

Abstract
Systemic lupus erythematosus (SLE) is a prototype systemic autoimmune disease that results from a break in immune tolerance to self-antigens, leading to multi-organ destruction. Autoantibody deposition and inflammatory cell infiltration in target organs such as kidneys and brain lead to complications of this disease. Dysregulation of cellular and humoral immune response elements, along with organ-defined molecular aberrations, form the basis of SLE pathogenesis. Aberrant T lymphocyte activation due to signaling abnormalities, linked to defective gene transcription and altered cytokine production, are important contributors to SLE pathophysiology. A better understanding of signaling and gene regulation defects in SLE T cells will lead to the identification of specific novel molecular targets and predictive biomarkers for therapy.
Introduction
Systemic lupus erythematosus (SLE) is a chronic debilitating autoimmune disease of unknown etiology that mainly afflicts women in the childbearing years (1, 2). The disease follows an unpredictable course of flares and remission with no predictive biomarkers for either phase. Current steroid-based immunosuppressive therapies are not specific and have undesirable adverse effects, rendering patients immunocompromised and susceptible to infections. SLE pathophysiology involves abnormal immune cell activation, leading to autoantibody and immune complex deposition in target organs such as the skin, joints, kidneys, and brain with potentially fatal complications.
There is increasing interest in the role of T cells in the pathophysiology of the disease, as they display an interesting phenotype. T cells have the ability to provide excessive help to B cells, but fail to raise proper cytotoxic responses to fend off infections. At the cytokine level, they fail to produce sufficient amounts of IL-2, although they produce increased amounts of IL-17 and IL-10. An understanding of the molecular events that occur inside the SLE T cells following antigen (autoantigen) engagement has been considered mandatory to resolving their aberrant function. It is also expected that correction of abnormal signaling molecules should correct T cell function and limit subsequent pathology that leads to clinical manifestations.
In this Review, cell signaling and gene regulation abnormalities in T cells from patients with SLE and lupus-prone mice will be presented with emphasis on how they contribute to aberrant T cell function and how they can be explored as therapeutic targets.
Altered response to antigen/autoantigen
T cells recognize antigen through the TCR in conjunction with the CD3-defined complex of transmembrane proteins (ε, δ, γ, and ζ) to instigate a signaling process, which, along with input from coreceptors and receptors for cytokines, dictates effector cell function. In SLE T cells, the TCR/CD3 complex is “rewired” whereby the CD3ζ chain is reduced and replaced by the homologous Fc receptor common g subunit (FcRγ) chain (ref. 3 and Figure 1). Unlike CD3ζ, which recruits ζ-associated protein kinase 70 kDa (ZAP70) to relay the signal, FcRγ recruits the spleen tyrosine kinase (Syk). Because FcRγ/Syk transfers a manyfold stronger signal than CD3ζ/ZAP70, the SLE T cell exhibits early and heightened signaling events and probably responds sufficiently when it meets low-avidity autoantigens to which a normal T cell would not respond. Pharmacologic inhibition of Syk in lupus-prone MRL/lpr mice results in significant reduction of autoimmunity and organ (kidney and skin) pathology even if treatment is initiated after the onset of the disease. Silencing or pharmacologic inhibition of Syk in T cells from patients with SLE corrects aberrant signaling (4), and replacement of CD3ζ normalizes IL-2 production (5).
Figure 1. Altered TCR/CD3 complex and lipid raft composition in SLE T cells.
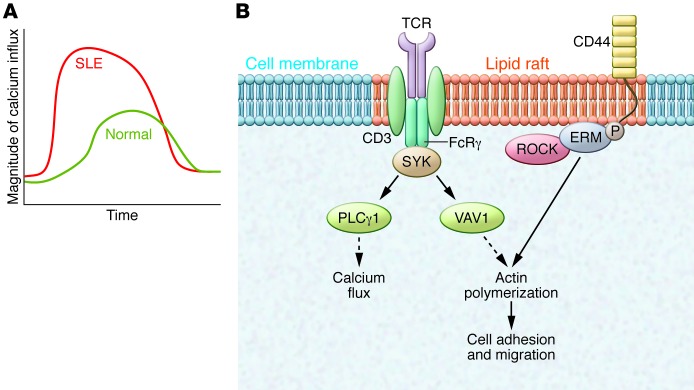
(A) Engagement of the CD3/TCR complex in SLE T cells leads to a heightened and earlier proximal signaling response characterized by increased free intracytoplasmic calcium concentration and cytosolic protein tyrosine phosphorylation. The graph shows the magnitude and kinetics of intracellular calcium flux in normal and SLE T cells. (B) Lipid rafts in SLE T cells are preclustered and display altered arrangement of signaling molecules. The TCR/CD3 complex undergoes rewiring to express FcRγ and Syk kinase in place of CD3ζ and ZAP70, respectively, sending a stronger downstream signal and increasing intracellular calcium flux. Signaling through VAV1 leads to actin polymerization and cellular migration. SLE T cells express higher levels of the surface adhesion molecule CD44. Upon activation of CD44, ROCK phosphorylates the ERM proteins, thereby inducing actin polymerization to increase adhesion and migration.
Exploration of mechanisms that account for the decreased expression of CD3ζ in SLE T cells has proved informative, because several pathways can be targeted to increase CD3ζ levels and correct T cell function. For example, transcription (6), mRNA stability (7), alternative splicing (8), proteasome degradation (9), caspase cleavage (10), and mTOR-dependent degradation (11) can all be targeted to treat SLE through the correction of excessive early signaling events. The decrease in CD3ζ is obviously a downstream event, but it cannot be ignored because it may contribute to the ability of T cells to home to tissues inappropriately and cause inflammation. This is inferred by observations in CD3ζ-deficient mice in which T cells accumulate in multiple organs, particularly when challenged with alloantigens or polyinosinic:polycytidylic acid (poly I:C) (12).
T cell activation, differentiation, function, and death are regulated by reactive oxygen intermediates (ROIs) and ATP synthesis. Mitochondrial transmembrane potential is a critical regulator of ROI and ATP generation. Aberrant, persistent mitochondrial hyperpolarization, increased ROI production (or reduced glutathione levels), and ATP depletion in SLE T cells mediate spontaneous apoptosis and decreased activation-induced apoptosis. In addition, oxidative modifications of self-antigens can trigger autoimmunity. Correlations between these modified serum proteins and disease activity and organ damage have been observed in SLE (13). The serine/threonine kinase mTOR is a sensor of mitochondrial membrane potential that activates numerous downstream substrates and is aberrantly increased in T cells from patients with SLE (11). Therapeutic targeting of mTOR with rapamycin in SLE patients leads to normalized T cell activation, calcium fluxing, and clinical improvement (14). Inhibition of mTOR with epigallocatechin-3-gallate (EGCG) attenuates inflammation in mesangial cells from lupus-prone MRL/lpr mice (15). Importantly, N-acetylcysteine reduced disease activity in SLE patients in a pilot double-blind, placebo-controlled trial (16), emphasizing that the study of T cell biochemistry substantially benefits the treatment of SLE patients.
Lipid rafts are sphingolipid/cholesterol/GM1-rich regions within the cell membrane that are scattered throughout the cell surface and contain the TCR/CD3 complex and associated signaling molecules. Whereas lipid rafts are uniformly distributed across the cell membrane of T cells from healthy individuals, these rafts appear to be preclustered and aggregated in SLE T cells, implying that SLE T cells are in an activated state (17). Dissolution of aggregated lipid rafts in SLE T cells ex vivo corrects TCR/CD3-mediated signaling (18, 19), and it has been proposed that statins, which inhibit 3-hydroxy-3-methygluteryl CoA reductase, may dissolve lipid rafts and could potentially be repurposed to treat SLE, as would drugs that inhibit glycosphingolipid metabolism (20). It is premature to link the dyslipidemia that SLE patients frequently experience (21) to the formation of aggregated lipid rafts on the surface of T cells. In support of these propositions, dissolution of lipid rafts delays disease onset in MRL/lpr mice, while acceleration of lipid raft formation accelerates disease manifestation (22).
Inappropriate lymphocyte homing to tissues
T cells are present in inflamed tissues including the kidneys and the skin in SLE patients, aided by their increased ability to adhere to tissue elements. Their presence in the tubulo-interstitial space in the kidney tissue of patients with lupus nephritis has been linked to decreased preservation of renal function (1), and therefore T cells contribute to the inflammatory process in the tissue damage. As we discuss below (23), T cells produce IL-17, among other cytokines. T cells from lupus patients express increased amounts of CD44 (24), as do T cells from CD3ζ-deficient mice (12). CD44+ T cells, specifically those expressing the v3 and v6 isoforms, are present in the kidneys of patients with lupus nephritis (25). CD44 binds better to its ligand hyaluronic acid if linked to the phosphorylated form of ezrin/radixin/moeisin (ERM), which is phosphorylated by the Rho-associated protein kinase (ROCK) in both humans (26) and mice (27). Pharmacologic inhibition of ROCK in MRL/lpr mice has been clearly shown to limit both autoimmunity and renal disease (27), and therefore, ROCK inhibitors should be investigated in clinical trials.
Excessive help to B cells
It has been long established that T cells provide help to B cells to produce anti-dsDNA antibodies. Both CD4+ and CD3+CD4–CD8– (double-negative) T cells, which are expanded in SLE patients (28), can help B cells, and this has provided the rationale to treat SLE patients with biologics designed to disrupt this interaction. The CD40 ligand/CD40 (CD40L/CD40) duo is among the dominant co-stimulatory pairs of molecules, and CD40L is increased on the surface of SLE T cells (29, 30). The increased expression of CD40L on the cell membrane of T cells is enabled by increased transcription of CD40L, which is caused by hypomethylation of the CD40L gene and increased binding of nuclear factor of activated T cells (NFAT) to its promoter (31, 32). NFAT is activated by the tyrosine phosphatase calcineurin, the activity of which is facilitated by increased intracytoplasmic calcium levels following engagement of the TCR/CD3 complex. Interestingly, cyclosporin, which inhibits calcineurin, has not been a focus in SLE treatment because of its nephrotoxicity. However, dipyridamole, a drug that prevents calcineurin-mediated dephosphorylation of NFAT, limits the expression of CD40L in T cells from patients with SLE and lupus-prone mice, prevents kidney and skin pathology (33), and can be repurposed as an adjuvant in the treatment of SLE. Thus, dipyridamole appears to perform an “inside” job in reducing CD40L, obviating the use of CD40L-blocking biologics, which have been linked to thrombotic events when used in patients with SLE (34).
Inducible T cell co-stimulator (ICOS) is another molecule that provides co-stimulatory signals upon interaction with its ligand ICOSL and is an essential co-stimulatory receptor for follicular Th (Tfh) cells. Using lupus-prone, ICOS-deficient mice (35) or mice overexpressing ICOS (36), it has been shown that ICOS is necessary for extrafollicular B cell differentiation, IL-21 production, and CD4 helper function. Increased numbers of circulating Tfh cells have been reported in patients with active SLE (37). The ICOS signaling pathway requires the PI3K p110δ, and Tfh cell development requires the presence of p110δ.
Transgenic overexpression of PI3K or expression of an activity-increasing p85 mutant leads to an autoimmune phenotype in mice (38). Deletion of PI3Kδ impairs antigen receptor signaling in both T cells and B cells (39), and PI3Kγ deficiency impairs T cell activation and migration of macrophages and neutrophils (40). Deletion of PI3Kγ in lupus-prone mice ameliorates disease (40), and pharmacologic inhibition of PI3Kγ with AS605240 leads to a significant decrease in glomerulonephritis and prolonged survival (41). Interestingly, the levels and activity of PI3Kδ were increased in PBMCs from patients with SLE and conferred resistance to activation-induced T cell death, which is a known mechanism for the persistence of autoreactive cells (42). GS9029, a highly potent and selective pharmacologic inhibitor of PI3Kδ, was used to treat MRL/lpr mice, resulting in limited macrophage infiltration into kidneys, significantly reduced pathology, and prolonged survival in mice, even when treatment was initiated after disease onset (43). PI3Kδ inhibitors are currently being tested in clinical trials for hematologic malignancies, which will generate sufficient information on its potential side effects prior to trials in SLE patients. Finally, T cell–specific AKT transgenic mice exhibit early mortality, high levels of autoantibodies and glomerulonephritis, and central and peripheral resistance to apoptosis similar to Bcl-xL T cell transgenic mice (44, 45), suggesting that AKT is a therapeutic target. In sum, it appears the PI3K/AKT pathway participates in the aberrant function of the SLE T cell and offers a number of potential therapeutic targets.
Limited production of IL-2
IL-2 is an important cytokine responsible not only for normal T cell activation and proliferation but also for activation-induced cell death, which is required for the deletion of autoreactive cells. Additionally, IL-2 is critical for cell-mediated immunity against infectious agents; infections account for the majority of deaths among SLE patients. Finally, Tregs depend on IL-2 for their survival and maintenance (46). IL2 mRNA is decreased in T cells from SLE patients and several lupus-prone mouse strains (47). Since its stability is normal, the levels of IL-2 are determined at the level of transcription (47).
IL2 transcription is complex, and the IL2 promoter region includes binding elements for a large number of transcription factors including NF-κB, NFAT, activator protein 1 (AP1), OCT1, cAMP response element binding protein (CREB), and cAMP response element modulator (CREM).
Both NF-κB and AP1 are decreased in SLE T cells and contribute to decreased IL2 transcription (48, 49). It appears that an imbalance between the transcription factors CREB and CREMα is also important in determining the rate of IL2 transcription. Both transcription factors recognize a CRE site at the –180 bp position within the core IL2 promoter. Phosphorylation leads to the activation of these proteins, and the relative amounts of p-CREB versus p-CREM determine binding to the promoter and the consequent activation or suppression of IL-2 expression, as p-CREB activates while p-CREM represses IL2 transcription. In SLE T cells, decreased PKA activity leads to dephosphorylation of CREB (50) and shifts the balance toward increased p-CREM levels. More importantly, increased levels and activity of the serine threonine protein phosphatase 2A (PP2A) account for the dephosphorylation of p-CREB (Figure 2), which allows the repressor CREMα to bind to the –180 bp site of the promoter. In addition, calcium calmodulin kinase IV (CAMKIV), which is increased in the nuclei of SLE T cells, phosphorylates CREM to increase p-CREM levels and therefore suppresses IL2 transcription (51). More recently it was shown that binding of CREMα occurs in a number of sites across the IL2 locus, where it recruits histone deacetylase 1 (HDAC1), DNA methyltransferase 1 (DNMT1), and G9α, which act together to close the locus and limit the production of IL2 mRNA (52). Recent work has suggested the role of serine/arginine-rich splicing factor 1 (SRSF1), a splicing regulator whose expression was reduced in T cells from several patients with SLE, more so in patients with worse disease. Importantly, forced expression of SRSF1 into SLE T cells rescues IL-2 production through a transcription-related mechanism (53).
Figure 2. Role of serine threonine phosphatase PP2A in SLE T cell pathophysiology.
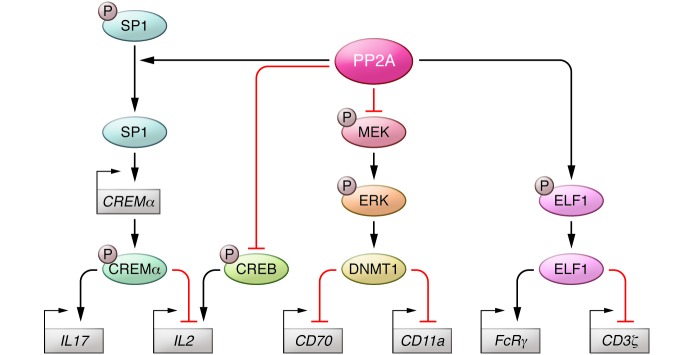
PP2A dephosphorylates and activates SP1 to increase expression of CREMα, which enhances expression of IL-17 and represses expression of IL-2. IL-2 expression is increased by binding of phospho-CREB, which is also dephosphorylated/deactivated by PP2A. Phospho-CREB and phospho-CREMα both bind to the –180 bp position within the IL2 promoter; thus, the ratio of these proteins determine overall IL-2 expression. PP2A dephosphorylates and activates the transcriptional enhancer ELF1, which increase expression of CD3ζ and decreases expression of FcRγ.PP2A dephosphorylates MEK1/2 to block activity of the methyltransferase DNMT1, leading to hypomethylation and increased expression of CD70 and CD11a.
Increased production of IL-17
Recently IL-17 has received much attention as an important contributor to inflammation and autoimmunity (54). Increased amounts of IL-17 cytokine as well as an increased proportion of IL-17–producing CD4+ T cells have been observed in the serum and peripheral blood of SLE patients (55). Increased numbers of IL-17–producing, double-negative T cells circulate in the peripheral blood of patients with SLE and in the kidneys of patients with lupus nephritis (23).
Under appropriate priming conditions, especially in the presence of inflammatory signals such as IL-6, IL-23, IL-21, and TGF-β, CD4 T cells undergo specific differentiation to become IL-17 producers, also known as Th17 cells (56, 57). Transcriptional activation of IL17 is mediated by the retinoid-related orphan receptor γt (RORγt) and RORα proteins, of which RORγt is essential for IL-17 production and is expressed only in Th17-differentiated cells (58). While IL-21 is important for the induction of IL-17, maintenance of IL-17 production requires IL-23. Genetic deficiency of the IL-23 receptor in lupus-prone C57BL/6/lpr mice leads to reduced frequency of double-negative T cells, reduced IL-17 production, and amelioration of kidney disease (59). IL-17–deficient mice were protected from the development of pristane-induced lupus, autoantibody production, and glomerulonephritis (60). IL-6, IL-21, and IL-23 activate STAT3, which can directly bind and activate IL17 and IL21 (61). STAT3 expression in SLE T cells is high and contributes to the increased chemokine-directed migratory capacity of these inflammatory cells (62). Besides its proinflammatory role, IL-17 also activates anti-dsDNA antibody production, as evidenced by high antibody production in PBMCs from patients with lupus nephritis (63). Lastly, epigenetic modifications invoked by STAT3 appear to account for the increased production of IL-10 in patients with SLE (64, 65).
Mice overexpressing CREMα in T cells predictably produce less IL-2 (66) and interestingly produce more IL-17. CREMα was found to bind to the IL17 promoter and to enhance its activity in vitro. In addition to the promoter, CREMα binds to additional sites across the IL17 locus but, unlike the IL2 locus, keeps the IL17 locus open because it fails to recruit HDAC1, G9α, and DNMT1 (67). Thus it appears that a single transcription factor, CREMα, accounts for both increased IL-17 production and decreased IL-2 production (Figure 3). Furthermore, the production of CREMα is determined at the transcriptional level, mainly through the increased binding of the transcription factor SP1. The production of SP1 seems to be influenced by hormones such as estrogen (68), which provides a molecular link to the preponderance of female SLE patients. We should note that SP1 is activated after it becomes dephosphorylated by PP2A (69). IL-17 production is further controlled by interferon regulatory factor 4 (IRF4), which is phosphorylated by ROCK2, a protein that is increased in lupus-prone mice (27).
Figure 3. Epigenetic role of CREMα in the transcriptional regulation of IL2, IL17A, and CD8.
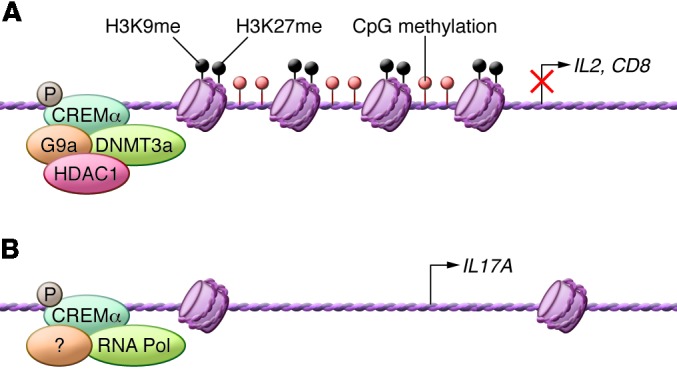
(A) Through binding to the promoters of IL2 and CD8 (and other sites throughout their loci), CREMα recruits DNMT3a, HDAC1, and H3K27, which render these loci inaccessible. (B) In contrast, binding of CREMα to the IL17A promoter does not result in the recruitment of locus-closing molecules.
Collectively, IL-17 production in SLE patients and lupus-prone mice is controlled by a number of transcription factors, epigenetic changes, and input from additional cytokines such as IL-23. This information and the fact that anti–IL-17 antibodies are useful in the treatment of psoriasis, psoriatic and rheumatoid arthritis, and uveitis (70, 71) strongly suggest that blockade of the IL-23/IL-17 axis should be evaluated in the treatment of SLE.
Aberrant distribution of T cell subsets
CD3+CD4–CD8– double-negative T cells.
As mentioned above, double-negative T cells are expanded in SLE patients and in lupus-prone mice. Because they share a number of genes with CD8+ T cells, it has been proposed that at least a percentage of them must derive from activated CD8+ T cells in the periphery (72). Loss of CD8 in activated T cells in both SLE patients and MRL/lpr mice appears to be caused by CREMα, which binds to the CD8 locus, recruits HDAC1, G9α, and DNMT1, and closes the chromatin (73). Because the expression of CREMα is regulated by S6 kinase, a canonical substrate of mTORC1 (74), it is possible that the expression of CD8 is also regulated through this pathway. In addition, there is evidence that mTORC1 is prominently activated in double-negative T cells (16) and, when blocked, limits their numbers in patients with SLE (75).
Effector T cells.
Effector CD4+ T cells are expanded in SLE patients and produce less IL-2 and more IL-17 than CD4+ T cells in healthy controls (67). It has been shown that CREMα limits the production of IL-2 and enhances the production of IL-17 in order to expand effector T cells in SLE patients (67). Thus, CREMα alone explains the aberrant numbers of potentially pathogenic T cell subsets.
Invariant NK T cells.
NK T cells are a unique T cell lineage, and those bearing an invariant TCR recognize certain glycolipid antigens in the context of CD1-d (non-classical MHC) molecules (76). Invariant NK T cells play an immunoregulatory role through their cytokine production and cytotoxicity and are implicated in the development of autoimmune disease. Reduced numbers of NKT cells are observed in the peripheral blood of patients with SLE, and NKT cell deficiency is inversely correlated with disease activity. In addition, proliferation and cytokine production are impaired (77–79).
Compromised Treg function
Tregs are responsible for the control of the immune response in the periphery, and defective function or decreased numbers contribute to the pathogenesis of several autoimmune diseases including type 1 diabetes and multiple sclerosis. The number and/or function of Tregs in the peripheral blood of patients with SLE are reportedly decreased and therefore have been considered culprits in the peripheral control of the autoimmune effector response (80). Decreased production of IL-2 has been linked to the poor numbers and function of Tregs in SLE patients. The ability of low doses of IL-2 to control hepatitis C–linked vasculitis (81) and graft-versus-host disease (82) by augmenting Treg numbers and function has indicated that therapeutic enhancement of Tregs could be used for the treatment of SLE.
We and others have been searching for abnormally expressed kinases and phosphatases in patients with SLE and lupus-prone mice that would account for defective Treg function. CaMKIV is found at increased levels in the nuclei of SLE T cells following engagement of the CD3/TCR complex (83). CaMKIV expression is increased in T cells from lupus-prone MRL/lpr mice, and its inhibition with the small drug KN-93 leads to the suppression of nephritis and skin disease (51). Genetic deletion improved survival, restored IL-2 production, curbed excessive T cell activation, and increased Treg numbers and function. Silencing CaMKIV in SLE T cells increased the expression of FOXP3 upon activation and induction with TGF-β (84). Furthermore, genetic deficiency of CaMKIV or inhibition with KN-93 decreased the frequency of IL-17–producing CD4 and double-negative T cells and ameliorated EAE and lupus-like disease in mice. At the biochemical level CaMKIV was found to act though the AKT/mTOR and CREMα pathways (85).
mTOR is present in two different complexes, mTORC1 and mTORC2, which have distinct components and downstream functions (86). mTORC1 is important for Th1 and Th17 differentiation, and mTORC2 is essential for Th2 differentiation in mice. Both complexes suppress FOXP3 expression and thereby inhibit Treg differentiation. Accordingly, inhibition of mTOR with rapamycin enhances Treg generation in vitro and in vivo (87–89). Recent work has demonstrated that compared with healthy controls, T cells from patients with SLE exhibited increased mTORC1 activity (mainly in double-negative T cells) and reduced mTORC2 activity. mTORC1 promoted a Th17 phenotype in CD4+ T cells, IL-4 production in double-negative T cells, and a reduction in Tregs (87). In a prospective clinical trial, mTORC1 blockade with rapamycin reversed the expansion of double-negative T cells, reduced IL-4 production, and significantly improved disease in patients with SLE (75). In this longitudinal study, the expansion of double-negative cells correlated with disease activity and mTORC1 activation preceded disease flares by approximately four months. These results suggest that mTOR is a driver and predictor of disease and a potential biomarker and therapeutic target for SLE.
Treg cells do not proliferate in vitro and in this state are considered anergic. Anergic Tregs display active mTOR, and blockade with rapamycin makes them proliferate and amplifies their function (90). Leptin, a molecule known to be linked to autoimmunity (91), promotes mTOR activity (92). This observation, along with the fact that leptin promotes IL-17 production by inducing RORγt expression (93), makes leptin a treatment target in lupus.
Epigenetic control of molecules important in SLE
Epigenetic events are important in activation, differentiation, and function of T cells, and dysregulation of these events can lead to altered gene expression, function, and autoimmune response (94, 95). Epigenetic changes can activate or suppress gene expression by regulating chromatin conformation and the accessibility of DNA to transcription factors and RNA polymerase. DNA CpG methylation and histone modifications are the most well-known epigenetic mechanisms (64). Hypermethylation of DNA suppresses gene expression via inactivation of chromatin, while hypomethylation of DNA leads to chromatin activation and increased gene expression. Patients treated with procainamide or hydralazine, which decrease DNA methylation, developed lupus-like disease, providing direct evidence for the role of epigenetics in the pathogenesis of autoimmunity. SLE is characterized by hypomethylation of numerous genes, including CD11A, perforin, CD70, and CD40L, resulting in their consequent aberrant overexpression and contribution to disease pathophysiology (96). A number of cytokine genes are overexpressed in CD4+ T cells in a chromatin-dependent manner including IL4, IL10, IL13, and IL17 (64). Recently it has been shown that the IL10 gene is hypomethylated in SLE T cells and allows for chromatin remodeling and p-STAT3 recruitment to its promoter to increase IL-10 expression (65). The promoter of the serine/threonine phosphatase PP2A is hypomethylated, resulting in its overexpression and aberrant activity in T cells from SLE patients (97). The expression and activity of DNMT1, which is responsible for DNA methylation, were reduced in SLE T cells, accounting for the observed global hypomethylation (98). PP2A, which is overexpressed in SLE T cells, decreases the activity of DNMT1 through dephosphorylation of ERK and MEK and consequently increases expression of the methylation-sensitive genes CD70 and CD11a (99). Growth arrest and DNA damage–induced 45a (GADD45a) induces DNA hypomethylation in CD4+ T cells in SLE patients and contributes to lupus-like autoimmunity (100). As discussed above, CREMα causes epigenetic alterations of the IL2, IL17, and IL10 loci. Study of the epigenetic control of gene expression in SLE will eventually explain how epigenetic alterations mediate SLE pathogenesis, which is not explained by genetics.
Conclusions
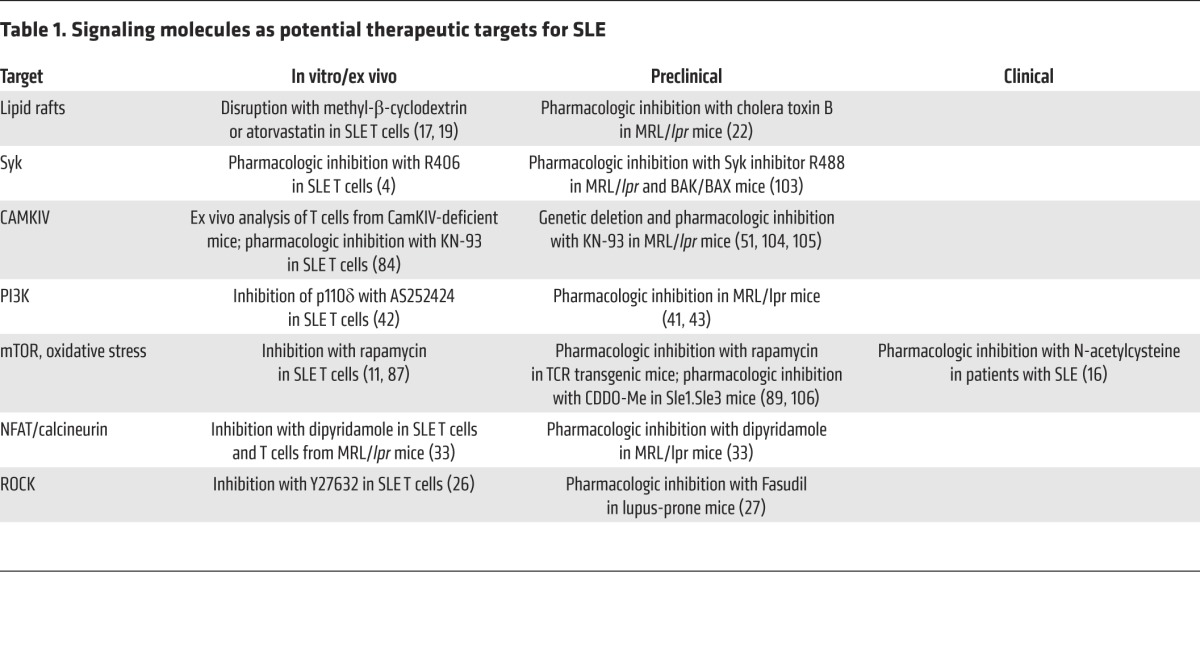
The development of novel, disease-specific, and side effect–free drugs for SLE has met unprecedented obstacles for a number of reasons. The T cell in patients with SLE presents a fascinating phenotype, which is now better understood after gaining insight into the underlying cell signaling and gene regulation. The preclustering of lipid rafts with altered composition explained why T cells are over-reactive, whereas the increased expression of CaMKIV in the nucleus explained the increased binding of CREMα to the IL2 and IL17 loci, which confers specific epigenetic changes that lead to decreased IL-2 and increased IL-17 production. Increased expression of PP2A contributes to the dephosphorylation of a number of molecules including Elf-1, SP1, MEK, and CREB and thus serves as a central node in aberrant T cell function. The concept that the two dozen or so regulatory subunits of the PP2A holoenzyme may regulate specific cell functions offers additional options for therapeutic targeting. As outlined in this Review, the ongoing efforts to understand what goes on inside the T cell have identified a number of viable therapeutic targets (Table 1) that could potentially be manipulated with small-molecule–based drugs. The use of small molecules should supersede that of biologics, as the clinical utility can be maximized by titrating dosages. Eventually, small-molecule drugs and/or gene targeting to correct the expression of molecules only in patients in whom they are abnormally expressed should be expected to meet success. Along these lines a gene expression array has been developed (101, 102) to determine the aberrant expression of signaling molecules in patients with SLE, pointing to the possibility that patients with SLE can be stratified along their molecular profiles.
Table 1. Signaling molecules as potential therapeutic targets for SLE.
Acknowledgments
The authors acknowledge funding support from NIH grants K01AR060781, R01AI42269, R37AI49954, R01AI068787, R01AI085567, and R01AR064350.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(6):2220–2227. doi:10.1172/JCI78087.
References
- 1.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 2.Moulton VR, Tsokos GC. Abnormalities of T cell signaling in systemic lupus erythematosus. Arthritis Res Ther. 2011;13(2):207. doi: 10.1186/ar3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krishnan S, Farber DL, Tsokos GC. T cell rewiring in differentiation and disease. J Immunol. 2003;171(7):3325–3331. doi: 10.4049/jimmunol.171.7.3325. [DOI] [PubMed] [Google Scholar]
- 4.Krishnan S, et al. Differential expression and molecular associations of Syk in systemic lupus erythematosus T cells. J Immunol. 2008;181(11):8145–8152. doi: 10.4049/jimmunol.181.11.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nambiar MP, et al. Reconstitution of deficient T cell receptor zeta chain restores T cell signaling and augments T cell receptor/CD3-induced interleukin-2 production in patients with systemic lupus erythematosus. Arthritis Rheum. 2003;48(7):1948–1955. doi: 10.1002/art.11072. [DOI] [PubMed] [Google Scholar]
- 6.Juang YT, Tenbrock K, Nambiar MP, Gourley MF, Tsokos GC. Defective production of functional 98-kDa form of Elf-1 is responsible for the decreased expression of TCR ζ-chain in patients with systemic lupus erythematosus. J Immunol. 2002;169(10):6048–6055. doi: 10.4049/jimmunol.169.10.6048. [DOI] [PubMed] [Google Scholar]
- 7.Moulton VR, Kyttaris VC, Juang YT, Chowdhury B, Tsokos GC. The RNA-stabilizing protein HuR regulates the expression of ζ chain of the human T cell receptor-associated CD3 complex. J Biol Chem. 2008;283(29):20037–20044. doi: 10.1074/jbc.M710434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moulton VR, Tsokos GC. Alternative splicing factor/splicing factor 2 regulates the expression of the ζ subunit of the human T cell receptor-associated CD3 complex. J Biol Chem. 2010;285(17):12490–12496. doi: 10.1074/jbc.M109.091660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nambiar MP, et al. Abnormal expression of various molecular forms and distribution of T cell receptor ζ chain in patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46(1):163–174. doi: 10.1002/1529-0131(200201)46:1<163::AID-ART10065>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 10.Krishnan S, et al. Increased caspase-3 expression and activity contribute to reduced CD3ζ expression in systemic lupus erythematosus T cells. J Immunol. 2005;175(5):3417–3423. doi: 10.4049/jimmunol.175.5.3417. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez DR, et al. Activation of mammalian target of rapamycin controls the loss of TCRζ in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol. 2009;182(4):2063–2073. doi: 10.4049/jimmunol.0803600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng GM, Beltran J, Chen C, Terhorst C, Tsokos GC. T cell CD3ζ deficiency enables multiorgan tissue inflammation. J Immunol. 2013;191(7):3563–3567. doi: 10.4049/jimmunol.1300634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol. 2013;9(11):674–686. doi: 10.1038/nrrheum.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54(9):2983–2988. doi: 10.1002/art.22085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peairs A, et al. Epigallocatechin-3-gallate (EGCG) attenuates inflammation in MRL/lpr mouse mesangial cells. Cell Mol Immunol. 2010;7(2):123–132. doi: 10.1038/cmi.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai ZW, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012;64(9):2937–2946. doi: 10.1002/art.34502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krishnan S, et al. Alterations in lipid raft composition and dynamics contribute to abnormal T cell responses in systemic lupus erythematosus. J Immunol. 2004;172(12):7821–7831. doi: 10.4049/jimmunol.172.12.7821. [DOI] [PubMed] [Google Scholar]
- 18.Deng GM, Tsokos GC. Cholera toxin B accelerates disease progression in lupus-prone mice by promoting lipid raft aggregation. J Immunol. 2008;181(6):4019–4026. doi: 10.4049/jimmunol.181.6.4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jury EC, Isenberg DA, Mauri C, Ehrenstein MR. Atorvastatin restores Lck expression and lipid raft-associated signaling in T cells from patients with systemic lupus erythematosus. J Immunol. 2006;177(10):7416–7422. doi: 10.4049/jimmunol.177.10.7416. [DOI] [PubMed] [Google Scholar]
- 20.McDonald G, et al. Normalizing glycosphingolipids restores function in CD4+ T cells from lupus patients. J Clin Invest. 2014;124(2):712–724. doi: 10.1172/JCI69571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinicato NA, da Silva Cardoso PA, Appenzeller S. Risk factors in cardiovascular disease in systemic lupus erythematosus. Curr Cardiol Rev. 2013;9(1):15–19. doi: 10.2174/157340313805076304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng GM, Tsokos GC. Cholera toxin B accelerates disease progression in lupus-prone mice by promoting lipid raft aggregation. J Immunol. 2008;181(6):4019–4026. doi: 10.4049/jimmunol.181.6.4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crispin JC, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181(12):8761–8766. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crispin JC, et al. Expression of CD44 variant isoforms CD44v3 and CD44v6 is increased on T cells from patients with systemic lupus erythematosus and is correlated with disease activity. Arthritis Rheum. 2010;62(5):1431–1437. doi: 10.1002/art.27385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen RA, et al. T cells and in situ cryoglobulin deposition in the pathogenesis of lupus nephritis. Clin Immunol. 2008;128(1):1–7. doi: 10.1016/j.clim.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, et al. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol. 2007;178(3):1938–1947. doi: 10.4049/jimmunol.178.3.1938. [DOI] [PubMed] [Google Scholar]
- 27.Biswas PS, et al. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J Clin Invest. 2010;120(9):3280–3295. doi: 10.1172/JCI42856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shivakumar S, Tsokos GC, Datta SK. T cell receptor α/β expressing double-negative (CD4–/CD8–) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J Immunol. 1989;143(1):103–112. [PubMed] [Google Scholar]
- 29.Koshy M, Berger D, Crow MK. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes. J Clin Invest. 1996;98(3):826–837. doi: 10.1172/JCI118855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desai-Mehta A, Lu L, Ramsey-Goldman R, Datta SK. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest. 1996;97(9):2063–2073. doi: 10.1172/JCI118643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kyttaris VC, Wang Y, Juang YT, Weinstein A, Tsokos GC. Increased levels of NF-ATc2 differentially regulate CD154 and IL-2 genes in T cells from patients with systemic lupus erythematosus. J Immunol. 2007;178(3):1960–1966. doi: 10.4049/jimmunol.178.3.1960. [DOI] [PubMed] [Google Scholar]
- 32.Jeffries MA, Sawalha AH. Epigenetics in systemic lupus erythematosus: leading the way for specific therapeutic agents. Int J Clin Rheumtol. 2011;6(4):423–439. doi: 10.2217/ijr.11.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kyttaris VC, Zhang Z, Kampagianni O, Tsokos GC. Calcium signaling in systemic lupus erythematosus T cells: a treatment target. Arthritis Rheum. 2011;63(7):2058–2066. doi: 10.1002/art.30353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sidiropoulos PI, Boumpas DT. Lessons learned from anti-CD40L treatment in systemic lupus erythematosus patients. Lupus. 2004;13(5):391–397. doi: 10.1191/0961203304lu1032oa. [DOI] [PubMed] [Google Scholar]
- 35.Odegard JM, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205(12):2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vinuesa CG, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435(7041):452–458. doi: 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 37.Simpson N, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010;62(1):234–244. doi: 10.1002/art.25032. [DOI] [PubMed] [Google Scholar]
- 38.Borlado LR, et al. Increased phosphoinositide 3-kinase activity induces a lymphoproliferative disorder and contributes to tumor generation in vivo. FASEB J. 2000;14(7):895–903. doi: 10.1096/fasebj.14.7.895. [DOI] [PubMed] [Google Scholar]
- 39.Okkenhaug K, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297(5583):1031–1034. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- 40.Barber DF, et al. Class IB-phosphatidylinositol 3-kinase (PI3K) deficiency ameliorates IA-PI3K-induced systemic lupus but not T cell invasion. J Immunol. 2006;176(1):589–593. doi: 10.4049/jimmunol.176.1.589. [DOI] [PubMed] [Google Scholar]
- 41.Barber DF, et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med. 2005;11(9):933–935. doi: 10.1038/nm1291. [DOI] [PubMed] [Google Scholar]
- 42.Suarez-Fueyo A, Barber DF, Martinez-Ara J, Zea-Mendoza AC, Carrera AC. Enhanced phosphoinositide 3-kinase Δ activity is a frequent event in systemic lupus erythematosus that confers resistance to activation-induced T cell death. J Immunol. 2011;187(5):2376–2385. doi: 10.4049/jimmunol.1101602. [DOI] [PubMed] [Google Scholar]
- 43.Suarez-Fueyo A, et al. Inhibition of PI3Kδ reduces kidney infiltration by macrophages and ameliorates systemic lupus in the mouse. J Immunol. 2014;193(2):544–554. doi: 10.4049/jimmunol.1400350. [DOI] [PubMed] [Google Scholar]
- 44.Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB. Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur J Immunol. 2003;33(8):2223–2232. doi: 10.1002/eji.200324048. [DOI] [PubMed] [Google Scholar]
- 45.Parsons MJ, Jones RG, Tsao MS, Odermatt B, Ohashi PS, Woodgett JR. Expression of active protein kinase B in T cells perturbs both T and B cell homeostasis and promotes inflammation. J Immunol. 2001;167(1):42–48. doi: 10.4049/jimmunol.167.1.42. [DOI] [PubMed] [Google Scholar]
- 46.Miyara M, Ito Y, Sakaguchi S. TREG-cell therapies for autoimmune rheumatic diseases. Nat Rev Rheumatol. 2014;10(9):543–551. doi: 10.1038/nrrheum.2014.105. [DOI] [PubMed] [Google Scholar]
- 47.Lieberman LA, Tsokos GC. The IL-2 defect in systemic lupus erythematosus disease has an expansive effect on host immunity. J Biomed Biotechnol. 2010;2010:740619. doi: 10.1155/2010/740619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong HK, Kammer GM, Dennis G, Tsokos GC. Abnormal NF-κB activity in T lymphocytes from patients with systemic lupus erythematosus is associated with decreased p65-RelA protein expression. J Immunol. 1999;163(3):1682–1689. [PubMed] [Google Scholar]
- 49.Kyttaris VC, Juang YT, Tenbrock K, Weinstein A, Tsokos GC. Cyclic adenosine 5′-monophosphate response element modulator is responsible for the decreased expression of c-fos and activator protein-1 binding in T cells from patients with systemic lupus erythematosus. J Immunol. 2004;173(5):3557–3563. doi: 10.4049/jimmunol.173.5.3557. [DOI] [PubMed] [Google Scholar]
- 50.Mishra N, Khan IU, Tsokos GC, Kammer GM. Association of deficient type II protein kinase A activity with aberrant nuclear translocation of the RII β subunit in systemic lupus erythematosus T lymphocytes. J Immunol. 2000;165(5):2830–2840. doi: 10.4049/jimmunol.165.5.2830. [DOI] [PubMed] [Google Scholar]
- 51.Ichinose K, Juang YT, Crispin JC, Kis-Toth K, Tsokos GC. Suppression of autoimmunity and organ pathology in lupus-prone mice upon inhibition of calcium/calmodulin-dependent protein kinase type IV. Arthritis Rheum. 2011;63(2):523–529. doi: 10.1002/art.30085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hedrich CM, Rauen T, Tsokos GC. cAMP-responsive element modulator (CREM)α protein signaling mediates epigenetic remodeling of the human interleukin-2 gene: implications in systemic lupus erythematosus. J Biol Chem. 2011;286(50):43429–43436. doi: 10.1074/jbc.M111.299339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moulton VR, Grammatikos AP, Fitzgerald LM, Tsokos GC. Splicing factor SF2/ASF rescues IL-2 production in T cells from systemic lupus erythematosus patients by activating IL-2 transcription. Proc Natl Acad Sci U S A. 2013;110(5):1845–1850. doi: 10.1073/pnas.1214207110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 55.Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin Immunol. 2008;127(3):385–393. doi: 10.1016/j.clim.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 56.Wilson NJ, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8(9):950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 57.Chen Z, Tato CM, Muul L, Laurence A, O’Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007;56(9):2936–2946. doi: 10.1002/art.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ivanov, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 59.Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. 2010;184(9):4605–4609. doi: 10.4049/jimmunol.0903595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amarilyo G, Lourenco EV, Shi FD, La Cava A. IL-17 promotes murine lupus. J Immunol. 2014;193(2):540–543. doi: 10.4049/jimmunol.1400931. [DOI] [PubMed] [Google Scholar]
- 61.Chen Z, O’Shea JJ. Regulation of IL-17 production in human lymphocytes. Cytokine. 2008;41(2):71–78. doi: 10.1016/j.cyto.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harada T, Kyttaris V, Li Y, Juang YT, Wang Y, Tsokos GC. Increased expression of STAT3 in SLE T cells contributes to enhanced chemokine-mediated cell migration. Autoimmunity. 2007;40(1):1–8. doi: 10.1080/08916930601095148. [DOI] [PubMed] [Google Scholar]
- 63.Dong G, et al. IL-17 induces autoantibody overproduction and peripheral blood mononuclear cell overexpression of IL-6 in lupus nephritis patients. Chin Med J (Engl). 2003;116(4):543–548. [PubMed] [Google Scholar]
- 64.Hedrich CM, Crispin JC, Tsokos GC. Epigenetic regulation of cytokine expression in systemic lupus erythematosus with special focus on T cells. Autoimmunity. 2014;47(4):234–241. doi: 10.3109/08916934.2013.801462. [DOI] [PubMed] [Google Scholar]
- 65.Hedrich CM, et al. Stat3 promotes IL-10 expression in lupus T cells through trans-activation and chromatin remodeling. Proc Natl Acad Sci U S A. 2014;111(37):13457–13462. doi: 10.1073/pnas.1408023111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lippe R, et al. CREMα overexpression decreases IL-2 production, induces a T(H)17 phenotype and accelerates autoimmunity. J Mol Cell Biol. 2012;4(2):121–123. doi: 10.1093/jmcb/mjs004. [DOI] [PubMed] [Google Scholar]
- 67.Hedrich CM, et al. cAMP response element modulator alpha controls IL2 and IL17A expression during CD4 lineage commitment and subset distribution in lupus. Proc Natl Acad Sci U S A. 2012;109(41):16606–16611. doi: 10.1073/pnas.1210129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moulton VR, Holcomb DR, Zajdel MC, Tsokos GC. Estrogen upregulates cyclic AMP response element modulator α expression and downregulates interleukin-2 production by human T lymphocytes. Mol Med. 2012;18:370–378. doi: 10.2119/molmed.2011.00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Juang YT, et al. Transcriptional activation of the CREM promoter in human T cells is regulated by PP2A mediated dephosphorylation of SP-1 reflects disease activity in patients with systemic lupus erythematosus. J Biol Chem. 2011;286(3):1795–1801. doi: 10.1074/jbc.M110.166785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hueber W, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2(52):52ra72. doi: 10.1126/scitranslmed.3001107. [DOI] [PubMed] [Google Scholar]
- 71.Genovese MC, et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis. 2013;72(6):863–869. doi: 10.1136/annrheumdis-2012-201601. [DOI] [PubMed] [Google Scholar]
- 72.Crispin JC, Tsokos GC. Human TCR-α β+ CD4–CD8– T cells can derive from CD8+ T cells and display an inflammatory effector phenotype. J Immunol. 2009;183(7):4675–4681. doi: 10.4049/jimmunol.0901533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hedrich CM, et al. cAMP responsive element modulator (CREM) α mediates chromatin remodeling of CD8 during the generation of CD3+CD4–CD8– T cells. J Biol Chem. 2014;289(4):2361–2370. doi: 10.1074/jbc.M113.523605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de Groot RP, Ballou LM, Sassone-Corsi P. Positive regulation of the cAMP-responsive activator CREM by the p70 S6 kinase: an alternative route to mitogen-induced gene expression. Cell. 1994;79(1):81–91. doi: 10.1016/0092-8674(94)90402-2. [DOI] [PubMed] [Google Scholar]
- 75.Lai ZW, et al. Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus. J Immunol. 2013;191(5):2236–2246. doi: 10.4049/jimmunol.1301005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Godo M, Sessler T, Hamar P. Role of invariant natural killer T (iNKT) cells in systemic lupus erythematosus. Curr Med Chem. 2008;15(18):1778–1787. doi: 10.2174/092986708785132988. [DOI] [PubMed] [Google Scholar]
- 77.Oishi Y, et al. Selective reduction and recovery of invariant Vα24JαQ T cell receptor T cells in correlation with disease activity in patients with systemic lupus erythematosus. J Rheumatol. 2001;28(2):275–283. [PubMed] [Google Scholar]
- 78.Gabriel L, Morley BJ, Rogers NJ. The role of iNKT cells in the immunopathology of systemic lupus erythematosus. Ann N Y Acad Sci. 2009;1173:435–441. doi: 10.1111/j.1749-6632.2009.04743.x. [DOI] [PubMed] [Google Scholar]
- 79.Cho YN, et al. Numerical and functional deficiencies of natural killer T cells in systemic lupus erythematosus: their deficiency related to disease activity. Rheumatology (Oxford). 2011;50(6):1054–1063. doi: 10.1093/rheumatology/keq457. [DOI] [PubMed] [Google Scholar]
- 80.Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev. 2008;223:371–390. doi: 10.1111/j.1600-065X.2008.00637.x. [DOI] [PubMed] [Google Scholar]
- 81.Saadoun D, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. 2011;365(22):2067–2077. doi: 10.1056/NEJMoa1105143. [DOI] [PubMed] [Google Scholar]
- 82.Koreth J, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365(22):2055–2066. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Juang YT, et al. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. J Clin Invest. 2005;115(4):996–1005. doi: 10.1172/JCI22854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koga T, Ichinose K, Mizui M, Crispin JC, Tsokos GC. Calcium/calmodulin-dependent protein kinase IV suppresses IL-2 production and regulatory T cell activity in lupus. J Immunol. 2012;189(7):3490–3496. doi: 10.4049/jimmunol.1201785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koga T, et al. CaMK4-dependent activation of AKT/mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124(5):2234–2245. doi: 10.1172/JCI73411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Delgoffe GM, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12(4):295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kato H, Perl A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4–CD8– double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J Immunol. 2014;192(9):4134–4144. doi: 10.4049/jimmunol.1301859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105(12):4743–4748. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 89.Kang J, Huddleston SJ, Fraser JM, Khoruts A. De novo induction of antigen-specific CD4+CD25+Foxp3+ regulatory T cells in vivo following systemic antigen administration accompanied by blockade of mTOR. J Leuk Biol. 2008;83(5):1230–1239. doi: 10.1189/jlb.1207851. [DOI] [PubMed] [Google Scholar]
- 90.Battaglia M, Stabilini A, Tresoldi E. Expanding human T regulatory cells with the mTOR-inhibitor rapamycin. Methods Mol Biol. 2012;821:279–293. doi: 10.1007/978-1-61779-430-8_17. [DOI] [PubMed] [Google Scholar]
- 91.Amarilyo G, Iikuni N, Shi FD, Liu A, Matarese G, La Cava A. Leptin promotes lupus T-cell autoimmunity. Clin Immunol. 2013;149(3):530–533. doi: 10.1016/j.clim.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 92.Procaccini C, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33(6):929–941. doi: 10.1016/j.immuni.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu Y, Liu Y, Shi FD, Zou H, Matarese G, La Cava A. Cutting edge: Leptin-induced RORgammat expression in CD4+ T cells promotes Th17 responses in systemic lupus erythematosus. J Immunol. 2013;190(7):3054–3058. doi: 10.4049/jimmunol.1203275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu A, La Cava A. Epigenetic dysregulation in systemic lupus erythematosus. Autoimmunity. 2014;47(4):215–219. doi: 10.3109/08916934.2013.844794. [DOI] [PubMed] [Google Scholar]
- 95.Guo Y, Sawalha AH, Lu Q. Epigenetics in the treatment of systemic lupus erythematosus: Potential clinical application. Clin Immunol. 2014;155(1):79–90. doi: 10.1016/j.clim.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 96.Patel DR, Richardson BC. Dissecting complex epigenetic alterations in human lupus. Arthritis Res Ther. 2013;15(1):201. doi: 10.1186/ar4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sunahori K, Juang YT, Tsokos GC. Methylation status of CpG islands flanking a cAMP response element motif on the protein phosphatase 2Ac α promoter determines CREB binding and activity. J Immunol. 2009;182(3):1500–1508. doi: 10.4049/jimmunol.182.3.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990;33(11):1665–1673. doi: 10.1002/art.1780331109. [DOI] [PubMed] [Google Scholar]
- 99.Sunahori K, Nagpal K, Hedrich CM, Mizui M, Fitzgerald LM, Tsokos GC. The catalytic subunit of protein phosphatase 2A (PP2Ac) promotes DNA hypomethylation by suppressing the phosphorylated mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)/phosphorylated ERK/DNMT1 protein pathway in T-cells from controls and systemic lupus erythematosus patients. J Biol Chem. 2013;288(30):21936–21944. doi: 10.1074/jbc.M113.467266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li Y, et al. Overexpression of the growth arrest and DNA damage-induced 45α gene contributes to autoimmunity by promoting DNA demethylation in lupus T cells. Arthritis Rheum. 2010;62(5):1438–1447. doi: 10.1002/art.27363. [DOI] [PubMed] [Google Scholar]
- 101.Grammatikos AP, Ghosh D, Devlin A, Kyttaris VC, Tsokos GC. Spleen tyrosine kinase (Syk) regulates systemic lupus erythematosus (SLE) T cell signaling. PLoS One. 2013;8(8):e74550. doi: 10.1371/journal.pone.0074550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grammatikos AP, et al. A T cell gene expression panel for the diagnosis and monitoring of disease activity in patients with systemic lupus erythematosus. Clin Immunol. 2014;150(2):192–200. doi: 10.1016/j.clim.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Deng GM, Liu L, Bahjat FR, Pine PR, Tsokos GC. Suppression of skin and kidney disease by inhibition of spleen tyrosine kinase in lupus-prone mice. Arthritis Rheum. 2010;62(7):2086–2092. doi: 10.1002/art.27452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ichinose K, et al. Cutting edge: Calcium/Calmodulin-dependent protein kinase type IV is essential for mesangial cell proliferation and lupus nephritis. J Immunol. 2011;187(11):5500–5504. doi: 10.4049/jimmunol.1102357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Koga T, et al. KN-93, an inhibitor of calcium/calmodulin-dependent protein kinase IV, promotes generation and function of Foxp3 regulatory T cells in MRL/lpr mice. Autoimmunity. 2014;47(7):445–450. doi: 10.3109/08916934.2014.915954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wu T, et al. Targeting multiple signaling axes oxidative stress using a synthetic triterpenoid prevents murine lupus nephritis. Arthritis Rheumatol. 2014;66(11):3129–3139. doi: 10.1002/art.38782. [DOI] [PMC free article] [PubMed] [Google Scholar]