

Abstract
BACKGROUND. Individuals treated with the cholesteryl ester transfer protein (CETP) inhibitor anacetrapib exhibit a reduction in both LDL cholesterol and apolipoprotein B (ApoB) in response to monotherapy or combination therapy with a statin. It is not clear how anacetrapib exerts these effects; therefore, the goal of this study was to determine the kinetic mechanism responsible for the reduction in LDL and ApoB in response to anacetrapib.
METHODS. We performed a trial of the effects of anacetrapib on ApoB kinetics. Mildly hypercholesterolemic subjects were randomized to background treatment of either placebo (n = 10) or 20 mg atorvastatin (ATV) (n = 29) for 4 weeks. All subjects then added 100 mg anacetrapib to background treatment for 8 weeks. Following each study period, subjects underwent a metabolic study to determine the LDL-ApoB-100 and proprotein convertase subtilisin/kexin type 9 (PCSK9) production rate (PR) and fractional catabolic rate (FCR).
RESULTS. Anacetrapib markedly reduced the LDL-ApoB-100 pool size (PS) in both the placebo and ATV groups. These changes in PS resulted from substantial increases in LDL-ApoB-100 FCRs in both groups. Anacetrapib had no effect on LDL-ApoB-100 PRs in either treatment group. Moreover, there were no changes in the PCSK9 PS, FCR, or PR in either group. Anacetrapib treatment was associated with considerable increases in the LDL triglyceride/cholesterol ratio and LDL size by NMR.
CONCLUSION. These data indicate that anacetrapib, given alone or in combination with a statin, reduces LDL-ApoB-100 levels by increasing the rate of ApoB-100 fractional clearance.
TRIAL REGISTRATION. ClinicalTrials.gov NCT00990808.
FUNDING. Merck & Co. Inc., Kenilworth, New Jersey, USA. Additional support for instrumentation was obtained from the National Center for Advancing Translational Sciences (UL1TR000003 and UL1TR000040).
Keywords: Cardiology, Therapeutics, Vascular Biology
Introduction
Cholesteryl ester transfer protein (CETP) is a hydrophobic plasma protein that promotes the bidirectional transfer of cholesteryl esters (CE) and triglycerides (TG) between and among HDL particles and atherogenic apolipoprotein B–containing (ApoB-containing) lipoproteins, including the predominantly TG-rich VLDL, intermediate-density lipoprotein (IDL), and LDL particles (1–3). Genetic deficiency of CETP is associated with elevated HDL cholesterol (HDL-C) and reduced LDL-C (1), and common variants at the CETP locus are associated with HDL-C and LDL-C in inverse directions (3). Pharmacologic inhibition of CETP activity in humans raises HDL-C levels and generally reduces LDL-C levels (4–7).
The mechanism by which CETP inhibition reduces LDL-C remains unknown. A study of ApoB kinetics during administration of the CETP inhibitor torcetrapib (120 mg), with or without atorvastatin (ATV), to subjects with dyslipidemia (8) suggested that in dyslipidemic subjects, torcetrapib monotherapy reduced LDL ApoB by increasing the fractional catabolic rate (FCR) and that torcetrapib administered with ATV may have reduced production of LDL ApoB. However, none of these changes were statistically significant. Thus, the study was underpowered for detecting changes in many of the ApoB kinetic parameters and led to no firm conclusions regarding the mechanisms responsible for the lowering of ApoB.
Anacetrapib is an orally active, potent, and highly selective CETP inhibitor currently in late-stage clinical development (9). Early studies conducted in healthy and dyslipidemic subjects demonstrated that treatment with anacetrapib led to dose-dependent decreases in LDL-C (up to 40%) and plasma ApoB-100 (referred to herein as ApoB) (up to 30%) as well as dose-dependent increases in HDL-C (up to 139%) (4, 10). Adding anacetrapib to existing statin treatment also resulted in further reductions of LDL-C and increases in HDL-C over 1.5 years of treatment (6). While no head-to-head comparisons have been conducted, anacetrapib appears to have more robust LDL-C–lowering effects compared with those of torcetrapib through an as-yet-undefined mechanism (6, 11). Therefore, a sufficiently powered examination of the effects of anacetrapib treatment on the kinetics of ApoB has the potential to clarify the kinetic mechanism of LDL reduction with potent CETP inhibition.
The current study was conducted to comprehensively evaluate the effects of CETP inhibition with anacetrapib, either alone or in combination with ATV, on lipoprotein metabolism in humans. Here, we report the effects of anacetrapib on the kinetics of ApoB in VLDL, IDL, and LDL. We also present data on the kinetics of plasma proprotein convertase subtilisin/kexin type 9 (PCSK9), a protein known to play a major role in regulating the turnover of ApoB-containing lipoproteins, and the effects of CETP inhibition on its metabolism. We examined the relationship between improvement in ApoB metabolism and changes in the lipoprotein composition, size, and abundance and activity of proteins that regulate the metabolism of those particles, including PCSK9.
Results
Demographics of study subjects.
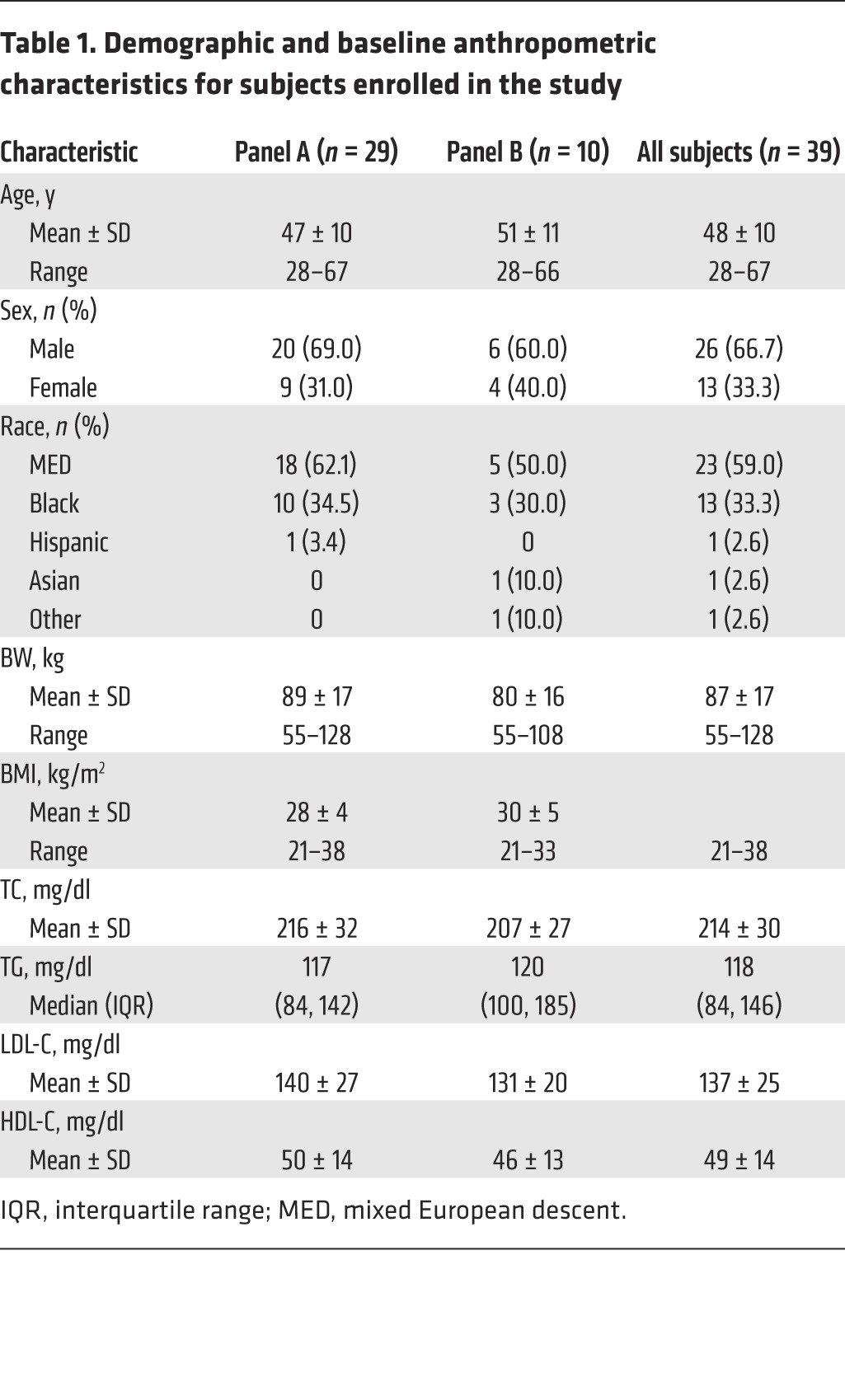
A total of 46 subjects were enrolled in the study. Of these, 39 ultimately completed all study procedures, while 7 subjects discontinued the study (1 for protocol-stipulated discontinuation for low LDL during statin treatment, 2 voluntary withdrawals, 1 dropout, 2 protocol deviations, and 1 relocation during the study). The flow chart of study subjects is shown in Figure 1. Subject demographics for completers are shown in Table 1. There were 26 men and 13 women with an average age of 48 years and an average BMI of 30 kg/m2. Fifty-nine percent were of mixed European descent and 33% were black. At screening, the mean total cholesterol (TC), LDL-C, HDL-C, and TG levels were 214, 137, 49, and 118 mg/dl, respectively.
Figure 1. CONSORT flow diagram showing the disposition of subjects participating in the current study.

*Patients are only counted once: Lost to follow-up, n = 28; lipids too low or too high, n = 35; qualified but declined, n = 8; cardiovascular risk factors, n = 11; high BMI, n = 5; taking excluded medication, n = 2; noncompliant during screening, n = 2; creatinine clearance too low, n = 4; kidney disease, n = 1; severe allergy, n = 2; glucose too high, n = 3; other, n = 17.
Table 1. Demographic and baseline anthropometric characteristics for subjects enrolled in the study.
Changes in plasma and lipoprotein lipid and ApoB concentrations.
Following screening, subjects were randomized to 1 of 2 background treatments: ATV, 20 mg (panel A) or placebo (panel B) for 4 weeks. By design, there were 29 subjects randomized to panel A and 10 subjects to panel B. At the end of period 1, the subjects in panel A had a mean TC, LDL-C, and HDL-C levels of 163, 90, and 48 mg/dl, respectively. Median plasma TG and mean ApoB levels were 89 and 71 mg/dl, respectively (Table 2). Subjects in panel B had a mean TC, LDL-C, and HDL-C levels of 209, 134, and 43 mg/dl, respectively. Median plasma TG and ApoB levels were 120 and 102 mg/dl, respectively (Table 2).
Table 2. Plasma lipid and ApoB values: mean baseline values, percentage change from baseline, and between-period differences for subjects in the total population and by panel.
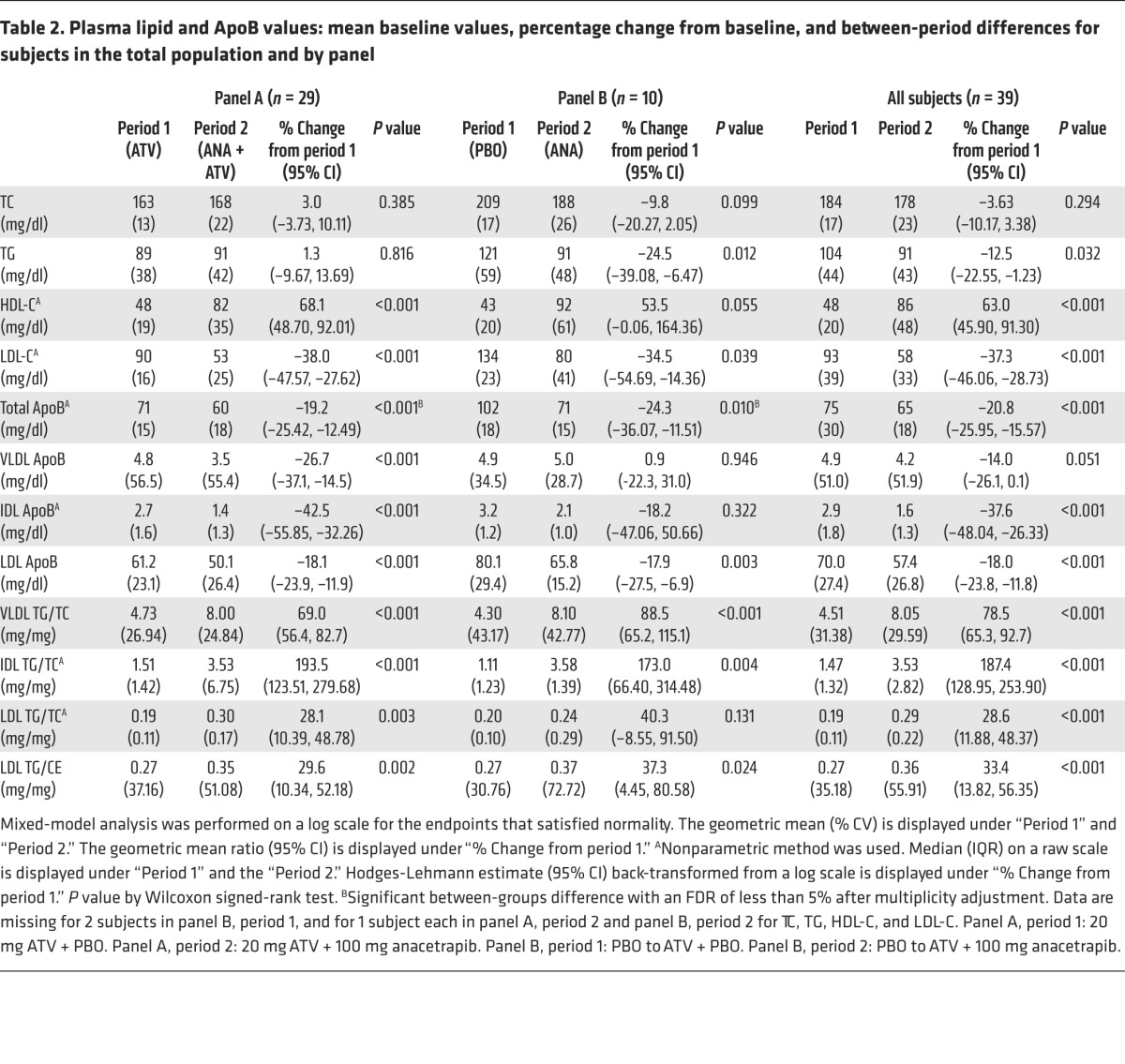
During period 2, subjects in both panels received anacetrapib (100 mg/day) in addition to their existing background treatment for 8 weeks. At the end of period 2, HDL-C levels were significantly increased in both panels, whereas significant reductions in LDL-C and total ApoB were observed in both panels (Table 2). In panel A (on a background of ATV), LDL-C was decreased by 38%, total ApoB by 19%, and LDL-ApoB by 18%. In addition, in panel B (no statin), LDL-C was decreased by 35%, total ApoB by 24%, and LDL-ApoB by 18% (Table 2). There were also changes in the lipid composition of the ApoB-containing lipoprotein fractions, with significant increases in the TG/C ratio of VLDL, IDL, and LDL following anacetrapib treatment in both panels (Table 2). In addition, the mean ratio of TG/CE in LDL was significantly increased by 30% in panel A and by 37% in panel B.
Effects of anacetrapib on ApoB metabolism.
At the end of each study period, the subjects underwent a lipoprotein kinetic study to measure the production rate (PR) and FCR of ApoB in LDL (in panel A, these were the coprimary endpoints). Following treatment with anacetrapib, the subjects in panel A (on a background of ATV) had a significant 18% reduction in LDL ApoB pool size (PS) compared with that seen with ATV treatment alone (Table 3 and Figure 2). The mechanism responsible for the reduction in LDL ApoB was an 18% increase in the FCR of LDL-ApoB (P = 0.001), whereas there was no significant change in the LDL-ApoB PR in response to anacetrapib. Subjects receiving anacetrapib on a placebo background (panel B) had a comparable 18% reduction in the LDL-ApoB PS (Table 3 and Figure 2), which was accompanied by an increase in the LDL-ApoB FCR and no change in the LDL ApoB PR in response to anacetrapib monotherapy.
Table 3. ApoB kinetic parameters: mean baseline values, percentage change from baseline, and between-period differences for subjects in the total population and by panel.
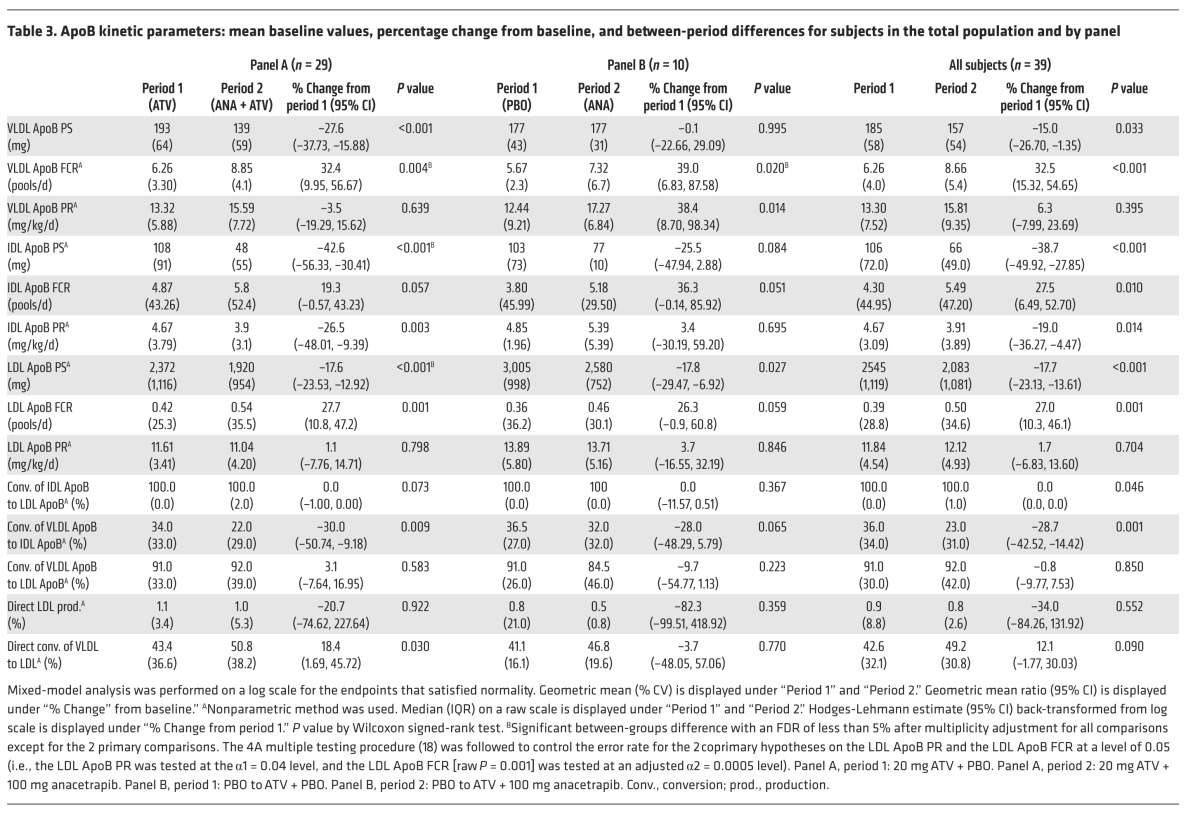
Figure 2. ApoB kinetics in lipoprotein fractions after treatment with anacetrapib.

VLDL-, IDL-, and LDL ApoB kinetic parameters for subjects in panel A (n = 29) and panel B (n = 10) at the end of each treatment period. The geometric mean is shown as a bar with unadjusted raw P values generated using a linear mixed-effects model containing fixed effects for panel and treatment-within-panel and random effects for subject-within-panel, unless indicated by an asterisk, in which case median values are indicated by a bar, and P values obtained by a Wilcoxon signed-rank test are shown. ANA, anacetrapib; PBO, placebo.
We also examined changes in VLDL- and IDL ApoB kinetics, which were both exploratory endpoints in this study. The subjects in Panel A (ATV background) had significant reductions of 28% and 43% in the apoB PS of VLDL and IDL with anacetrapib compared to placebo (Table 3 and Figure 2). The mechanism responsible for the reduction in VLDL ApoB was an increased FCR of 32%, with no change in the PR of VLDL ApoB. The FCR of IDL-ApoB was increased by 19%, while the IDL-ApoB PR was reduced by 26%, both potentially contributing to the decrease in IDL-ApoB PS.
Subjects receiving anacetrapib on a placebo background (panel B) had no significant changes in the VLDL ApoB PS due to parallel increases in the VLDL ApoB FCR and PR. There was a nonsignificant trend toward a reduction in the IDL-ApoB PS of 25% (P = 0.084) compared with that seen with placebo (Table 3 and Figure 2). There was an increase in the FCR of IDL-ApoB of borderline significance (P = 0.051) and no change in the PR of IDL-ApoB in response to anacetrapib monotherapy. ApoB kinetic parameters for individual subjects are shown in Supplemental Tables 1–3.
Changes in factors with a potential to influence ApoB metabolism.
To explore potential mechanisms responsible for the increased clearance of VLDL-, IDL-, and LDL ApoB seen in response to anacetrapib treatment, we examined candidate factors known to be associated with ApoB clearance. There were no significant changes in the post-heparin plasma lipoprotein lipase (LPL) or hepatic lipase (HL) concentrations or activity in response to anacetrapib in either the monotherapy- or statin-treated subjects (Table 4).
Table 4. Factors known to be associated with ApoB clearance: mean baseline values, percentage change from baseline, and between-period differences for subjects in the total population and by panel.
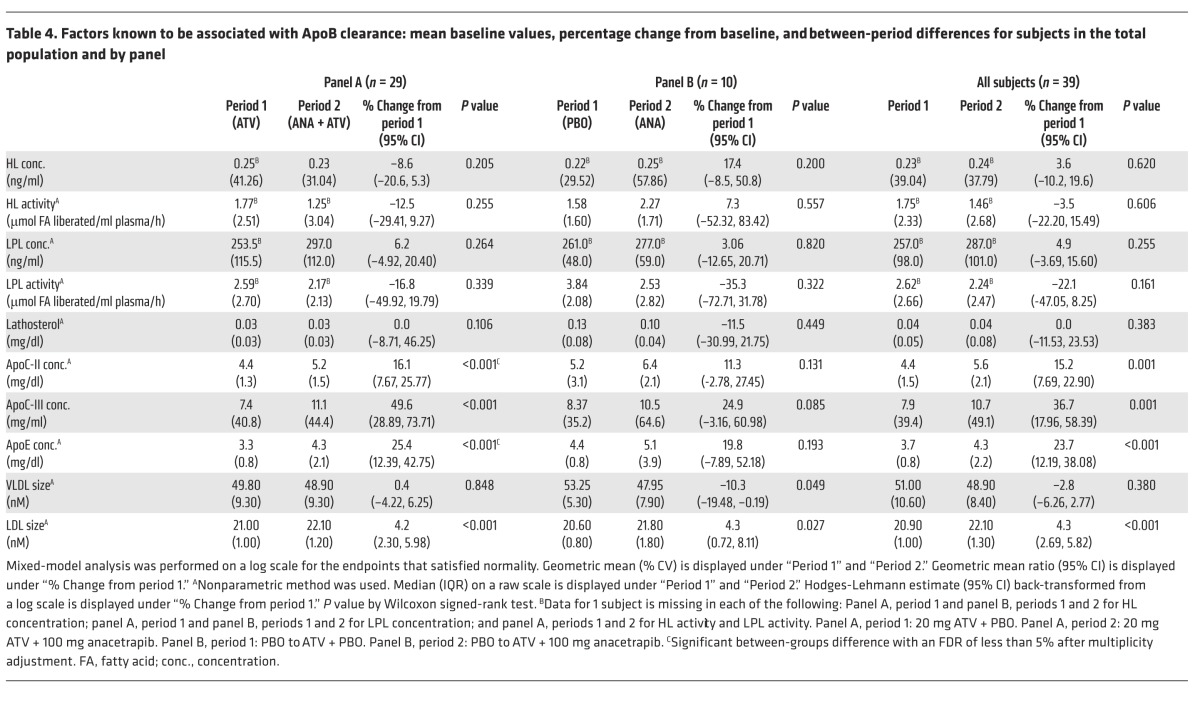
We also measured plasma lathosterol, a marker of hepatic cholesterol synthesis. Cholesterol synthesis and LDL receptor (LDLR) expression are both under the control of SREBP2; therefore, changes in lathosterol might be expected to reflect parallel changes in hepatic LDLR expression. There was no significant change in plasma lathosterol in response to treatment with anacetrapib in either the monotherapy- or ATV-treated subjects (Table 4). We also examined changes in ApoE, ApoC-II, and ApoC-III, plasma apolipoproteins that are known to influence the metabolism of ApoB-containing lipoproteins. There were no significant changes in plasma ApoE, ApoC-II, or ApoC‑III concentrations in the subjects treated with anacetrapib monotherapy (panel B) compared with those receiving placebo (Table 4). There were, however, significant increases in the plasma ApoE (25%), ApoC-II (16%), and ApoC-III (50%) concentrations (Table 4) in ATV-treated subjects in response to anacetrapib administration compared with those receiving ATV alone (panel A).
The size of ApoB-containing lipoproteins can influence their metabolism. We found that there was no change in VLDL size in panel A subjects in response to anacetrapib treatment compared with those receiving placebo, while subjects in panel B had VLDL levels that were modestly smaller (P = 0.049) on anacetrapib treatment compared with those who received statin-only baseline therapy (Table 4). Subjects in both treatment groups had a significant increase of about 4% in the overall size of LDL (Table 4) due to a decrease in the amount of smaller-sized LDL particles (Supplemental Table 4).
Effect of anacetrapib on PCSK9 metabolism.
There was a significant reduction of 19% in plasma PCSK9 concentrations in subjects treated with anacetrapib monotherapy, while there was no significant change in plasma PCSK9 concentrations in response to anacetrapib in statin-treated subjects (Table 5). To determine the mechanism responsible for the reduction in PCSK9 seen in response to anacetrapib monotherapy, we used a novel method to determine the production and clearance rates of PCSK9 (Figure 3 and ref. 12). We found that there was a trend (P = 0.066) toward an increase in the FCR of PCSK9 in subjects treated with anacetrapib monotherapy, while there was no significant change in the PCSK9 PR in these subjects. Statin-treated subjects had no significant changes in the PCSK9 FCR or PR following anacetrapib treatment, consistent with the absence of change in plasma PCSK9 concentrations in this group. PCSK9 kinetic data for individual subjects are shown in Supplemental Table 5.
Table 5. PCSK9-associated kinetic parameters: mean baseline values, percentage of change from baseline, and between-period differences for subjects in the total population and by panel.
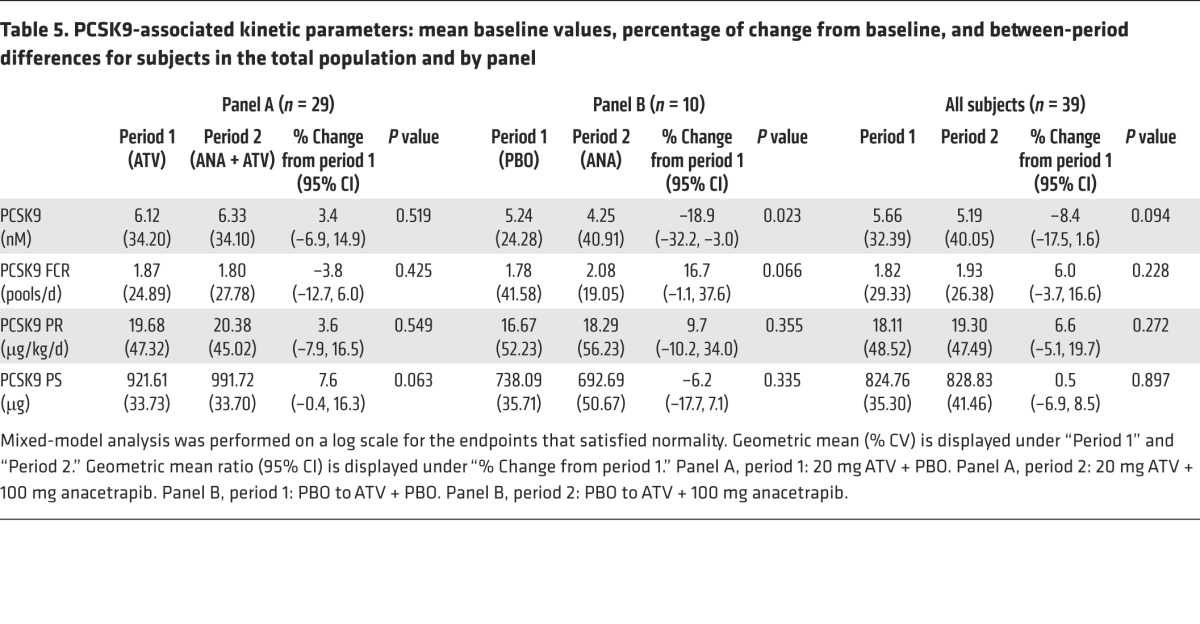
Figure 3. PCSK9 kinetics after treatment with anacetrapib.

PCSK9 kinetic parameters for subjects in panel A (n = 29) and panel B (n = 10) at the end of each treatment period. The geometric mean is shown as a bar. None of the comparisons achieved a raw P value of less than 0.05 on the basis of the linear mixed-effects models containing fixed effects for panel and treatment-within-panel and random effects for subject-within-panel.
Safety summary.
Thirty-four subjects reported a total of 17 nonserious clinical adverse events (AEs) that were considered to be drug related by the study investigator, all of which were mild-to-moderate in intensity and resolved by the end of the study. Of these, 9 AEs, including headache, back pain, constipation, and nausea, were reported by subjects who received ATV and/or placebo; 8 AEs, including headache, dyspepsia, muscle spasm, nausea, pain, gastrointestinal reflux, and influenza, were reported by subjects who received anacetrapib and ATV. There was 1 serious AE of accidental overdose of anacetrapib, with no other associated AEs. Four subjects had 5 laboratory AEs that were considered related to treatment with both anacetrapib and ATV, including elevated alkaline phosphatase (n = 1), elevated blood glucose (n = 1), and glucosuria (3 incidents reported by 2 subjects).
Discussion
CETP inhibitors were initially developed as a novel pharmacological approach to increase HDL-C (13). In studies in animals and humans, it became apparent that CETP inhibition also led to reduced levels of LDL-C (5, 13, 14). This effect on LDL-C was consistent with observations that humans genetically deficient in CETP have reduced LDL-C levels (1) and that common genetic variants at the CETP locus are significantly associated with LDL-C levels in an inverse direction to their association with HDL-C levels (3).
The mechanism responsible for the reduction in LDL-C in response to inhibiting CETP has been uncertain. A lipoprotein kinetic study in CETP-deficient subjects indicated that the LDL-ApoB FCR was more rapid than that measured in normal control subjects (15). Subsequently, a kinetic study performed in the setting of torcetrapib treatment suggested enhanced LDL-ApoB FCR in the monotherapy group and reduced LDL-ApoB PR when administered with ATV (8). However, the number of subjects in these subgroups was small, and the data for LDL FCR or PR were not statistically significant. Therefore, we decided to carry out an appropriately powered study of the effects of the potent CETP inhibitor anacetrapib on ApoB metabolism in humans.
In the current study, we determined the effect of 100 mg anacetrapib, the dose being used in the ongoing cardiovascular outcomes trial of anacetrapib, on ApoB metabolism as a monotherapy and in combination with a statin in mildly hyperlipidemic subjects. We found that anacetrapib reduced LDL-C levels by increasing the LDL-ApoB FCR. This was seen in both subjects treated with anacetrapib monotherapy as well as in those who received anacetrapib in combination with a statin. This finding appears to point to a common mechanism driving enhanced LDL-ApoB clearance in response to anacetrapib monotherapy and to anacetrapib given in combination with a statin.
A potential mechanism responsible for an increase in the LDL-ApoB FCR in response to anacetrapib is increased hepatic LDLR expression. This could occur in response to reduced cholesterol delivery to regulatory cholesterol pools in liver (Figure 4A), which might result from targeting of cholesterol delivered to liver via HDL particles for biliary secretion as cholesterol or bile acids (16) rather than entry into a common pool of free cholesterol that regulates the transcriptional activity of SREBP2. Alternatively, there could be reduced delivery of HDL-C to liver via scavenger receptor class B type I (SR-BI). Less cholesterol delivered by ApoB-containing lipoproteins to a hepatic regulatory pool would stimulate SREBP2 activity and increase expression of the LDLR gene. While we were unable to measure this directly, we did measure biomarkers that reflect changes in the hepatic regulatory cholesterol pools in response to anacetrapib. Plasma lathosterol is a cholesterol precursor that has been shown to reflect cholesterol synthesis rates (17), which are also regulated by SREBP2. Treatment with ezetimibe, an inhibitor of intestinal cholesterol absorption, leads to reduced delivery of cholesterol to the hepatic regulatory pool, increasing both cholesterol synthesis and hepatic LDLR expression in an animal model (18). In humans, ezetimibe use is associated with an increase in plasma lathosterol and LDL-ApoB clearance that is consistent with reduced delivery of cholesterol to the hepatic regulatory pool (19). However, we did not observe any effect of anacetrapib treatment on plasma lathosterol concentrations, suggesting that the hepatic regulatory cholesterol pool did not change in response to CETP inhibition. While it is possible that there was increased hepatic-derived lathosterol that was counterbalanced by a reduction in lathosterol derived from peripheral tissues (a situation which would imply reduced cholesterol delivery to liver and excess cholesterol in extrahepatic tissues), we consider this scenario unlikely. Osono and colleagues showed that variable expression of CETP in mice had no effect on cholesterol synthesis in extrahepatic tissues (20). In addition, Castro-Perez et al. showed that transport of cholesterol from macrophages to feces was enhanced in hamsters treated with anacetrapib, suggesting that cholesterol delivery from the periphery to liver functions normally in the context of CETP inhibition (16). Dong et al. recently reported on mouse studies in which CETP inhibitors, including anacetrapib, increased hepatic cholesterol levels, which led to reduced activation of SREBP2 and downregulation of LDLRs in mice and cultured human hepatocytes (21). However, there was an increase, rather than a decrease, in LDL-C levels, making these findings of uncertain relevance to CETP inhibition in humans.
Figure 4. Potential mechanisms responsible for the increase in the LDL ApoB FCR observed in response to anacetrapib treatment.

(A) A reduction in the cholesterol content of the regulatory pool of intracellular cholesterol activates SREBP2, leading to increased transcription of LDLR, PCSK9, and cholesterol synthetic genes. Increased LDLR transcription increases the amount of LDLR at the cell surface of hepatocytes (indicated by “Y”), leading to an increase in LDL clearance. This scenario should be accompanied by an increase in the PCSK9 PR and cholesterol synthesis as reflected by lathosterol levels. (B) Reduced levels of PCSK9 circulating in plasma results in less targeting of the LDLR for degradation and an increase in LDL recycling. This would increase the amount of LDLR at the cell surface of hepatocytes and lead to an increase in LDL clearance. (C) A decrease in hepatic oxysterols reduces activation of the LXR, leading to reduced transcription of MYLIP, the gene that encodes IDOL, and APOA1. Reduced IDOL at the plasma membrane attenuates LDLR degradation and increases LDL recycling, leading to an increased number of LDLRs at the hepatocyte cell surface and, consequently, an increase in LDL clearance. This scenario should be accompanied by a reduced ApoA-I PR. (D) An increase in the TG/cholesterol ratio as well as an increase in LDL particle size as seen by NMR increases the affinity of LDL for the LDLR, leading to a greater degree of LDL binding to the LDLR and an increase in overall LDL clearance.
Another biomarker reflecting the hepatic cholesterol regulatory pools and directly affecting LDL catabolism is PCSK9. PCSK9 production is known to increase in response to reduced hepatic cholesterol levels, also via activation of SREBP2, resulting in increased PCSK9 levels in plasma (22). The PCSK9 PR should parallel the synthesis of LDLRs. Although we saw a modest reduction in plasma PCSK9 levels during anacetrapib monotherapy, there was no change in PCSK9 concentrations when anacetrapib was added to ATV. We developed a method for determining the kinetics of PCSK9 in humans. Using this approach, we found no statistically significant effect of anacetrapib on the PR of PCSK9 in subjects who were either on anacetrapib monotherapy or anacetrapib plus ATV. Together with the data for lathosterol, these results support the hypothesis that there was no substantial change in the hepatic regulatory cholesterol pools or SREBP2 activation in response to anacetrapib and that this is unlikely to be a mechanism for upregulation of the hepatic LDLR and increased LDL ApoB catabolism.
A reduction in circulating PCSK9 levels could influence the FCR of LDL-ApoB (Figure 4B and ref. 22). Plasma PCSK9 levels were modestly but significantly reduced in subjects treated with anacetrapib monotherapy. Rhesus macaques also had lower PCSK9 levels following a 10-day treatment with anacetrapib (23). Our PCSK9 kinetics studies suggested a modestly increased FCR for PCSK9, though this was not statistically significant. A portion (at least 40%) of circulating PCSK9 has been found to be associated with LDL (24). It is currently unknown whether LDL-associated PCSK9 is functional in promoting LDLR degradation. PCSK9 is required to bind to the EGF receptor homology domain (EGF-A) of the LDLR to promote degradation. PCSK9 has also been reported to bind some of the repeats within the N-terminal ligand-binding domain (LBD) of the LDLR, although this has not been unequivocally confirmed in any of the reported crystal structures at both neutral and acidic pHs (25, 26). These findings led Yamamoto et al. to propose a model whereby PCSK9 requires interaction with both the EGF-A and LBD domains of the LDLR to promote degradation (27). While LDL-associated PCSK9 is capable of binding to the LDLR (24), this may only occur through interaction with either the EGF-A domain or the LBD, but not both domains. Thus, it is possible that there is increased fractional clearance of LDL-associated PCSK9 without enhanced LDLR degradation. We did not observe a reduction in plasma PCSK9 levels with anacetrapib treatment in statin-treated subjects; the basis for this difference is unknown. Clearly, this topic requires further investigation.
A third possible mechanism that could increase the LDL-ApoB FCR is reduced expression of inducible degrader of the LDLR (IDOL) (Figure 4C). IDOL is an E3 ubiquitin ligase that has been shown to promote degradation of the LDLR through a lysosome-dependent pathway (28). IDOL expression is known to be induced by oxysterol-mediated activation of the liver X receptor (LXR). Attenuation of LXR activity by anacetrapib resulting in reduced IDOL activity could lead to an increase in LDLR activity and the LDL-ApoB FCR. While plasma oxysterol levels were not measured in the current study, anacetrapib has been shown to reduce hepatic cholesterol levels in hamsters treated with 60 mg/kg anacetrapib for 2 weeks (16). This was associated with increased cholesterol and cholic acid excretion in feces. However, in humans, the CETP inhibitor torcetrapib was shown to have no effect on fecal bile acids (29), suggesting that there is no change in hepatic oxysterol levels following CETP inhibition. Consistent with this concept, we found no significant effect of anacetrapib treatment on the PR of the known LXR targets CETP and ApoA-I (30). Thus, decreased LDLR degradation via IDOL is unlikely to be the basis for an increased LDL-ApoB FCR.
A fourth mechanism that could increase the LDL ApoB FCR in response to CETP inhibition with anacetrapib would be a change in LDL lipid composition, size, and/or apolipoprotein content that increased its affinity for the LDLR or other receptors (Figure 4D). We found that LDL was relatively enriched in TG and depleted of cholesterol and CE during CETP inhibition. LDL enriched in TG is generally larger and has relatively high affinity for the LDLR (31). We measured LDL size by NMR and found that the average LDL size was increased to a degree similar to that reported in response to torcetrapib treatment (5). Krauss et al. showed that average LDL size measured by ion mobility is slightly reduced following anacetrapib treatment (150 mg/day) in association with a relative decrease in larger particles and an increase in smaller-sized LDL particles (32). Further work needs to be done to resolve the discrepancy in particle sizing by these 2 methods. It is worth noting that CETP deficiency is associated with polydisperse LDL, with the presence of both large and small LDL subspecies noted (33). Krauss et al. reported that anacetrapib treatment (150 mg/day) increased ApoE in the smaller LDL subfraction and C-III in the smallest LDL subfraction, with no change in the ApoE or C‑III content of the larger LDL subfractions (32). This finding is consistent with the concept that following anacetrapib treatment, there is preferential clearance of the large, TG-enriched LDL particles over smaller LDL ones.
In the present study, we also found a significant (P < 0.001) reduction in the VLDL ApoB PS in subjects treated with anacetrapib in combination with a statin that was due to an increased VLDL ApoB FCR, with no change in the VLDL ApoB PR. The VLDL ApoB FCR was also increased in subjects treated with anacetrapib monotherapy. This was accompanied by a significant (P = 0.014) increase in the VLDL ApoB PR, resulting in no net change in the VLDL ApoB PS in this group. CETP inhibition with torcetrapib was associated with an increased VLDL ApoB FCR. Similar to what was observed in the current study, torcetrapib was associated with an increase in the VLDL ApoB PR when given as a monotherapy, but not when given in combination with a statin (8). The FCR of VLDL ApoB is known to be influenced by factors that regulate lipolysis and affinity for lipoprotein receptors including LPL, ApoC‑II, ApoC-III, ApoE, and VLDL lipid composition. We observed no changes in LPL or HL mass or activity during anacetrapib treatment on either background therapy. However, the TG enrichment of VLDL that we observed during anacetrapib treatment could have increased the efficiency of lipolysis in the absence of increased availability of LPL or HL; larger TG-rich, ApoB-containing lipoproteins are lipolyzed more efficiently (34). In addition, relative enrichment of VLDL with ApoE and/or reduced ApoC‑III content of VLDL could increase the rate of fractional removal of VLDL by the liver (35). Fractional removal of VLDL ApoB includes both direct clearance and conversion to IDL ApoB or LDL ApoB. While CETP deficiency has been associated with enhanced (though nonsignificant) conversion of VLDL ApoB to LDL ApoB (15), we observed no change in the conversion of VLDL to LDL, either directly or through IDL ApoB, in response to anacetrapib. It should be noted that that the ApoB-containing lipoproteins in this study were isolated using traditional density cutpoints. Because of changes in lipid composition in response to CETP inhibition, there may be changes in lipoprotein density, such that there may be overlap of lipoprotein fractions between traditional density cutpoints, which could complicate interpretation of the kinetics results for VLDL and IDL ApoB.
These in vivo human data definitively establish that anacetrapib treatment, as monotherapy and in combination with a statin, reduces LDL by increasing its FCR. This increase in LDL catabolism might be explained by enhanced clearance of TG-rich LDL particles generated by CETP inhibition. This mechanism reduces the total number of LDL particles and contributes to the reduction in LDL-C and ApoB levels. In addition, anacetrapib reduces the cholesterol content of LDL particles, which may contribute to the overall reduction in LDL-C levels. If anacetrapib is ultimately shown to reduce cardiovascular events (36), the reduction in LDL-C through these mechanisms is likely to to be found to contribute to that benefit.
Methods
Study subjects
Subjects were recruited from internal clinical trial databases and from advertisements in local newspapers and postings at each study site (Columbia University Medical Center and the University of Pennsylvania). Eligible subjects included nonsmoking or social smoking (fewer than 5 cigarettes over 3 months) men and women between 18 and 75 years of age with a BMI between 18.5 and 40 kg/m2. All subjects had an LDL-C level of 100 mg/dl or higher and 190 mg/dl or lower (subjects with 0–1 coronary heart disease [CHD] risk factor) or 160 mg/dl or lower (subjects with ≥2 CHD risk factors) at screening for statin-naive subjects, or following a 2- to 3-week washout period for subjects taking statins at screening. The initial upper limit for BMI (35 kg/m2) and lower limit for LDL (≥120 mg/dl) were modified to aid in recruitment. A requirement for subjects to have an LDL cholesterol level of 70 mg/dl or higher after 2 weeks on ATV treatment was removed to increase enrollment numbers. Subjects were also required to have a TG level of 400 mg/dl or lower at the screening visit. All subjects were counseled to maintain a diet consistent with the recommendations of the American Heart Association.
The female subjects were of non-childbearing potential (i.e., surgically sterilized or postmenopausal for at least 1 year) or of childbearing potential with a negative serum pregnancy test and who were using reliable birth control methods. Individuals bearing a clinically significant medical condition and those with a known history of alcohol and/or drug abuse or requiring concomitant lipid-altering medications were excluded from this study.
Study design
This was a multicenter, randomized, 2-panel, double-blind, placebo-controlled (for ATV background versus placebo), double-dummy, 2-period, fixed-sequence (for placebo or ATV alone, followed by placebo or ATV plus anacetrapib) phase I study in which 46 dyslipidemic subjects were enrolled between December 2009 and July 2011 (Merck & Co. Inc., protocol number 0859-026-03; NCT00990808). A copy of the study protocol can be found in the supplemental materials. Subjects were randomized to either panel A (ATV background) or panel B (placebo background) at a 3:1 ratio according to a computer-generated allocation schedule stratified by an LDL-C level lower than or greater than or equal to 160 mg/dl to ensure a balanced distribution of these subjects across panels. Each panel consisted of 2 treatment periods, with no washout period separating the treatments within each panel.
Panel A (n = 29).
In period 1, subjects in panel A received 20 mg/day ATV (Lipitor; Pfizer Inc.) coadministered with placebo to match 100 mg anacetrapib (Merck & Co. Inc.) for a minimum of 4 weeks (maximum of 5 weeks). In period 2, subjects in panel A added 100 mg anacetrapib once daily for 8 weeks (maximum of 9 weeks) to their existing ATV treatment regimen.
Panel B (n = 10).
In period 1, subjects received daily oral doses of placebo to match 20 mg ATV coadministered with placebo to match the 100 mg anacetrapib treatment regimen for a minimum of 4 weeks (maximum of 5 weeks). In period 2, subjects in panel B added 100 mg anacetrapib once daily for a minimum of 8 weeks (maximum of 9 weeks) to their existing placebo to match the ATV treatment regimen.
Subjects could be discontinued from the study for any AE that jeopardized the subject’s safety and/or well-being or deviation from protocol requirements. At the end of each treatment period, subjects were admitted to the corresponding clinical and translational research center (CTRC) before undergoing a lipoprotein kinetics study performed with bolus injections of [13C6]-phenylalanine (29.4 μmol/kg BW); [1,1,2,3,3-2H5]-glycerol (100 μmol/kg BW); and [5,5,5-2H3]-leucine (9-10 μmol/kg BW) (isotopes from Cambridge Isotope Laboratories), followed immediately by a constant infusion of [5,5,5-2H3]-leucine (9-10 μmol/kg BW/hour) over a 15-hour period under constantly fed conditions (i.e., small isocaloric meals consisting of ~18% fat were given every 2 hours [16 meals over 30 hours]). Feeding was started 8 hours before isotope administration to establish steady-state conditions. Blood samples were collected at 0 (prebolus), 20, and 40 minutes, and 1, 2, 4, 6, 8, 10, 12, 14, 15, 15.5, 16, 18, 21, 24, and 48 hours after infusion to determine the kinetics of ApoB in VLDL, IDL, and LDL, as well as the kinetics of plasma PCSK9.
In both treatment periods, the study medication was self-administered at home (in the evening with dinner). During the kinetics studies, the medication was administered by CTRC staff. Subjects continued to receive the study drug during the kinetics study protocols. Compliance was monitored through pill counts and random phone calls. Subjects received monetary compensation for study participation and study-related expenses.
Safety measurements
The safety and tolerability of the study medication were assessed by clinical evaluation of AEs and inspection of other safety parameters, including physical examinations, vital signs, and routine laboratory safety measurements (hematology, blood chemistry, and 12-lead ECGs) at prespecified times throughout the study. AEs were monitored throughout the study and evaluated in terms of intensity (mild, moderate, or severe), duration, severity, outcome, and relationship to the study drug. All subjects who took at least 1 dose of study medication (panel A [n = 35] or B [n = 11]) were included in the safety and tolerability analyses.
Biochemical and immunologic assays
Blood for biochemical measurements was collected at the end of each treatment period following a 12-hour fast. TC and TG were measured enzymatically on a Cobas Fara II autoanalyzer (Roche Diagnostic Systems) using Sigma-Aldrich reagents. LDL-C levels were determined using the Friedewald formula. The TG/TC ratio in VLDL, IDL, and LDL and the TG/CE ratio in LDL were measured in ultracentrifugally isolated lipoprotein fractions using enzymatic reagents. CE levels were determined by subtracting free cholesterol from the TC measurement. ApoB, ApoC-II, ApoC-III, and ApoE were measured with immunoturbidimetric assays using Wako reagents. ApoB in the VLDL and IDL fractions was measured using the AssayMax Human ApoB ELISA kit (AssayPro). LDL ApoB was determined by subtracting VLDL ApoB and IDL ApoB from plasma ApoB. Particle sizes and the concentration of lipoprotein subspecies were measured using NMR (LipoScience). Lathosterol was analyzed by gas chromatography–mass spectrometry (GC-MS) using selected ion monitoring of lathosterol (37). CETP mass was assayed using a double-antibody sandwich ELISA kit from Wako (catalog 997-29001; performed by Pharmaceutical Product Development LLC [PPD]).
LPL and HL concentrations and activity
Post-heparin plasma was collected 10–15 minutes after an i.v. injection of heparin (60 units/kg BW) at the end of each treatment period following the final blood draw of the lipoprotein kinetic study. Post-heparin LPL and HL concentrations were measured using commercially available ELISAs (ALPCO Diagnostics and Abnova, respectively). Total lipase activities were assayed in triplicate using radiolabeled TG emulsion as a substrate (38, 39). Results were expressed as μmol of free fatty acids (FFAs) released per hour. The contribution of HL was determined by including 1 mM NaCl in the assay; the activity of HL was subtracted from the total lipase activity to estimate LPL.
ApoB kinetics
ApoB kinetics were measured in the Metabolic Tracer Resource at the University of Pennsylvania as previously described (40, 41). Lipoprotein fractions were isolated by sequential ultracentrifugation from blood samples collected throughout the kinetics study. ApoB-100 was isolated by SDS-PAGE from the VLDL, IDL, and LDL fractions and acid hydrolyzed. The resulting amino acids as well as free [5,5,5-2H3]-leucine and [13C6]-phenylalanine in plasma were isolated by cation exchange chromatography and derivatized (41). The isotope enrichment with [5,5,5-2H3]-leucine and [13C6]-phenylalanine tracers in plasma and apolipoproteins was measured by GC-MS using an Agilent 7890A/5975 GC-MS system. ApoB kinetic parameters were determined by simultaneous fitting of the stable isotope–labeled leucine and phenylalanine tracer data to a multicompartmental model with a weighted least-squares approach using WinSAAM modeling software, version 3.0.7. The model was adapted from that used previously (40), with the addition of a VLDL remnant compartment. FCRs were calculated from kinetic parameters as the fraction of ApoB cleared from each lipoprotein class, either directly or by transfer to another lipoprotein class. PRs were calculated as the product of the FCR and the corresponding PS. PSs for VLDL-, IDL-, and LDL ApoB were calculated as the product of the average concentration of ApoB in each lipoprotein fraction, calculated from 5 time points during the kinetics study, and the plasma volume, assumed to be 4.5% of BW.
PCSK9 kinetics
PCSK9 concentration and kinetics were determined as previously described (12). Briefly, PCSK9 protein was immunopurified from 500 μl plasma using mAbs coupled to magnetic beads. mAbs against PCSK9 (clone 1B20) used for immunoaffinity purification were produced using standard techniques from hybridomas generated following the immunization of mice with recombinant human PCSK9 (12). Protein was eluted with formic acid, evaporated to dryness, and resuspended for tryptic digestion. Tryptic peptides were analyzed by microflow liquid chromatography–tandem mass spectrometry (LC-MS/MS). Isotope enrichment was determined by calculating the ratio of M3/M0 and subtracting the baseline for the peptide GTVSGTLIGLEFIR > LIGLEFIR.
PCSK9 kinetic parameters were determined by fitting the stable isotope–labeled leucine tracer data to a multicompartmental model with a weighted least-squares approach using WinSAAM, version 3.0.7. The multicompartmental model consisted of 3 compartments: a hepatic precursor, a synthetic delay, and plasma PCSK9. The hepatic precursor was represented by the plasma [5,5,5-2H3]-leucine enrichment. The FCR of PCSK9 was calculated from kinetic parameters as the fraction of PCSK9 cleared from plasma each day. The PCSK9 PR was calculated as the product of the FCR and the plasma PCSK9 PS. The PCSK9 PS was calculated as the product of the average plasma PCSK9 concentration, measured at 3 time points during the metabolic study, and the plasma volume, assumed to be 4.5% of BW.
Statistics
Collected site-specific data were reported to each study site, where data were reviewed and manually entered into a secure database. Following data entry, all data were adjudicated before analysis. All statistical analyses were conducted using SAS software (SAS Institute Inc.). Analysis was performed on log scale, and the estimates obtained were back-transformed using the formula 100*(exp(estimate)–1), to yield point estimates, 95% CIs, and between-treatment, 2-tailed P values for the true percentage change from the treatment in period 1. Data normality was first assessed. For normally distributed data, linear mixed-effects models containing fixed effects for panel and treatment-within-panel and random effect for subject-within-panel were used to assess the percentage change from period 1 treatment. The geometric mean and percentage of coefficient of variation (%CV) were also provided for each treatment. For non-normally distributed data, median and interquartile ranges were reported for individual treatment periods. Hodges-Lehmann estimates based on the Wilcoxon signed-rank test and corresponding P values were reported for treatment period differences (period 2 – period 1).
This study had 2 primary comparisons and additional between-treatment comparisons. Although the P values quoted in the text and shown in the tables are raw P values, various multiplicity adjustments were also performed in order to facilitate a balanced interpretation of these results. Significance for the 2 primary comparisons, the percentage of change in the LDL ApoB PR, and the percentage of change in the LDL ApoB FCR after administration of anacetrapib on a background of ATV therapy versus ATV alone were assessed using the 4A multiple-testing procedure (42), which controls for the FWER for the primary hypotheses at α = 0.05. Following this procedure, the LDL ApoB PR was tested at the α1 = 0.04 level, and the LDL ApoB FCR was tested at an adjusted α2 (0.0005) level based on both the observed raw P value for PR (0.798) and the upper limit of 95% CI of the correlation (0.690) between the 2 primary endpoints. Using this procedure, success for either comparison is sufficient. Using the 2-stage FDR method (43), the FDR for the comparisons of anacetrapib with ATV versus anacetrapib alone, anacetrapib with ATV versus ATV alone, and anacetrapib versus placebo was controlled at 5% for the following endpoints: kinetics of ApoB in VLDL, IDL, and LDL; conversions of VLDL ApoB to IDL ApoB, VLDL ApoB to LDL ApoB, and IDL ApoB to LDL ApoB; concentrations and activity of HL and LPL; and concentrations of ApoB, ApoC-II, ApoC-III, and ApoE. All other exploratory endpoints and comparisons were tested at the 0.05 level and were not subject to multiplicity adjustment. All available data were included, and no data were excluded from analysis.
The study was powered to detect absolute differences in the LDL ApoB 100 PR and the LDL ApoB-100 FCR for anacetrapib with ATV versus ATV alone. With 30 subjects, assuming a pooled, within-subject SD of 1.94 mg/kg/day and a significance level of 0.04 (2-tailed), there was 96.0% probability to detect a –1.4 mg/kg/day decrease in the LDL ApoB-100 PR. Assuming a pooled, within-subject SD of 0.05 pools/day and a significance level of 0.05 (2-tailed), an increase of 0.026 pools/day in the LDL ApoB-100 FCR could be detected with 80.0% probability.
Study approval
This study was conducted in accordance with the Principles of Good Clinical Practice and was approved by the IRBs of Columbia University Medical Center and the University of Pennsylvania. All study subjects provided written informed consent.
Supplementary Material
Acknowledgments
Editorial assistance was provided by Jennifer Rotonda of Merck & Co. Inc. The authors would like to acknowledge Ellie Coromilas (Columbia University Medical Center [CUMC], New York, New York, USA) and Vishala Patel, Phyllis May, and Julia Doveikis (University of Pennsylvania) for technical and laboratory assistance and the nursing staff of the Dietary Assessment Unit and Investigational Drug Service at the Clinical and Translational Research Center (CTRC) of the University of Pennsylvania. This study was funded by Merck & Co. Inc. Additional support for instrumentation for this work at the Perelman School of Medicine at the University of Pennsylvania was obtained from the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR000003. Further support for instrumentation for this work at the Irving Center for Clinical Research at the College of Physicians & Surgeons of Columbia University was obtained from the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR000040. The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Role of funding source: The sponsor was involved in the study design, collection, analysis, and interpretation of data, writing of the manuscript, and decision to submit the manuscript for publication.
Conflict of interest: Patricia Jumes, Amy O. Johnson-Levonas, Yang Liu, Michael E. Lassman, and David E. Gutstein are employees of Merck & Co. Inc. (Merck) and own stock and/or hold stock options in the company. John A. Wagner was an employee of Merck and owned stock and held stock options in the company. John S. Millar received grant support and honoraria from Merck. Gissette Reyes-Soffer reports having received a grant from Merck during the conduct of the study. Richard L. Dunbar and Daniel S. Donovan report having received grants from Merck during the conduct of the study. Henry N. Ginsberg reports having received grants from Merck during the conduct of the study and personal fees from Merck outside the submitted work. Daniel J. Rader is a member of the Merck Scientific Advisory Board.
Reference information:J Clin Invest. 2015;125(6):2510–2522. doi:10.1172/JCI80025.
References
- 1.Inazu A, et al. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N Engl J Med. 1990;323(18):1234–1238. doi: 10.1056/NEJM199011013231803. [DOI] [PubMed] [Google Scholar]
- 2.Tall AR. Plasma cholesteryl ester transfer protein. J Lipid Res. 1993;34(8):1255–1274. [PubMed] [Google Scholar]
- 3.Thompson A, et al. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008;299(23):2777–2788. doi: 10.1001/jama.299.23.2777. [DOI] [PubMed] [Google Scholar]
- 4.Bloomfield D, et al. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am Heart J. 2009;157(2):352–360. doi: 10.1016/j.ahj.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 5.Brousseau ME, et al. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N Engl J Med. 2004;350(15):1505–1515. doi: 10.1056/NEJMoa031766. [DOI] [PubMed] [Google Scholar]
- 6.Cannon CP, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363(25):2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 7.Stein EA, Roth EM, Rhyne JM, Burgess T, Kallend D, Robinson JG. Safety and tolerability of dalcetrapib (RO4607381/JTT-705): results from a 48-week trial. Eur Heart J. 2010;31(4):480–488. doi: 10.1093/eurheartj/ehp601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millar JS, et al. Effects of the cholesteryl ester transfer protein inhibitor torcetrapib on apolipoprotein B100 metabolism in humans. Arterioscler Thromb Vasc Biol. 2006;26(6):1350–1356. doi: 10.1161/01.ATV.0000219695.84644.56. [DOI] [PubMed] [Google Scholar]
- 9.Gutstein DE, et al. Anacetrapib, a novel CETP inhibitor: pursuing a new approach to cardiovascular risk reduction. Clin Pharmacol Ther. 2012;91(1):109–122. doi: 10.1038/clpt.2011.271. [DOI] [PubMed] [Google Scholar]
- 10.Krishna R, et al. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomised placebo-controlled phase I studies. Lancet. 2007;370(9603):1907–1914. doi: 10.1016/S0140-6736(07)61813-3. [DOI] [PubMed] [Google Scholar]
- 11.Barter PJ, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 12.Lassman ME, et al. Practical immunoaffinity-enrichment LC-MS for measuring protein kinetics of low-abundance proteins. Clin Chem. 2014;60(9):1217–1224. doi: 10.1373/clinchem.2014.222455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamoto H, Yonemori F, Wakitani K, Minowa T, Maeda K, Shinkai H. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature. 2000;406(6792):203–207. doi: 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- 14.de Grooth GJ, et al. Efficacy and safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in humans: a randomized phase II dose-response study. Circulation. 2002;105(18):2159–2165. doi: 10.1161/01.CIR.0000015857.31889.7B. [DOI] [PubMed] [Google Scholar]
- 15.Ikewaki K, et al. Increased catabolic rate of low density lipoproteins in humans with cholesteryl ester transfer protein deficiency. J Clin Invest. 1995;96(3):1573–1581. doi: 10.1172/JCI118196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castro-Perez J, et al. Anacetrapib promotes reverse cholesterol transport and bulk cholesterol excretion in Syrian golden hamsters. J Lipid Res. 2011;52(11):1965–1973. doi: 10.1194/jlr.M016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kempen HJ, Glatz JF, Gevers Leuven JA, van der Voort HA, Katan MB. Serum lathosterol concentration is an indicator of whole-body cholesterol synthesis in humans. J Lipid Res. 1988;29(9):1149–1155. [PubMed] [Google Scholar]
- 18.Telford DE, Sutherland BG, Edwards JY, Andrews JD, Barrett PH, Huff MW. The molecular mechanisms underlying the reduction of LDL apoB-100 by ezetimibe plus simvastatin. J Lipid Res. 2007;48(3):699–708. doi: 10.1194/jlr.M600439-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Chan DC, Watts GF, Gan SK, Ooi EM, Barrett PH. Effect of ezetimibe on hepatic fat, inflammatory markers, and apolipoprotein B-100 kinetics in insulin-resistant obese subjects on a weight loss diet. Diabetes Care. 2010;33(5):1134–1139. doi: 10.2337/dc09-1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osono Y, Woollett LA, Marotti KR, Melchior GW, Dietschy JM. Centripetal cholesterol flux from extrahepatic organs to the liver is independent of the concentration of high density lipoprotein-cholesterol in plasma. Proc Natl Acad Sci U S A. 1996;93(9):4114–4119. doi: 10.1073/pnas.93.9.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong B, Singh AB, Fung C, Kan K, Liu J. CETP inhibitors downregulate hepatic LDL receptor and PCSK9 expression in vitro and in vivo through a SREBP2 dependent mechanism. Atherosclerosis. 2014;235(2):449–462. doi: 10.1016/j.atherosclerosis.2014.05.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Browning JD, Horton JD. Fasting reduces plasma proprotein convertase, subtilisin/kexin type 9 and cholesterol biosynthesis in humans. J Lipid Res. 2010;51(11):3359–3363. doi: 10.1194/jlr.P009860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roddy TP, et al. Effects of anacetrapib on plasma lipids, apolipoproteins and PCSK9 in healthy, lean rhesus macaques. Eur J Pharmacol. 2014;740:410–416. doi: 10.1016/j.ejphar.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Kosenko T, Golder M, Leblond G, Weng W, Lagace TA. Low density lipoprotein binds to proprotein convertase subtilisin/kexin type-9 (PCSK9) in human plasma and inhibits PCSK9-mediated low density lipoprotein receptor degradation. J Biol Chem. 2013;288(12):8279–8288. doi: 10.1074/jbc.M112.421370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seidah NG, Awan Z, Chretien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res. 2014;114(6):1022–1036. doi: 10.1161/CIRCRESAHA.114.301621. [DOI] [PubMed] [Google Scholar]
- 26.Lo SP, et al. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 2011;12(12):1300–1305. doi: 10.1038/embor.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto T, Lu C, Ryan RO. A two-step binding model of PCSK9 interaction with the low density lipoprotein receptor. J Biol Chem. 2011;286(7):5464–5470. doi: 10.1074/jbc.M110.199042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325(5936):100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brousseau ME, et al. Effects of cholesteryl ester transfer protein inhibition on high-density lipoprotein subspecies, apolipoprotein A-I metabolism, and fecal sterol excretion. Arterioscler Thromb Vasc Biol. 2005;25(5):1057–1064. doi: 10.1161/01.ATV.0000161928.16334.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reyes-Soffer G, et al. Effects of anacetrapib treatment on CETP metabolism. Arterioscler Thromb Vasc Biol. 2013;33:A120. [Google Scholar]
- 31.Campos H, Arnold KS, Balestra ME, Innerarity TL, Krauss RM. Differences in receptor binding of LDL subfractions. Arterioscler Thromb Vasc Biol. 1996;16(6):794–801. doi: 10.1161/01.ATV.16.6.794. [DOI] [PubMed] [Google Scholar]
- 32.Krauss RM, et al. Changes in lipoprotein subfraction concentration and composition in healthy individuals treated with the CETP inhibitor anacetrapib. J Lipid Res. 2012;53(3):540–547. doi: 10.1194/jlr.M018010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakai N, et al. Detection of two species of low density lipoprotein particles in cholesteryl ester transfer protein deficiency. Arterioscler Thromb. 1991;11(1):71–79. doi: 10.1161/01.ATV.11.1.71. [DOI] [PubMed] [Google Scholar]
- 34.Streja DA. Triglyceride removal from very low density lipoproteins in vivo as a function of their triglyceride content. Atherosclerosis. 1979;32(1):57–67. doi: 10.1016/0021-9150(79)90147-3. [DOI] [PubMed] [Google Scholar]
- 35.Windler E, Chao Y, Havel RJ. Regulation of the hepatic uptake of triglyceride-rich lipoproteins in the rat. Opposing effects of homologous apolipoprotein E and individual C apoproteins. J Biol Chem. 1980;255(17):8303–8307. [PubMed] [Google Scholar]
- 36.Hewing B, Fisher EA. Rationale for cholesteryl ester transfer protein inhibition. Curr Opin Lipidol. 2012;23(4):372–376. doi: 10.1097/MOL.0b013e328353ef1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lutjohann D, et al. Profile of cholesterol-related sterols in aged amyloid precursor protein transgenic mouse brain. J Lipid Res. 2002;43(7):1078–1085. doi: 10.1194/jlr.M200071-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Nilsson-Ehle P, Schotz MC. A stable, radioactive substrate emulsion for assay of lipoprotein lipase. J Lipid Res. 1976;17(5):536–541. [PubMed] [Google Scholar]
- 39.Hocquette JF, Graulet B, Olivecrona T. Lipoprotein lipase activity and mRNA levels in bovine tissues. Comp Biochem Physiol B Biochem Mol Biol. 1998;121(2):201–212. doi: 10.1016/S0305-0491(98)10090-1. [DOI] [PubMed] [Google Scholar]
- 40.Nagashima K, et al. Effects of the PPARγ agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J Clin Invest. 2005;115(5):1323–1332. doi: 10.1172/JCI23219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Millar JS, et al. Complete deficiency of the low-density lipoprotein receptor is associated with increased apolipoprotein B-100 production. Arterioscler Thromb Vasc Biol. 2005;25(3):560–565. doi: 10.1161/01.ATV.0000155323.18856.a2. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Mehrotra DV. An efficient method for accommodating potentially underpowered primary endpoints. Stat Med. 2008;27(26):5377–5391. doi: 10.1002/sim.3369. [DOI] [PubMed] [Google Scholar]
- 43.Mehrotra DV, Heyse JF. Use of the false discovery rate for evaluating clinical safety data. Stat Methods Med Res. 2004;13(3):227–238. doi: 10.1191/0962280204sm363ra. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.