

Abstract
Telomere shortening in normal human cells causes replicative senescence, a p53-dependent growth arrest state, which is thought to represent an innate defence against tumour progression. However, although it has been postulated that critical telomere loss generates a ‘DNA damage' signal, the signalling pathway(s) that alerts cells to short dysfunctional telomeres remains only partially defined. We show that senescence in human fibroblasts is associated with focal accumulation of γ-H2AX and phosphorylation of Chk2, known mediators of the ataxia-telangiectasia mutated regulated signalling pathway activated by DNA double-strand breaks. Both these responses increased in cells grown beyond senescence through inactivation of p53 and pRb, indicating that they are driven by continued cell division and not a consequence of senescence. γ-H2AX (though not Chk2) was shown to associate directly with telomeric DNA. Furthermore, inactivation of Chk2 in human fibroblasts led to a fall in p21waf1 expression and an extension of proliferative lifespan, consistent with failure to activate p53. Thus, Chk2 forms an essential component of a common pathway signalling cell cycle arrest in response to both telomere erosion and DNA damage.
Keywords: Chk2, DNA double-strand breaks, γ-H2AX, senescence, telomeres
Introduction
The TTAGGG repeat sequence at chromosome telomeres forms a protective T-loop structure, which is stabilised by telomere-binding proteins (Griffith et al, 1999) and is believed to prevent chromosome ends from being recognised as DNA breaks (de Lange, 2002). Most human somatic cells lack the telomere-synthesising enzyme telomerase and consequently suffer telomere shortening with each round of DNA synthesis, owing to the so-called ‘end-replication' problem (Harley et al, 1990). Progressive telomere erosion with ongoing proliferation eventually results in one or more telomeres becoming critically short, or even absent (Baird et al, 2003), which triggers cell cycle arrest and replicative senescence. While the causal role of telomere erosion in this process has been firmly established by the ability of telomerase to prevent senescence in normal human fibroblasts (Bodnar et al, 1998), the exact biochemical links between dysfunctional telomeres and cell cycle arrest have remained elusive.
One attractive hypothesis is that telomere erosion, either directly via unmasking of chromosome ends, or indirectly via chromosome bridge/breakage cycles, generates a signal equivalent to DNA double-strand breaks (DSBs). Consistent with this, we and others have shown that, like classical DNA damage-induced proliferative arrest, telomere-driven senescence is dependent on functional p53 (Gire and Wynford-Thomas, 1998) and is associated with a similar pattern of p53 phosphorylation (Webley et al, 2000). Also, experimental telomere uncapping through inactivation of the telomeric protein TRF2 led to a senescence response that is mainly dependent on a functional ataxia-telangiectasia mutated (ATM) and p53 pathway (Karlseder et al, 1999). While this is consistent with a model in which dysfunctional telomeres generate an exposed end recognised as a DNA DSB, none of these findings provide direct evidence that physiological telomere shortening leads to a DNA damage response. Here, we identify the DNA damage checkpoint protein kinase Chk2 as a key mediator of replicative senescence.
Chk2 is a component of the DNA damage checkpoint signalling pathway that responds to damage caused, for example, by ionising irradiation (IR), and to a lesser extent by ultraviolet radiation or replication blocks by hydroxyurea (Bartek et al, 2001). Direct phosphorylation of Chk2 by ATM at Thr68 within the SQ/TQ cluster domain is required for activation and is also a prerequisite for subsequent phosphorylation at other Chk2 regulatory sites (Matsuoka et al, 2000; Melchionna et al, 2000; Lee and Chung, 2001). Activated Chk2 in turn directly phosphorylates and regulates the functions of p53 (Chehab et al, 2000; Takai et al, 2002) and BRCA1 (Lee et al, 2000) tumour suppressors or the CDC25 family phosphatase (Matsuoka et al, 1998; Falck et al, 2001), resulting in checkpoint activation and cell cycle arrest in G1 (Chehab et al, 2000), S (Falck et al, 2001) and G2/M phase (Matsuoka et al, 1998; Hirao et al, 2000). Consistent with these multiple roles in protecting genome integrity, Chk2 is itself a tumour suppressor, mutations in Chk2 have been identified in a variant form of Li–Fraumeni syndrome, a highly penetrant familial cancer phenotype normally associated with p53 mutations, and in sporadic human cancers (Bell et al, 1999).
Here we report that two ATM substrates, Chk2 and H2AX, are key components of a telomere loss/DNA damage-signalling pathway that is activated in response to telomere erosion in human fibroblasts. Furthermore, we show that Chk2 function is essential for normal senescence-associated growth arrest, consistent with a causal role in activating p53.
Results
Phosphorylation and activation of Chk2 at senescence
Normal human foreskin fibroblasts (HFF) were serially passaged until they reached replicative senescence (at around population doubling (PD) of 87) as defined previously (Gire and Wynford-Thomas, 1998) by cell cycle arrest (bromodeoxyuridine (BrdU) labelling index <1%), flattened morphology and accumulation of senescence-associated β-galactosidase (SA-βgal) activity (Dimri et al, 1995). As expected, senescent HFF showed upregulation of the cyclin-dependent kinase inhibitor p21waf1 (p21) with the concomitant presence of pRb in its hypophosphorylated, growth-suppressive form (Figure 1A) (Stein et al, 1999). Chk2 protein at senescence displayed an electrophoretic mobility shift (Figure 1A), a characteristic phosphorylation-induced reduction in mobility strictly associated with its activation (Matsuoka et al, 2000). However, the extent of the shift in senescent cells was significantly less pronounced compared to the shift produced in young cells exposed to the radio-mimetic drug, bleomycin (Figure 1A). Phosphatase treatment ablated the altered mobility, indicating that Chk2 is modified by phosphorylation at senescence (data not shown). Furthermore, similar results were obtained in senescent IMR90, another fibroblast strain, indicating that Chk2 mobility shift is not unique to normal foreskin fibroblasts (Figure 1B).
Figure 1.
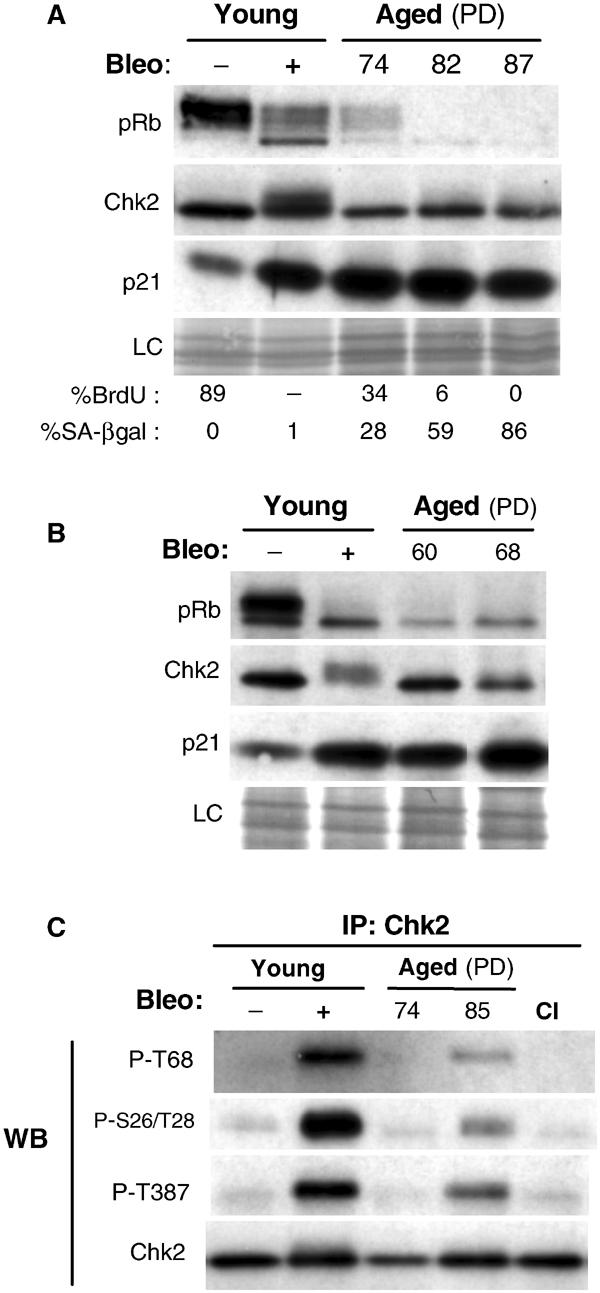
Chk2 activation at replicative senescence. (A) Abundance and status of pRb, Chk2 and p21 of fibroblasts at different PDs up to senescence. Western blot analysis of young fibroblasts at PD35 untreated (−) or treated (+) with bleomycin (bleo) (used here as positive control) and ageing fibroblasts at PD74, PD82 and PD87 (cell population fully senescent). Loading control (LC) is the scan of amido-black-stained membrane, which showed equal amounts of protein transferred. The phosphorylation status of Rb is used to monitor cell cycle regulation. The percentages of cells positive for BrdU (24 h labelling) and SA-βgal are shown below. The standard deviations were 1–3%. Positive cells were scored by microscopic observation after indirect immunostaining of BrdU incorporation and histochemical assessment of β-galactosidase activity at pH 6. (B) Similar analysis for young and aged IMR90 fibroblasts at PD60 and PD68 (cell population fully senescent). (C) Chk2 phosphorylation at senescence. Phosphorylation at Thr68, Ser26/Thr28 and Thr387 was examined with phospho-specific antibodies on immunoblots of Chk2 immunoprecipitates from lysates of young fibroblasts untreated (−) or treated (+) with bleomycin (bleo), and of aged and young contact-inhibited (CI) fibroblasts. Recovery of immunoprecipitated Chk2 was monitored by immunoblotting with total Chk2. IP, immunoprecipitation; WB, Western blot.
Damage-dependent activation of Chk2 is accompanied by the phosphorylation of a cluster of SQ/TQ sites within the N-terminus of Chk2, which are consensus targets for members of the ATM/ATR kinase family (Matsuoka et al, 2000; Melchionna et al, 2000). The predominant phosphorylated site is Thr68 and this event promotes oligomerisation and entails further autophosphorylation at Thr383 and/or Thr387 on the catalytic domain, which is required for full activation of Chk2 (Lee and Chung, 2001; Xu et al, 2002). Therefore, we investigated whether these modifications, which function as a surrogate marker for Chk2 activation (Bartek et al, 2001), could be detected in cells at senescence. In addition, we studied Thr26/Ser28, two other Chk2 regulatory sites, phosphorylation of which correlates with activation (Matsuoka et al, 2000; Xu et al, 2002).
Phosphorylation of Chk2 at Thr68 and at Thr387 could be readily detected, using phospho-specific antibodies, by Western blot analysis of young HFF treated with bleomycin, but was only barely detectable in lysates of senescent HFF (data not shown). However, a significant increase in phosphorylation of Chk2 at Thr68, Thr26/Ser28 and Thr387 could be detected by immunoblotting of Chk2 immunoprecipitates from senescent fibroblasts compared to young HFF (Figure 1C). In contrast, another form of cell cycle arrest, quiescence induced by contact inhibition, did not lead to any increase in Chk2 phosphorylation (Figure 1C). As anticipated from the low magnitude of the electrophoretic changes and levels of phosphorylation of Chk2 observed at senescence, we were unable to demonstrate directly an increase in Chk2 activity at senescence using an in vitro kinase assay.
Forced telomere shortening amplifies Chk2 activation
We next sought to formally exclude the possibility that Chk2 activation might be merely a consequence rather than a cause of senescence. To test this, we examined whether Chk2 activation was abolished in cells that had escaped senescence through inactivation of the essential senescence effectors p53 and pRb, using a retrovirus encoding the human papilloma virus (HPV-16) genes E6 and E7. Such cells continue to proliferate beyond the senescence limit until they reach a second proliferative barrier termed ‘crisis', by which time extensive loss of telomeric sequences results in genomic catastrophe and massive cell death (Zhu et al, 1999). Young HFF expressing E6/E7 failed to show either induction of endogenous p21 (Figure 2A) or cell cycle arrest (data not shown) after bleomycin treatment. However, the level of expression and the degree of phosphorylation of Chk2 following bleomycin exposure were not altered in these cells compared to normal HFF (Figure 2A), indicating that activation of Chk2 does not depend on p53 and pRb function.
Figure 2.
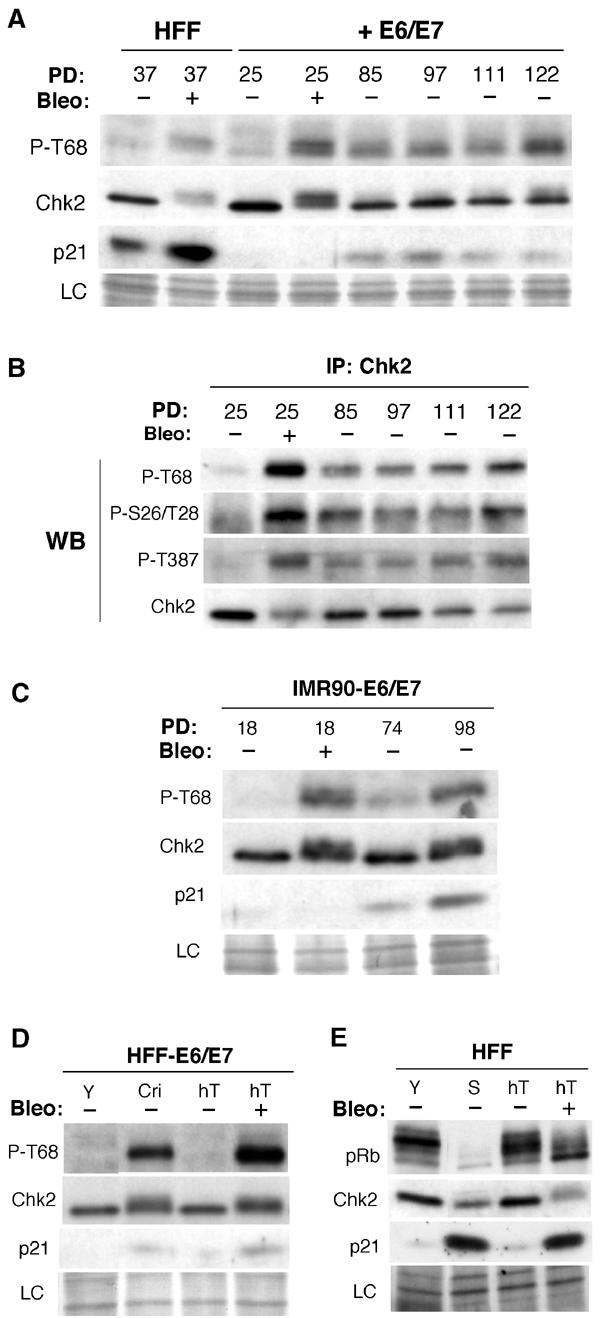
Chk2 activation in fibroblasts expressing E6/E7 and/or hTERT. (A) Immunoblots for abundance of phospho-Thr68-Chk2, Chk2 and p21 of fibroblasts expressing HPV-E6/E7 grown beyond senescence up to near crisis. Young HFF and those expressing HPV-E6/E7 (+E6/E7) were untreated (−) or treated (+) with bleomycin (bleo). Serially passaged HFF expressing E6/E7 were analysed at PD85 up to PD122. Reduction of the endogenous level of p21, a direct transcriptional target of p53, served as a marker for the activity of E6 in E6/E7 cells. Loading control (LC) is the scan of the amido-black-stained membrane. (B) Chk2 phosphorylation of fibroblasts expressing HPV-E6/E7 grown beyond senescence up to near crisis. Phosphorylation at Thr68, Ser26/Thr28 and Thr387 was examined with phospho-specific antibodies on immunoblots of Chk2 immunoprecipitates from lysates of young fibroblasts expressing E6/E7 at PD25 untreated (−) or treated (+) with bleomycin, and of E6/E7 cells serially passaged and analysed at PD85 up to PD122. Phospho-specific immunoblots are from independent blot of the same lysates. Recovery of immunoprecipitated Chk2 was monitored by immunoblotting with total Chk2 (lower panel). IP, immunoprecipitation; WB, Western blot. (C) Similar analysis as in (A) for young and IMR90 fibroblasts expressing E6/E7 at the indicated PD. (D) hTERT expression prevents Chk2 activation in fibroblasts expressing E6/E7. Immunoblots for Chk2 phosphorylated at Thr68 and total levels of Chk2, p21 in E6/E7 cells, young (Y) and at crisis (Cri) compared to immortalised E6/E7 cells through ectopic expression of hTERT (hT) untreated (−) or treated (+) with bleomycin (bleo). (E) hTERT expression prevents Chk2 activation in fibroblasts. Immunoblots for Rb, Chk2 and p21 in young (Y) and senescent (S) cells compared to immortalised fibroblasts through ectopic expression of hTERT (hT) untreated (−) or treated (+) with bleomycin (bleo). Loading control (LC) is the scan of the amido-black-stained membrane.
Serial passage of HFF expressing E6/E7 beyond the point where they would normally enter senescence was associated with a progressive increase in Chk2 electrophoretic mobility shift and increased phosphorylation at Thr68 (Figure 2A), consistent with increasing activation. Similarly, immunoprecipitated Chk2 from these cells, in contrast to young HFF expressing E6/E7, was recognised by anti-phospho-Thr68, anti-phospho-Ser26/Thr28 and anti-phospho-Thr387 antibodies (Figure 2B). Similar results were obtained with IMR90 cells expressing E6/E7 (Figure 2C and data not shown).
Thus, Chk2 activation seems to be a direct result of events provoked by continued cell division rather than from the senescence state itself. Furthermore, the continued proliferation of E6/E7 cells, despite evidence of increasing Chk2 activation, indicates that in the absence of p53 and pRb pathways these cells are unable to arrest in response to signals elicited by eroded telomeres. The increasing phosphorylation of Chk2 observed in such cells is consistent with the ongoing process of continual telomere erosion, increasing the proportion of critically short telomeres beyond the small number per cell thought to be present at normal senescence (Hemann et al, 2001). Thus, these data place Chk2 upstream of p53 and pRb and downstream of telomere erosion.
Telomerase expression prevents Chk2 activation
We also wished to exclude the possibility that the observed activation of Chk2 might be related to the time the cells have spent in culture, for example, as a consequence of accumulation of physiological stress-induced cell ‘damage' rather than telomere erosion per se. We therefore examined the effect of forced expression of the catalytic subunit of telomerase (hTERT), which is sufficient to arrest or reverse telomere erosion (Stewart et al, 2003) and allows escape from both senescence and crisis in normal HFF (Bodnar et al, 1998), or those expressing E6 and E7 (Zhu et al, 1999).
In HFF expressing E6/E7, which had escaped crisis through expression of hTERT, phosphorylation of Chk2 at Thr68 was no longer detectable and its mobility resembled that of young fibroblasts, although the ability to be induced by bleomycin was retained (Figure 2D). Moreover, in normal HFF immortalised by hTERT, no changes in Chk2 immunoreactivity or mobility could be detected in cultures, which had been passaged well beyond the point where the corresponding hTERT-negative normal cells would have senesced (Figure 2E).
These data suggest therefore that Chk2 activation is specifically induced by telomere erosion, and not as a response to cumulative ‘stress' during extended in vitro culture, a conclusion consistent with recent experiments in Saccharomyces cerevisiae where Rad53p, a homologue of human Chk2, is phosphorylated in response to short telomeres, brought about by inactivation of telomerase (AS and Greider, 2003).
Focal accumulation of phosphorylated histone H2AX (γ-H2AX) in senescent and post-senescent cells
The Thr68 phosphorylated form of Chk2 accumulates in senescing cells (Figure 1C), suggesting that senescence invokes a DNA damage response signalling pathway that includes at least ATM. We next examined by immunofluorescence the presence of another ATM substrate, γ-H2AX, which is the earliest known cellular response to DNA damage and localises at the sites of DSB (Rogakou et al, 1999).
In the absence of DNA damage, the nuclear γ-H2AX appeared to exist as two distinct populations in proliferating young fibroblasts. In approximately 70% of the cells, γ-H2AX staining was weak or diffuse, whereas in the remainder we detected one to two discrete γ-H2AX foci per nuclei (Figure 3A and B). As described earlier for various cell lines (Rogakou et al, 1999; Ward et al, 2001), exposure of HFF to bleomycin caused the appearance of numerous large γ-H2AX foci in most of the cells. Although there was a marked cell-to-cell heterogeneity in the number of observed foci per nuclei, 50% of nuclei had at least 30 foci (Figure 3A). Notably, such foci were also evident in senescent HFF, although significantly less abundant even without any bleomycin exposure (Figure 3A). In 30–35% of the cells, three to five large γ-H2AX foci were observed, and in 20–25% more than five (Figure 3A). Furthermore, as shown in Figure 3B, there was a clear inverse correlation between the proportion of nuclei containing γ-H2AX foci and DNA replication (BrdU labelling index) as late-passage HFF approach senescence. Finally, in HFF expressing hTERT grown beyond senescence, the occurrence of γ-H2AX was comparable to young proliferating cells (data not shown), reinforcing the importance of telomere erosion as opposed to cumulative nonspecific ‘damage' in inducing these changes.
Figure 3.
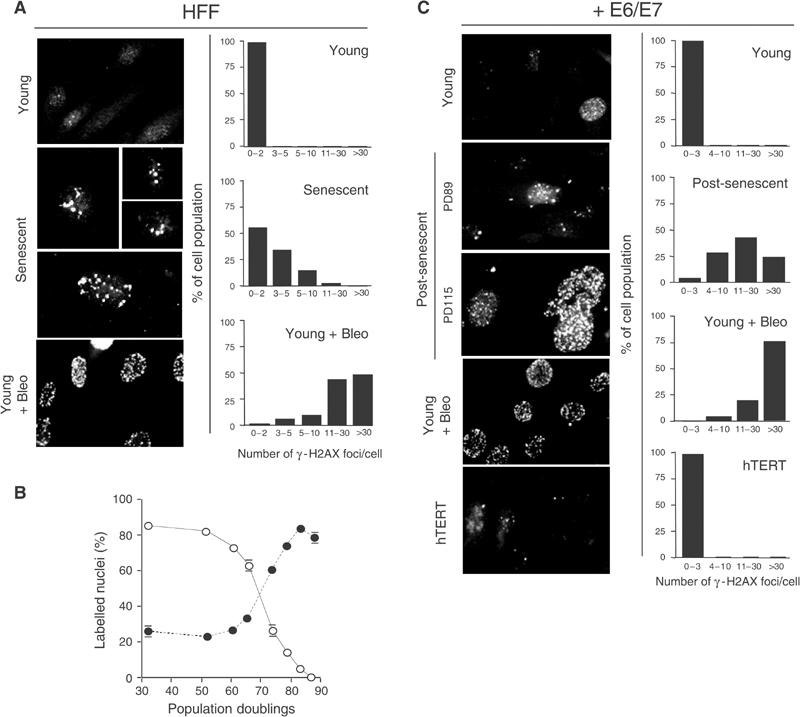
Accumulation of phosphorylated H2AX (γ-H2AX) foci in senescent and E6/E7-expressing fibroblasts grown beyond senescence. (A, C) Immunofluorescence images of fibroblasts (HFF) and fibroblasts expressing E6/E7 (+E6/E7) immunostained with anti-γ-H2AX antibodies. (A) Young at PD39 and senescent at PD82 fibroblasts. (C) Young E6/E7 cells at PD43, post-senescent E6/E7 cells at PD89 and PD115 and immortalised through ectopic expression of hTERT (hTERT). Treatment of young cells with bleomycin is indicated (young+bleo). Right panels show quantification of γ-H2AX foci. The number of γ-H2AX foci per nucleus was counted for each sample and nuclei were categorised as indicated. Note that quantification shown for post-senescent E6/E7 cells refers to cells at PD115. Panels are representative of four independent experiments. (B) Age-dependent increase in the incidence of γ-H2AX foci in HFF cultures. The percentage of cells with γ-H2AX foci (closed symbols) versus the percentage of cells incorporating BrdU (open symbols) is shown at the indicated PD. Bars: standard deviation.
To further corroborate the essential role of continued cell division in inducing γ-H2AX foci, we performed similar analysis on HFF expressing E6/E7 grown beyond senescence. In these cultures, both the percentage of nuclei containing γ-H2AX foci and the mean number of foci per nucleus increased until, at crisis, levels were reached similar to those seen after exposure of young E6/E7 cells to bleomycin (Figure 3C). Again, as with normal HFF, expression of hTERT in HFF expressing E6/E7 was sufficient to abolish γ-H2AX foci formation associated with their extended lifespan (Figure 3C).
Phosphorylated H2AX, but not Chk2, associates with telomeric DNA
The DNA damage signal documented here could arise either indirectly from rupture of dicentric chromosomes at mitosis (themselves the result of telomeric fusion) and/or directly from eroded chromosome ends (assuming that telomeres at senescence resemble damaged DNA).
To address this, we assessed whether γ-H2AX accumulates at telomeres by performing chromatin immunoprecipitation (ChIP) assay on in vivo cross-linked chromatin (Loayza and De Lange, 2003) from young and post-senescent E6/E7 cells, since we had already shown that the latter show significant Chk2 activation (Figure 2) and massive γ-H2AX foci accumulation (Figure 3).
ChIPs performed using antibodies to γ-H2AX showed that telomeric DNA association of γ-H2AX was undetectable in young cells expressing E6/E7 (PD25), was readily detectable at PD96 and increased further (approximately five- to six-fold) by PD111 (Figure 4A and B). These data provide direct evidence that γ-H2AX binds directly to telomeres in late-passage cells as a result of progressive telomere shortening. On the contrary, parallel ChIPs assays with antibodies to Chk2 revealed no detectable association. These latter results may suggest, as shown in a recent study using live-cell imaging (Lukas et al, 2003), that Chk2 does not stably reside at DSB sites despite interacting transiently with DSB to become phosphorylated by ATM.
Figure 4.
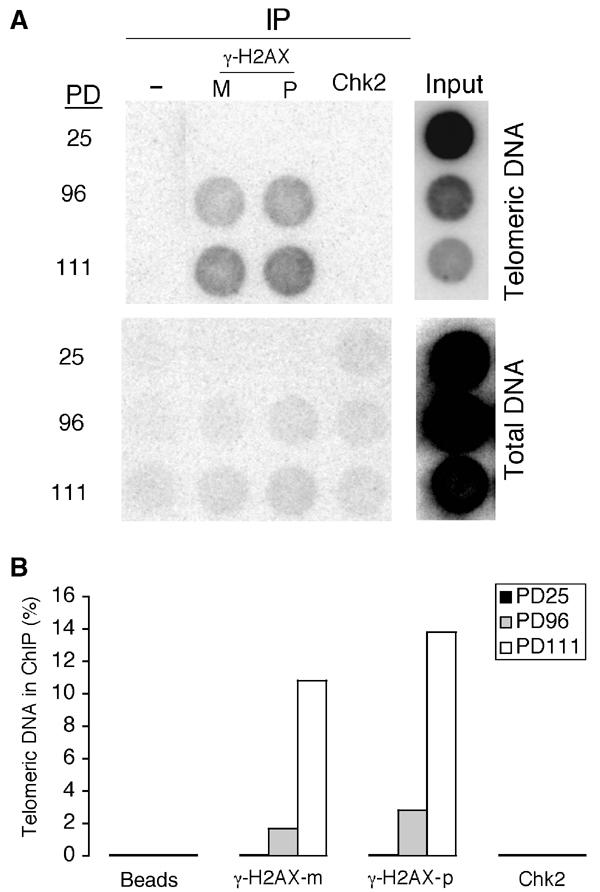
Association of γ-H2AX, but not Chk2, with telomeric DNA in post-senescent cells as detected by ChIP. (A) Dot blot analysis of DNA co-immunoprecipitated using the indicated antibodies or beads only (control) of crosslinked chromatin from cells expressing E6/E7, young (at PD25) and post-senescent (at PD96 and PD117). We used two antibodies to H2AX, a monoclonal antibody (m) and a polyclonal antibody (p). Duplicate dot blots were hybridised with a telomeric or Alu probe and membranes were exposed to PhosphorImager. (B) Quantification of the ChIP experiments shown in (A). The histograms give a quantitation of the signal recovered in each immunoprecipitate and detected by PhosphorImager as a percentage of the total amount of telomeric DNA used in each immunoprecipitation.
Inactivation of Chk2 leads to an extension of proliferative lifespan
Given the above evidence suggesting Chk2 activation in replicative senescence, we next set out to test whether it is necessary for senescence-associated cell cycle arrest. Chk2−/− mouse embryonic fibroblasts are available and have been shown to be deficient in their ability to maintain a cell cycle arrest in response to ionising radiation (Takai et al, 2002). Unfortunately, we did not consider these to be a suitable model system, in view of the now well-recognised differences in the genetic control of senescence between murine and human fibroblasts (Drayton and Peters, 2002), notably the lack of dependence on telomere erosion in the former. Therefore, we used instead a dominant-negative mutant of Chk2 (Chk2DN), which had been shown to be non-activatable and failed to undergo the mobility shift in response to ionising radiation (Matsuoka et al, 1998; Chehab et al, 2000).
We constructed a retroviral vector, which in young HFF was able to confer stable high levels of expression of Chk2DN (six- to ten-fold above endogenous wild-type Chk2) (Figure 5A). Expression of mutant Chk2DN blocked the mobility shift of endogenous Chk2 and significantly reduced the activating phosphorylation at Thr387 in response to bleomycin, therefore confirming the dominant-negative effect of the construct (Figure 5A). The observation that Thr387 phosphorylation was not totally abolished probably reflects cell–cell heterogeneity in Chk2DN expression, since immunocytochemical analysis showed that, in pooled drug-selected cultures, 10–15% of cells fail to express high Chk2DN levels (data not shown).
Figure 5.
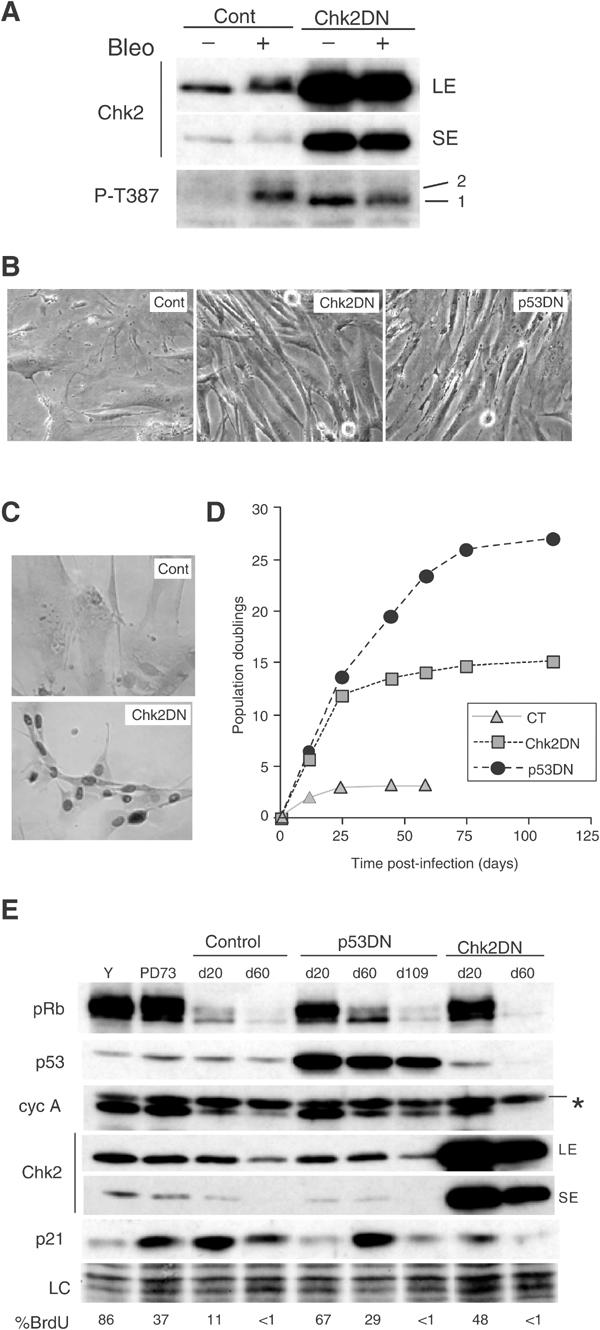
Senescence-associated replicative arrest impaired by dominant-negative Chk2 expression. (A) Validation of the retroviral vector encoding dominant-negative Chk2 in young HFF. Young HFF at PD28 were infected by a retroviral vector encoding Chk2DN (Chk2DN) or neo-only (control) and pooled drug-resistant cell populations were untreated (−) or treated (+) with bleomycin (bleo). Immunoblot analysis of Chk2 activation indicated by appearance of a slower migrating species using an antibody to total Chk2 (upper panel) and by band intensity using a phospho-specific antibody to Thr387-Chk2 (lower panel). Two exposures for Chk2 detection are shown in order to better visualise the absence of mobility shift in cells expressing Chk2DN after DNA damage (LE: long exposure; SE: short exposure). The line numbered 2 depicts the slower shifted Thr387 band. Note that Thr387 antibody revealed a faster migrating form of Chk2 in cells expressing Chk2DN, which probably reflects basal phosphorylation at Thr387 (line numbered 1). (B) Cell morphology of early colonies of fibroblasts stably expressing Chk2DN compared with HFF expressing p53DN and neo-only. Near-senescent HFF were infected with virus encoding Chk2DN, p53DN and neo-only (Cont), drug selected and photographed on day 20 post-infection. (C) Stable expression of mutant Chk2 in early colonies as revealed by increased nuclear Chk2 protein content. Indirect immunoperoxidase immunostaining of Chk2 protein. Only background signal was observed in senescent controls. (D) Growth curves of near-senescent HFF cultures infected with Chk2DN or p53DN. Near-senescent HFF were infected with retrovirus expressing the indicated proteins, drug-selected, serially passaged over the indicated period of time and cell number determined at each passage, as described in Materials and methods. (E) Immunoblot analysis of post-senescent HFF expressing Chk2DN. Cell lysates were prepared from culture of HFF at the time of infection (PD73) and of HFF expressing Chk2DN or p53DN that had entered into an extended lifespan phase (at day 20 post-infection) and were terminally growth arrested. Comparison was carried out with young (y) and age-matched control culture of HFF. Expression of pRb, p53, Chk2, cyclin A and p21 as indicated was analysed by Western blotting. Loading control (LC) is the scan of the amido-black-stained membrane. The percentages of BrdU-positive cells within these same populations are shown at the bottom. Procedure as in Figure 1A. The asterisk indicates a nonspecific band.
Near-senescent HFF cultures were infected either with retroviral vectors encoding Chk2DN, neo-only (negative control) or dominant-negative p53 mutant (p53DN, positive control). Near-senescent cells were used in order to minimise the possibility of selection for additional unknown genetic events, and to facilitate quantitation of lifespan extension (Bond et al, 1994). Chk2DN expression gave rise to rapidly expanding colonies, as described previously for mutant p53 (Bond et al, 1994), with morphology similar to young HFF, while control populations showed early cessation of growth (Figure 5B). Immunocytochemical analysis confirmed high expression of Chk2 in 85–90% of cells derived from near-senescent HFF infected with Chk2DN (Figure 5C). Assessment by colony formation efficiency and fluorescence microscopy after co-infection of a retroviral vector encoding GFP indicated that 60–70% of p53DN and 40–50% of the Chk2DN-infected cells initially divide successfully.
The extent of proliferative lifespan extension conferred by the retroviral vectors was monitored by growth curves (Figure 5D) and by serial BrdU labelling analyses (Figure 5E). As expected (Bond et al, 1994), p53DN expression conferred an additional ∼PD20 over neo-only controls, terminating in a second state of viable, proliferative arrest similar to senescence (Figure 5D and data not shown). Chk2DN also extended the lifespan, although to a lesser extent (PD10–12), and again ending in a senescence-like state with low BrdU index (Figure 5D and E), large and flattened morphology. Chk2DN-arrested cells stained intensively for acidic β-galactosidase and remained viable for at least 2 months (data not shown). Immunoblot analysis confirmed that growth arrest was not due to loss of expression of the Chk2DN mutant (Figure 5E).
To gain insight into the molecular mechanism by which Chk2 extends lifespan, we analysed the expression of several cell cycle regulators in pooled drug-selected cultures. By day 20 post-infection, in HFF expressing Chk2DN, we observed a reduction of p21 protein level, and an increase in the hyperphosphorylated form of Rb and in cyclin A expression compared to age-matched control HFF (Figure 5E). As expected, similar results were obtained following expression of p53DN (Figure 5E). Subsequent cessation of growth by day 60, in both Chk2DN and p53DN cultures, was associated with reduction in the hyperphosphorylated form of Rb and reduction of cyclin A expression.
To complement the dominant-negative approach, we next used siRNA transfection (Elbashir et al, 2001) to ‘knock down' transiently the expression of Chk2. Transfection of senescent fibroblasts with Chk2 siRNA resulted in a significant (approximately 55–70%) reduction in Chk2 protein levels compared to control siRNA (Figure 6), without affecting the expression of other DNA-damage-associated protein kinases, notably Chk1 (Figure 6). Knockdown of endogenous Chk2 protein led to short-term responses equivalent to those seen with the dominant-negative Chk2, namely reversion to a ‘young' morphology, reduced SA-βgal expression and increased BrdU incorporation, associated with reduced p21 expression, and increased pRb hyperphosphorylation and cyclin A expression (Figure 6B and C). As expected from the half-life of siRNA, culture reverted to a senescent phenotype several days after the last siRNA transfection (data not shown).
Figure 6.
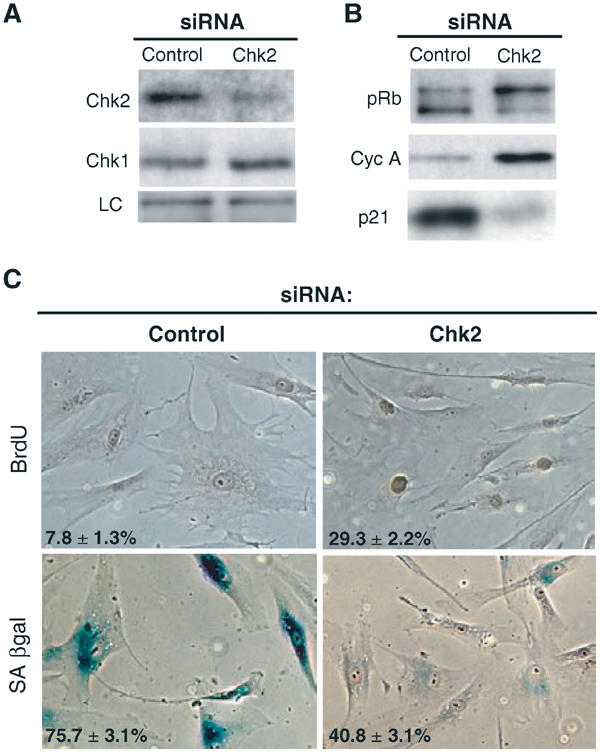
Abrogation of senescence-associated replicative arrest by Chk2 siRNA. (A) Senescent fibroblasts at PD80 were repeatedly transfected with Chk2 siRNA (Chk2) or with control siRNA (control). After 7 days, cells were collected and immunoblotted with anti-Chk2, anti-Chk1 and anti-β-tubulin antibodies used as an internal control (LC). (B) Immunoblots for pRb, cyclin A and p21 of senescent fibroblasts transfected with control siRNA or Chk2 siRNA at day 7. (C) BrdU incorporation and SA-βgal activity of senescent fibroblasts transfected with control siRNA or Chk2 siRNA at day 7. The percentage of cells incorporating BrdU (24 h labelling) and staining positive for SA-βgal is indicated in the bottom left of each photograph. Procedure as in Figure 1A. Representative photographs of stained field are shown.
From these data, we conclude that Chk2 is essential (though not necessarily sufficient) for induction of senescence-associated replicative arrest in human fibroblasts.
Discussion
Previous studies have shown that short telomeres or disruption of telomere structure can activate a checkpoint response (Lee et al, 1998; Karlseder et al, 1999) that requires p53 (Chin et al, 1999; Karlseder et al, 1999) and in some cases ATM (Karlseder et al, 1999). Consistent with this, we show that phosphorylation of Chk2 at Thr68, the principal site for activation by ATM in response to DNA DSBs (Melchionna et al, 2000), occurs during replicative senescence. However, the role of ATM itself is difficult to address since ATM participates directly in telomere length maintenance, as demonstrated by accelerated telomere loss and increased chromosomal end-to-end fusions in ATM-deficient mice and human cells (Metcalfe et al, 1996; Wong et al, 2003). This role could outweigh the effect of losing the ATM-mediated signalling pathway. We also observed that Chk2 is phosphorylated at Thr26 or Ser28 regulatory sites which do not appear to be ATM substrates in vitro (Matsuoka et al, 2000), suggesting the existence of an additional, ATM-independent pathway for the regulation of Chk2 at senescence. This is consistent with the observation that senescence in ATM-deficient human fibroblasts is not delayed and is associated with an induction of p53 DNA-binding activity (Vaziri et al, 1997). Potential alternative kinases that may act as transducers of the damage signal include ATR, which can phosphorylate Chk2 at Thr26 in vitro (Matsuoka et al, 2000).
Inactivation of Chk2 resulted in temporary escape from senescence, strongly suggesting that the growth arrest in senescent HFF cells is primarily due to a DNA damage-induced signal. As observed here and previously in HFF expressing mutant p53 (Bond et al, 1994), escape from growth arrest is not accompanied by a complete loss of p21 expression, suggesting that partial abrogation of p53 function may be sufficient to allow fibroblasts to initially overcome senescence. Consistent with this, p21 levels are only transiently induced in late-passage HFF expressing p53DN, as it is the case for replicative senescence of normal fibroblasts (Stein et al, 1999).
The similar functional consequences of disrupting Chk2 or p53 function observed here provide a plausible explanation for the finding of CHK2 gene mutation as an apparent alternative to p53 mutation in the Li–Fraumeni inherited cancer predisposition syndrome (Bell et al, 1999).
Despite the striking similarities, however, the smaller extent of lifespan extension conferred by Chk2DN compared to p53DN raises the possibility that the absence of Chk2 can be compensated by activation of other kinases capable of activating the p53 pathway. These include ATM, which can phosphorylate p53 directly (on serine 15) in response to DNA damage, as well as Chk2 (on threonine 18 or serine 20) (Bartek et al, 2001). Indeed, we have previously shown that p53 is phosphorylated at several sites including not only threonine 18 but also serine 15 in senescent fibroblasts (Webley et al, 2000).
Senescent HFF cultures accumulate γ-H2AX, a marker of DNA DSBs. We cannot exclude the involvement of nontelomeric chromosomal breaks in this response because chromosome fusion and breakage events following critical telomere shortening occur at senescence, as evidenced by a relatively high incidence of di-centric chromosomes in ageing cell populations (Benn, 1976). Furthermore, as telomere erosion continues in cells expressing E6/E7, these secondary events likely trigger the more conventional DNA DSBs damage response. However, our ChIPs data strongly suggest that an additional, if not the major, source of the damage signal in cells undergoing telomere erosion is the shortened telomeres themselves.
It is still not clear how telomere erosion leads to a state that resembles DNA damage. Estimates of the number and critical length of shortened telomeres needed to trigger senescence vary widely (Baird et al, 2003), and it appears that telomere structure, not length per se, is the prerequisite for normal telomere function (Karlseder et al, 2002). The single-stranded 3′ telomeric overhang is normally sequestered within a protective T-loop structure, which is believed to mask telomeres from being recognised as classical DNA breaks (Griffith et al, 1999). Destabilisation of the T-loop and ensuing deprotection of chromosome ends in cells lacking TRF2 lead to the telomeric recruitment of known DNA damage proteins (Oh et al, 2003; Takai et al, 2003), and a cell cycle arrest with all the hallmarks of senescence (van Steensel et al, 1998). Hence, exposure of this 3′ overhang and/or its subsequent processing to a DSB may be the key consequence of critical telomere shortening at replicative senescence (Stewart et al, 2003). Consistent with this, overexpression of TRF2 delays the onset of senescence, presumably by increasing the stability of the loop structure (Karlseder et al, 2002).
Other studies published while this work was under revision (d'Adda di Fagagna et al, 2003; Sedelnikova et al, 2004) have described an accumulation of γ-H2AX in senescent fibroblasts with a telomeric (d'Adda di Fagagna et al, 2003) and a nontelomeric (Sedelnikova et al, 2004) origin for the majority of the γ-H2AX foci. Failure to detect telomeric accumulation in the latter study may have been due to the limitation of the in situ telomere labelling technique used in detecting very short telomeres.
On the basis of present and previously published data (Karlseder et al, 1999; Takai et al, 2003), we propose a model (Figure 7) in which chromosomal breaks and dysfunctional telomere elicit an ATM-dependent and -independent phosphorylation of H2AX and Chk2. Activated Chk2 would in turn phosphorylate p53, thereby making an essential contribution to its activation, and hence induction of the cell cycle inhibitor p21.
Figure 7.
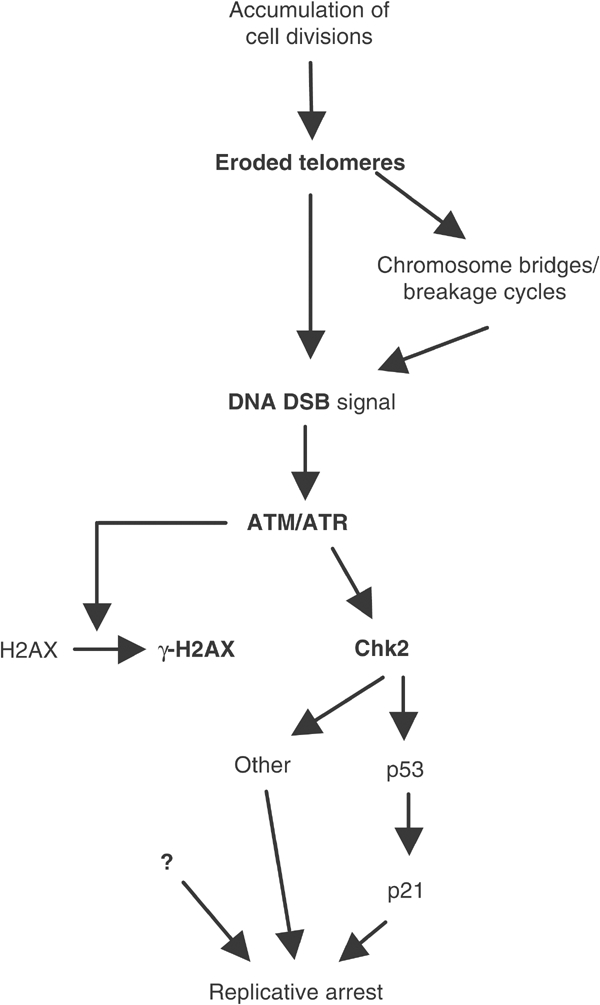
A model connecting telomere damage signalling and senescence-associated replicative arrest. As a consequence of cumulative cell divisions, a DNA DSB signal is generated either directly via exposure of critically shortened telomere or indirectly via chromosomal breaks, which may occur as a consequence of chromosome fusion and breakage following critical telomere shortening. These events elicit an ATM-dependent and -independent phosphorylation of targets including H2AX and Chk2. Activated Chk2 could further propagate the signal via a downstream substrate such as p53, which would in turn activate the transcription of the cell cycle inhibitor p21, leading to replicative arrest. Other intermediates and co-factors in the signalling pathway from the senescence-associated DNA damage to p53 activation are undoubtedly necessary.
The functional significance of senescence-induced H2AX phosphorylation remains undetermined, although a likely possibility is that upon relocalising to sites of DNA damage H2AX phosphorylation is involved in the recruitment of DNA damage signalling factors (Rogakou et al, 1999).
Our data add further support to the postulate that cells have evolved a DNA damage-like response in response to loss of telomere function to preserve the integrity of the genome. This is consistent with the long-standing observation that human fibroblasts undergo sustained growth arrest following γ-irradiation and display a virtually indistinguishable phenotype from senescent fibroblasts (Di Leonardo et al, 1994), and with the evidence that genotoxic agents and telomere dysfunction elicit additive or synergistic effects (Goytisolo et al, 2000; Wong et al, 2000). The latter observations may prove to have important implications for combined use of telomerase inhibitors with conventional chemotherapy for cancer treatment.
Materials and methods
Cell culture and growth arrest analysis
Normal HFF (kindly provided by Dr Jacques Piette, Montpellier) and IMR90 lung fibroblasts (purchased from ATCC) were cultured as described (Gire and Wynford-Thomas, 1998; Stein et al, 1999). Senescence, as defined by the criteria described previously (Gire and Wynford-Thomas, 1998), occurred at an estimated PD of 85–87 for HFF and 69–71 for IMR90 cells. Crisis was defined as the period when cultures could no longer be passaged and exhibited widespread cell death, and occurs at an estimated PD of 133–135 for HFF and 111–115 for IMR90. hTERT-infected cells were grown for at least PD100 beyond the lifespan of the parental control-infected cells to ensure that they were immortalised.
BrdU incorporation assays were performed as described (Gire and Wynford-Thomas, 1998). The senescent phenotype was scored by determining the percentage of population exhibiting an SA-βgal activity as described (Dimri et al, 1995). At least 350 cells were counted for each sample; each experiment was performed at least twice.
Cells were treated with bleomycin at 10 μg/ml for 10 h.
Retroviral infection
The retroviral vectors used were: pBabe-p53V143A (Bond et al, 1994) and HPV16-E6/E7 genes in pLXSN, packaged in PA317 cells (a kind gift of Dr Denise Galloway, Seattle). The pLPC-hTERT was a kind gift from Geron Corporation. pLXSN was used to clone dominant-negative Chk2 from pcDNA3-D347A-Chk2 (a kind gift of Dr Jiri Bartek, Copenhagen).
Retroviral gene transfer experiments have been described (Bond et al, 1994). Cells infected with a retroviral vector carrying only the drug resistance genes were used as controls. Pools of infected cell were selected in medium containing 1.5 μg/ml puromycin for pLPC and pBabe-based vectors or 400 μg/ml G418 for pLXSN-derived vectors. A PD of 0 was arbitrarily given to the first confluent dish under selection. Cumulative PDs per passage were calculated as log 2 (number of cells at the time of subculture/number of cells plated).
Near-senescent cultures of HFF (PD71–73) were infected with retroviral vector expressing pBabe-p53V143A or pLXSN-Chk2D347A or neo-only with an equivalent titre. The infection efficiency was between 15 and 25%, as determined in parallel infection of near-senescent cultures using viruses expressing green fluorescent protein. PD of cultures stably expressing p53DN were calculated from cumulative cell number at each passage. PD of cultures expressing Chk2DN was determined by serial cell counting by microscopic examination of individual colonies of cultures seeded at clonal density (as described in Bond et al, 1994).
siRNA transfection
The 21-nt base-pair siRNA duplexes were purchased from Dharmacon (Lafayette Co). The sequence of the Chk2 oligonucleotides was: 5′-GAACCUGAGGAGCCUACCCdTdT-3′ and 5′-GGGUAGGCUCCUCAGGUUCdTdT-3′; the control directed against the pBS cloning vector was 5′-GACCCGCGCCGAGGUGAAGUU-3′ and 5′-CUUCACCUCGGCGCGGGUCUU-3′. Fibroblasts were plated in a six-well plate 24 h prior to transfection at a concentration of 2 × 105 cells per well. Cells were transfected three times, 72 h apart, using Oligofectamine according to the manufacturer's instructions (Invitrogen, Carlsbad, CA): 7 μl of 20 μM siRNA duplex was mixed with 200 μl of Opti-MEM (Invitrogen), while in a second tube 7 μl of Oligofectamine was mixed with 54 μl of Opti-MEM. The two mixtures above were combined, gently mixed and added to the cells. Cells were harvested 7 days after the first transfection.
Cell extracts, Western blotting and immunoprecipitation analysis
Cells were lysed and analysed by immunoblotting as described previously (Stein et al, 1999). Amido-black staining of the membrane confirmed equal loading of the samples. The primary antibodies used were: Chk2 phosphorylated at Thr68 (described in Lukas et al (2003); lot 2), Chk2 phosphorylated at Thr387 (described in Lee and Chung (2001); lot 1), Chk2 phosphorylated at Thr26/Ser28 (described in Xu et al (2002); a kind gift of Vincent Bussmann), all from Cell Signaling Technology; Chk2 (sc-9064), p21 (sc-397) and p53 (sc-26/clone DO1) from Santa-Cruz; Chk2 (clone 7) from Upstate Biotechnology, cyclin A from Novocastra and Rb (14001A) from Pharmingen. To immunoprecipitate Chk2, 200 μg of whole-cell lysates was incubated with 1.5 μg of anti-Chk2 antibody overnight at 4°C. Gel electrophoresis, protein transfer and signal detection were performed as described (Stein et al, 1999).
Chromatin immunoprecipitations
Cells were harvested, crosslinked in 1% formaldehyde for 1 h at room temperature, washed with PBS and lysed in 1% SDS, 10 mM EDTA, 50 mM Tris–HCl, pH 8.0, at a density of 107 cells/ml. Immunoprecipitations of crosslinked chromatin were carried out as described in Loayza and De Lange (2003), with the following antibodies: rabbit anti-γ-H2AX, mouse anti-γ-H2AX (clone S139), all from Upstate Biotechnology, and rabbit polyclonal anti-Chk2 (sc-9064). Reversal of crosslinks and DNA purification were performed according to Loayza and De Lange (2003), and the input corresponding to one-quarter of the amount of lysates used for immunoprecipitation was treated alongside the immunoprecipitation samples at this step. The DNA precipitate was dissolved in 100 μl of water, denatured in 0.4 N NaOH/25 mM EDTA and dot blotted on Hybond membranes. The DNA was fixed on the membrane and hybridised at 46°C overnight in SSPE/1% SDS buffer with either a terminal transferase-labelled telomeric DNA or an Alu repeat probe (Loayza and De Lange, 2003). Filters were washed twice at 46°C with 2 × SSPE/0.01% SDS, and twice with 0.2 × SSPE, then exposed to PhosphorImager and quantitated using ImageQuant software. ChIPs yield was calculated as the percentage of telomeric association for each immunoprecipitation after normalisation of data to the corresponding ratio of the Alu probe (to measure nonspecific DNA binding). Therefore, values take into account reductions in telomere length.
Immunofluorescence labelling
Cells were fixed in 3.7% formaldehyde and indirect immunofluorescence was carried out as described (Stein et al, 1999). Mouse anti-γ-H2AX antibody (1/500) and goat anti-mouse antibody conjugated with Texas red (Molecular Probes, Eugene, OR) were used. DNA was counterstained with Hoescht dye. Images were captured by using a DHRB Leica fluorescence microscope equipped with a MicroMax 1300Y/HS black and white CCD camera (Roper) driven by software Metamorph (Universal Imaging Corporation). Foci were scored by eye at a magnification of × 630 and at least 100 nuclei per condition were examined.
Acknowledgments
We are grateful to J Piette, D Galloway, J Bartek and the Geron Coorporation for providing reagents. We thank P Travo Head of our Integrated Imaging Facility. VG thanks W Edwards for his support, and members of the CRBM and IGMM institutes for ideas, discussion and help. We thank E Fabbrizzio, J Pueschberty and E Brun for advice on ChIP experiments. This work was supported mainly by funding from the Association pour la Recherche sur le Cancer (ARC) and Ligue Nationale contre le Cancer. VG is the recipient of a fellowship from ARC and Fondation de France.
References
- AS I, Greider CW (2003) Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol Biol Cell 14: 987–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird DM, Rowson J, Wynford-Thomas D, Kipling D (2003) Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat Genet 33: 203–207 [DOI] [PubMed] [Google Scholar]
- Bartek J, Falck J, Lukas J (2001) CHK2 kinase—a busy messenger. Nat Rev Mol Cell Biol 2: 877–886 [DOI] [PubMed] [Google Scholar]
- Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA (1999) Heterozygous germ line hCHK2 mutations in Li–Fraumeni syndrome. Science 286: 2528–2531 [DOI] [PubMed] [Google Scholar]
- Benn PA (1976) Specific chromosome aberrations in senescent fibroblast cell lines derived from human embryos. Am J Hum Genet 28: 465–473 [PMC free article] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279: 349–352 [DOI] [PubMed] [Google Scholar]
- Bond JA, Wyllie FS, Wynford TD (1994) Escape from senescence in human diploid fibroblasts induced directly by mutant p53. Oncogene 9: 1885–1889 [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Appel M, Halazonetis TD (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev 14: 278–288 [PMC free article] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA (1999) p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97: 527–538 [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194–198 [DOI] [PubMed] [Google Scholar]
- de Lange T (2002) Protection of mammalian telomeres. Oncogene 21: 532–540 [DOI] [PubMed] [Google Scholar]
- Di Leonardo A, Linke SP, Clarkin K, Wahl GM (1994) DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev 8: 2540–2551 [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke H, Campisi J (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92: 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayton S, Peters G (2002) Immortalisation and transformation revisited. Curr Opin Genet Dev 12: 98–104 [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498 [DOI] [PubMed] [Google Scholar]
- Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J (2001) The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410: 842–847 [DOI] [PubMed] [Google Scholar]
- Gire V, Wynford-Thomas D (1998) Reinitiation of DNA synthesis and cell division in senescent human fibroblasts by microinjection of anti-p53 antibodies. Mol Cell Biol 18: 1611–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goytisolo FA, Samper E, Martin-Caballero J, Finnon P, Herrera E, Flores JM, Bouffler SD, Blasco MA (2000) Short telomeres result in organismal hypersensitivity to ionizing radiation in mammals. J Exp Med 192: 1625–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T (1999) Mammalian telomeres end in a large duplex loop. Cell 97: 503–514 [DOI] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345: 458–460 [DOI] [PubMed] [Google Scholar]
- Hemann MT, Strong MA, Hao LY, Greider CW (2001) The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 107: 67–77 [DOI] [PubMed] [Google Scholar]
- Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW (2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287: 1824–1827 [DOI] [PubMed] [Google Scholar]
- Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T (1999) p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 283: 1321–1325 [DOI] [PubMed] [Google Scholar]
- Karlseder J, Smogorzewska A, de Lange T (2002) Senescence induced by altered telomere state, not telomere loss. Science 295: 2446–2449 [DOI] [PubMed] [Google Scholar]
- Lee CH, Chung JH (2001) The hCds1 (Chk2)-FHA domain is essential for a chain of phosphorylation events on hCds1 that is induced by ionizing radiation. J Biol Chem 276: 30537–30541 [DOI] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner II JW, Greider CW, DePinho RA (1998) Essential role of mouse telomerase in highly proliferative organs. Nature 392: 569–574 [DOI] [PubMed] [Google Scholar]
- Lee JS, Collins KM, Brown AL, Lee CH, Chung JH (2000) hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 404: 201–204 [DOI] [PubMed] [Google Scholar]
- Loayza D, De Lange T (2003) POT1 as a terminal transducer of TRF1 telomere length control. Nature 424: 1013–1018 [DOI] [PubMed] [Google Scholar]
- Lukas C, Falck J, Bartkova J, Bartek J, Lukas J (2003) Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol 5: 255–260 [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282: 1893–1897 [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ (2000) Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci USA 97: 10389–10394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchionna R, Chen XB, Blasina A, McGowan CH (2000) Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat Cell Biol 2: 762–765 [DOI] [PubMed] [Google Scholar]
- Metcalfe JA, Parkhill J, Campbell L, Stacey M, Biggs P, Byrd PJ, Taylor AM (1996) Accelerated telomere shortening in ataxia telangiectasia. Nat Genet 13: 350–353 [DOI] [PubMed] [Google Scholar]
- Oh H, Wang SC, Prahash A, Sano M, Moravec CS, Taffet GE, Michael LH, Youker KA, Entman ML, Schneider MD (2003) Telomere attrition and Chk2 activation in human heart failure. Proc Natl Acad Sci USA 100: 5378–5383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 146: 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC (2004) Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol 6: 168–170 [DOI] [PubMed] [Google Scholar]
- Stein GH, Drullinger LF, Soulard A, Dulic V (1999) Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol 19: 2109–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Ben-Porath I, Carey VJ, O'Connor BF, Hahn WC, Weinberg RA (2003) Erosion of the telomeric single-strand overhang at replicative senescence. Nat Genet 33: 492–496 [DOI] [PubMed] [Google Scholar]
- Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, Anderson CW, Appella E, Nakanishi M, Suzuki H, Nagashima K, Sawa H, Ikeda K, Motoyama N (2002) Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J 21: 5195–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T (2003) DNA damage foci at dysfunctional telomeres. Curr Biol 13: 1549–1556 [DOI] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A, de Lange T (1998) TRF2 protects human telomeres from end-to-end fusions. Cell 92: 401–413 [DOI] [PubMed] [Google Scholar]
- Vaziri H, West MD, Allsopp RC, Davison TS, Wu YS, Arrowsmith CH, Poirier GG, Benchimol S (1997) ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J 16: 6018–6033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Wu X, Chen J (2001) Threonine 68 of Chk2 is phosphorylated at sites of DNA strand breaks. J Biol Chem 276: 47755–47758 [DOI] [PubMed] [Google Scholar]
- Webley K, Bond JA, Jones CJ, Blaydes JP, Craig A, Hupp T, Wynford-Thomas D (2000) Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Mol Cell Biol 20: 2803–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KK, Chang S, Weiler SR, Ganesan S, Chaudhuri J, Zhu C, Artandi SE, Rudolph KL, Gottlieb GJ, Chin L, Alt FW, DePinho RA (2000) Telomere dysfunction impairs DNA repair and enhances sensitivity to ionizing radiation. Nat Genet 26: 85–88 [DOI] [PubMed] [Google Scholar]
- Wong KK, Maser RS, Bachoo RM, Menon J, Carrasco DR, Gu Y, Alt FW, DePinho RA (2003) Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature 421: 643–648 [DOI] [PubMed] [Google Scholar]
- Xu X, Tsvetkov LM, Stern DF (2002) Chk2 activation and phosphorylation-dependent oligomerization. Mol Cell Biol 22: 4419–4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Wang H, Bishop JM, Blackburn EH (1999) Telomerase extends the lifespan of virus-transformed human cells without net telomere lengthening. Proc Natl Acad Sci USA 96: 3723–3728 [DOI] [PMC free article] [PubMed] [Google Scholar]