

Abstract
To identify kinases that regulate integrin recycling, we have immunoprecipitated αvβ3 integrin from NIH 3T3 fibroblasts in the presence and absence of primaquine (a drug that inhibits receptor recycling and leads to accumulation of integrins in endosomes) and screened for co-precipitating kinases. Primaquine strongly promoted association of αvβ3 integrin with PKD1, and fluorescence microscopy indicated that integrin and PKD1 associate at a vesicular compartment that is downstream of a Rab4-dependent transport step. PKD1 association was mediated by the C-terminal region of the β3 integrin cytodomain, and mutants of β3 that were unable to recruit PKD1 did not recycle in a PDGF-dependent fashion. Furthermore, suppression of endogenous PKD1 levels by RNAi, or overexpression of catalytically inactive PKD1 inhibited PDGF-dependent recycling of αvβ3 from early endosomes to the plasma membrane and blocked recruitment of αvβ3 to newly formed focal adhesions during cell spreading. These data indicate that PKD1 influences cell migration by directing vesicular transport of the αvβ3 integrin heterodimer.
Keywords: focal adhesion, integrin, PKD1, Rab4, recycling
Introduction
Integrins are heterodimeric transmembrane receptors that mediate interactions between cells and the extracellular matrix (ECM). Integrins transmit signals across the plasma membrane, and engagement of integrins with the ECM activates numerous signalling pathways that influence a range of biological processes, including cell proliferation, migration and apoptosis (Schwartz and Baron, 1999). Correspondingly, intracellular signalling pathways can influence integrin function by increasing the affinity for ligand (affinity modulation) (Shattil, 1999) and initiating clustering of active integrins into macromolecular structures such as focal adhesions/complexes and fibrillar adhesions (Zamir and Geiger, 2001). The rho subfamily of GTPases have a well-established role in regulating integrin clustering, and phosphatidylinositol 3-kinases (PI(3)Ks), H-ras (Kinashi et al, 2000), PKCs (Kolanus and Seed, 1997) and more recently ERK (Roberts et al, 2003) (possibly via the activation of calpain) are also implicated in the activation of integrins. Clearly any kinase found to associate with an integrin would be of interest in determining the pathways that regulate integrin function. Indeed PKC-α, src family kinases, pp125FAK (Eliceiri et al, 2002), ERK (Ahmed et al, 2002; Roberts et al, 2003) and ILK (Delcommenne et al, 1998) have all been shown to associate physically with integrins and to regulate their function.
Kinases are now known to influence the distribution and function of integrins by controlling their transport through the endocytic and receptor recycling pathways. Internalisation of β1 integrins is regulated by PKC-α (Ng et al, 1999), and PKC-ɛ (Ivaska et al, 2002) has a role in controlling an endosomal transport step that permits recycling of integrin to the plasma membrane. In our laboratory we have found that integrins recycle in two time domains, and that the signalling pathways that regulate these processes are divergent (Roberts et al, 2001). Under basal conditions (30 min of serum starvation), internalised α5β1 and αvβ3 heterodimers are transported through early endosomes to the perinuclear recycling compartment, and return to the plasma membrane by a pathway that is regulated by Rab11 and PKB/Akt via the inhibition of GSK-3β (Roberts et al, 2004). This is termed ‘long-loop' recycling. Following stimulation with PDGF, αvβ3 (but not α5β1) is rapidly recycled directly to the plasma membrane without the involvement of Rab11, thus engaging in a ‘short-loop' cycle of endocytosis and recycling. As the PKB/GSK-3β axis has only minimal effect on ‘short-loop' recycling of αvβ3 (Roberts et al, 2004), we are interested in elucidating the kinase cascades responsible for this event.
The association of kinases with integrins can be either constitutive (i.e. bound to the heterodimer at all times and at numerous locations including endosomes and focal adhesions), as is the case for Src family kinases (Arias-Salgado et al, 2003) and Syk (Woodside et al, 2001), or regulated. Kinases mediating growth factor-stimulated recycling might be predicted to associate with the integrin at the cytoplasmic face of an intracellular vesicle, and then dissociate following its delivery to the plasma membrane. To identify signalling moieties that form such transient associations with integrins, we have treated cells with primaquine, a drug that inhibits receptor recycling and leads to accumulation of integrin in endosomes (Roberts et al, 2001), and then screened for kinases that co-precipitate with αvβ3 integrin. We have found that addition of primaquine strongly promotes association of αvβ3 with PKD1, a kinase related to PKC that is proposed to play a role in the regulation of fission of transport vesicles from the trans-Golgi network (TGN). In this paper, we present data that support a critical role for PKD1 in regulating recycling of αvβ3 integrin via the ‘short loop', and on recruitment of the integrin to newly forming focal adhesions.
Results
Association of kinases with integrins following addition of primaquine
We have previously shown that the function of αvβ3 integrin is influenced by a growth factor-regulated recycling pathway directing the integrin to the plasma membrane (Roberts et al, 2001). We hypothesised that kinases responsible for this would be likely to associate with the heterodimer at an endosomal compartment, and then dissociate following delivery to the plasma membrane. As the receptor recycling inhibitor primaquine causes cycling integrins to accumulate rapidly within endosomes, we have sought to identify kinases that associate with αvβ3 integrin on the cytoplasmic face of these endosomal vesicles following the treatment of cells with this drug. NIH 3T3 fibroblasts were serum-starved for 30 min and then challenged with PDGF in the presence and absence of primaquine. Cells were lysed in a buffer containing 0.5% (v/v) Triton X-100 and 0.25% (v/v) Igepal CA-630, and αvβ3 heterodimers were isolated by immunoprecipitation. These immunoprecipitates were analysed using the Kinexus 1.2 protein kinase screen that tests for numerous signalling kinases (Pelech and Zhang, 2002). A selection of kinases co-immunoprecipitated with αvβ3, and prominent among these were PKC-ζ, PKC-β and ERK1 (Figure 1A). A brief treatment (5 min) with primaquine altered the profile of αvβ3-associated kinases such that there was a substantial enrichment of PKD1/PKCμ and, to a lesser extent, PKCɛ. We verified these results by Western blotting and confirmed that PKD1 associated with αvβ3 only in the combined presence of PDGF and primaquine, and that this kinase did not co-immunoprecipitate with α5β1 (Figure 1B). Moreover, we found that PKC-ζ associated with both αvβ3 and α5β1 heterodimers and that this was not augmented by primaquine (Figure 1B), indicating that this aPKC was a less promising candidate for a specific regulator of growth factor-regulated αvβ3 recycling than PKD1 and PKCɛ. These analyses also revealed that GFP-PKD1 associated with αvβ3 immunoprecipitates in a fashion that was dependent on the prior incubation of cells with PDGF and primaquine (Figure 1C), indicating that this GFP-tagged kinase behaves similarly to the endogenous kinase.
Figure 1.
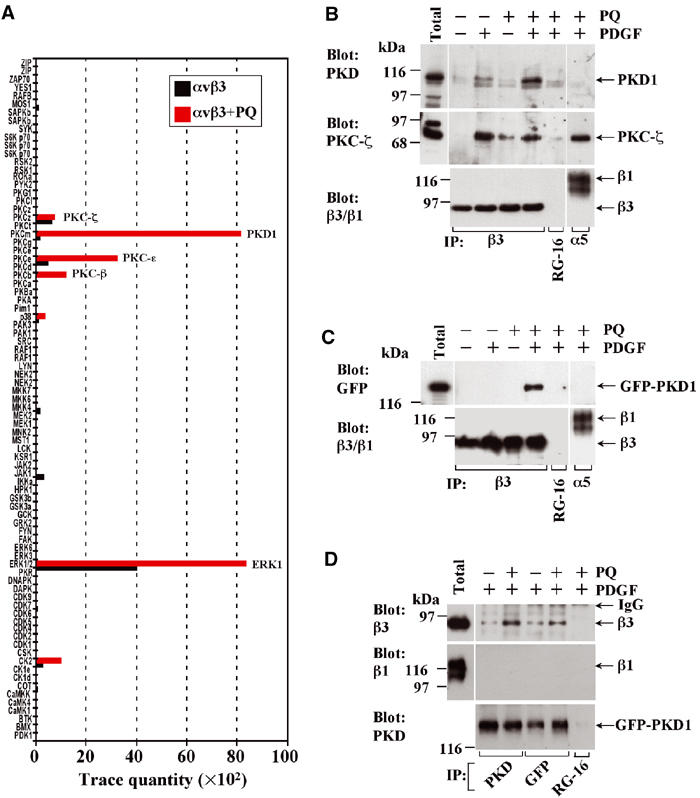
Association of kinases with integrins following addition of primaquine. NIH 3T3 fibroblasts were transfected with αvβ3 or α5β1 integrin alone (A, B), or αvβ3 in combination with GFP-PKD1 (C, D). Cells were serum-starved, challenged with 10 ng/ml PDGF-BB for 10 min in the absence and presence of 0.6 mM primaquine (PQ), and lysed in a buffer containing 0.5% (v/v) Triton X-100 and 0.25% (v/v) Igepal CA-630. Primaquine was added 5 min following PDGF addition and 5 min prior to lysis. Lysates were immunoprecipitated (IP) using magnetic beads coupled to monoclonal antibodies against human β3 or α5 integrin (A–C), PKD or GFP (D), or control antibody (RG-16; B–D). Immobilised material was analysed using the Kinexus 1.2 protein kinase screen (A) or Western blotting (B–D).
To further confirm this association, we expressed GFP-PKD1 and immunoprecipitated the kinase from cells that had been treated with PDGF and primaquine using antibodies recognising either PKD1 or GFP. Western blotting of these immunoprecipitates indicated that β3 (but not β1) integrin co-precipitated with GFP-PKD1 but not with a control antibody and that addition of primaquine promoted this association (Figure 1D).
Inhibition of receptor recycling promotes the accumulation of PKD1 and αvβ3 integrin at intracellular vesicles
We studied the intracellular distribution of αvβ3 and GFP-PKD1 using fluorescence microscopy. Following treatment with PDGF, GFP-PKD1 was observed in a diffuse cytoplasmic distribution and did not appreciably colocalise with αvβ3 (Figure 2A). However, 5 min following the addition of primaquine, the integrin and PKD1 became closely colocalised in a set of subplasmalemmal vesicles (Figure 2B). It is notable that this brief exposure to primaquine was sufficient to disassemble focal adhesions completely, highlighting the dynamic nature of these structures and the speed with which integrins are trafficking through the endosomal pathway (the internalisation of αvβ3 integrin has a t1/2 of between 5 and 10 min; Roberts et al, 2001). A 10 min exposure to primaquine induced more extensive colocalisation of αvβ3 with PKD1 in vesicles that were distributed throughout the cell (Figure 2C). Longer exposure to primaquine caused cell-rounding and detachment at a rate that correlated with the internalisation of matrix receptors required for cell adhesion (not shown).
Figure 2.
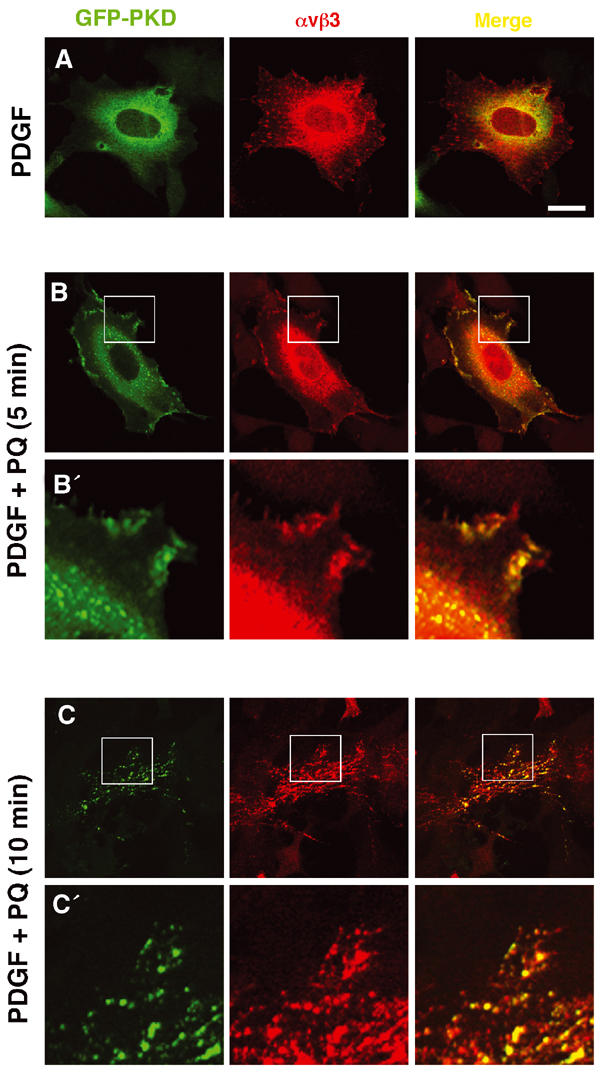
Colocalisation of PKD1 and αvβ3 integrin at intracellular vesicles following inhibition of receptor recycling. NIH 3T3 fibroblasts were transfected with αvβ3 integrin in combination with GFP-PKD1. Cells were serum-starved and challenged with 10 ng/ml PDGF-BB in the absence (A) and presence (B, C) of 60 μM primaquine (PQ) for 5 min (B) or 10 min (C). Following fixation, cells were permeabilised in 0.2% (v/v) Triton X-100 and stained for β3 integrin by indirect immunofluorescence using a Texas red-conjugated secondary antibody (red) and GFP-PKD1 was visualised directly (green). Colocalisation of the two fluorophores is shown in yellow in the merged panels. The areas encompassed by the white boxes in (B) and (C) are enlarged four-fold in (B′) and (C′), respectively. Images are presented as a single confocal optical slice centred approximately 0.5 μm above the plane of the substratum. Bar, 10 μm.
Role of the C-terminal region of β3 cytodomain in recruitment of PKD1
A number of studies have shown that the cytodomains of integrin β subunits are responsible for recruiting signalling kinases and cytoskeletal proteins to the heterodimer (LaFlamme et al, 1997). As initial pull-down experiments indicated that GST-β3737–762 (Figure 3A) was able to recruit PKD1 (Figure 3B), we proceeded to further define regions of the cytodomain responsible for this association. We employed an assay in which GST-cytodomain fusion proteins were immobilised on the surface of microtitre wells. These were then incubated with cell lysates, and bound PKD1 was detected by ELISA. Using this assay, we found that GST-β3727–748 did not bind to PKD1, whereas GST-β3749–762 bound to the kinase to the same extent as GST-β3747–762 (Figure 3C). This indicated that the PKD1-binding site resided exclusively within the C-terminal 14 amino acids of the integrin.
Figure 3.
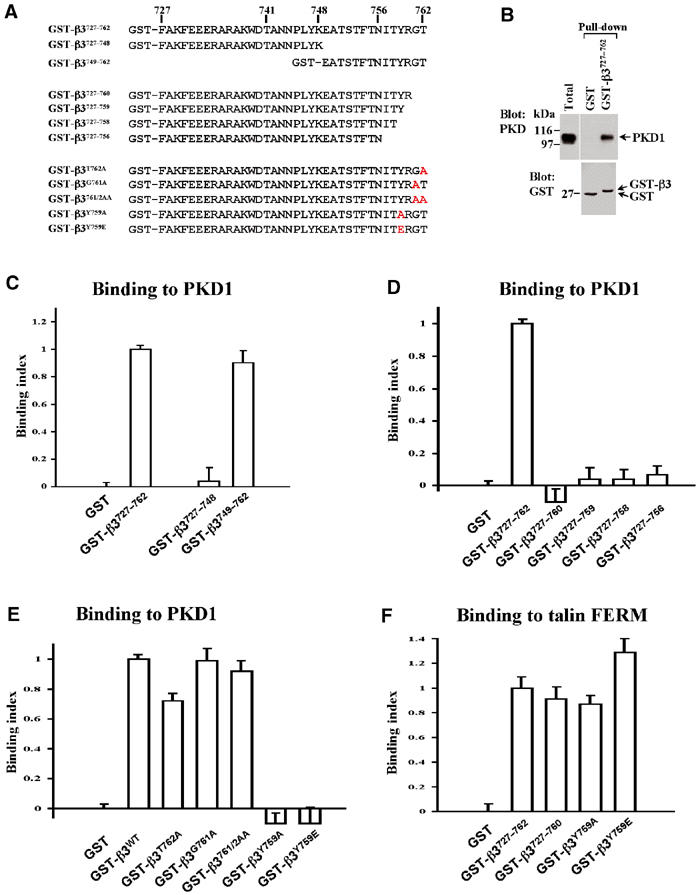
Association of PKD1 with the β3 integrin cytodomain. (A) Description and nomenclature of the GST-β3 integrin cytodomain fusion proteins employed for the present study. GST fusion proteins were expressed in E. coli strain BL-21 and purified as described previously (Woods et al, 2002). (B) Lysates from NIH 3T3 fibroblasts were incubated with magnetic beads conjugated to GST, or GST-β3727–762. Immobilised material was analysed by Western blotting with anti-PKD1 (upper panel), and the loading of the GST fusion proteins was confirmed by Western blotting for GST (lower panel). (C–F) GST and GST-cytodomain fusion proteins were immobilised on the surface of microtitre wells. These were then incubated with either cell lysate (C–E) or biotinylated talin-FERM domain (residues 86–410; mouse sequence) (F). Bound PKD1 was detected by ELISA using antibodies recognising PKD1 (C–E) and binding of the biotinylated talin-FERM domain was detected with HRP-conjugated streptavidin (F).
To highlight individual residues necessary for recruiting PKD1, we constructed a series of β3 cytodomain mutants in which amino acids at or near the C-terminus were either deleted or replaced with alanines (Figure 3A). Although removal of Gly761 and Thr762 from the C-terminus ablated PKD1 binding (Figure 3D), substitution of these residues for alanine did not significantly reduce kinase recruitment (Figure 3E). Replacement of Tyr759 with alanine, however, ablated association with PKD1 (Figure 3E). Tyr759 is known to be phosphorylated in vivo and this reduces binding of β3 integrins to certain cytoplasmic ligands, such as Shc (Cowan et al, 2000). We, therefore, investigated the effect of a phosphomimetic substitution at this position. Indeed, replacement of Tyr759 with glutamic acid completely ablated kinase binding (Figure 3E), indicating the requirement for an unphosphorylated tyrosine at this position for efficient recruitment of PKD1. Taken together, these data indicate that (a) the PKD1-binding site resides exclusively within the C-terminal 14 amino acids of the β3 cytodomain, (b) an unphosphorylated Tyr at position 759 is essential for PKD1 recruitment and (c) Tyr759 must be flanked C-terminally by three amino acids. However, the presence of Gly761 and Thr762 is not mandatory, as alanines at these positions will suffice.
Talin is now established to be recruited to focal adhesions via an interaction between its FERM domain and the membrane proximal NXXY motif in the β3 integrin cytodomain (Ulmer et al, 2003). We found that β3 mutants that did not recruit PKD1 (β3727–760, β3Y759A and β3Y759E), bound strongly to the talin FERM domain (Figure 3F), indicating that these mutations disrupted neither the overall structure and/or folding of the cytodomain nor the function of the membrane proximal NXXY motif.
Catalytically inactive PKD1 associates with αvβ3 in the absence of primaquine and traps the integrin in intracellular vesicles
Rey et al (2001) have reported that following agonist stimulation kinase-dead PKDs associate with cellular membranes and become trapped, presumably due to a requirement for substrate or autophosphorylation in membrane dissociation of the kinase. We employed a mutant of PKD1, PKDD733A, that is catalytically inactive, and determined the ability of this to associate with immunoprecipitated αvβ3 integrin. We found that PKDD733A had a greatly increased ability to co-immunoprecipitate with αvβ3 and, interestingly, did so in the absence of primaquine (Figure 4A and B). We studied the localisation of GFP-PKDD733A with respect to αvβ3 integrin by fluorescence microscopy. Consistent with the immunoprecipitation data, we found that GFP-PKDD733A colocalised closely with αvβ3 in a set of subplasmalemmal vesicles. These often appeared to have a tubular morphology and were observable in both the absence (Figure 5A) and presence (Figure 5B) of primaquine. In some cells, PKDD733A and αvβ3 integrin colocalised in vesicles that were more evenly distributed throughout the cell (Figure 5C), similar to that observed following longer (10 min) exposures to primaquine (Figure 2C). To confirm that association of PKD and αvβ3 was not restricted to a single immortalised cell line, we investigated the distribution of PKD1 and αvβ3 in primary cultured mouse embryo fibroblasts (MEFs). Indeed, GFP-PKDD733A and αvβ3 integrin colocalised closely in MEFs, and this was seen to occur in subplasmalemmal vesicles (Figure 5D), indicating that these proteins are associated with each other in these primary cultured cells. We have also performed these experiments with another mutant of PKD1, PKDS744/748A, that lacks kinase activity due to the substitution of critical phosphorylation sites in the kinase activation loop. We found this to behave similarly to PKDD733A in both αvβ3 immunoprecipitation and colocalisation experiments (not shown).
Figure 4.
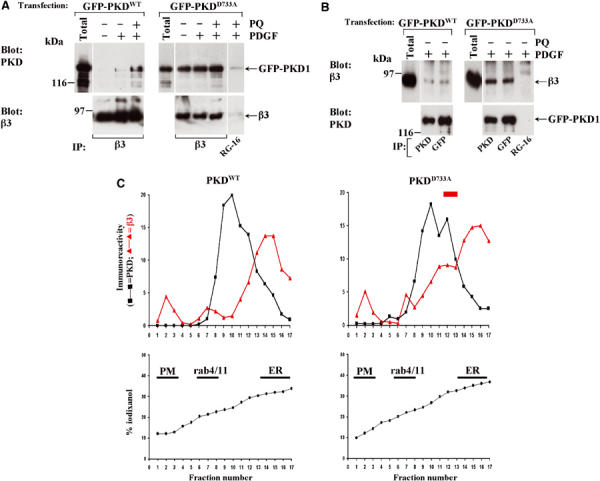
Catalytically inactive PKD1 associates with αvβ3 in the absence of primaquine. (A, B) NIH 3T3 fibroblasts were transfected with αvβ3 integrin in combination with GFP-PKD1 or GFP-PKDD733A. Cells were serum-starved, challenged with 10 ng/ml PDGF-BB for 10 min in the absence and presence of 0.6 mM primaquine (PQ), lysed and immunoprecipitated (IP) using magnetic beads coupled to monoclonal antibodies against human β3 integrin, PKD1, GFP or control antibody (RG-16). Immobilised material was analysed by Western blotting. (C) Cells were transfected with PKD1 or PKDD733A and fractionated on 10–40% (v/v) iodixanol gradients. The linearity of the gradient was confirmed by refractometry (•). The sedimentation profile of PKD1 (▪) and β3 integrin (▴; red line) was determined by Western blotting followed by densitometric scanning. These profiles represent the mean values from a pool of four experiments. The sedimentation positions of intracellular compartments were determined by Western blotting with antibodies against the following markers: calreticulin (ER), Rab11 and Rab11 (endosomes) and biotinylated cell surface proteins (plasma membrane).
Figure 5.
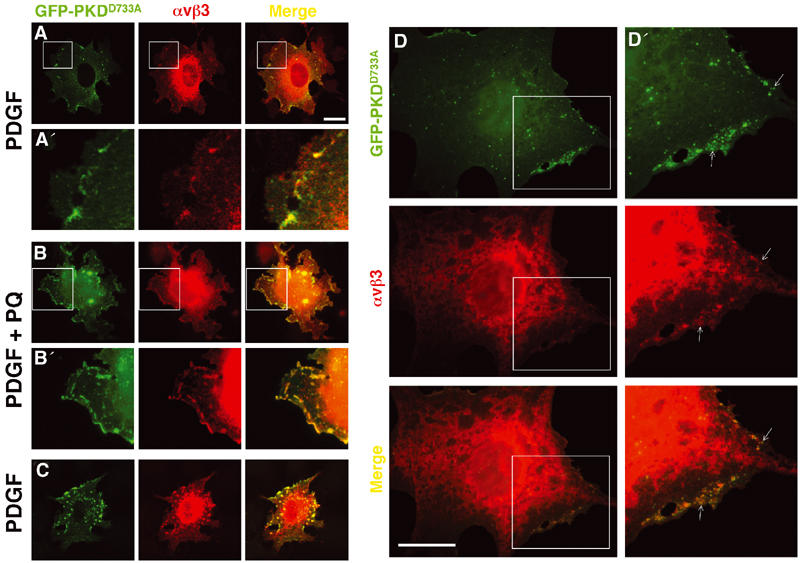
Colocalisation of αvβ3 integrin with catalytically inactive GFP-PKDD733A. NIH 3T3 fibroblasts (A–C) or primary cultured MEFs (D) were transfected with αvβ3 integrin in combination with GFP-PKDD733A. Cells were serum-starved and challenged with 10 ng/ml PDGF-BB in the absence (A, C, D) and presence (B) of 60 μM primaquine (PQ). Cells were stained for β3 integrin (red) and GFP-PKD1 was visualised directly (green). Colocalisation of the two fluorophores is shown in yellow in the merged panels. The areas encompassed by the white boxes are enlarged four-fold in (A′) and (B′) and two-fold in (D′). Bar, 10 μm.
To obtain biochemical data supporting the colocalisation of PKD1 and αvβ3 at an endomembrane compartment, we employed iodixanol density gradient centrifugation. On these gradients, αvβ3 integrin was seen to reside primarily in three membrane fractions: the plasma membrane (PM), Rab4- and Rab11-positive endosomes and the endoplasmic reticulum (ER) and these did not co-sediment appreciably with overexpressed wild-type PKD1 (PKDWT) (Figure 4C, left panels). In contrast, following expression of PKDD733A, a proportion (≈20–30%) of cellular αvβ3 co-sedimented with the inactive kinase in a membrane fraction that was buoyant near the mid-point of the gradient (Figure 4C, right panels, red bar). Taken together, these data indicate that PKD1 is recruited to αvβ3 at a subplasmalemmal compartment that is distinct from the plasma membrane, the Rab4- or Rab11-positive endosomes and the ER, and that dissociation of the integrin–kinase complex requires the catalytic activity of PKD1.
PDGF-dependent αvβ3 recycling is dependent on PKD1
We used RNA interference (RNAi) to suppress cellular levels of PKD1. Expression of the PKD1-RNAi vector (mu6pro-PKD1) reduced levels of PKD1 by approximately six-fold, whereas empty vector (mu6pro) and a control hairpin RNA (mu6pro-CON) were ineffective in this regard (Figure 6A). We therefore determined the effect of mu6pro-PKD1 on recycling of αvβ3 from early endosomes to the plasma membrane using the ELISA-based method that we have employed previously (Roberts et al, 2001). Indeed, RNAi of PKD1 strongly opposed the ability of PDGF to stimulate integrin delivery to the plasma membrane (Figure 6B).
Figure 6.
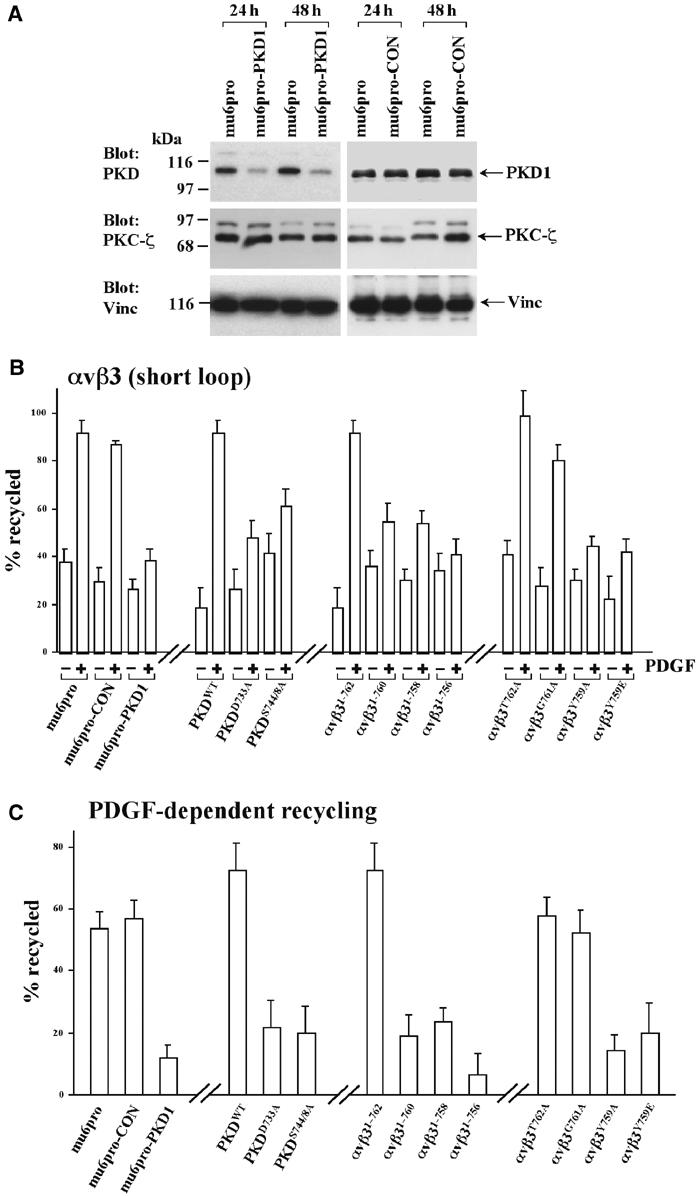
PDGF-stimulated recycling of αvβ3 integrin requires association with active PKD1. (A) NIH 3T3 fibroblasts were transfected with mu6pro, mu6pro-CON or mu6pro-PKD1 using the Amaxa ‘Nucleofector'. Cells were lysed 24 and 48 h following transfection and the lysates were probed for the presence of PKD1, PKC-ζ and vinculin (as a loading control) by Western blotting. (B, C) Cells were transfected with αvβ3 integrin or αvβ3 with mutated β3 subunits, in combination with the mu6pro RNAi vectors or PKD1 constructs as indicated. ‘Short-loop' recycling of αvβ3 in the presence and absence of 10 ng/ml PDGF-BB was determined as described previously (Roberts et al, 2001). Values are mean±s.e.m., n>5. To evaluate the PDGF-dependent component of recycling, the data from (B) are presented following subtraction of the appropriate basal values (C).
Catalytically inactive PKD1s display an enhanced ability to bind to αvβ3 at a vesicular compartment, indicating the possibility that these mutant kinases might oppose recycling of the integrin. Expression of PKDWT had little effect on the basal and PDGF-stimulated components of αvβ3 recycling (Figure 6B). In contrast, expression of catalytically inactive PKD1s, PKDD733A and PKDS744/748A, tended to raise the basal levels of recycling, but inhibited PDGF-driven integrin recycling (Figure 6B). To evaluate the PDGF-dependent component of recycling, we have presented the data from Figure 6B following subtraction of the appropriate basal values. This analysis revealed that either RNAi of PKD1, or expression of PKDD733A and PKDS744/748A suppressed this value by approximately 70% (Figure 6C).
The data in Figure 3 indicate that truncation of the β3 cytodomain or substitution of Tyr759 opposed the ability of the integrin to recruit PKD1. We tested the behaviour of αvβ3 heterodimers with mutated β subunits in the recycling assay. From this analysis, it was clear that the ability of an integrin to recycle in response to PDGF correlated closely with its capacity to bind PKD1. In this regard, αvβ3T762A and αvβ3G761A (which bind to PKD1) recycled efficiently following addition of PDGF (Figure 6C). In contrast, αvβ31–760, αvβ3Y759A and αvβ3Y759E (which do not bind to PKD1) had reduced PDGF-dependent recycling (Figure 6C).
In addition to ‘short-loop' recycling, αvβ3 also recycles in a ‘long loop', which proceeds via the perinuclear (Rab11-positive) recycling compartment. We have found that ‘long-loop' recycling of αvβ3 is not reduced by truncation of the β3 integrin subunit to Asn756 (a construct that does not associate with PKD1), nor by expression of PKDD733A (not shown). These data indicate that recruitment of active PKD1 is necessary for ‘short-loop' PDGF-driven recycling from early endosomes, but that this kinase is unlikely to contribute to ‘long-loop' recycling of integrins.
PKD and αvβ3 associate downstream of a Rab4-regulated step
As PDGF-stimulated recycling of αvβ3 requires Rab4 (Roberts et al, 2001), we tested the effect of expression of dominant-negative mutants of Rab4 on the association of αvβ3 with PKD1. Rab4S22N and Rab4N121I both abolished the ability of PKD1 to co-immunoprecipitate with αvβ3 (Figure 7A). Moreover, we were unable to detect convincing colocalisation of αvβ3 and PKDD733A following expression of Rab4S22N (Figure 7B). By contrast, expression of dominant-negative Rab11 and treatment of cells with wortmannin (both manipulations that target ‘long-loop' recycling) do not inhibit co-immunoprecipitation of integrin and PKD1 (Figure 7A).
Figure 7.
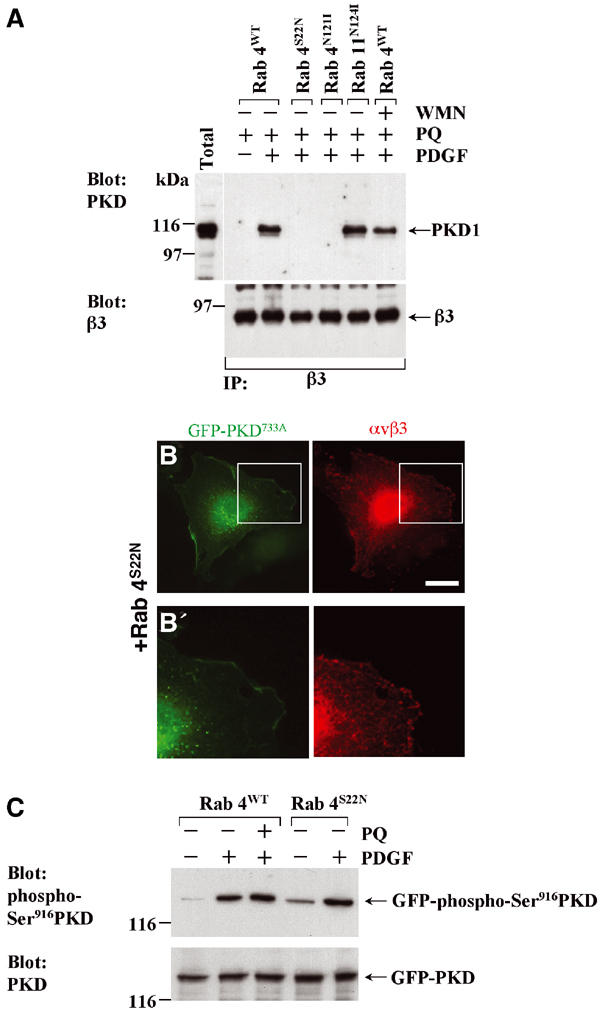
Association of αvβ3 with PKD1 requires the activity of Rab4. (A) NIH 3T3 fibroblasts were transfected with αvβ3 integrin in combination with Rab4, Rab4S22N, Rab4N121I or Rab11N124I. Cells were serum-starved in the absence and presence of 100 nM wortmannin (WMN), challenged with 10 ng/ml PDGF-BB in the presence of 0.6 mM primaquine (PQ), lysed and immunoprecipitated (IP) using magnetic beads coupled to monoclonal antibodies against human β3 integrin. Immobilised material was analysed by Western blotting. (B) Cells were transfected with αvβ3 integrin in combination with GFP-PKDD733A and Rab4S22N, serum-starved and challenged with 10 ng/ml PDGF-BB. Cells were stained for β3 integrin (red) and GFP-PKD1 was visualised directly (green). The areas encompassed by the white box in (B) is enlarged four-fold in (B′). Bar, 10 μm. (C) Cells were transfected with GFP-PKD1 in combination with Rab4 or Rab4S22N. Following serum starvation, the cells were challenged with 10 ng/ml PDGF in the absence or presence of 0.6 mM primaquine. The cellular content of GFP-PKD1 and phospho-Ser916GFP-PKD1 was determined by Western blotting.
To determine whether this effect of dominant-negative Rab4s was due to a requirement for Rab4 in PKD activation, we probed Western blots with an antibody that detects PKD only when phosphorylated at Ser916. As this residue is an autophosphorylation site, its phosphorylation can be taken as measure of activation of cellular PKD (Matthews et al, 1999a). PDGF clearly induced PKD autophosphorylation, as reported previously, and Rab4S22N did not oppose this (Figure 7C). These data suggest that Rab4 does not have a role in the activation of the PKD per se, but acts to mediate the transport of αvβ3 to a compartment where association with PKD1 is initiated. It should also be noted that primaquine did not alter cellular levels of phospho-Ser916PKD (Figure 7C) and is therefore unlikely to block integrin recycling by inhibiting PKD1, or any other signalling pathway leading to its activation.
A role for PKD1 in the delivery of αvβ3 to newly forming focal adhesions
Expression of a kinase inactive form of PKD1 has been recently shown to compromise fibroblast motility (Prigozhina and Waterman-Storer, 2004). As overexpression of inactive forms of kinases can result in ‘sideways' inhibition of other pathways under control of the same upstream kinases (Lawlor and Alessi, 2001), we wished to confirm the role of PKD1 in cell migration using RNAi. Cells expressing the control RNAi vector closed the wound by 24 h (Figure 8A and B). In contrast, cells expressing mu6pro-PKD1 had reduced migration, and were only able to achieve ≈50% wound closure after 24 h (Figure 8A and B). We therefore examined the distribution of αvβ3 on the surface of migrating cells following suppression of PKD1. In cells transfected with control vector, migrating cells displayed a typical fan-like morphology with αvβ3 localised to focal adhesions at the leading edge (Figure 8C). Following suppression of PKD1, migrating cells had altered morphology with αvβ3 being no longer concentrated at the leading edge, but present in focal adhesions that were distributed more uniformly on the lower surface of the cell (Figure 8C). Taken together, these data indicated that following RNAi of PKD1 cells migrated with reduced efficiency, and that this may be due to their inability to transport αvβ3 to newly forming focal adhesions at the leading edge.
Figure 8.
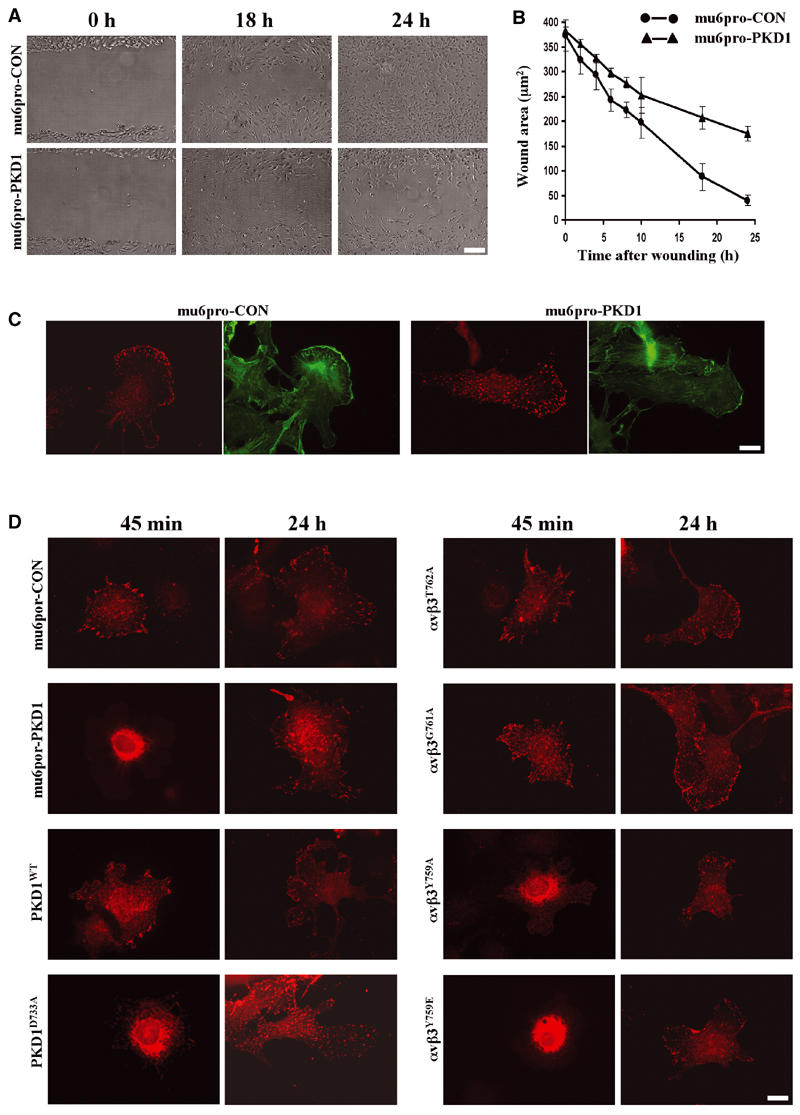
Incorporation of αvβ3 integrin into newly forming focal adhesions requires association with active PKD1. (A, B) NIH 3T3 fibroblasts were transfected with mu6pro-CON or mu6pro-PKD1 using the Amaxa Nucleofector and allowed to grow to confluence over 48 h. Following wounding, the cells were allowed to migrate in DMEM supplemented with 1% (v/v) FCS and 10 ng/ml PDGF-BB. Photographs of the wounds were taken at various times post-wounding and the degree of wound closure was determined by delineation of the wound envelope using the ‘NIH image' software (values are mean±s.e.m., n=10 fields). (C) Confluent monolayers of NIH 3T3 fibroblasts expressing αvβ3 integrin in combination with mu6pro-CON or mu6pro-PKD1 were wounded as for (A) and the cells were allowed to migrate for 4 h. Surface αvβ3 was visualised by indirect immunofluorescence (red) and cells were counterstained with phalloidin (green). Surface-only αvβ3 staining was obtained by addition of the primary antibody prior to detergent permeabilisation. Bar, 10 μm. (D) NIH 3T3 fibroblasts were transfected with αvβ3 integrin or αvβ3 with mutated β3 subunits, in combination with the PKD1-RNAi vector (mu6pro-PKD1) or appropriate control vectors (mu6pro, mu6pro-CON), PKD1 or catalytically inactive PKD as indicated. Following trypsinisation, cells were allowed to adhere to fibronectin-coated coverslips for 45 min or 24 h. β3 integrin was visualised by indirect immunofluorescence (red). Bar, 10 μm.
Work by Zaidel-Bar et al (2003) has shown that proteins are recruited to focal adhesions during cell migration in a defined sequence: with αvβ3 integrin and paxillin arriving early while zyxin and vinculin are recruited as relative latecomers. As the data in Figure 8C indicate that PKD1 might be required for transport of αvβ3 to nascent focal adhesions during migration, we examined recruitment of αvβ3 to focal adhesions formed shortly after attachment of cells to fibronectin. In cells expressing the control RNAi vector or wild-type PKD1, αvβ3 was recruited to focal adhesions within 45 min of plating (Figure 8D). In contrast, if PKD1 were suppressed by RNAi or if cells were expressing PKDD733A, little αvβ3 was recruited to newly formed focal adhesions, and most of the integrin was visible in the body of the cell (Figure 8D).
We tested the ability of αvβ3 heterodimers with mutated β subunits to be incorporated into newly forming focal adhesions. This revealed that the ability of an integrin to be recruited to newly formed focal adhesions correlated closely with its capacity to bind PKD1. For instance, αvβ3T762A and αvβ3G761A (which bind to PKD1) were rapidly incorporated into newly formed focal adhesions (Figure 8D). In contrast, αvβ3Y759A and αvβ3Y759E (that do not associate with PKD1) remained in the perinuclear region 45 min following adhesion of cells to fibronectin (Figure 8D). It should be noted that this reduction in recruitment of αvβ3 to nascent focal adhesions is not due to blockade of focal adhesion assembly per se as (a) under all these conditions αvβ3 was seen in focal adhesions if the cells were allowed to spread for 24 h (Figure 8D), and (b) the level of paxillin in newly formed focal adhesions is unaffected by expression of PKDD733A or RNAi of PKD1 (not shown).
These data support the proposal that association of active PKD1 with αvβ3, and its consequent recycling via a PKD1-dependent pathway is necessary to recruit rapidly this integrin into newly forming focal adhesions during cell spreading and migration.
Discussion
Previous studies from our laboratory indicate that αvβ3 is rapidly transported from early endosomes to the plasma membrane in response to PDGF (Roberts et al, 2001). To identify signalling moieties that mediate this, we screened for kinases immunoprecipitating specifically with αvβ3 following blockade of integrin recycling. We report that PKD1 was particularly abundant in these αvβ3 immunoprecipitates, and fluorescence microscopy confirmed that αvβ3 and PKD1 associate at a vesicular compartment that is likely to be downstream of a Rab4-dependent transport step. The interaction between PKD1 and αvβ3 involved the C-terminal region of the β3 cytodomain and mutants of β3 that were unable to recruit PKD1 were not recycled to the plasma membrane in response to PDGF. Suppression of endogenous PKD1 by RNAi or expression of catalytically inactive PKDs also blocked PDGF-driven αvβ3 recycling, and opposed the delivery of αvβ3 to newly forming focal adhesions/complexes.
PKD1·αvβ3 association
Kinases that associate with integrin β subunit cytoplasmic tails play a role in regulating integrin function. Indeed, a number of studies have reported PKCα to be localised at sites of integrin contact with the ECM, and also to vesicles involved in integrin trafficking (Ng et al, 1999). More recently, association of β1 integrins with PKCα has been shown to involve a direct interaction between the kinase and regions of the β1 cytodomain that correspond to the membrane proximal and distal NPXY motifs (Parsons et al, 2002). A number of factors led us to propose that PKD1 associates with β3 integrin in a different way from that reported for β1 and PKCα. Firstly, we do not find PKD1 to co-immunoprecipitate with α5β1 integrin. Secondly, our data show that mutation of the extreme membrane distal NXXY motif of β3 is sufficient to abrogate PKD binding (PKCα requires the disruption of both membrane proximal and distal NPXY motifs), and thirdly it is necessary to use n-octyl-β-D-glucopyranoside as a detergent to demonstrate β1 integrin·PKCα binding (not shown), whereas the β3 integrinPKD1 complex survives cell lysis in the somewhat harsher combination of Triton X-100 and Igepal CA-630.
Other kinases bind directly to integrin β cytodomains and these include ILK (Delcommenne et al, 1998), pp125FAK (Eliceiri et al, 2002), and those of the Src (Arias-Salgado et al, 2003) and Syk (Woodside et al, 2001) families. ILK and pp125FAK bind to β1 and β3 (and possibly other) integrins, whereas p60src and Syk require the extreme C-terminal 14 amino acids of β3 (and not β1), and this association survives lysis in Igepal CA-630-containing buffers (Arias-Salgado et al, 2003). Thus the region of β3 that associates with p60src and Syk appears to be similar to that required for recruiting PKD1. However, the substitution of Tyr759 for Phe in the β3 cytodomain has no effect on the interaction of β3 integrin with c-Src (Arias-Salgado et al, 2003), indicating that the precise determinants for binding this tyrosine kinase differ from those that permit recruitment of PKD1. Additionally, it must be noted that studies on the p60src·β3 integrin complex have been performed in platelets that express high levels of Src family kinases (Obergfell et al, 2002), or in CHO cells following overexpression of these kinases (Arias-Salgado et al, 2003). The fibroblasts employed for the present study do not express high levels of src family kinases, and accordingly we do not find p60src to be detectable in αvβ3 immunoprecipitates unless the kinase is overexpressed (not shown). Preliminary data suggest that bindings of PKD1 and p60src to β3 are mutually exclusive, and it will be interesting to determine the respective contributions of these two kinases to β3 integrin function in a cell type that expresses significant quantities of both PKD1 and p60src.
The data presented in this paper do not resolve whether the association of PKD1 and αvβ3 is direct. Proteins that are capable of binding directly to the β3 cytodomain may serve to link PKD to the integrin. In this regard, the PKC-adaptor protein, RACK1, certainly binds to PKCɛ to mediate its association with β1 integrin (Besson et al, 2002), and it is possible that RACK may bind to PKD1. However, due to the fact that RACK binds to a membrane proximal Kθθθ motif that is absent in our GST-β3 integrin cytodomain fusion proteins (Liliental and Chang, 1998), this is unlikely to be the mechanism by which αvβ3 recruits PKD1.
PKD1, integrin traffic and cell migration
Previous studies from our laboratory show that the PKB/GSK3β axis has a strong influence on ‘long-loop' recycling of integrins, but only minimal effect on ‘short-loop' recycling of αvβ3 in response to PDGF (Roberts et al, 2004). Conversely, PKD1 appears to be responsible for the majority of the ‘short-loop' recycling, but does not regulate the ‘long loop', indicating that there are key differences in the cellular machinery that controls these two recycling pathways. So how might PDGF act via PKD1 to regulate αvβ3 recycling? It is clear that PDGF invokes the activity of both phospholipase C and PKC to activate PKD1 in fibroblasts (Rey et al, 2001). Indeed, we find that treatment with either U73122 (phospholipase C inhibitor) or GF109203X (PKC inhibitor) completely inhibits recruitment of PKD1 to αvβ3, and these agents correspondingly block the ability of PDGF to drive integrin recycling (not shown). This is not surprising, as it has been known for some time that the PLC/DAG/PKC pathway plays a major role in PKD activation, and that PKC is required for this. More recently, it has been shown that PKCɛ activates PKD (Waldron and Rozengurt, 2003), and the fact that we find PKCɛ to co-precipitate with αvβ3 is consistent with a role for this isoform of PKC in activating PKD1 and recruiting it to an integrin-recycling vesicle. So what might this compartment be? Although the PKD1·αvβ3 integrin compartment has a subplasmalemmal tubulo-vesicular morphology, we have not been able to demonstrate convincing colocalisation of PKD with a number of endosomal markers (Rab GTPases, EEA1 and the transferrin receptor), even following primaquine treatment. PKD has been shown to have a role in regulating transport from the TGN to the plasma membrane (Liljedahl et al, 2001). Prigozhina and Waterman-Storer (2004) have recently proposed that PKD1-mediated transport from the TGN to the leading edge of migrating cells delivers a regulatory molecule key to cell migration, and these authors suggest that this may be an integrin. As we provide clear evidence of a role for PKD1 in the return of endocytosed integrin to the plasma membrane, it is possible that αvβ3 recycles via a Rab4-dependent step that delivers it to the TGN or another post-Golgi compartment that is involved with anterograde vesicular transport.
PKD1 has been localised to sites of active matrix proteolysis during the invasion of breast cancer cells into gelatin (Bowden et al, 1999). The soluble matrix protease, MMP-2, and its cognate activator, MT1-MMP, have been shown to bind to αvβ3 integrin (Deryugina et al, 2001) and, moreover, MT1-MMP is known to recycle to the plasma membrane via the TGN (Wang et al, 2004). This raises the exciting possibility that PKD1 may act to target not only αvβ3 integrin but also matrix metalloproteases to invadopodia and other sites of matrix invasion during angiogenesis and metastasis.
In conclusion, our studies have identified an association between an integrin (αvβ3) and a kinase (PKD1) that occurs in a compartment likely to represent a vesicular recycling intermediate. We propose that PKD1 regulates the vesicular transport of the integrin, which, in turn, influences its incorporation into focal adhesions. These data highlight the need to characterise the signalling machinery involved in integrin recycling, in order to fully understand complex cellular processes such as invasion and metastasis.
Materials and methods
Materials
Monoclonal mouse anti-human β3 integrin (clone VI-PL2) and α5 integrin (clone VC5) were purchased from Pharmingen (San Diego, CA). Polyclonal rabbit anti-PKCμ (sc-639) was from Santa Cruz Biotechnology (Santa Cruz, CA), and anti-phospho-Ser916PKD1 was as described by Matthews et al (1999a). The sample preparation and parameters of the Kinexus protein kinase screen were as described by Pelech and Zhang (2002). Texas red-conjugated anti-mouse immunoglobulins were from Southern Biotechnology (Birmingham, AL). Magnetic sheep anti-mouse IgG Dynabeads (Dynal, Oslo, Norway) and bovine serum albumin (BSA) were from First Link (UK) Ltd. NHS-SS-biotin (21331) and enhanced chemiluminescence reagents (ECL) were from Pierce and Warriner Ltd (Chester, Cheshire, UK). Cell culture medium and Maxisorb 96-well plates were from Life Technologies (Rockville) and fetal calf serum (FCS) was from Sera-Q (Tunbridge Wells, Kent, UK). Fugene 6 transfection reagent was from Roche Diagnostics GmbH (Mannheim, Germany). PDGF-BB (100-14B) was from PreproTech Inc. (Rocky Hill, NJ). Streptavidin-conjugated horseradish peroxidase (HRP) was from Amersham Biosciences (Little Chalfont, Bucks). Primaquine diphosphate (16039-3) was from Aldrich Chem. Co. (Milw., WI). Iodixanol was purchased as Optiprep from Axis-Shield PoC AS (Oslo, Norway). All other reagents were purchased from Sigma Chemical Co (Poole, Dorset, UK).
Expression plasmids
αv, β3, α5 and β1 integrins, and Rab4, S22NRab4, N121IRab4 and N124IRab11 were in pcDNA3 as described by Roberts et al (2001). GFP-PKD1, GFP-PKDD733A and GFP-PKDS744/748A were in the pEF-plink2-GFPC3 expression vector and are as described in Matthews et al (1999b). The mU6pro vector was used for the expression of RNA duplexes (Vojtek et al, 2003). A gene-specific sequence exclusive to murine PKD1 was targeted (available on request). All plasmids were purified by CsCl banding prior to transfection.
Cell culture and transfection
NIH 3T3 mouse fibroblasts and primary cultures of MEFs were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% (v/v) FCS and 100 U/ml penicillin, 100 μg/ml streptomycin and 0.25 μg/ml amphotericin B at 37°C with 10% CO2. Cells were grown to 50% confluence, fed with fresh DMEM containing 10% (v/v) FCS and transfected using Fugene 6 according to the manufacturer's instructions. The ratio of Fugene 6 to DNA was maintained at 3 μl Fugene 6:1 μg DNA. Immunoprecipitations, integrin recycling and cell-spreading assays were carried out 24 h post-transfection.
For the RNAi studies, transfections were carried out using the Amaxa ‘Nucleofector' system. Briefly, cells were grown to 80% confluence, removed by trypsinisation, washed in PBS and resuspended in Amaxa solution ‘R' together with 10 μg DNA of mu6pro, mu6pro-CON or mu6pro-PKD1. Following electroporation (in the ‘Nucleofector'; program T-20), the cells were re-plated in eight-well dishes. All RNAi experiments were carried out 48 h post-transfection.
Expression and purification of GST-integrin cytodomain fusion proteins
PCR-amplified DNA fragments corresponding to the indicated regions of the human sequence of β3 integrin were subcloned into the EcoRI–XhoI site of the pGEX-4T-1 vector. GST fusion proteins were expressed in Escherichia coli strain BL-21 and purified as described previously (Woods et al, 2002).
Immunoprecipitations and pull-downs
Cells were grown to 90% confluence, serum-starved for 30 min and treated with PDGF (10 ng/ml) for 10 min with or without the inclusion of primaquine (0.6 mM) where appropriate. Primaquine was added 5 min following PDGF addition and 5 min prior to lysis. Following this, cells were washed twice in ice-cold PBS, lysed and subjected to immunoprecipitation or pull-downs as described previously (Roberts et al, 2003).
Microtitre ELISA
GST-integrin cytodomain fusion proteins were bound at saturating concentrations to wells of microtitre plates (Immunol. 2; Dynatech Labs, Chantilly, VI) in 0.05 M Na2CO3 pH 9.6 at 4°C and the wells were blocked with PBS containing 0.1% (v/v) Tween 20 (PBS-T). For detection of binding to PKD1, lysates of NIH 3T3 fibroblasts (50 μl) were prepared as described above for immunoprecipitation, and were added to the wells and incubated for 1 h. After three washes with PBS-T, PKD1 was detected by serial incubations with rabbit anti-PKD and HRP-conjugated anti-rabbit IgG, followed by chromogenic reaction with ortho-phenylenediamine. For measurement of binding to talin, 50 μl of PBS-T containing biotinylated talin-FERM domain (residues 86–410) at 50 μM (a generous gift from Prof. DR Critchley and Mr BC Patel, University of Leicester) was added to the blocked microtitre wells and incubated at 4°C overnight. Bound talin-FERM domain was detected by incubation with streptavidin-conjugated HRP in PBS-T containing 1% (w/v) BSA for 1 h at 4°C.
Iodixanol gradient centrifugation
Cells were grown to 60% confluence, lysed and fractionated on a linear gradient of 10–40% iodixanol as described previously (Woods et al, 2002). Proteins were precipitated from the gradient fractions with 20% TCA and the migration positions of PKD1, β3 integrin and markers of intracellular compartments were determined by Western blotting with antibodies against the following markers: calreticulin (ER), Rab11 and Rab11 (endosomes) and biotinylated cell surface proteins (plasma membrane).
Integrin recycling assay and immunofluorescence
These were performed as described previously by Roberts et al (2001, 2003).
Cell migration assay
NIH 3T3 fibroblasts were transfected with mu6pro-CON or mu6pro-PKD1 using the Amaxa Nucleofector, plated on 3 cm tissue culture plates and allowed to grow to confluence over 48 h. Using a 1 mm thick pipette tip, crosses were scraped into the confluent monolayer. The cells were washed with PBS and allowed to migrate in DMEM supplemented with 1% (v/v) FCS and 10 ng/ml PDGF-BB at 37°C. Photographs of the wounds were taken at 0, 2, 4, 6, 8, 10, 18 and 24 h post-wounding. Wound closure was determined by quantification of these images using the ‘NIH image' software.
Cell spreading assays
Glass coverslips were coated overnight at 4°C with fibronectin (Sigma, F-1141) at a concentration of 10 μg/ml and then blocked with 2% (w/v) BSA. Cells were transfected with αvβ3 or αvβ3 with mutated β3 subunits in conjunction with the mu6pro RNAi vectors or GFP-PKD constructs. At 24 h following transfection, the cells were harvested by trypsinisation and collected by centrifugation in the presence of 20 μg/ml soyabean trypsin inhibitor. Cell suspensions were added to ligand-coated coverslips in serum-free DMEM containing 10 ng/ml PDGF-BB for 45 min or in DMEM containing 10% (v/v) FCS for 24 h. Attached cells were fixed and examined by immunofluorescence as described above.
Acknowledgments
This work was generously supported by grants from the Wellcome Trust and Cancer Research UK. We gratefully acknowledge Dave Critchley for critical reading and helpful comments on the manuscript. We thank Kul Sikand and Sam Wattam for their invaluable assistance with the fluorescence microscopy. We also thank Doreen Cantrell and Ulrica Marklund for the generous gift of cDNAs for PKD1, and Bipin Patel for the generous gift of biotinylated talin-FERM domain.
References
- Ahmed N, Niu J, Dorahy DJ, Gu X, Andrews S, Meldrum CJ, Scott RJ, Baker MS, Macreadie IG, Agrez MV (2002) Direct integrin alphavbeta6-ERK binding: implications for tumour growth. Oncogene 21: 1370–1380 [DOI] [PubMed] [Google Scholar]
- Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ (2003) Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc Natl Acad Sci USA 100: 13298–13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson A, Wilson TL, Yong VW (2002) The anchoring protein RACK1 links protein kinase Cepsilon to integrin beta chains. Requirements for adhesion and motility. J Biol Chem 277: 22073–22084 [DOI] [PubMed] [Google Scholar]
- Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC (1999) An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene 18: 4440–4449 [DOI] [PubMed] [Google Scholar]
- Cowan KJ, Law DA, Phillips DR (2000) Identification of shc as the primary protein binding to the tyrosine-phosphorylated beta 3 subunit of alpha IIbbeta 3 during outside-in integrin platelet signaling. J Biol Chem 275: 36423–36429 [DOI] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S (1998) Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA 95: 11211–11216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryugina EI, Ratnikov B, Monosov E, Postnova TI, DiScipio R, Smith JW, Strongin AY (2001) MT1-MMP initiates activation of pro-MMP-2 and integrin alphavbeta3 promotes maturation of MMP-2 in breast carcinoma cells. Exp Cell Res 263: 209–223 [DOI] [PubMed] [Google Scholar]
- Eliceiri BP, Puente XS, Hood JD, Stupack DG, Schlaepfer DD, Huang XZ, Sheppard D, Cheresh DA (2002) Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J Cell Biol 157: 149–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivaska J, Whelan RD, Watson R, Parker PJ (2002) PKC epsilon controls the traffic of beta1 integrins in motile cells. EMBO J 21: 3608–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinashi T, Katagiri K, Watanabe S, Vanhaesebroeck B, Downward J, Takatsu K (2000) Distinct mechanisms of alpha 5beta 1 integrin activation by Ha-Ras and R-Ras. J Biol Chem 275: 22590–22596 [DOI] [PubMed] [Google Scholar]
- Kolanus W, Seed B (1997) Integrins and inside-out signal transduction: converging signals from PKC and PIP3. Curr Opin Cell Biol 9: 725–731 [DOI] [PubMed] [Google Scholar]
- LaFlamme SE, Homan SM, Bodeau AL, Mastrangelo AM (1997) Integrin cytoplasmic domains as connectors to the cell's signal transduction apparatus. Matrix Biol 16: 153–163 [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 114: 2903–2910 [DOI] [PubMed] [Google Scholar]
- Liliental J, Chang DD (1998) Rack1, a receptor for activated protein kinase C, interacts with integrin beta subunit. J Biol Chem 273: 2379–2383 [DOI] [PubMed] [Google Scholar]
- Liljedahl M, Maeda Y, Colanzi A, Ayala I, Van Lint J, Malhotra V (2001) Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell 104: 409–420 [DOI] [PubMed] [Google Scholar]
- Matthews S, Iglesias T, Cantrell D, Rozengurt E (1999a) Dynamic re-distribution of protein kinase D (PKD) as revealed by a GFP-PKD fusion protein: dissociation from PKD activation. FEBS Lett 457: 515–521 [DOI] [PubMed] [Google Scholar]
- Matthews SA, Rozengurt E, Cantrell D (1999b) Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/protein kinase Cmu. J Biol Chem 274: 26543–26549 [DOI] [PubMed] [Google Scholar]
- Ng T, Shima D, Squire A, Bastiaens PI, Gschmeissner S, Humphries MJ, Parker PJ (1999) PKCalpha regulates beta1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J 18: 3909–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obergfell A, Eto K, Mocsai A, Buensuceso C, Moores SL, Brugge JS, Lowell CA, Shattil SJ (2002) Coordinate interactions of Csk, Src, and Syk kinases with [alpha]IIb[beta]3 initiate integrin signaling to the cytoskeleton. J Cell Biol 157: 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M, Keppler MD, Kline A, Messent A, Humphries MJ, Gilchrist R, Hart IR, Quittau-Prevostel C, Hughes WE, Parker PJ, Ng T (2002) Site-directed perturbation of protein kinase C–integrin interaction blocks carcinoma cell chemotaxis. Mol Cell Biol 22: 5897–5911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelech S, Zhang H (2002) Plasticity of the kinomes in monkey and rat tissues. Sci STKE 2002: PE50. [DOI] [PubMed] [Google Scholar]
- Prigozhina NL, Waterman-Storer CM (2004) Protein kinase D-mediated anterograde membrane trafficking is required for fibroblast motility. Curr Biol 14: 88–98 [DOI] [PubMed] [Google Scholar]
- Rey O, Young SH, Cantrell D, Rozengurt E (2001) Rapid protein kinase D translocation in response to G protein-coupled receptor activation. Dependence on protein kinase C. J Biol Chem 276: 32616–32626 [DOI] [PubMed] [Google Scholar]
- Roberts M, Barry S, Woods A, van der Sluijs P, Norman J (2001) PDGF-regulated rab4-dependent recycling of alphavbeta3 integrin from early endosomes is necessary for cell adhesion and spreading. Curr Biol 11: 1392–1402 [DOI] [PubMed] [Google Scholar]
- Roberts MS, Woods AJ, Dale TC, Van Der Sluijs P, Norman JC (2004) Protein kinase B/Akt acts via glycogen synthase kinase 3 to regulate recycling of alpha v beta 3 and alpha 5 beta 1 integrins. Mol Cell Biol 24: 1505–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts MS, Woods AJ, Shaw PE, Norman JC (2003) ERK1 associates with alpha(v)beta 3 integrin and regulates cell spreading on vitronectin. J Biol Chem 278: 1975–1985 [DOI] [PubMed] [Google Scholar]
- Schwartz MA, Baron V (1999) Interactions between mitogenic stimuli, or, a thousand and one connections. Curr Opin Cell Biol 11: 197–202 [DOI] [PubMed] [Google Scholar]
- Shattil SJ (1999) Signaling through platelet integrin alpha IIb beta 3: inside-out, outside-in, and sideways. Thromb Haemost 82: 318–325 [PubMed] [Google Scholar]
- Ulmer TS, Calderwood DA, Ginsberg MH, Campbell ID (2003) Domain-specific interactions of talin with the membrane-proximal region of the integrin beta3 subunit. Biochemistry 42: 8307–8312 [DOI] [PubMed] [Google Scholar]
- Vojtek AB, Taylor J, DeRuiter SL, Yu JY, Figureueroa C, Kwok RP, Turner DL (2003) Akt regulates basic helix–loop–helix transcription factor–coactivator complex formation and activity during neuronal differentiation. Mol Cell Biol 23: 4417–4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron RT, Rozengurt E (2003) Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J Biol Chem 278: 154–163 [DOI] [PubMed] [Google Scholar]
- Wang X, Ma D, Keski-Oja J, Pei D (2004) Co-recycling of MT1-MMP and MT3-MMP through the trans-Golgi network. Identification of DKV582 as a recycling signal. J Biol Chem 279: 9331–9336 [DOI] [PubMed] [Google Scholar]
- Woods AJ, Roberts MS, Choudhary J, Barry ST, Mazaki Y, Sabe H, Morley SJ, Critchley DR, Norman JC (2002) Paxillin associates with poly(A)-binding protein 1 at the dense endoplasmic reticulum and the leading edge of migrating cells. J Biol Chem 277: 6428–6437 [DOI] [PubMed] [Google Scholar]
- Woodside DG, Obergfell A, Leng L, Wilsbacher JL, Miranti CK, Brugge JS, Shattil SJ, Ginsberg MH (2001) Activation of Syk protein tyrosine kinase through interaction with integrin beta cytoplasmic domains. Curr Biol 11: 1799–1804 [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, Ballestrem C, Kam Z, Geiger B (2003) Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J Cell Sci 116: 4605–4613 [DOI] [PubMed] [Google Scholar]
- Zamir E, Geiger B (2001) Molecular complexity and dynamics of cell–matrix adhesions. J Cell Sci 114: 3583–3590 [DOI] [PubMed] [Google Scholar]