

Abstract
A transition from fetal hemoglobin (HbF) to adult hemoglobin (HbA) normally occurs within a few months after birth. Increased production of HbF after this period of infancy ameliorates clinical symptoms of the major disorders of adult β-hemoglobin: β-thalassemia and sickle cell disease. The transcription factor BCL11A silences HbF and has been an attractive therapeutic target for increasing HbF levels; however, it is not clear to what extent BCL11A inhibits HbF production or mediates other developmental functions in humans. Here, we identified and characterized 3 patients with rare microdeletions of 2p15-p16.1 who presented with an autism spectrum disorder and developmental delay. Moreover, these patients all exhibited substantial persistence of HbF but otherwise retained apparently normal hematologic and immunologic function. Of the genes within 2p15-p16.1, only BCL11A was commonly deleted in all of the patients. Evaluation of gene expression data sets from developing and adult human brains revealed that BCL11A expression patterns are similar to other genes associated with neurodevelopmental disorders. Additionally, common SNPs within the second intron of BCL11A are strongly associated with schizophrenia. Together, the study of these rare patients and orthogonal genetic data demonstrates that BCL11A plays a central role in silencing HbF in humans and implicates BCL11A as an important factor for neurodevelopment.
Keywords: Development, Genetics, Hematology, Neuroscience
Introduction
The switch from fetal hemoglobin (HbF) to adult hemoglobin (HbA) expression that occurs during the months following birth is of considerable therapeutic interest, since elevated HbF ameliorates the clinical symptoms in β-thalassemia and sickle cell disease (SCD) (1, 2). Genome-wide association and functional follow-up studies in cell and animal models have shown that BCL11A, a multiple zinc-finger–containing transcription factor, is an important silencer of HbF expression (3, 4). This has resulted in a concerted effort to develop targeted approaches to induce HbF by inhibiting BCL11A (1, 2). However, the extent to which BCL11A silences HbF and its other functions in vivo in humans is unknown. BCL11A plays a key dosage-dependent role in the immune system in mouse models (5, 6), and recent studies implicate it as an autism spectrum disorder (ASD) and developmental delay (DD) candidate gene (7, 8).
To address the in vivo role of BCL11A in humans, we sought to study patients with small deletions involving this gene. A microdeletion syndrome of the 2p15-p16.1 region has been described in rare patients and consists of a number of features, including an ASD, DD, hypotonia, fine motor dysfunction, and facial dysmorphism (OMIM 612513) (9, 10). Most such deletions are large and involve a number of genes. We identified 3 patients with small de novo deletions that only removed BCL11A and 1–2 adjacent genes. Analysis of these patients, along with orthogonal genetic data, allowed us to assess the in vivo role of BCL11A. We demonstrated that BCL11A plays a key role in both silencing HbF and in human neurodevelopment.
Results and Discussion
We identified 3 patients with small de novo deletions of the 2p15-p16.1 region that only removed BCL11A and 1–2 adjacent genes (Figure 1A). Patient 1 had an approximately 440 kb deletion (chr2: 60,689,727–61,128,229 in hg19 coordinates) (10), Patient 2 had an approximately 1 Mb deletion (chr2: 60,029,857–61,059,383), and Patient 3 had an approximately 875 kb deletion (chr2: 59,958,420–60,834,298). BCL11A was the only deleted gene shared in all 3 patients, while PAPOLG and MIR4432 were each deleted in 2 of the 3 patients. PAPOLG has been suggested to encode a protein that mediates posttranscriptional 3′ adenylation of specific RNAs, although it does not have a known physiologic role (11). MIR4432 encodes a microRNA that has been identified from deep RNA sequencing of B lymphocytes (12). We noted that both BCL11A and PAPOLG were expressed in developing red blood cell (erythroid) precursors from humans, although BCL11A was expressed at higher levels than PAPOLG (Figure 1B and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI81163DS1). MIR4432 was not detectable, and we did not identify any other RNAs that would be removed by these deletions in erythroid cells (Supplemental Figure 1), although we cannot entirely rule out effects on regulatory elements.
Figure 1. Involvement of BCL11A in the 2p microdeletion syndrome.

(A) A depiction of the 2p15-p16.1 region with coordinates shown (hg19). The position of the patient deletions are shown in orange (Patient 1), blue (Patient 2), and red (Patient 3), and RefSeq genes are shown below. The commonly deleted region of patients 1 and 2 is shown between dotted lines. (B) The common deleted region of patients 1 and 2 involving BCL11A and PAPOLG. RNA expression is shown below at various stages of human erythroid differentiation. This includes proerythroblasts (ProE), early basophilic erythroblasts (eBasoE), late basophilic erythroblasts (lBasoE), polychromatic erythroblasts (PolyE), and orthochromatic erythroblasts (OrthoE). The height of RNA peaks in each region demonstrates the number of reads per million at that site and the reads per kb per million (RPKM) mapped reads for BCL11A is shown in log2 scale.
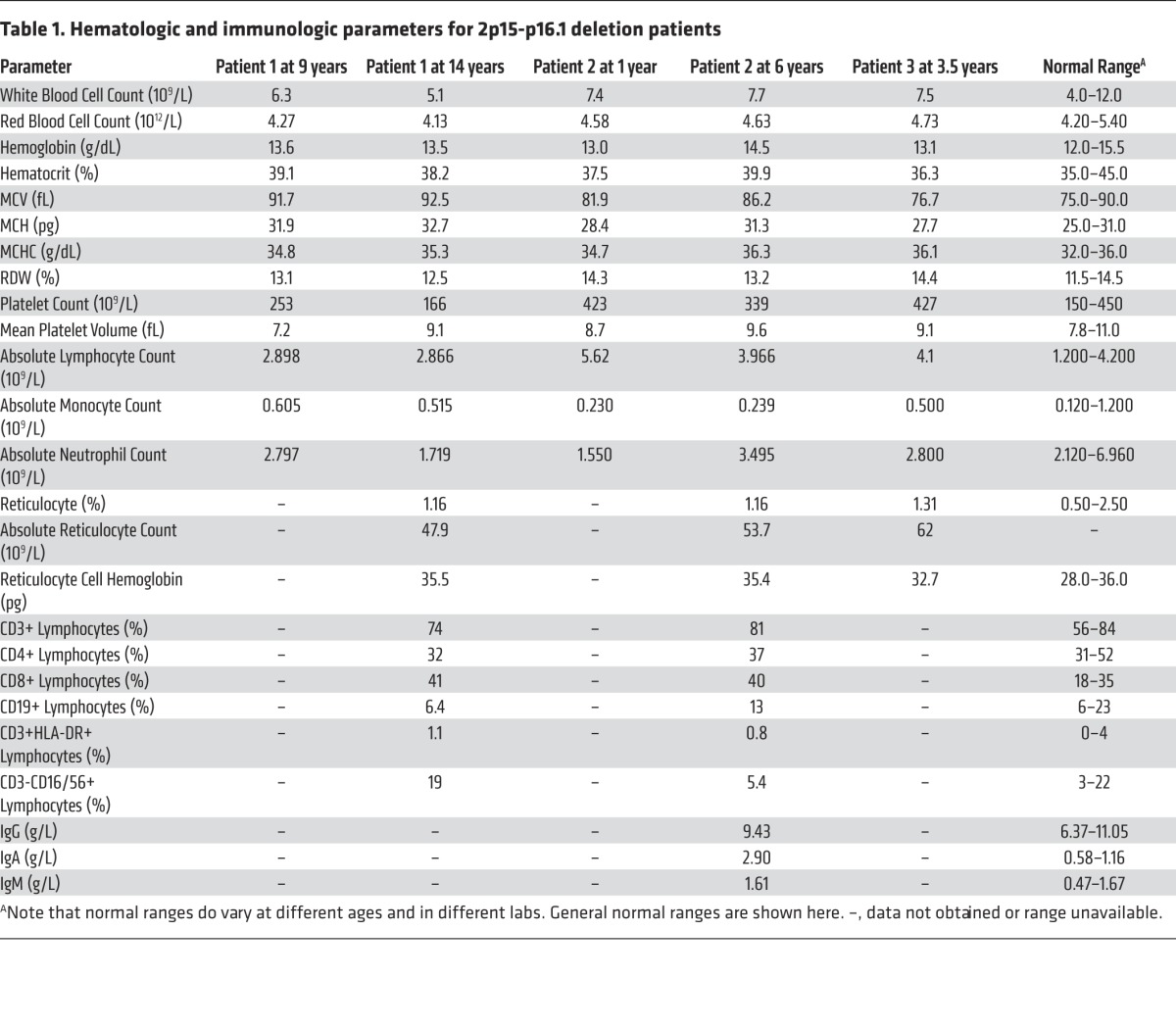
Analysis of mononuclear cell RNA from patients 1 and 2 and age-matched controls revealed that BCL11A was haploinsufficient in the patients, while PAPOLG was not significantly reduced compared with controls (Figure 2A). Concomitantly, we noted that there were higher mRNA levels of the HbF-encoding genes, HBG1 and HBG2 (Figure 2A). Consistent with this (though, at lower levels, consistent with known maturational and posttranscriptional regulation) (1, 13), we found that HbF was substantially elevated at 23.8%, 16.1%, and 29.7% in blood from patients 1–3, respectively (Figure 2B), whereas in normal age-matched controls, it would be < 1% (patients 1, 2, and 3 were 14, 6, and 3.5 years, respectively, when this test was done). Since PAPOLG was expressed in erythroid cells, we suppressed this gene using shRNAs in primary human erythroid cells and observed no change in the expression of the HbF-encoding genes (Supplemental Figure 2). Given the variation in HbF levels observed in the patients, we genotyped HbF-associated common variants in the remaining intact BCL11A gene locus in all of the patients. Patient 1 had 2 of 3 minor alleles associated with higher HbF levels (alleles A, T, and G at rs4671393, rs1427407, and rs7606173, respectively), while patients 2 and 3 had the reference alleles that are associated with lower HbF levels (alleles G, G, and G at rs4671393, rs1427407, and rs7606173, respectively) (14). This suggests that the variation in HbF levels between patients cannot be fully explained by common genetic variation at the remaining intact BCL11A locus. We assessed the loci containing the HBG1, HBG2, and HBB genes and found no deletions or mutations in the patients that would result in elevated HbF. The levels of HbF observed in the microdeletion patients would be sufficient to ameliorate symptoms in patients with β-thalassemia or SCD (1, 15). Importantly, there were no changes in blood counts or other hematologic parameters in the patients (Table 1). Lymphocyte subset levels in patients 1 and 2 and immunoglobulin levels in Patient 2 were tested and were normal (Table 1), suggesting that BCL11A haploinsufficiency does not impair immune function in humans, in contrast to its effects in mice (5, 6). None of the patients had a history of severe or unusual infections, supporting the observation that they all appeared to have normal immunologic function.
Figure 2. Persistence of HbF with BCL11A haploinsufficiency.

(A) Relative gene expression from qPCR analysis done for BCL11A and PAPOLG. In addition, the percentage of HBG1 and HBG2 are shown. The color-coding of various samples in all the panels is shown on the right, and independent replicates (n = 3 per individual) are plotted individually. ***P < 0.001. (B) Hemoglobin electrophoresis (patients 1 and 2) and high-performance liquid chromatography (HPLC; Patient 3) chromatograms with the level of different hemoglobin subtypes quantified from peripheral blood samples. The level of hemoglobin A (HbA), HbF, and HbA2 are shown below the chromatograms. In the HPLC chromatogram, the peaks for HbF and HbA2 are filled in, while HbA remains without any filling. The ordering of the labels below the chromatograms is in the order of peak positions. All comparisons were performed using the 2-tailed nonparametric Mann-Whitney U test.
Table 1. Hematologic and immunologic parameters for 2p15-p16.1 deletion patients.
All 3 patients exhibited common features, including an ASD, moderate to severe DD, hypotonia, and facial dysmorphism (with common features including an asymmetric face, telecanthus, strabismus, mild ptosis, and long eyelashes). We also noted that there were progressive neurological features in the older patients (1 and 2), including worsening of fine motor activity and coordination, as well as hyperactivity and aggression. Patients 1 and 2 had MRI scans of the brain performed without signs of structural abnormalities, with the exception of microcephaly (both had head circumferences < 3rd percentile for age). Patient 3 had a normal head size (25th percentile) but was noted to have a posterior fossa malformation on MRI. EEGs were also performed on patients 1 and 2 and showed no focal abnormalities in electrical activity.
Recent studies have implicated BCL11A as a potential DD and ASD candidate gene (7, 8). We aimed to evaluate whether the DD, ASD, and other features seen in the patients may be attributable to BCL11A or PAPOLG haploinsufficiency. We found that 4.4% of individuals in a healthy population of 6,503 harbored loss-of-function (LOF) mutations in PAPOLG, including 2 individuals with homozygous LOF mutations (Figure 3A and Supplemental Table 1). Given the observed high frequency of LOF variants in a sample of the general population, all of the observed phenotypes, which are rarely observed in the general population, are extremely unlikely to be due to LOF for PAPOLG. In contrast, no LOF alleles were found in BCL11A in this population (Figure 3A). Furthermore, orthogonal data revealed that it was among the most constrained in the human genome (ranked 106/15877, P value LOF = 2.26 × 10–6) (16). These findings are consistent with the neurologic phenotypes seen in Patient 3 and in a previously described patient (17), who had deletions involving BCL11A without disrupting the protein-coding region of PAPOLG. We do note that there have been distinct neurologic phenotypes seen in rare patients with 2p15-p16.1 microdeletions that do not disrupt the protein-coding region of BCL11A (18), suggesting that these deletions may either disrupt regulatory elements of BCL11A or that other genes in the region may also have neurologic functions.
Figure 3. Role of BCL11A in human neurodevelopment.

(A) The percentage of LOF alleles in BCL11A and PAPOLG from the 6,503 individuals in the Exome Sequencing Project. Error bars represent 95% confidence intervals around the percentage of individuals with one or more LOF alleles. Comparison of these frequencies is performed by Fisher’s exact test. (B) The expression of select genes in brain tissue at different developmental stages (from various brain regions that are aggregated here for simplicity). Data are plotted as the number of reads per kb per million (RPKM) for the genes shown. A locally weighted scatterplot smoothing regression was applied to expression of each gene. Results from this regression are plotted with 95% confidence intervals. (C) Regional association plot depicting data analyzed from a recent schizophrenia GWAS (22).
To better delineate a neurodevelopmental role for BCL11A, we examined its expression in 524 RNA sequencing data sets from numerous regions of the developing and adult human brain (Figure 3B). BCL11A was expressed at high levels and similar to other ASD/DD candidate genes, including CHD8 (19) and DYRK1A (20), during brain development and in adult brain tissue (Figure 3B and Supplemental Figures 3 and 4). We noted that PAPOLG was expressed at lower levels and KLF1, which is mutated in cases of persistent HbF without neurologic phenotypes (1), was not expressed in the human brain, illustrating the specificity of this expression data (Figure 3B). BCL11A has been suggested to have a role in neurogenesis in model systems, although the consequences of this have not been fully characterized (21). Our results from the rare patients with 2p15-p16.1 microdeletions and orthogonal data strongly implicate BCL11A as a high-confidence candidate gene underlying disorders of altered human neurodevelopment.
Finally, since ASD and DD are known to have connections with other neurodevelopmental disorders, we examined data from a recent study of common genetic variation underlying schizophrenia (22). While not initially identified, upon reanalysis, we noted that there were intronic SNPs in BCL11A that were significantly associated with schizophrenia and that were located close to or overlapping the common SNPs associated with HbF levels in humans (Figure 3C and ref. 14). In addition, SNPs in this region have also been implicated in attention deficit/hyperactivity disorder (ADHD), another condition thought to be due to underlying alterations in neurodevelopment (23). Therefore, by studying rare patients with BCL11A haploinsufficiency in concert with orthogonal genetic data of human neurodevelopmental disorders, we were able to strongly implicate BCL11A as a key gene whose function is necessary for normal human neurologic function and where alterations underlie a number of neuropsychiatric disorders.
Recent functional studies have raised hope that targeting BCL11A may be highly effective to induce HbF in patients with hemoglobin disorders (2). Our findings from rare patients with 2p15-p16.1 microdeletions demonstrate that haploinsufficiency of BCL11A is sufficient to allow persistence of HbF at a high enough level to ameliorate β-thalassemia or SCD. Indeed, the levels of HbF observed in these patients are similar to cases of elevated HbF due to mutations in the β-globin locus itself, which have been shown to result in a benign clinical course when acquired with β-thalassemia or SCD (1, 24). Moreover, we observe no immune dysfunction in these patients, in contrast to haploinsufficient mouse models (5, 6). By studying these rare patients in concert with orthogonal genetic data from neurodevelopmental disorders — including ASD, DD, schizophrenia, and ADHD — we are able to strongly implicate BCL11A as a key neurodevelopmental gene. This finding emphasizes the importance of using hematopoietic-specific or CNS-nonpenetrating approaches when attempting to target BCL11A therapeutically (2).
Methods
Further information can be found in Supplemental Methods.
Statistics.
All pairwise comparisons were performed using the 2-tailed nonparametric Mann-Whitney U test unless otherwise stated in the text. Differences were considered significant if the P value was less than 0.05.
Study approval.
All family members had provided written informed consent to participate in this study. The institutional review boards at Boston Children’s Hospital, Charles University in Prague, and University of Ottawa approved the study protocols.
Supplementary Material
Acknowledgments
We are grateful to the patients and their families for their willingness to participate in this study and to D. Nathan, A. Chakravarti, E. Benz, and C. Walsh for their advice and input. This article is dedicated to the memory of the late Professor Bill Wood.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(6):2363–2368. doi:10.1172/JCI81163.
This work was supported by grants from the NIH (U01 HL117720, R21 HL120791, and R01 DK103794) (to V.G. Sankaran) and from the Czech Ministry of Health (NT/14200 and 00064203) (to Z. Sedlacek). A. Basak is a Translational Research Development Scholar at Boston Children’s Hospital.
References
- 1.Sankaran VG, Orkin SH. The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med. 2013;3(1):a011643. doi: 10.1101/cshperspect.a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sankaran VG, Weiss MJ. Anemia: progress in molecular mechanisms and therapies. Nat Med. 2015;21(3):221–230. doi: 10.1038/nm.3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sankaran VG, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 4.Sankaran VG, et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460(7259):1093–1097. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Y, et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J Exp Med. 2012;209(13):2467–2483. doi: 10.1084/jem.20121846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ippolito GC, et al. Dendritic cell fate is determined by BCL11A. Proc Natl Acad Sci U S A. 2014;111(11):E998–E1006. doi: 10.1073/pnas.1319228111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coe BP, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46(10):1063–1071. doi: 10.1038/ng.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Rubeis S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajcan-Separovic E, et al. Clinical and molecular cytogenetic characterisation of a newly recognised microdeletion syndrome involving 2p15-16.1. J Med Genet. 2007;44(4):269–276. doi: 10.1136/jmg.2006.045013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hancarova M, Simandlova M, Drabova J, Mannik K, Kurg A, Sedlacek Z. A patient with de novo 0.45 Mb deletion of 2p16.1: the role of BCL11A, PAPOLG, REL, and FLJ16341 in the 2p15-p16.1 microdeletion syndrome. Am J Med Genet A. 2013;161A(4):865–870. doi: 10.1002/ajmg.a.35783. [DOI] [PubMed] [Google Scholar]
- 11.Kyriakopoulou CB, Nordvarg H, Virtanen A. A novel nuclear human poly(A) polymerase (PAP), PAP γ. J Biol Chem. 2001;276(36):33504–33511. doi: 10.1074/jbc.M104599200. [DOI] [PubMed] [Google Scholar]
- 12.Jima DD, et al. Deep sequencing of the small RNA transcriptome of normal and malignant human B cells identifies hundreds of novel microRNAs. Blood. 2010;116(23):e118–127. doi: 10.1182/blood-2010-05-285403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Exp Hematol. 2005;33(3):259–271. doi: 10.1016/j.exphem.2004.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42(12):1049–1051. doi: 10.1038/ng.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musallam KM, Sankaran VG, Cappellini MD, Duca L, Nathan DG, Taher AT. Fetal hemoglobin levels and morbidity in untransfused patients with beta-thalassemia intermedia. Blood. 2012;119(2):364–367. doi: 10.1182/blood-2011-09-382408. [DOI] [PubMed] [Google Scholar]
- 16.Samocha KE, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46(9):944–950. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peter B, Matsushita M, Oda K, Raskind W. De novo microdeletion of BCL11A is associated with severe speech sound disorder. Am J Med Genet A. 2014;164(8):2091–2096. doi: 10.1002/ajmg.a.36599. [DOI] [PubMed] [Google Scholar]
- 18.Chabchoub E, Vermeesch JR, de Ravel T, de Cock P, Fryns JP. The facial dysmorphy in the newly recognised microdeletion 2p15-p16.1 refined to a 570 kb region in 2p15. J Med Genet. 2008;45(3):189–192. doi: 10.1136/jmg.2007.056176. [DOI] [PubMed] [Google Scholar]
- 19.Bernier R, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158(2):263–276. doi: 10.1016/j.cell.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willsey AJ, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155(5):997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.John A, et al. Bcl11a is required for neuronal morphogenesis and sensory circuit formation in dorsal spinal cord development. Development. 2012;139(10):1831–1841. doi: 10.1242/dev.072850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinney A, et al. Genome-wide association study in German patients with attention deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(8):888–897. doi: 10.1002/ajmg.b.31246. [DOI] [PubMed] [Google Scholar]
- 24.Sankaran VG, et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365(9):807–814. doi: 10.1056/NEJMoa1103070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.