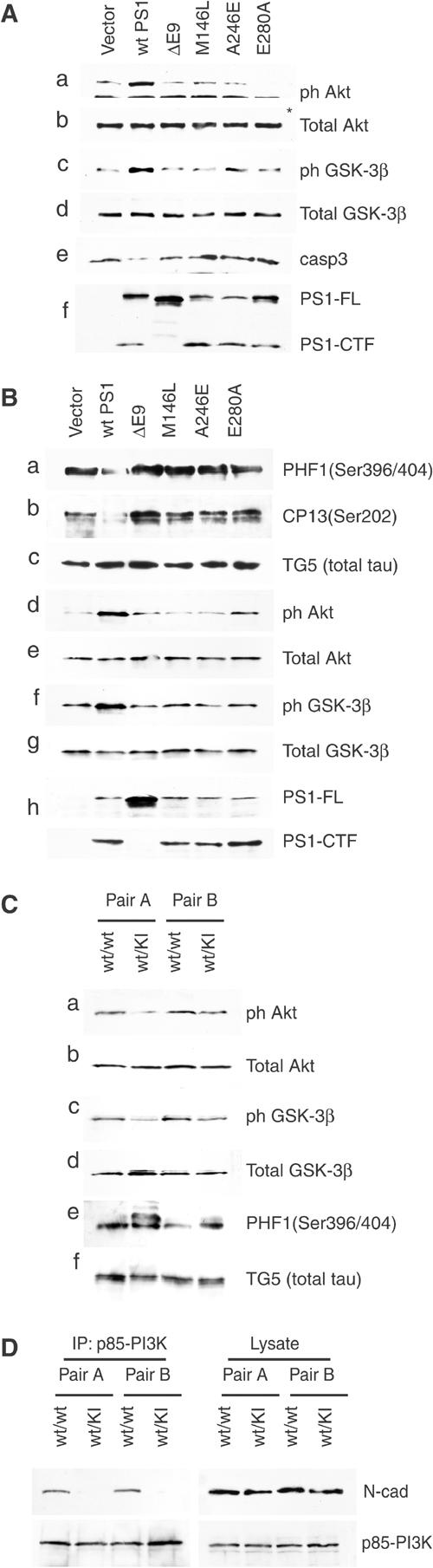
PS1 FAD mutants are defective in their ability to activate the PI3K–Akt pathway and to suppress phosphorylation of tau. (A) E-cadherin-expressing PS1−/− fibroblasts were infected with HSV vector (vector), with HSV recombinant viruses encoding WT PS1 (wt PS1) or with one of the PS1 FAD mutations shown on the top of each lane. Lysates prepared at 40 h postinfection were analyzed for PS1 expression (panel f), Akt phosphorylation (Ser473, panels a and b), GSK-3β phosphorylation (Ser9, panels c and d) or activated caspase-3 (panel e). Asterisk denotes nonspecific signal. (B) E-cadherin-expressing PS1−/− cells transiently expressing the longest human tau isoform were infected with HSV vector (vector), with HSV recombinant viruses encoding WT PS1 (wt PS1) or with one of the PS1 FAD mutants shown on the top of each lane. Lysates were analyzed for PS1 expression (panel h), tau phosphorylation (panels a–c) and phosphorylation of Akt and GSK-3β (panels d–g). (C) Brain lysates were prepared from two independent pairs of mice each pair consisting of a heterozygous PS1 I213T knock-in mouse (Nakano et al, 1999) and its WT littermate. Lysates were analyzed for phosphorylation of Akt (panels a and b), GSK-3β (panels c and d) and tau (panels e and f). Increased levels of multiple isoforms of overphosphorylated tau are detected in the knock-in mice. (D) Brain homogenates from two independent pairs of heterozygous PS1 I213T knock-in mice and their WT littermates (see C) were immunoprecipitated with anti-p85/PI3K antibodies and analyzed as shown.
To examine the effects of the FAD mutations on tau phosphorylation, PS1−/− cells expressing exogenous tau were infected with recombinant viruses carrying either WT PS1 or one of the above PS1 FAD mutants. Figure 8B show that, in contrast to WT PS1 and in accord with their inability to promote phosphorylation of Akt and GSK-3β (panels d–g), all FAD mutants were impaired in their ability to downregulate tau phosphorylation at epitopes found overphosphorylated in AD (panels a–c). The consistent effects of the WT and mutant PS1 on the phosphorylation of Akt, GSK-3β and tau (Figure 8B) support the suggestion that by activating the PI3K/Akt pathway, PS1 controls a number of downstream targets, indicating phosphorylation of tau.
To explore the consequences of FAD mutations in a physiologically more relevant system, we examined phosphorylation of Akt, GSK-3β and tau in the brains of adult gene-targeted (knock-in) mice heterozygous for the human PS1 FAD mutation I213T (Nakano et al, 1999). Knock-in systems mimic closely the gene dosage and expression of heterozygous FAD patients and are considered excellent in vivo FAD models. Figure 8C (panels a–d) shows that phosphorylation of both Akt and GSK-3β is reduced in the brains of knock-in mice. In agreement with the reduced phosphorylation, and hence increased activation, of GSK-3β, tau protein is overphosphorylated in the knock-in mice (panels e–f). Importantly, co-immunoprecipitation experiments showed that cadherin/PI3K association is reduced in the FAD mutant knock-in mice (Figure 8D), supporting the suggestion that this mutation may reduce Akt phosphorylation and signaling by interfering with the ability of PS1 to promote cadherin/PI3K association. Together, our data show that PS1 FAD mutants are impaired in their ability to stimulate the PI3K/Akt pathway and to suppress AD-related tau overphosphorylation and activation of apoptotic caspase-3.
Discussion
Our data reveal a novel PS1 function by which this protein stimulates PI3K/Akt signaling and promotes cell survival. This conclusion is supported by the following observations: (1) absence of PS1 results in low levels of phosphorylated Akt and increased apoptosis; (2) exogenous PS1 stimulates Akt phosphorylation and rescues PS1 null cells from apoptosis; (3) a constitutively active PI3K restores Akt activation and suppresses apoptosis induced by the absence of PS1; (4) pharmacological inhibition of either PI3K or Akt prevents the PS1-dependent Akt phosphorylation and caspase-3 inactivation, indicating that the PI3K/Akt pathway mediates the anti-apoptotic effects of PS1.
Cadherin–cadherin interactions initiate a cascade of signaling events that result in increased cadherin/PI3K association, activation of PI3K/Akt signaling and increased cell survival (Pece et al, 1999; Peluso et al, 2001; Kovacs et al, 2002; Tran et al, 2002; Yap and Kovacs, 2003). Our data that cadherin overexpression accelerates apoptosis of confluent PS1 null cells suggest that PS1 may be needed for the transmission of cadherin-dependent survival signals (Yap and Kovacs, 2003). In agreement with this suggestion, PS1 stimulates both cadherin/PI3K association and the PI3K/Akt cell survival pathway. Furthermore, anti-cadherin antibodies that block cadherin/PI3K association (Pece et al, 1999; Laprise et al, 2002; Tran et al, 2002) prevented the PS1-induced Akt phosphorylation (Figures 5B). In addition, Ca2+ switch experiments (Pece et al, 1999; Tran et al, 2002) showed that in the presence of PS1, calcium stimulates a parallel increase of both cadherin/PI3K complexes and Akt phosphorylation to steady-state levels. In contrast, in the absence of PS1, both cadherin/PI3K complexes and Akt phosphorylation remained low even in the presence of calcium. Together these data indicate involvement of cadherin function and cadherin/PI3K association in the PS1-induced activation of PI3K/Akt signaling. In agreement with a critical role of PS1 in the cadherin/PI3K association and Akt activation in vivo, PS1 knockout mice show decreased cadherin/PI3K complexes and reduced Akt phosphorylation. Our data do not exclude the possibility that cadherin-dependent juxtacrine signals may contribute to PS1-dependent PI3K signaling. However, PS1 stimulates cadherin homophilic adhesion (Baki et al, 2001), and recent work has shown that cadherin homophilic interactions can lead to PI3K activation independent of juxtacrine signaling (Kovacs et al, 2002).
Our data showing that PS1 stimulates Akt phosphorylation and signaling indicate that PS1 regulates the activity of substrates downstream of Akt (Brunet et al, 2001). One of these is GSK-3 kinase (Cross et al, 1995; Kaytor and Orr, 2002), whose activity has been shown to promote AD-like tau phosphorylation (Hanger et al, 1992; Hong et al, 1997; Pei et al, 1999; Lucas et al, 2001). Indeed, we show that by activating the PI3K/Akt pathway, PS1 promotes phosphorylation, and hence suppresses activity, of GSK-3. It thus decreases phosphorylation of transfected human tau at residues found to be hyperphosphorylated in AD brains. These observations suggest that PS1 may control phosphorylation of brain tau by stimulating the cadherin/PI3K/Akt/GSK-3 signaling. Strong support for a critical role of PS1 in the in vivo activation of the cadherin/PI3K/Akt signaling and tau phosphorylation is provided by PS1 knockout mice, which show decreased cadherin/PI3K association, reduced PI3K/Akt activity, indicated by the decreased phosphorylation of Akt and GSK-3, and increased tau phosphorylation at AD-related residues. In agreement with the decreased activity of the PI3K/Akt cell survival pathway, PS1 null mouse embryos die at birth showing increased neuronal death, probably by apoptosis, and serious deformities (Shen et al, 1997).
Since a large number of PS1 mutations are linked to FAD, we asked whether FAD mutations interfere with the PS1 function in the PI3K/Akt pathway. Our data show that, compared to WT PS1, all FAD mutants tested in this study are impaired in their ability to phosphorylate either Akt or its downstream target GSK-3β when introduced in a PS1 null background. In addition, in contrast to WT PS1, none of the FAD mutants was able to suppress overphosphorylation of human tau at amino acids found overphosphorylated in AD. Significantly, brains from heterozygous knock-in transgenic mice carrying FAD mutant PS1I213T, a system that closely models the gene dosage of FAD (Nakano et al, 1999), contain reduced levels of both phosphorylated Akt and GSK-3β and increased levels of tau phosphorylated at serine residues involved in AD. Furthermore, these mice contain reduced levels of cadherin/PI3K complexes (Figure 8D). These data suggest that a single mutant allele of PS1 is sufficient to cause a significant decrease in cadherin/PI3K/Akt signaling and an increase in the phosphorylation of AD-related tau epitopes even in the presence of a WT allele. Although our data with FAD mutants will need further verification by examining additional mutations in vitro and in vivo, they do indicate that PS1 FAD mutations may promote tau overphosphorylation by inhibiting the PS1-dependent PI3K/Akt/GSK-3 signaling. Since overphosphorylated tau is the main component of the NFTs of AD, PS1 FAD mutations may promote formation of brain NFTs by decreasing this signaling and thus increasing GSK-3 activity and tau phosphorylation. The decreased ability of PS1 FAD mutants to suppress apoptotic caspase-3 is consistent with the loss of PS1 function in the PI3K/Akt signaling and suggests that cells carrying these mutants may be more sensitive to apoptotic insults than wild type cells.
Many PS1 FAD mutations, including M146L, A246E, DE9 and E280A used here, stimulate the γ-secretase activity of PS1 resulting in increased production of Aβ peptides (gain of function with respect to γ-secretase; Murayama et al, 1999). In contrast, our data show that these PS1 mutations cause a loss of PS1 function in the PI3K/Akt signaling pathway. These observations provide further support for the conclusion that the ability of PS1 to activate the PI3K/Akt signaling and suppress apoptosis is a new function independent of the γ-secretase activity of PS1.
PI3K/Akt signaling controls the activity of a number of apoptotic substrates including caspases (Datta et al, 1999; Brunet et al, 2001) and downregulates the pro-apoptotic activity of neuronal GSK-3 (Pap and Cooper, 1998; Hetman et al, 2000; Cross et al, 2001; Lucas et al, 2001), a kinase implicated in AD (Hanger et al, 1992; Pei et al, 1999; Kaytor and Orr, 2002). By activating the PI3K/Akt signaling, PS1 may downregulate the activity of many pro-apoptotic factors and may thus promote cell survival. Recent reports suggest that GSK-3α activity stimulates Aβ production by a mechanism not involving γ-secretase activity (Phiel et al, 2003). Our data showing that PS1 suppresses the activity of GSK-3α suggest that the function of PS1 in the PI3K/Akt pathway may counteract production of Aβ, that is promoted by the γ-secretase activity of PS1. Thus, it may be important to ask whether PS1 FAD mutations compromise the ability of the cell to downregulate Aβ via the PI3K/Akt/GSK-3α pathway.
In summary, our results show that by promoting cadherin/PI3K association and PI3K/Akt signaling, PS1 prevents apoptosis, suppresses activity of GSK-3 and inhibits tau hyperphosphorylation. In contrast, PS1 FAD mutations inhibit the ability of PS1 to activate the PI3K/Akt cell survival pathway and may thus promote GSK-3 activity, hyperphosphorylation of tau and apoptosis. These findings support the hypothesis that loss of PS1 function in the PI3K/Akt pathway caused by FAD mutations may contribute to the AD pathology independent of the PS1 activity associated with the γ-secretase cleavage (Wolfe and Selkoe, 2002).
Materials and methods
Materials
Polyclonal antibody R222 against PS1/NTF and monoclonal 33B10 against PS1/CTF have been described (Georgakopoulos et al, 1999). Anti-phospho-Akt (Ser473) and anti-p85/PI3K polyclonal antibodies were from Pharmingen and Upstate Biotechnology, respectively. Anti-GSK-3β and anti-E-cadherin (cytoplasmic domain) antibodies were from BD Transduction Laboratories. Anti-activated caspase-3, anti-Akt, anti-phospho-GSK-3β (Ser9) and anti-phospho-GSK-3α/β (Ser21 of GSK-3α and Ser9 of GSK-3β) antibodies were from Cell Signaling Technology. Anti-tau antibody TG5 as well as anti-phospho-tau antibodies PHF1 and CP13 were a gift of Dr Peter Davies. EGFP-PS1 was prepared as described (Singh et al, 2001), human tau 441 cDNA was a gift of Dr Iqbal and mutant p110/PI3K cDNA (mutation K227E) was obtained from Dr Downward (Rodriguez-Viciana et al, 1996).
Cell culture, transfections and infections
Fibroblasts from PS1+/+ or PS1–/– mice were immortalized, stably transfected with human E-cadherin and cultured as described (Baki et al, 2001). Transient transfections were performed using Lipofectamine Plus and cells were collected 40 h later. For virus-mediated gene transfer, cDNAs were subcloned into vector pHSVPrPUC and replication-defective, recombinant HSV viruses were prepared as described (Bursztajn et al, 1998). Infections were performed in serum-free medium at a 0.8–1 multiplicity of infection. At 3 h postinfection, virus-containing medium was replaced with fresh virus-free medium plus 10% FBS and cells were harvested 36–40 h later. Approximately 60–70% of the cells were infected.
Treatment with inhibitors and calcium switch experiments
Treatment with PI3K inhibitor LY24002 (50 μM, Cell Signaling Technology), Akt inhibitor SH-6 (10 μM, Calbiochem), γ-secretase inhibitor L-685,458 (0.5 μM, Calbiochem), γ-secretase inhibitor Compound E (1 μM, Calbiochem) or LiCl (5 mM) was overnight in complete media. For treatment with function-blocking antibodies, cells were cultured overnight in complete medium plus anti-N-cadherin (40 μg/ml, GC-4, Sigma) or anti-E-cadherin (20 μg/ml, clone SHE78-7, Zymed Laboratories Inc.) antibodies and, 4 h before harvesting, the medium was replaced with fresh serum-free medium plus antibodies. For calcium switch experiments, confluent cells were incubated in serum-free medium for 4 h, treated with 4 mM EGTA for 40–50 min and then switched to serum-free, calcium-containing medium for the times shown.
Cell lysates, immunoprecipitation and immunoblotting
Cell lysates for Western blotting were prepared in SDS lysis buffer (100 mM Tris–HCl, 20 mM NaCl, 10 mM EGTA, 10 mM EDTA, 1% SDS+20 mM NaF+5 mM sodium orthovanadate) containing complete protease inhibitor cocktail (Boehringer Mannheim) and phosphatase inhibitor cocktail I (Sigma). For immunoprecipitation, cells were lysed in TNE buffer (Baki et al, 2001) containing 1% Triton X-100 plus protease inhibitor and phosphatase inhibitor cocktails and lysates were processed for immunoprecipitation as described (Georgakopoulos et al, 1999).
Tissue homogenates
WT and PS1−/− mouse embryos (embryonic day 17) and their WT littermates, or brains from 2-month-old heterozygous PS1 I213T knock-in mice (Nakano et al, 1999) and their WT littermates were homogenized in 1% Triton X-100 lysis buffer as above and processed for immunoprecipitation as described (Baki et al, 2001). For analysis of tau, brains were homogenized in TBS containing 10 mM NaF, 2 mM EGTA, 1 mM sodium vanadate, 1 mM PMSF, protease inhibitors and phosphatase inhibitor cocktail I. Heat-stable fractions were prepared by addition of NaCl (2%) and beta-mercaptoethanol (5%) to the homogenates, followed by boiling for 10 min. Samples were cooled on ice for 30 min and then centrifuged to collect supernatants.
Cell viability, DNA fragmentation, TUNEL and annexin assays and flow cytometry
Cell counts and viability were determined in triplicate, by trypan blue exclusion. Mean values were expressed as percentages of the mean value obtained for freshly confluent cells (0 point). DNA for fragmentation assays was isolated using the ‘apoptotic DNA ladder kit' (ROCHE) and samples were analyzed by agarose gel electrophoresis. For TUNEL staining, cells were cultured on coverslips and each day postconfluence PS1+/+ and −/− cells were fixed, permeabilized and subjected to TUNEL staining using the ‘in situ cell death detection kit, fluorescein' (ROCHE). Determination of early apoptotis by flow cytometry was performed using the annexin V–PE apoptosis detection kit following the manufacturer's directions (Pharmingen). Labeled cells were analyzed by three-color flow cytometry (EGFP, PE, 7AAD), using a FACS Calibur flow cytometer (Becton Dickinson) and CellQuest software. Annexin-negative cells were considered as nonapoptotic, whereas annexin-positive and 7AAD-negative cells were considered as early apoptotic.
Acknowledgments
We thank Drs Peter Davies and Khalid Iqbal for anti-tau antibodies and human tau cDNA, respectively. This work was supported by NIH grants AG-17926, AG-08200, NS-47229 and AG-05138 and the Alzheimer's Association.
References
- Baki L, Marambaud P, Efthimiopoulos S, Georgakopoulos A, Wen P, Cui W, Shioi J, Koo E, Ozawa M, Friedrich VL Jr, Robakis NK (2001) Presenilin-1 binds cytoplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates stability and function of the cadherin/catenin adhesion complex. Proc Natl Acad Sci USA 98: 2381–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K–Akt signaling pathway. Curr Opin Neurobiol 11: 297–305 [DOI] [PubMed] [Google Scholar]
- Bursztajn S, DeSouza R, McPhie DL, Berman SA, Shioi J, Robakis NK, Neve RL (1998) Overexpression in neurons of human presenilin-1 or a presenilin-1 familial Alzheimer disease mutant does not enhance apoptosis. J Neurosci 18: 9790–9799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TO, Rittenhouse SE, Tsichlis PN (1999) AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem 68: 965–1014 [DOI] [PubMed] [Google Scholar]
- Chan TO, Tsichlis PN (2001) PDK2: a complex tail in one Akt. Sci STKE 2001: PE1. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785–789 [DOI] [PubMed] [Google Scholar]
- Cross DA, Culbert AA, Chalmers KA, Facci L, Skaper SD, Reith AD (2001) Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J Neurochem 77: 94–102 [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13: 2905–2927 [DOI] [PubMed] [Google Scholar]
- Elder GA, Tezapsidis N, Carter J, Shioi J, Bouras C, Li HC, Johnston JM, Efthimiopoulos S, Friedrich VL Jr, Robakis NK (1996) Identification and neuron specific expression of the S182/presenilin I protein in human and rodent brains. J Neurosci Res 45: 308–320 [DOI] [PubMed] [Google Scholar]
- Fortini ME (2002) Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling. Nat Rev Mol Cell Biol 3: 673–684 [DOI] [PubMed] [Google Scholar]
- Georgakopoulos A, Marambaud P, Efthimiopoulos S, Shioi J, Cui W, Li HC, Schutte M, Gordon R, Holstein GR, Martinelli G, Mehta P, Friedrich VL Jr, Robakis NK (1999) Presenilin-1 forms complexes with the cadherin/catenin cell–cell adhesion system and is recruited to intercellular and synaptic contacts. Mol Cell 4: 893–902 [DOI] [PubMed] [Google Scholar]
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH (1992) Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett 147: 58–62 [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z (2000) Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci 20: 2567–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CS, Caromile L, Nomata Y, Mori H, Bredesen DE, Koo EH (1999) Contrasting role of presenilin-1 and presenilin-2 in neuronal differentiation in vitro. J Neurosci 19: 637–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Chen DC, Klein PS, Lee VM (1997) Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem 272: 25326–25332 [DOI] [PubMed] [Google Scholar]
- Irving NG, Miller CC (1997) Tau phosphorylation in cells transfected with wild-type or an Alzheimer's disease mutant Presenilin 1. Neurosci Lett 222: 71–74 [DOI] [PubMed] [Google Scholar]
- Kaytor MD, Orr HT (2002) The GSK3 beta signaling cascade and neurodegenerative disease. Curr Opin Neurobiol 12: 275–278 [DOI] [PubMed] [Google Scholar]
- Kovacs EM, Ali RG, McCormack AJ, Yap AS (2002) E-cadherin homophilic ligation directly signals through Rac and phosphatidylinositol 3-kinase to regulate adhesive contacts. J Biol Chem 277: 6708–6718 [DOI] [PubMed] [Google Scholar]
- Laprise P, Chailler P, Houde M, Beaulieu JF, Boucher MJ, Rivard N (2002) Phosphatidylinositol 3-kinase controls human intestinal epithelial cell differentiation by promoting adherens junction assembly and p38 MAPK activation. J Biol Chem 277: 8226–8234 [DOI] [PubMed] [Google Scholar]
- Leroy K, Boutajangout A, Richardson J, Octave JN, Lovestone S, Anderton BH, Brion JP (2003) Mutant presenilin 1 proteins induce cell death and reduce tau-dependent processes outgrowth. Neurosci Lett 353: 226–230 [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20: 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK (2002) A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21: 1948–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK (2003) A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114: 635–645 [DOI] [PubMed] [Google Scholar]
- Murayama O, Tomita T, Nihonmatsu N, Murayama M, Sun X, Honda T, Iwatsubo T, Takashima A (1999) Enhancement of amyloid beta 42 secretion by 28 different presenilin 1 mutations of familial Alzheimer's disease. Neurosci Lett 265: 61–63 [DOI] [PubMed] [Google Scholar]
- Nakano Y, Kondoh G, Kudo T, Imaizumi K, Kato M, Miyazaki JI, Tohyama M, Takeda J, Takeda M (1999) Accumulation of murine amyloidbeta42 in a gene-dosage-dependent manner in PS1 ‘knock-in' mice. Eur J Neurosci 11: 2577–2581 [DOI] [PubMed] [Google Scholar]
- Pap M, Cooper GM (1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem 273: 19929–19932 [DOI] [PubMed] [Google Scholar]
- Pece S, Chiariello M, Murga C, Gutkind JS (1999) Activation of the protein kinase Akt/PKB by the formation of E-cadherin-mediated cell–cell junctions. Evidence for the association of phosphatidylinositol 3-kinase with the E-cadherin adhesion complex. J Biol Chem 274: 19347–19351 [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF (1999) Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol 58: 1010–1019 [DOI] [PubMed] [Google Scholar]
- Peluso JJ, Pappalardo A, Fernandez G (2001) E-cadherin-mediated cell contact prevents apoptosis of spontaneously immortalized granulosa cells by regulating Akt kinase activity. Biol Reprod 64: 1183–1190 [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS (2003) GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature 423: 435–439 [DOI] [PubMed] [Google Scholar]
- Pigino G, Pelsman A, Mori H, Busciglio J (2001) Presenilin-1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci 21: 834–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J (1996) Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J 15: 2442–2451 [PMC free article] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S (1997) Skeletal and CNS defects in presenilin-1-deficient mice. Cell 89: 629–639 [DOI] [PubMed] [Google Scholar]
- Singh N, Talalayeva Y, Tsiper M, Romanov V, Dranovsky A, Colflesh D, Rudamen G, Vitek MP, Shen J, Yang X, Goldgaber D, Schwarzman AL (2001) The role of Alzheimer's disease-related presenilin 1 in intercellular adhesion. Exp Cell Res 263: 1–13 [DOI] [PubMed] [Google Scholar]
- Sperber BR, Leight S, Goedert M, Lee VM (1995) Glycogen synthase kinase-3 beta phosphorylates tau protein at multiple sites in intact cells. Neurosci Lett 197: 149–153 [DOI] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95: 29–39 [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B (1998) Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc Natl Acad Sci USA 95: 9637–9641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS (1996) Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17: 181–190 [DOI] [PubMed] [Google Scholar]
- Tran NL, Adams DG, Vaillancourt RR, Heimark RL (2002) Signal transduction from N-cadherin increases Bcl-2. Regulation of the phosphatidylinositol 3-kinase/Akt pathway by homophilic adhesion and actin cytoskeletal organization. J Biol Chem 277: 32905–32914 [DOI] [PubMed] [Google Scholar]
- Vestling M, Wiehager B, Tanii H, Cowburn RF (2001) Akt activity in presenilin 1 wild-type and mutation transfected human SH-SY5Y neuroblastoma cells after serum deprivation and high glucose stress. J Neurosci Res 66: 448–456 [DOI] [PubMed] [Google Scholar]
- Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP (1999) Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci 19: 5360–5369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS, Selkoe DJ (2002) Biochemistry, intramembrane proteases—mixing oil and water. Science 296: 2156–2157 [DOI] [PubMed] [Google Scholar]
- Yap AS, Kovacs EM (2003) Direct cadherin-activated cell signaling: a view from the plasma membrane. J Cell Biol 160: 11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]