

Abstract
Mitogen-activated protein kinase (MAPK) cascades are involved in a variety of cellular responses including proliferation, differentiation, and apoptosis. We have developed an expression screening method to detect in vivo substrates of MAPKs in mammalian cells, and identified a membrane protein, linker for activation of T cells (LAT), as an MAPK target. LAT, an adapter protein essential for T-cell signaling, is phosphorylated at its Thr 155 by ERK in response to T-cell receptor stimulation. Thr 155 phosphorylation reduces the ability of LAT to recruit PLCγ1 and SLP76, leading to attenuation of subsequent downstream events such as [Ca2+]i mobilization and activation of the ERK pathway. Our data reveal a new role for MAPKs in a negative feedback loop in T-cell activation via threonine phosphorylation of LAT.
Keywords: LAT, MAP kinase, negative feedback, T cell
Introduction
Signal transduction systems in eukaryotic cells rely on a delicate balance between phosphorylation and dephosphorylation of intracellular molecules, enabling the cells to respond to extracellular stimuli rapidly and precisely. Among signaling molecules mediating such phosphorylation/dephosphorylation processes, the mitogen-activated protein kinase (MAPK) cascade is a conserved eukaryotic signaling module consisting of three components, MAPK, MAPK kinase (MAPKK), and MAPKK kinase (MAPKK-K); MAPKK-K phosphorylates and activates MAPKK, which in turn phosphorylates and activates MAPK (Ahn et al, 1992; Nishida and Gotoh, 1993; Cobb and Goldsmith, 1995). Activated MAPK in general translocates into nucleus, and regulates gene expression through direct or indirect phosphorylation of various transcription factors including AP-1 (Karin, 1995; Treisman, 1996; Yang et al, 1998; Hazzalin and Mahadevan, 2002). In mammalian cells, several distinct subgroups of MAPK family have been identified, including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 (Davis, 1994; Kyriakis and Avruch, 1996; Ip and Davis, 1998; Schaeffer and Weber, 1999).
ERK subgroup is implicated in diverse cellular processes such as proliferation, transformation, and cell differentiation, while JNK and p38 are activated primarily under stress conditions like apoptosis and inflammation (Robinson and Cobb, 1997; Ip and Davis, 1998; Lewis et al, 1998). Activation of these signaling cascades elicits variable responses in different cell types, presumably due to cell type-specific downstream targets. Although in vitro studies identified several proteins as putative targets for MAPKs, it still remains unclear whether these proteins are phosphorylated under physiological conditions. Moreover, conventional approaches using biochemical techniques have faced difficulties in identifying substrates when partially phosphorylated. Considering that protein kinases must interact with their substrates, interaction-based cloning by yeast two-hybrid system is a useful method for screening potential substrates (Yang et al, 1992). However, it is unable to identify signaling targets that require scaffolding proteins when interacting with their cognate protein kinases. Thus, development of an alternative approach is necessary to uncover physiological targets especially unique to distinct cell types.
Accumulating evidence suggests that MAPK family plays a pivotal role in regulating T-cell functions. JNK and p38 are synergistically activated by stimulation via T-cell receptor (TCR) and costimulatory molecule such as CD28, while ERK can be fully activated by engagement of the TCR alone (Su et al, 1994; Matsuda et al, 1998). Coordinated activation of MAPK family leads to T-cell activation and differentiation. Among them, it has been well established that the ERK signaling pathway is essential for T-cell functions (Alberola-Ila et al, 1995; Crompton et al, 1996; Sharp et al, 1997; Pagès et al, 1999). On the other hand, knockout studies have revealed that the JNK pathway has rather inhibitory effect on T-cell activation at least in some circumstances (Dong et al, 2000). In this regard, JNK-mediated phosphorylation of an NFAT protein, an essential transcription factor for T-cell activation, has been implicated as a responsible pathway in such an inhibitory effect (Chow et al, 1997). However, the MAPK targets involved in T-cell function remain elusive.
To identify key molecules phosphorylated by MAPK family members in T-cell activation, we have developed an expression screening method that utilizes a GAL4-dependent transcriptional activation system to monitor in vivo phosphorylation status. In a screening using a constitutively active form of MEKK1, we cloned linker for activation of T cells (LAT), a membrane protein, as a novel target of MAPK family.
Results
cDNA cloning of MAPK substrates
Activation domains of transcription factors are often activated through phosphorylation. Conversely, enhancement of transcriptional activity may serve as a measure of phosphorylation status. To detect MAPK-mediated in vivo phosphorylation, we took advantage of a GAL4-dependent transcriptional activation system, which consists of a GAL4 DNA binding domain (GAL4DB)-fused cDNA library derived from Jurkat T lymphocytes and a GFP reporter plasmid containing a GAL4 upstream activating sequence (GAL4 UAS) (Figure 1A). Jurkat cells were cotransfected with a constitutively active form of MEKK1 (ΔMEKK1) (an upstream activator of the MAPK pathways; Lange-Carter et al, 1993; Minden et al, 1994), the library and the reporter plasmid expressing GFP. When an appropriate clone is expressed, it is phosphorylated by MAPK(s) driven by ΔMEKK1. The presence of a phosphorylated residue(s) mimics a transcriptional activation domain consisting of negatively charged residues (Karin and Hunter, 1995), which may recruit coactivator proteins like p300/CBP (Mayr and Montminy, 2001; Sharrocks, 2001) and increases the GAL4-dependent GFP expression through binding to the GAL4 UAS. GFP-expressing cells were isolated by sorting on a fluorescence activated cell sorter. Resultant clones were rescreened using another reporter plasmid expressing luciferase in the presence or absence of ΔMEKK1, and DNA sequences were determined. We obtained several independent clones encoding Elk-1, a previously characterized MAPK substrate (Yang et al, 1998), confirming the feasibility of this method for identifying physiological substrate(s) (data not shown).
Figure 1.

Cloning of LAT as a putative target of the MAPK pathway. (A) Outline of the screening method. GFP-positive cells were sorted on a FACS Vantage (Becton Dickinson). The resultant clones were subjected to a second screening with pFR-Luc (Stratagene) along with pRL-SV40 (Promega), and assayed for luciferase activity using the Dual-Luc assay system (Promega). (B) Schematic diagram of clone #5–8 and LAT. Potential MAPK phosphorylation sites are indicated. (C) Phosphoamino acid analysis of LAT. 32P-labeled LAT molecules were immunoprecipitated from TCR-stimulated Jurkat cell lysates (human) or AE7 cell lysates (mouse), and subjected to phosphoamino acid analysis. (D) MAPK-dependent activation of clone #5–8. Jurkat cells were transiently transfected with clone #5–8, pFR-Luc, and pRL-SV40 along with pFC-MEKK1 (an expression vector for ΔMEKK1, Stratagene) and/or pSRαMyc-MKP5. Luciferase activities measured in triplicate are expressed as fold increase.
One of the clones (termed #5–8) encoded the C-terminal fragment of LAT (Figure 1B). LAT was initially identified as a prominent tyrosine-phosphorylated protein and revealed to be a critical adapter molecule required for TCR-mediated signaling (Weber et al, 1998; Zhang et al, 1998a). It has been shown that the tyrosine-phosphorylated LAT associates with multiple molecules such as PLCγ1 (Finco et al, 1998; Zhang et al, 1998a, 2000). However, whether LAT is modified by a Ser- and/or Thr-phosphorylation remains unknown. Since our data raised the possibility that LAT is phosphorylated by the ΔMEKK1-induced Ser/Thr kinase cascade, we performed a phosphoamino acid analysis of [32P]LAT in response to TCR stimulation. We found that LAT is indeed phosphorylated at both Ser and Thr residues in addition to Tyr under physiological conditions (Figure 1C). Although ΔMEKK1 can activate the NF-κB pathway as well as the MAPK pathways (Karin and Delhase, 1998), coexpression of MKP5 (Tanoue et al, 1999), a negative regulator of MAPK family, completely diminished the effect of ΔMEKK1 on clone #5–8, demonstrating the involvement of MAPK(s) (Figure 1D). Consistent with these results, the amino-acid sequence of LAT contains four possible MAPK phosphorylation sites (Figure 1B).
LAT is phosphorylated by MAPK in vitro and in vivo
To examine which of these sites is phosphorylated, glutathione S-transferase (GST)/LAT fusion proteins with mutated phosphorylation sites (Ala substitutions) were incubated with MAPK. The S84A mutant, in which Ser 84 was changed to Ala, was phosphorylated by JNK to the same extent as wild-type LAT in the presence of [γ-32P]ATP (Figure 2A), suggesting that JNK phosphorylates LAT at Thr(s). In accordance with these findings, the phosphorylated LAT protein was recognized by a monoclonal antibody (mAb) against the phospho-Thr-Pro motif (Figure 2B). Mutational analysis revealed that T155 but not T94 or T140 is the site of JNK-mediated phosphorylation (Figure 2B). ERK also phosphorylated LAT at T155 (Figure 2C), whereas p38, which was able to phosphorylate ATF2, failed to induce threonine phosphorylation of LAT (Figure 2D). These results indicate that LAT is directly phosphorylated by ERK and JNK at the same site, T155.
Figure 2.

Phosphorylation of LAT at Thr 155 by JNK and ERK. (A) GST-LAT proteins were incubated with active JNK (+) or inactive JNK (−) in the presence of [γ-32P]ATP at 30°C for 30 min. (B) GST-LAT proteins were phosphorylated with active JNK (lanes 2–5) or inactive JNK (lane 1), and subjected to immunoblot with an anti-phospho-Thr-Pro (Cell Signaling) antibody. (C) GST-LAT proteins were phosphorylated with active ERK, and subjected to immunoblot with the anti-phospho-Thr-Pro antibody. (D) GST-LAT or GST-ATF2 proteins were phosphorylated with active (+) or inactive (−) p38, and subjected to immunoblot with the anti-phospho-Thr-Pro antibody (upper) or anti-phospho-ATF2 antibody (lower). (E) LAT proteins were immunoprecipitated from transiently transfected COS7 cells, and subjected to immunoblot with the anti-phospho-Thr-Pro antibody or the antibody against LAT. (F) GFP-fused mouse LAT proteins were immunoprecipitated from transiently transfected Jurkat cells, which are cotransfected with (+) or without (−) ΔSESE, and subjected to immunoblot with the anti-phospho-Thr-Pro antibody or the antibody against GFP.
Given that activated MAPKs translocate to the nucleus whereas LAT functions as a transmembrane protein (Zhang et al, 1998a, 1998b), we examined whether full-length LAT is phosphorylated by MAPKs in vivo. COS7 cells were transfected with a LAT expression vector along with activators for MAPK (ΔSESE, a constitutively active form of ERK activator MEK1 for ERK (Gotoh et al, 1994); MKK7-JNK1 and ΔMEKK1 for JNK), and immunoprecipitated LAT proteins were assayed for Thr phosphorylation. Both JNK and ERK phosphorylate LAT at Thr in vivo although ERK does so less efficiently (Figure 2E), whereas both ΔMEKK1-induced JNK and ΔSESE-induced ERK failed to induce the Thr phosphorylation in the T155A mutant (data not shown), demonstrating that MAPKs in vivo also target the T155 of LAT. Consistently, ERK failed to phosphorylate murine LAT, which lacks the phosphorylation site corresponding to human LAT T155 (Figure 2F).
ERK is responsible for LAT phosphorylation at T155 during T-cell activation
To investigate whether LAT phosphorylation at T155 occurs under physiological conditions, we reconstituted the LAT-deficient Jurkat cell line, J.CaM2 (Finco et al, 1998), with either wild-type (referred to here as J.CaM2-WT) or T155A mutant LAT (referred to here as J.CaM2-T155A). TCR induced LAT phosphorylation at Thr only in J.CaM2-WT but not in J.CaM2-T155A (Figure 3A), confirming that LAT molecules are phosphorylated at T155 in response to TCR stimulation. It should be noted that T155 phosphorylation occurred as early as 2 min and was maximal by 5 min. Since TCR stimulation without CD28 engagement led to activation of ERK but not JNK in Jurkat cells (Su et al, 1994; Matsuda et al, 1998) (Figure 3B), it is likely that ERK mediates TCR-induced LAT phosphorylation at T155. Accordingly, pretreatment with PD98059 (Alessi et al, 1995), a specific inhibitor of the ERK signaling pathway, abolished TCR-induced Thr phosphorylation of LAT (Figure 3C).
Figure 3.

LAT phosphorylation at T155 during T-cell activation. (A) J.CaM2 cells transfected with WT or T155A mutant LAT were stimulated with anti-CD3ɛ mAb 2Ad2A2 for the indicated times. Immunoprecipitated LAT was subjected to immunoblot with 4G10 or the mAb against phospho-Thr-Pro. (B) Jurkat cells were stimulated with 2Ad2A2 for the indicated times (αCD3) or with pervanadate for 10 min (PV). The lysates were subjected to immunoblot analysis with an mAb against phospho-ERK (Cell Signaling) (p-ERK2) and the anti-ERK2 antibody (ERK2). The lysates were also subjected to immunoprecipitation with anti-JNK1 antibody followed by the kinase assay using ATF2 as a substrate (p-ATF2). Immunoprecipitation of JNK1 was confirmed with immunoblot (JNK). (C) Jurkat cells were stimulated with (+) or without (−) 2Ad2A2 for 3 min in the presence or absence of 50 μM PD98059. Immunoprecipitated LAT was subjected to immunoblot with the mAb against phospho-Thr-Pro (upper panel) or 4G10 (an mAb against phospho-Tyr) (lower panel).
Attenuation of TCR-induced Ca2+ influx by LAT phosphorylation at T155
While T155 is present in the human LAT but not in the murine LAT, T155 would be important if its phosphorylation modulates the function of LAT in human T lymphocytes. Since LAT is required for TCR-induced Ca2+ influx (Finco et al, 1998; Zhang et al, 2000), we examined whether LAT phosphorylation at T155 affects its function. [Ca2+]i mobilization in response to TCR stimulation was markedly enhanced in J.CaM2-T155A compared with J.CaM2-WT (Figure 4A). To confirm the result, we established another J.CaM2 cell line expressing Myc-tagged LAT molecules (WT #2A expressing wild-type LAT; T155A #1A and T155A #2A expressing T155A mutant LAT), and essentially the same results were obtained (Figure 4B). Tyr phosphorylation of ZAP70, a Tyr kinase responsible for Tyr phosphorylation of LAT, was observed at comparable levels in all transfectants (Figure 4C). In addition, subcellular fractionation revealed that T155A LAT as wild-type LAT localized to specialized subdomains of the plasma membrane known as lipid rafts (Figure 4D), which is essential for LAT function (Zhang et al, 1998b). These data indicate that enhanced [Ca2+]i mobilization in J.CaM2-T155A results from a lack of LAT phosphorylation at T155, but not from a difference in TCR signal intensity or subcellular distribution.
Figure 4.

Effects of LAT phosphorylation at T155 on LAT function. (A) The cells were assayed for TCR-induced Ca2+ influx. [Ca2+]i levels were measured on a flow cytometer (right), and calculated as described in Materials and methods (left). (B) J.CaM2 cells stably expressing Myc-tagged LAT were assayed for TCR-induced Ca2+ influx (left). LAT expression levels were confirmed by immunoblot with the anti-LAT antibody and an anti-ZAP70 mAb (Transduction Labs.) (right). It should be noted that wild-type LAT is more expressed compared to T155A mutants. (C) The cells were stimulated with (+) or without (−) 2Ad2A2 for 2 min. ZAP70 and LAT were immunoprecipitated, and subjected to immunoblot with 4G10. (D) Raft fractions were prepared as described in Materials and methods. Existence of LAT was detected with the anti-LAT antibody.
Conversely, LAT phosphorylation at T155, presumably mediated by ERK, may attenuate TCR-induced Ca2+ influx. To examine the hypothesis, J.CaM2-WT cells were stimulated via TCR in the presence of PD98059, a specific inhibitor of ERK pathway. Pretreatment with PD98059 enhanced TCR-induced Ca2+ influx (Figure 5A). In contrast, PD98059 had only a marginal effect on Ca2+ influx in J.CaM2-T155A (Figure 5B). These data also indicate that enhanced [Ca2+]i mobilization in J.CaM2-T155A compared to J.CaM2-WT is not due to a quantitative difference like variable expression levels of LAT molecules but due to a qualitative difference in the TCR-induced signaling pathway(s). Essentially the same results were obtained with human peripheral T cells in the presence of PD98059 (Figure 5C) or U0126, another inhibitor of the ERK pathway (DeSilva et al, 1998) (Figure 5D), excluding the possibility that the negative effect of the ERK pathway on Ca2+ influx is an artifact of Jurkat cell lines. In contrast, U0126 had little effect on TCR-induced Ca2+ influx in mouse splenic T cells, whose LAT does not contain the Thr residue corresponding to human LAT T155 (Figure 5E). Furthermore, inhibitors of the ERK pathway such as U0126 had little effect on TCR-induced Tyr phosphorylation of PLCγ1 (Figure 5F) or ZAP-70 (data not shown), excluding the possibility that the ERK pathway generally down-modulates the TCR signaling pathway. These results, taken together, demonstrate that ERK-mediated LAT phosphorylation at T155 negatively regulates TCR-mediated [Ca2+]i mobilization in human T lymphocytes.
Figure 5.
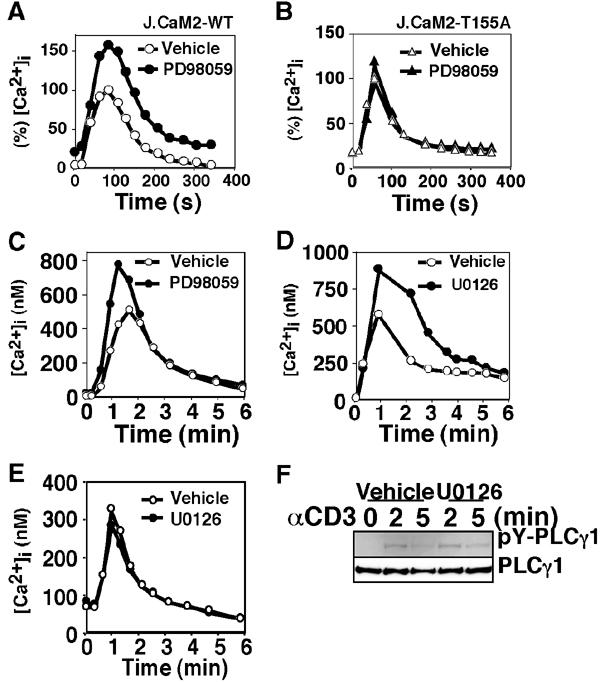
Effect of LAT phosphorylation at T155 on TCR-induced Ca2+ influx. J.CaM2-WT cells (A), J.CaM2-T155A cells (B), human peripheral T cells (C, D), or mouse splenic T cells (E) were assayed for Ca2+ influx. The cells were pretreated with or without the indicated inhibitor (50 μM PD98059 or 10 μM U0126) for 10 min. The data (A, B) are shown as % [Ca2+]i relative to the peak concentration of the vehicle-treated cells (170 nM for J.CaM2-WT; 331 nM for J.CaM2-T155A). (F) J.CaM2-WT cells were stimulated with 2Ad2A2 in the presence or absence of U0126 for the indicated times. The lysates were subjected to immunoprecipitation with an anti-PLCγ1 mAb (Upstate Biotechnology), followed by immunoblot analysis with 4G10 (upper) and the anti-PLCγ1 mAb (lower).
Mechanisms of ERK-mediated negative feedback
Finally, we examined the mechanism by which LAT phosphorylation at T155 decreases TCR-induced Ca2+ influx. We first hypothesized that LAT phosphorylation at T155 results in the recruitment of a Tyr phosphatase, which would terminate TCR-induced signaling events. Since SHP1 is involved in dephosphorylation of LAT in the TCR-mediated signaling pathway (Kosugi et al, 2001), we examined whether T155 phosphorylation affects the interaction between LAT and SHP1 using an in vitro pull-down assay. Consistent with recent observations (Kosugi et al, 2001), association between LAT and SHP1 was independent of Tyr phosphorylation of LAT (Figure 6A, bottom, compare lane 1 and lane 2). Thr phosphorylation of LAT had little effect on its interaction with SHP1 (Figure 6A, bottom, compare lane 2 and lane 3), excluding the above possibility.
Figure 6.
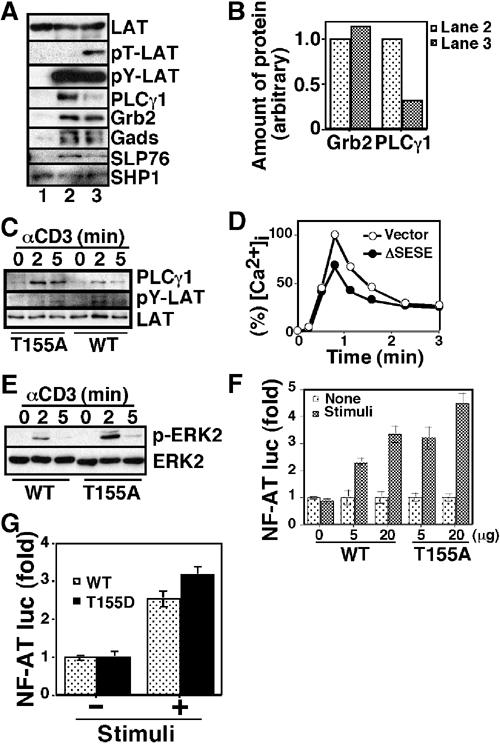
Effect of LAT phosphorylation at T155 on signalsome formation. (A) Jurkat cell lysates were subjected to pull-down assay using mock-treated (lane 1), tyrosine-phosphorylated (lane 2), or tyrosine and threonine doubly phosphorylated (lane 3) GST-LAT molecules. Precipitated GST-LAT proteins and their phosphorylation status were confirmed by immunoblot analysis with the anti-LAT antibody (LAT), the mAb against phospho-Thr-Pro (pT-LAT), and 4G10 (pY-LAT), respectively. Associated proteins were probed with the anti-PLCγ1 mAb, an anti-Grb2 mAb (Transduction Labs.), an anti-Gads antibody (Upstate Biotechnology), an anti-SLP76 antibody (Upstate Biotechnology), and an anti-SHP1 antibody (Upstate Biotechnology). (B) Immunoreactive bands in (A) were quantified, and amounts of PLCγ1 and Grb2 relative to tyrosine-phosphorylated LAT (pY-LAT) were indicated. (C) J.CaM2-WT cells (WT) or J.CaM2-T155A cells (T155A) were stimulated with 2Ad2A2 for indicated times, and then the lysates were subjected to immunoprecipitation with the anti-LAT antibody followed by immunoblot analysis with the anti-PLCγ1 mAb (PLC-γ1) and 4G10 (pY-LAT). (D) Jurkat cells were transiently transfected with an expression vector for human CD8 along with (ΔSESE) or without (vector) an expression vector for ΔSESE. The transfected cells were purified by an anti-human CD8 mAb conjugated with MicroBeads (Miltenyi Biotech GmbH) using an autoMACS (Miltenyi Biotech GmbH), and assayed for Ca2+ influx. The data are shown as % [Ca2+]i relative to the peak concentration of the vector-transfected cells (558 nM). (E) J.CaM2-WT cells (WT) or J.CaM2-T155A cells (T155A) were stimulated with 2Ad2A2 for the indicated times. The lysates were subjected to immunoblot analysis with an mAb against phospho-ERK (upper) and the anti-ERK2 antibody (lower). (F, G) J.CaM2 cells were transiently transfected with NF-AT luc along with the indicated amount of LAT expression vectors, followed by incubation for 40 h. The cells were stimulated with or without 2Ad2A2 in combination with PMA for 7 h, and assayed for luciferase activities.
It has been shown that formation of a multiprotein complex with signaling molecules such as Grb2, Gads, and PLCγ1 is essential for LAT function (Zhang et al, 2000; Lin and Weiss, 2001). We thus investigated whether T155 phosphorylation of LAT affects association of these molecules. PLCγ1 associated with LAT in a Tyr phosphorylation-dependent manner, and T155 phosphorylation markedly decreased their interaction (Figures 6A and B, compare lane 2 and lane 3). In contrast, T155 phosphorylation had marginal effect on the Tyr phosphorylation-induced interaction with Gads or Grb2 (Figures 6A and B). Consistent with recent reports revealing that the interaction between the SH3 domain of PLCγ1 and the proline-rich domain of SLP76 stabilizes the complex consisting of LAT, PLCγ1, and SLP76 (Yablonski et al, 2001a, 2001b), T155 phosphorylation resulted in a slight decrease of the association between SLP76 and the Tyr-phosphorylated LAT. Gads, which directly binds to both LAT and SLP76, may mediate the residual interaction. Therefore, attenuation of [Ca2+]i mobilization is likely due to the decreased formation of a LAT-based signalsome upon T155 phosphorylation. Accordingly, more PLCγ1 was associated with T155A mutant in response to TCR stimulation compared with wild-type LAT (Figure 6C). Moreover, TCR-induced association between PLC-γ1 and LAT was more transient in J.CaM2-WT compared to J.CaM2-T155A. It may reflect the time course of T155 phosphorylation in J.CaM2-WT (Figure 3A). One can argue that there is discrepancy between the effect of T155 phosphorylation on association with PLC-γ1: almost no PLC-γ1 was brought down using in vitro T155-phosphorylated GST-LAT, whereas a modest decrease in PLC-γ1 association was observed with wild-type LAT upon stimulation compared with T155A mutant in vivo. However, it is likely due to different T155 phosphorylation levels between in vitro and in vivo system since endogenous LAT molecules were only partially phosphorylated at T155 within 2 min after TCR stimulation (Figure 3A). Conversely, only a partial phosphorylation at T155 may cause a drastic effect on LAT function.
To further confirm that ERK-mediated T155 phosphorylation of LAT leads to attenuation of TCR-induced Ca2+ influx, we examined the effect of ΔSESE, an upstream activator of ERK (Gotoh et al, 1994). As shown in Figure 6D, expression of ΔSESE in Jurkat cells resulted in a reduction of TCR-induced Ca2+ influx. Since PLCγ1 and SLP76 play a pivotal role in TCR-induced activation of the Ras/ERK signaling pathway in addition to [Ca2+]i mobilization (Yablonski et al, 1998), we investigated whether T155 phosphorylation affects ERK activation. TCR-induced ERK activation was indeed more prominent in J.CaM2-T155A than J.CaM2-WT (Figure 6E). Furthermore, TCR-induced CD69 upregulation, which is mediated through the Ras pathway, was also augmented in J.CaM2-T155A (data not shown). These results indicate that ERK-mediated LAT phosphorylation at T155 constitutes a negative feedback loop in TCR-mediated signaling pathways including the activation of ERK itself.
We further examined whether T155 phosphorylation has any impact on T-cell activation at a transcription level. It is well established that IL-2 gene expression is induced in response to TCR in the presence of costimulatory signaling. We thus examined the transcriptional activation of the distal NF-AT element of IL-2 promoter, a critical cis-element for IL-2 gene expression, which is regulated downstream of the calcium signaling pathway (Northrop et al, 1993). To avoid any bias resulting from clonal variation, we transiently transfected the NF-AT/luciferase reporter construct (referred to here as NF-AT luc) with the expression vector for either wild-type or T155A mutant LAT. TCR-induced NF-AT activation was augmented in T155A-transfected J.CaM2 cells compared with wild-type LAT-transfected cells, demonstrating that a loss of T155 phosphorylation results in an enhancement of TCR signaling events (Figure 6F). We then examined whether a negatively charged amino acid mimics the phosphorylated T155. In contrast to our expectation, however, T155D where Asp was substituted for T155 rather augments the TCR-induced NFAT activation like T155A mutant (Figure 6G). These results suggest that phosphorous group at T155 plays a critical role in down-modulation of LAT function.
Discussion
In this study, we established a screening method for detecting in vivo substrates of the MAPK family and identified LAT as a physiological target of MAPKs. Since we have developed this method based on a GAL4-dependent transcriptional activation system, it is initially expected that resultant clones will belong to transcription factors that can be activated through MAPK phosphorylation. Indeed, several independent clones turned out to encode Elk-1, a well-known substrate of MAPK, indicating that our screening method is able to identify physiological substrates for MAPK family. Contrary to our expectation, we identified LAT, which is an adapter protein functioning at the plasma membrane. We do not know the exact reason as to why LAT phosphorylation at T155 increases the transcriptional activity of fused GAL4DB. One possible explanation is that the negative charge created by T155 phosphorylation serves as an interaction site with coactivator protein like p300/CBP as is the case with CREB (Mayr and Montminy, 2001), resulting in transcriptional activation. In this regard, we cannot formally exclude the possibility that LAT has a transcriptional activation domain yet to be identified and functions as a transcription factor under physiological conditions.
To identify target molecules of MAPK family in the context of T-lymphocyte signaling, we utilized ΔMEKK1 as an upstream activator of MAPK family and Jurkat T lymphocyte as a screening environment. Since ΔMEKK1 can activate both ERK and JNK signaling pathways (Lange-Carter et al, 1993; Minden et al, 1994), introduction of ΔMEKK1 as an activator would widen the window of potential targets. On the other hand, ΔMEKK1 is also implicated in the activation of the NF-κB pathway (Karin and Delhase, 1998). It is thus reasonable to expect that resultant clones are not exclusively restricted to substrates downstream of MAPK family. Indeed, we obtained a second clone encoding an unknown protein that has no potential MAPK phosphorylation site (Ser-Pro or Thr-Pro) (data not shown). Whether it is activated in the NF-κB signaling pathway remains to be determined. To exclude such an adverse side effect, we can modify the assay system using specific activators for cognate MAPK family members: MKK7-JNK1 for identification of JNK substrates and ΔSESE for ERK. This technique can be easily applied to other cell types such as nerve cells. Given that cell-specific target molecules may be identified in the context of cell-specific protein expression, it seems likely that our assay system has an advantage to identify cell-specific substrates over conventional approaches including yeast two-hybrid system (Yang et al, 1992; Fukunaga and Hunter, 1997).
We have demonstrated that ERK-mediated LAT phosphorylation at T155 attenuates TCR-induced signaling events such as [Ca2+]i mobilization. The data here and those of other studies (Finco et al, 1998; Yablonski et al, 1998; Zhang et al, 2000; Lin and Weiss, 2001) collectively suggest that ERK constitutes a negative feedback loop in T-cell activation in the following mechanism: LAT becomes phosphorylated by ZAP70 on multiple tyrosine residues in response to TCR stimulation allowing PLCγ1 to be recruited with the aid of SLP76; PLCγ1 activates the Ras/ERK pathway through generation of IP3 and diacylglycerol from PIP2 (Dower et al, 2000), and activated ERK in turn phosphorylates LAT at T155; T155 phosphorylation of LAT decreases the affinity of PLCγ1 and SLP76 for LAT, and attenuates the downstream signaling events including ERK activation. Thus, T155 phosphorylation may function as a molecular switch disconnecting the linkage between PLCγ1 and tyrosine-phosphorylated LAT. Although it remains to be examined whether T155 phosphorylation induces an alteration in the conformation of LAT molecules, T155 phosphorylation had little or no effect on the association with SHP1 or Gads, suggesting that LAT phosphorylation at T155 causes slight conformational change, if any, only around the site critical for PLCγ1 interaction, presumably Y132. Alternatively, phosphorylated T155 site may recruit a phospho-Ser/Thr recognition molecule such as 14-3-3 family proteins, WW domain containing proteins like Pin1, and FHA domain containing proteins like Rad53p (Yaffe and Elia, 2001) leading to interference with binding of LAT and its cognate partners. Given that T155D mutation, whose negative charged amino acid may mimic the conformation change induced by T155 phosphorylation, had no inhibitory effect on LAT function, the latter possibility seems more likely.
Several studies on the insulin signaling pathway have shown that ERK is directly or indirectly involved in desensitization of insulin-induced Ras activation (Langlois et al, 1995; Zhao et al, 1998). Insulin receptor transmits signals for cell proliferation and gene regulation through activation of Ras mediated by the guanine nucleotide exchange factor SOS. SOS is constitutively bound to the adapter protein Grb2 and insulin stimulation induces translocation of SOS–Grb2 complex to the plasma membrane where Ras is activated. Activation of the Ras–ERK pathway results in phosphorylation of SOS and subsequent dissociation of SOS from Grb2. In contrast to insulin- and other growth factor-mediated Ras activation pathway, RasGRP rather than SOS–Grb2 complex is responsible for Ras activation in the T-cell signaling pathway (Dower et al, 2000). Given that RasGRP is activated in the presence of diacylglycerol produced by PLC-γ1, disconnecting LAT–PLCγ1 association by ERK-mediated phosphorylation instead of targeting SOS is a reasonable system to desensitize Ras pathway in human T cells. Our data demonstrate that ERK-mediated LAT phosphorylation decreases the affinity toward PLC-γ1 as early as 2 min after TCR stimulation as compared with J.CaM2-T155A cells, consistent with the augmented Ca2+ response in J.CaM2-T155A cells. Since LAT molecules phosphorylated at T155 accumulate gradually after stimulation, one would expect that more pronounced effects appear at later time points. Although we often observed higher [Ca2+]i levels at later time points in cells lacking T155 phosphorylation than those in untreated cells (see Figures 4A and B, 5A and D), such enhancement seems weaker than it would be if T155 phosphorylation is the only negative feedback mechanism. It is thus reasonable to assume that another desensitizing mechanism(s) is also operative at various levels. For example, down-modulation toward PLC-γ1 and IP3 receptors together with LAT threonine phosphorylation may regulate the Ca2+ response at later time points in T-cell activation. Alternatively, the function of LAT may be further modulated through phosphorylation at serine and/or threonine residue(s) other than T155 since murine LAT, which lacks the phosphorylation site corresponding to T155, was still phosphorylated at both serine and threonine residues in response to TCR stimulation (Figure 1C). Interestingly, human caspase 9, an essential component of the apoptotic pathway, has also been revealed to be regulated through a similar kind of multisite phosphorylation: Akt-mediated phosphorylation at Ser 196, which is specific for human caspase 9, and ERK-mediated phosphorylation at Thr 125, which is conserved among mammals (Cardone et al, 1998; Allan et al, 2003). Existence of these pathways would explain a loss of prolonged Ca2+ response, which could be expected in JCaM2-T155A.
We also showed that JNK is able to phosphorylate T155 of LAT. Whether JNK is involved in the attenuation of T-cell activation under physiological conditions remains unclear. However, the recent report that IL-2 production is augmented in JNK-deficient T cells raises the possibility that JNK also contributes to the negative feedback loop (Dong et al, 2000). The desensitization of signal transduction pathways after stimulation seems important in coordinating the complex network of events leading to appropriate gene expression. Sustained cell activation may cause damaging responses such as apoptosis, oncogenesis, and autoimmunity. LAT–ERK pathway is likely to be one of the molecular mechanisms underlying the fine-tuning of signal transduction pathways. Interestingly, it has been shown that treatment of T cells with PD98059 enhances production of Th2-type cytokines such as IL-4 and IL-13 although its mechanism remains obscure (Dumont et al, 1998). According to our observations, the effect of PD98059 is presumably due to augmentation of the TCR-induced [Ca2+]i mobilization resulting from the inhibition of LAT phosphorylation at T155. Consistently, it has been demonstrated that activation of the Ca2+/calcineurin pathway regulates T-cell differentiation toward Th2 (Yamashita et al, 2000). Thus, the LAT–ERK pathway may also affect T-cell differentiation in response to TCR stimulation. Interestingly, it has recently been shown that disruption of the interaction between LAT and PLC-γ1 also results in the induction of Th2 differentiation (Aguado et al, 2002; Sommers et al, 2002). TCR signaling with inappropriate intensity or duration may cause deleterious dysfunction in the immune system. Whether a similar mechanism is involved in other signal transduction systems such as B-cell activation remains to be elucidated.
Materials and methods
Screening
The cDNA fragment corresponding to GAL4DB was excised from pGBT9 (Clontech), and inserted into the HindIII and EcoRI sites of pEB6CAG (Tanaka et al, 1999) with a Kozak sequence (CCACC) 5′ to the start ATG to construct pEB6CAG-GAL4. Double-stranded cDNA was synthesized from Jurkat poly(A)+ RNA with an oligo(dT) primer using a cDNA synthesis kit (Amersham Pharmacia), and inserted unidirectionally into the EcoRI and NotI sites of pEB6CAG-GAL4 to construct the pEB6CAG-GAL4/library. To construct pGAL4UAS-GFP, a PCR fragment corresponding to GAL4UAS was obtained using pFR-Luc (Stratagene) as a template, and inserted into the EcoRI site of pEGFP-1 (Clontech). Nucleotide sequencing was performed by an automated sequencer (PRISM310, ABI). An expression vector for ΔMEKK1 (pFC-MEKK1), a constitutively active form of MEKK1, was purchased from Stratagene. Jurkat cells (107 cells) were electroporated with 20 μg of the pEB6CAG-GAL4/library, 4 μg of pGAL4UAS-GFP, and 1 μg of pFC-MEKK1 at 320 V, 960 μF using a Gene Pulser Electroporator (Bio-Rad). So far, we have obtained five independent clones: three clones encoding Elk-1, one encoding an unknown protein, and clone #5–8.
Plasmids and transfectants
A human LAT cDNA corresponding to residues 34–233 was inserted into a GST expression plasmid pGEX-4T-1 (Amersham Pharmacia) to produce GST-fused LAT molecules. Full-length human LAT cDNA was cloned into pAhygro as described (Ishiai et al, 2000). Phosphorylation-deficient mutants were created by PCR-based site-directed mutagenesis (Imai et al, 1991). The T94/140/155A mutant replaces all potential threonine phosphorylation sites (T94, T140, and T155) with Ala, whereas the T94/140A mutant has Ala substitutions at both T94 and T140. The expression vector for either wild-type or T155A mutant LAT was transfected into J.CaM2 cells (kindly provided by A Weiss) by electroporation at 320 V, 960 μF. The stable transfectants were selected in the presence of 400 μg/ml hygromycin. In some experiments, Myc-tagged wild-type or T155A mutant LAT cDNA was cloned into pEB6CAG, and transfected into J.CaM2 cells followed by selection with 1 mg/ml G418. A cDNA for mouse LAT was kindly provided by Dr A Kosugi. A cDNA for the fusion protein of mouse MKK7γ2 (Moriguchi et al, 1997) and HA-tagged human JNK1 (MKK7-JNK1) was constructed using a PCR-based method, and inserted into the NotI site of pEF/myc/cyto (Invitrogen). The expression of MKK7-JNK1 led to constitutive activation of the JNK signaling pathway (data not shown).
Phosphoamino acid analysis
Following SDS–PAGE, 32P-labeled LAT protein was extracted from the gel with 50 mM ammonium carbonate containing 0.1% SDS and 0.5% β-ME, and precipitated with trichloroacetate in the presence of 20 μg RNaseA. The sample was hydrolyzed in 6 N HCl at 110°C for 2 h, lyophilized, and re-suspended with pH 3.5 buffer solution (5% acetic acid and 0.5% pyridine). The sample was then spotted with standards (0.5 μg each of pSer, pThr, and pTyr) on a cellulose thin-layer chromatography plate, and subjected to electrophoresis at 1.0 kV for 30 min in a pH 3.5 buffer solution.
Pull-down assay
GST-LAT (5 μg) was incubated at 30°C for 20 min in a buffer solution containing 100 μM ATP, 10 mM MgCl2, and 5 mM MnCl2 with (to yield tyrosine-phosphorylated GST-LAT) or without (to yield mock-treated GST-LAT) active ZAP70 molecules, which were obtained from pervanadate-treated Jurkat cell lysates. For both threonine- and tyrosine-phosphorylated GST-LAT, GST-LAT was incubated at 30°C for 15 min in a buffer solution containing 100 μM ATP and 10 mM MgCl2 with active ERK2 obtained from pervanadate-treated Jurkat cell lysates, and further incubated with active ZAP70 under the conditions described above. For GST pull-down assays, pervanadate-treated Jurkat cell lysates were precleared with glutathione-Sepharose for 30 min at 4°C, and the cleared lysates were incubated for 1 h at 4°C with the appropriate GST-LAT bound to glutathione-Sepharose. Beads were washed twice with PBS containing 0.05% Tween 20, and bound proteins were eluted by boiling with Laemmli's sample buffer solution.
Immunoprecipitation
Active JNK and active ERK were obtained from Jurkat cells stimulated with 10 μg/ml anisomycin for 40 min and 10 ng/ml PMA for 10 min, respectively. Inactive JNK was obtained from mock-treated Jurkat cells. The cells were lysed in a buffer solution containing 20 mM Tris–HCl (pH 7.5), 12.5 mM β-glycerophosphate, 2 mM EGTA, 10 mM NaF, 1 mM benzamidine, 1% Triton X-100, 2 mM dithiothreitol (DTT), 1 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride, and 1% aprotinin. Cell extracts were clarified by centrifugation at 15 000 g for 10 min. For immunoprecipitation, the supernatants were incubated with antibodies for 30 min at 4°C. After the addition of protein A–Sepharose (Amersham Pharmacia), the lysates were incubated for an additional 2 h. The beads were then washed, and subjected to kinase assays and/or immunoblot analysis. Antibodies against JNK1 (C-20), ERK2 (C-14), and ZAP70 (LR) were purchased from Santa Cruz, and an antibody against LAT was kindly provided by LE Samelson.
Ca2+ mobilization
Peripheral T cells of a normal adult donor were obtained by Ficoll–Paque PLUS (Amersham) density gradient centrifugation, followed by incubation with IL-2 (100 U/ml) containing medium for 6 days. Cells (106 cells) were loaded with 5 μg/ml Fluo-3 AM (Molecular Probes) at 37°C for 30 min, and then activated by anti-CD3ɛ mAb, 2Ad2A2 (Reinherz et al, 1982) ascites (1:100). [Ca2+]i levels were measured on a FACScan by fluorescence intensity of Fluo-3, and calculated according to the manufacturer's instructions (Becton Dickinson).
Raft preparation
J.CaM2-WT (WT) or J.CaM2-T155A (T155A) cells (4 × 107) were washed with PBS containing 5 mM EDTA, suspended with 400 μl of TNEV buffer solution (25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM sodium vanadate, 12.5 mM β-glycerophosphate, 1% aprotinin, and 2 mM DTT) containing 0.5% Triton X-100. The lysates were gently mixed with 400 μl of 85% sucrose in TNEV buffer solution and placed in the bottom of a centrifugation tube. The samples were then overlaid with 2.6 ml of 30% sucrose and 1.4 ml of 5% sucrose in TNEV buffer solution, and centrifuged for 16 h at 200 000 g in an SW50.1 rotor (Beckman) at 4°C. Following centrifugation, 12 0.4 ml fractions (excluding the pellet) were collected from the top of the gradient.
Luciferase assay
To examine transcriptional activation of the NF-AT promoter, we employed luciferase assay system as described previously (Matsuda et al, 1998, 2000). J.CaM2 cells were transiently cotransfected with LAT expression vectors and an NF-AT luc (kindly provided by GR Crabtree) in combination with pRL-TK (Promega) for normalization by electroporation at 320 V, 960 μF. Luciferase activities in cell lysates were measured in triplicate on a luminometer (LB9507; Berthold), using the Dual-Luc assay system (Promega).
Acknowledgments
This work was supported by a grant from the Kato Memorial Bioscience Foundation (to SM), a Grant-in-Aid for Scientific Research (B) (11694312, 12770168 and 12877056), a National Grant-in-Aid for the Establishment of a High-Tech Research Center in a private University, a Grant for the promotion of the advancement of education and research in graduate schools, and a Scientific Frontier Research Grant from the Ministry of Education, Culture, Sports, Science and Technology, Japan. We thank GR Crabtree, T Kurosaki, A Kosugi, and EL Reinherz for reagents, and LE Samelson, LK Clayton, and A Weiss for valuable discussions and critical reading of the manuscript.
References
- Aguado E, Richelme S, Nunez-Cruz S, Miazek A, Mura AM, Richelme M, Guo XJ, Sainty D, He HT, Malissen B, Malissen M (2002) Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science 296: 2036–2040 [DOI] [PubMed] [Google Scholar]
- Ahn NG, Seger R, Krebs EG (1992) The mitogen-activated protein kinase activator. Curr Opin Cell Biol 4: 992–999 [DOI] [PubMed] [Google Scholar]
- Alberola-Ila J, Forbush KA, Seger R, Krebs EG, Perlmutter RM (1995) Selective requirement for MAP kinase activation in thymocyte differentiation. Nature 373: 620–623 [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR (1995) PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem 270: 27489–27494 [DOI] [PubMed] [Google Scholar]
- Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR (2003) Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol 5: 647–654 [DOI] [PubMed] [Google Scholar]
- Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282: 1318–1321 [DOI] [PubMed] [Google Scholar]
- Chow CW, Rincon M, Cavanagh J, Dickens M, Davis RJ (1997) Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science 278: 1638–1641 [DOI] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ (1995) How MAP kinases are regulated. J Biol Chem 270: 14843–14846 [DOI] [PubMed] [Google Scholar]
- Crompton T, Glimour KC, Owen MJ (1996) The MAP kinase pathway controls differentiation from double-negative to double-positive thymocyte. Cell 86: 243–251 [DOI] [PubMed] [Google Scholar]
- Davis RJ (1994) MAPKs: new JNK expands the group. Trends Biochem Sci 19: 470–473 [DOI] [PubMed] [Google Scholar]
- DeSilva DR, Jones EA, Favata MF, Jaffee BD, Magolda RL, Trzaskos JM, Scherle PA (1998) Inhibition of mitogen-activated protein kinase kinase blocks T cell proliferation but does not induce or prevent anergy. J Immunol 160: 4175–4181 [PubMed] [Google Scholar]
- Dong C, Yang DD, Tournier C, Whitmarsh AJ, Xu J, Davis RJ, Flavell RA (2000) JNK is required for effector T-cell function but not for T-cell activation. Nature 405: 91–95 [DOI] [PubMed] [Google Scholar]
- Dower NA, Stang SL, Bottorff DA, Ebinu JO, Dickie P, Ostergaard HL, Stone JC (2000) RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat Immunol 1: 317–321 [DOI] [PubMed] [Google Scholar]
- Dumont FJ, Staruch MJ, Fischer P, DaSilva C, Camacho R (1998) Inhibition of T cell activation by pharmacologic disruption of the MEK1/ERK MAP kinase or calcineurin signaling pathways results in differential modulation of cytokine production. J Immunol 160: 2579–2589 [PubMed] [Google Scholar]
- Finco TS, Kadlecek T, Zhang W, Samelson LE, Weiss A (1998) LAT is required for TCR-mediated activation of PLCγ1 and the Ras pathway. Immunity 9: 617–626 [DOI] [PubMed] [Google Scholar]
- Fukunaga R, Hunter T (1997) MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J 16: 1921–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh Y, Matsuda S, Takenaka K, Hattori S, Iwamatsu A, Ishikawa M, Kosako H, Nishida E (1994) Characterization of recombinant Xenopus MAP kinase kinases mutated at potential phosphorylation sites. Oncogene 9: 1891–1898 [PubMed] [Google Scholar]
- Hazzalin CA, Mahadevan LC (2002) MAPK-regulated transcription: a continuously variable gene switch? Nat Rev Mol Cell Biol 3: 30–40 [DOI] [PubMed] [Google Scholar]
- Imai Y, Matsushima Y, Sugimura T, Terada M (1991) A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res 19: 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip YT, Davis RJ (1998) Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr Opin Cell Biol 10: 205–219 [DOI] [PubMed] [Google Scholar]
- Ishiai M, Kurosaki M, Inabe K, Chan AC, Sugamura K, Kurosaki T (2000) Involvement of LAT, Gads, and Grb2 in compartmentation of SLP-76 to the plasma membrane. J Exp Med 192: 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M (1995) The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem 270: 16483–16486 [DOI] [PubMed] [Google Scholar]
- Karin M, Delhase M (1998) JNK or IKK, AP-1 or NF-kappaB, which are the targets for MEK kinase 1 action? Proc Natl Acad Sci USA 95: 9067–9069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Hunter T (1995) Transcriptional control by protein phosphorylation: signal transmission from the cell surface to the nucleus. Curr Biol 5: 747–757 [DOI] [PubMed] [Google Scholar]
- Kosugi A, Sakakura J, Yasuda K, Ogata M, Hamaoka T (2001) Involvement of SHP-1 tyrosine phosphatase in TCR-mediated signaling pathways in lipid rafts. Immunity 14: 669–680 [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J (1996) Protein kinase cascades activated by stress and inflammation. BioEssays 18: 567–577 [DOI] [PubMed] [Google Scholar]
- Lange-Carter CA, Pleiman CM, Gardner AM, Blumer KJ, Johnson GL (1993) A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science 260: 315–319 [DOI] [PubMed] [Google Scholar]
- Langlois WJ, Sasaoka T, Saltiel AR, Olefsky JM (1995) Negative feedback regulation and desensitization of insulin- and epidermal growth factor-stimulated p21ras activation. J Biol Chem 270: 25320–25323 [DOI] [PubMed] [Google Scholar]
- Lewis TS, Shapiro PS, Ahn NG (1998) Signal transduction through MAP kinase cascades. Adv Cancer Res 74: 49–139 [DOI] [PubMed] [Google Scholar]
- Lin J, Weiss A (2001) Identification of the minimal tyrosine residues required for linker for activation of T cell function. J Biol Chem 276: 29588–29595 [DOI] [PubMed] [Google Scholar]
- Matsuda S, Moriguchi T, Koyasu S, Nishida E (1998) T lymphocyte activation signals for interleukin-2 production involve activation of MKK6-p38 and MKK7-SAPK/JNK signaling pathways sensitive to cyclosporin A. J Biol Chem 273: 12378–12382 [DOI] [PubMed] [Google Scholar]
- Matsuda S, Shibasaki F, Takehana K, Mori H Nishida E, Koyasu S (2000) Two distinct action mechanisms of immunophilin–ligand complexes for the blockade of T cell activation. EMBO Rep 1: 428–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2: 599–609 [DOI] [PubMed] [Google Scholar]
- Minden A, Lin A, McMahon M, Lange-Carter C, Derijard B, Davis RJ, Johnson GL, Karin M (1994) Differential activation of ERK and JNK mitogen activated protein kinases by Raf-1 and MEKK. Science 266: 1719–1723 [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Toyoshima F, Masuyama N, Hanafusa H, Gotoh Y, Nishida E (1997) A novel SAPK/JNK kinase, MKK7, stimulated by TNFalpha and cellular stresses. EMBO J 16: 7045–7053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida E, Gotoh Y (1993) The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem Sci 18: 128–131 [DOI] [PubMed] [Google Scholar]
- Northrop JP, Ullman KS, Crabtree GR (1993) Characterization of the nuclear and cytoplasmic components of the lymphoid-specific nuclear factor of activated T cells (NF-AT) complex. J Biol Chem 268: 2917–2923 [PubMed] [Google Scholar]
- Pagès G, Guérin S, Gral D, Bonino F, Smith A, Anjuere F, Auberger P, Pouysségur J (1999) Defective thymocyte maturation in p44 MAP kinase (erk 1) knockout mice. Science 286: 1374–1377 [DOI] [PubMed] [Google Scholar]
- Reinherz EL, Meuer S, Fitzgerald KA, Hussey RE, Levine H, Schlossman SF (1982) Antigen recognition by human T lymphocytes is linked to surface expression of the T3 molecular complex. Cell 30: 735–743 [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Cobb MH (1997) Mitogen-activated protein kinase pathways. Curr Opin Cell Biol 9: 180–186 [DOI] [PubMed] [Google Scholar]
- Schaeffer HJ, Weber MJ (1999) Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol 19: 2435–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp LL, Schwarz DA, Bott CM, Marshall CJ, Hedrick SM (1997) The influence of the MAPK pathway on T cell lineage commitment. Immunity 7: 609–618 [DOI] [PubMed] [Google Scholar]
- Sharrocks AD (2001) The Ets-domain transcription factor family. Nat Rev Mol Cell Biol 2: 827–837 [DOI] [PubMed] [Google Scholar]
- Sommers CL, Park CS, Lee J, Feng C, Fuller CL, Grinberg A, Hildebrand JA, Lacana E, Menon RK, Shores EW, Samelson LE, Love PE (2002) A LAT mutation that inhibits T cell development yet induces lymphoproliferation. Science 296: 2040–2043 [DOI] [PubMed] [Google Scholar]
- Su B, Jacinto E, Hibi M, Kallunki T, Karin M, Ben-Neriah Y (1994) JNK is involved in signal integration during costimulation of T lymphocytes. Cell 77: 727–736 [DOI] [PubMed] [Google Scholar]
- Tanaka J, Miwa Y, Miyoshi K, Ueno A, Inoue H (1999) Construction of Epstein–Barr virus-based expression vector containing mini-oriP. Biochem Biophys Res Commun 264: 938–943 [DOI] [PubMed] [Google Scholar]
- Tanoue T, Moriguchi T, Nishida E (1999) Molecular cloning and characterization of a novel dual specificity phosphatase, MKP-5. J Biol Chem 274: 19949–19956 [DOI] [PubMed] [Google Scholar]
- Treisman R (1996) Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol 5: 392–399 [DOI] [PubMed] [Google Scholar]
- Weber JR, Ørstavik S, Torgersen KM, Danbolt NC, Berg SF, Ryan JC, Taskén K, Imboden JB, Vaage JT (1998) Molecular cloning of the cDNA encoding pp36, a tyrosine-phosphorylated adaptor protein selectively expressed by T cells and natural killer cells. J Exp Med 187: 1157–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonski D, Kuhne MR, Kadlecek T, Weiss A (1998) Uncoupling of nonreceptor tyrosine kinases from PLCr1 in an SLP-76-deficient T cell. Science 281: 413–416 [DOI] [PubMed] [Google Scholar]
- Yablonski D, Kadlecek T, Weiss A (2001a) Identification of a phospholipase C-γ1 (PLC-γ1) SH3 domain-binding site in SLP76 required for T-cell receptor-mediated activation of PLC-γ1 and NFAT. Mol Cell Biol 21: 4208–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonski D, Kuhne MR, Kadlecek T, Weiss A (2001b) Uncoupling of nonreceptor tyrosine kinases from PLC-γ1 in an SLP-76-deficient T cell. Science 281: 413–416 [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Elia AEH (2001) Phosphoserine/threonine-binding domains. Curr Opin Cell Biol 13: 131–138 [DOI] [PubMed] [Google Scholar]
- Yamashita M, Katsumata M, Iwashima M, Kimura M, Shimizu C, Kamata T, Shin T, Seki N, Suzuki S, Taniguchi M, Nakayama T (2000) T cell receptor-induced calcineurin activation regulates T helper type 2 cell development by modifying the interleukin 4 receptor signaling complex. J Exp Med 191: 1869–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Whitmarsh AJ, Davis RJ, Sharrocks AD (1998) Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J 17: 1740–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Hubbard EJ, Carlson M (1992) A protein kinase substrate identified by the two-hybrid system. Science 257: 680–682 [DOI] [PubMed] [Google Scholar]
- Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE (1998a) LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92: 83–92 [DOI] [PubMed] [Google Scholar]
- Zhang W, Trible RP, Samelson LE (1998b) LAT palmytoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation. Immunity 9: 239–246 [DOI] [PubMed] [Google Scholar]
- Zhang W, Trible RP, Zhu M, Liu SK, McGlade CJ, Samelson LE (2000) Association of Grb2, Gads, and phospholipase C-γ1 with phosphorylated LAT tyrosine residues. J Biol Chem 275: 23355–23361 [DOI] [PubMed] [Google Scholar]
- Zhao H, Okada S, Pessin JE, Koretzky GA (1998) Insulin receptor-mediated dissociation of Grb2 from Sos involves phosphorylation of Sos by kinase(s) other than extracellular signal-regulated kinase. J Biol Chem 273: 12061–12067 [DOI] [PubMed] [Google Scholar]