

Abstract
Rodent cells are notable for their inability to support normal assembly of HIV particles. In this report, we address possible causes for this defect by considering the hypothesis that mRNA-associated events occurring in the nucleus can regulate the activity of their encoded proteins in the cytoplasm. We show that altering the RNA nuclear export element used by HIV gag-pol mRNA from the Rev response element to the constitutive transport element restores both the trafficking of Gag to cellular membranes and efficient HIV assembly in murine cells. These results suggest that two phases of the HIV life cycle, RNA export and capsid assembly, that have hitherto been regarded as distinct are, in fact, linked. Thus, protein function and fate may depend upon the full and precise history of its encoding mRNA.
Keywords: HIV-1 assembly, mRNA export, species barrier
Introduction
The nuclear history of a messenger RNA can dramatically affect its fate in the cytoplasm. This is illustrated by the role of pre-mRNA splicing in the determination of messenger ribonucleoprotein (mRNP) particle composition (Dreyfuss et al, 2002; Reed and Hurt, 2002). Specifically, splicing results in a complex of proteins known as the exon junction complex (EJC) being assembled 20–24 nucleotides upstream of the exon–exon boundary. Multiple components of the EJC have the potential to affect the cytoplasmic fate of mRNA in a number of ways: for example, in human cells, the EJC marks the RNA for efficient translation or, in the presence of a premature termination codon, for nonsense mediated decay (Dreyfuss et al, 2002; Maquat, 2004; Nott et al, 2004), while in Drosophila cells, the EJC marks oskar mRNA for proper cytosolic localization (Hachet and Ephrussi, 2004; Palacios et al, 2004). Similar to splicing, RNA export is also capable of modulating mRNP composition. For example, in Chironomus tentans Balbiani ring mRNA export, the mRNP is formed cotranscriptionally as multiple different RNA binding proteins assemble on the elongating transcript (Daneholt, 2001). Some of these RNA binding proteins, such as hrp36 and hrp84, are associated with the RNA from transcription through to translation at the polysome, whereas others, such as hrp23 and hrp45, dissociate from the mRNP coincident with export through the nuclear pore complex. Thus, post-transcriptional events that are initiated in the nucleus can dictate mRNA fate in the cytoplasm, potentially through the regulation of mRNP composition.
While many proteins are important for the nuclear export of RNA and proteins, NXF1 (also called Tap) and Crm1 (also called exportin 1) are the best characterized (Lei and Silver, 2002; Cullen, 2003). Most cellular mRNA export is mediated by a heterodimer of NXF1 and its partner p15 that is recruited to spliced RNA by multiple, redundant factors including the EJC and SR proteins such as Srp20 and 9G8 (Stutz and Izaurralde, 2003). Crm1, a member of the karyopherin family of nuclear transport receptors, is unrelated to NXF1/p15 and mediates the nuclear export of leucine-rich nuclear export signal (NES)-containing proteins by binding to them in the nucleus and escorting them to the cytoplasm. In addition to its role in protein export, Crm1 also mediates the export of several classes of RNA such as 5S ribosomal RNA, U snRNAs and some mRNAs through the action of NES-containing RNA binding proteins (Lei and Silver, 2002; Cullen, 2003). Although the inhibition of NXF1 (or Mex67p in yeast) function prevents the bulk of poly A+ RNA export in metazoans and yeast (Segref et al, 1997; Tan et al, 2000; Herold et al, 2001; Wilkie et al, 2001), fine regulation of mRNA export is likely to be more complex. For instance, a recent study in yeast showed that the export of different mRNA populations was differentially sensitive to the loss of Mex67p or the EJC component Yra1 (Hieronymus and Silver, 2003). In higher eukaryotes, most mRNAs appear to use the NXF1 nuclear export pathway (Herold et al, 2003), but under specific physiological conditions such as cellular stress, a minority of transcripts may utilize Crm1-dependent export pathways (Gallouzi and Steitz, 2001). For example, during heat shock, AU-rich elements (AREs) in the 3′ untranslated regions (UTRs) of mRNAs such as c-fos act as cis-acting elements to recruit a cellular protein, HuR, to the transcript, which allows Crm1-dependent nuclear export. Extrapolating from these points, it would not be unexpected if cis-acting elements connected to specific mRNA nuclear export pathways could influence cytoplasmic mRNA fate, perhaps by modulating mRNP composition.
Characterization of retroviral RNA export has helped elucidate the mechanisms of nuclear export for both cellular RNAs and proteins. Most notably, work on human immunodeficiency virus (HIV) and Mason-Pfizer monkey virus (M-PMV) RNA export has resulted in the identification of the aforementioned export factors Crm1 and NXF1. All retroviruses must export unspliced (intron-containing) as well as spliced transcripts to express the full complement of viral proteins and to allow packaging of the unspliced genomic RNA into cytoplasmically assembled viral particles (Pollard and Malim, 1998). In the case of HIV, all intron-containing mRNAs contain a highly structured cis-acting element called the Rev response element (RRE). This is bound by the viral Rev protein, a product of fully spliced mRNA, which induces export through the recruitment of Crm1 via its leucine-rich NES. Accordingly, two viral components, Rev and the RRE are essential for unspliced HIV mRNA export (Pollard and Malim, 1998). All M-PMV transcripts include an extended stem–loop structure known as the constitutive transport element (CTE) (Bray et al, 1994). In contrast to the RRE, the CTE serves as the direct binding site for NXF1, which then mediates export through engagement of normal mRNA transport processes (Gruter et al, 1998). Thus, the RRE and CTE are well-defined RNA export elements that connect to distinct nuclear export pathways.
Cytoplasmic HIV unspliced transcripts are either packaged into virions or translated into the Gag or Gag-Pol polyproteins (Freed, 1998). Gag is a 55 kDa protein containing all the signals necessary to mediate viral assembly, being both necessary and sufficient to form virion-like particles with the same density and size as wild-type particles. Coincident with budding, p55Gag is proteolytically cleaved five times to produce p17Gag (matrix, MA), p24Gag (capsid, CA), p7Gag (nucleocapsid, NC), p6Gag and two short spacer peptides, a process that drives core condensation and is known as maturation. Several important assembly domains have been characterized within HIV Gag. MA contains the membrane targeting (M) domain, which consists of a myristoylated amino-terminal glycine and a neighboring patch of basic amino acids. NC contains a major interaction (I) domain, which allows Gag to bind RNA and thereby nucleate protein–protein interactions. p6Gag contains a late budding (L) domain, which is required for membrane fission and virus release. Following translation, Gag translocates to the plasma membrane where the bulk of Gag–Gag oligomerization takes place. However, most of the steps involved in HIV particle assembly remain ill defined, particularly with respect to the role(s) of cellular factors. One exception to this is L domain activity; recent work has demonstrated that p6Gag binds Tsg101 and AIP-1/ALIX, which recruit the ESCRT vacuolar protein sorting machinery to allow membrane fission and budding (Garrus et al, 2001; VerPlank et al, 2001; Martin-Serrano et al, 2003; Strack et al, 2003; von Schwedler et al, 2003).
The analysis of species-specific barriers in the HIV life cycle has been very influential in identifying human genes important for virus replication. For example, reconstitution of HIV entry or transcription in rodent cells helped verify the key roles of CD4 and chemokine receptors, or CyclinT1, respectively, in these processes (Maddon et al, 1986; Feng et al, 1996; Wei et al, 1998). Importantly, however, murine cells expressing the human CXCR4/CCR5, CD4 and CycT1 genes still fail to support significant HIV replication (Garber et al, 1998; Bieniasz and Cullen, 2000; Mariani et al, 2000). Work from several groups has shown that at least one additional block to HIV replication in rodent cells is manifested during virus assembly (Bieniasz and Cullen, 2000; Mariani et al, 2000; Koito et al, 2003). In particular, even when substantial expression of p55Gag is achieved in murine cells, both Gag processing and virus budding are severely diminished.
Here, we report a novel example of how RNA nuclear export elements can modulate post-translational events in the cytoplasm. Specifically, we show that changing the HIV gag-pol mRNA export pathway from RRE/Rev/Crm1 dependence to CTE/NXF1 dependence restores efficient virion budding in murine cells. These findings suggest that the activities of retroviral RNA export elements extend beyond translocation across the nuclear envelope and raise the possibility that RNA export pathway selection can modulate the cytosolic fate or function of proteins.
Results
There are multiple HIV replication blocks in murine cells including entry, transcriptional elongation, splicing, assembly and infectivity (Maddon et al, 1986; Trono and Baltimore, 1990; Malim et al, 1991; Atchison et al, 1996; Wei et al, 1998; Bieniasz and Cullen, 2000; Mariani et al, 2000; Zheng et al, 2003). To analyze the assembly block specifically, we constructed a subgenomic vector containing the gag-pol and vif genes plus the RRE (GPV-RRE in Figure 1). Because this vector contains the major 5′ splice donor and the splice acceptors for vif and vpr, Gag is produced from an unspliced, intron-containing transcript similar to that expressed by a provirus. Transient transfection of constructs such as this bypasses the blocks in both viral entry and transcriptional elongation (the latter because this vector contains the CMV-IE promoter). In murine cells, the natural genomic transcript of HIV is ‘overspliced' to yield the fully spliced transcripts for tat, rev and nef (Trono and Baltimore, 1990; Malim et al, 1991; Zheng et al, 2003); the GPV-RRE construct should not have this problem because it does not contain the splice acceptors that lie upstream of these genes.
Figure 1.
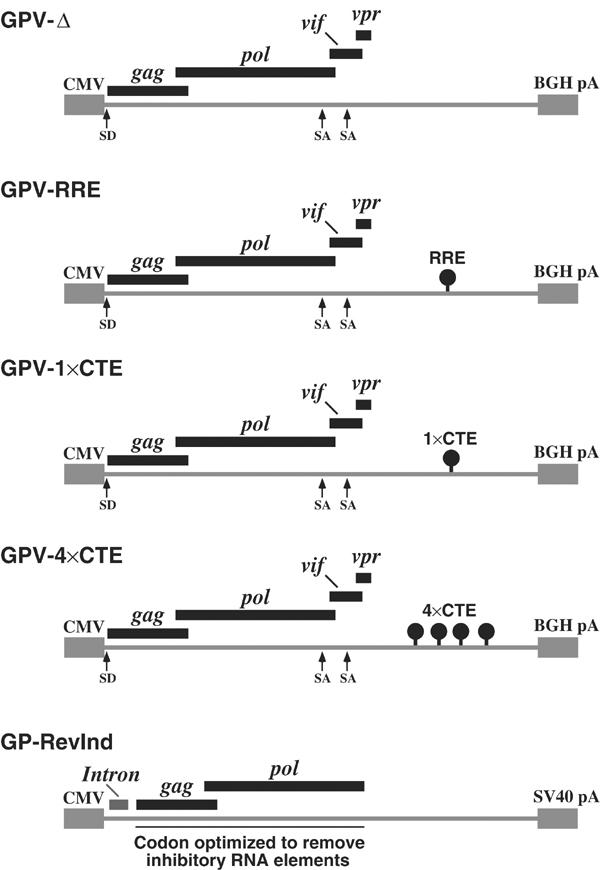
HIV Gag-Pol constructs. GPV-Δ, GPV-RRE, GPV-1 × CTE and GPV-4 × CTE were cloned into the pcDNA 3.1 vector with a CMV-IE promoter (CMV) and a bovine growth hormone polyadenylation signal (BGH pA). Splice donors (SD) and splice acceptors (SA) are marked. GP-RevInd was a codon-optimized Gag-Pol cloned into the pCI-Neo vector with the CMV-IE promoter (CMV) and the SV40 polyadenylation signal (SV40 pA) (Kotsopoulou et al, 2000). The vector contained an intron 5′ of the Gag-Pol coding region.
By examining Gag expression in cell lysates and corresponding culture supernatants, it is possible to determine if Gag processing and budding are occurring. For example, when human cells such as HeLa were transfected with GPV-RRE, Gag processing was clearly evident in the lysate with both the p55Gag precursor and processed p24Gag (CA) being present (Figure 2, lane 1). The substantial amount of p24Gag in the supernatant demonstrates that budding was also efficient in these cells (lane 1). In contrast, when transfected murine 3T3 cells were examined in the same way, p55Gag protein was expressed, but processing was very inefficient and budding was essentially absent (lane 3). These phenotypes parallel those previously described in a variety of rodent cells, including murine T cells (Bieniasz and Cullen, 2000; Mariani et al, 2000; 2001; Koito et al, 2003).
Figure 2.
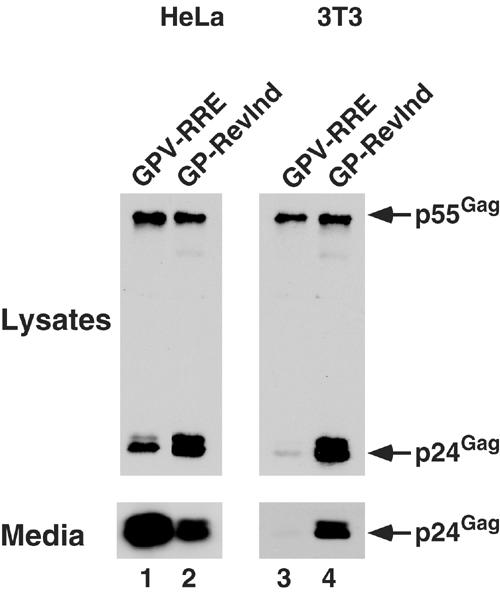
Rev-independent HIV Gag-Pol expression results in efficient Gag processing and budding in murine cells. HeLa (lanes 1 and 2) or 3T3 (lanes 3 and 4) cells were transfected with GPV-RRE+pcRev (lanes 1 and 3) or GP-RevInd+pcDNA 3.1 (lanes 2 and 4) expression vectors and then analyzed at ∼40 h for the presence of Gag proteins in whole-cell lysates and culture media by immunoprecipitation/immunoblot. The p24Gag content in the media from the 3T3 cells was also quantitated by ELISA, and GPV-RRE-transfected cells (lane 3) produced 0.88 ng/ml and the GP-RevInd-transfected cells (lane 4) produced 12 ng/ml.
It has previously been shown that a deficiency in trafficking to the plasma membrane underlies the inefficient processing and budding of HIV Gag in murine cells. Indirect immunofluorescence demonstrated that Gag was not localized at the plasma membrane in murine cells, and an electron microscopy analysis showed that budding structures were rarely found and cytoplasmic capsids were absent (Mariani et al, 2000; Chen et al, 2001; Koito et al, 2003). One possible reason for defective plasma membrane trafficking is that cellular factors necessary for Gag translocation may be defective in murine cells. Supporting this hypothesis, it has been shown that altering the membrane targeting signal by replacing the HIV MA region with murine leukemia virus (MLV) MA can partially restore budding in murine cells (Chen et al, 2001; Reed et al, 2002). In contrast, however, we had noted that certain expression vectors could produce wild-type Gag protein that is functional for robust budding in murine cells. For instance, an HIV RRE/Rev-independent Gag-Pol expression vector (Figure 1, GP-RevInd), in which codon optimization inactivated cis-acting inhibitory elements in gag and pol (Kotsopoulou et al, 2000), resulted in a 14-fold increase in virion particle production from transfected murine cells (Figure 2, lanes 3 and 4). These results indicated that the cellular factors necessary for the cytoplasmic transport of the HIV Gag protein are functional in murine cells. We therefore hypothesized that Gag translated from an mRNA analogous to that of a provirus is nonfunctional because it is unable to utilize the cellular factors necessary for trafficking to the plasma membrane and budding.
In the light of data developed in different systems regarding the potential regulatory effect(s) of nuclear history on RNA fate, we speculated that the mode of RNA nuclear export may be playing a crucial role in the functionality of gag-pol mRNAs and their translated products. In particular, HIV gag-pol mRNA normally utilizes the Crm1/Rev nuclear export pathway, whereas the Rev-independent gag-pol RNA has been shown to be Crm1 independent (Kotsopoulou et al, 2000) and most likely uses the NXF1/p15-dependent nuclear export pathway due to the presence of an EJC. To test the idea that RNA export pathways and HIV assembly may be connected, we constructed matched Gag-Pol-Vif constructs containing no export element, one copy of the M-PMV CTE (1 × CTE) or four tandem copies of the CTE (4 × CTE) (Wodrich et al, 2000) (see Figure 1) and compared these to the GPV-RRE construct. Importantly, the coding sequence of gag-pol was identical in all four constructs: thus, any changes in Gag membrane trafficking or virion budding would have had to be due to changes in the nuclear export element.
Each vector was transfected into HeLa (Figure 3A) or 3T3 (Figure 3B) cells and budding assays were performed as above. As expected, very little Gag was expressed in HeLa cells in the absence of an export element or with the RRE alone (Figure 3A, lanes 2 and 3). Also as expected, the addition of Rev to the RRE-containing vector resulted in efficient Gag expression, processing and particle budding (lane 4). A single copy of the CTE also stimulated significant Gag expression, but Gag processing and budding were both relatively inefficient (lane 5). Although the basis for this finding is not yet resolved, it was not entirely surprising since earlier work had demonstrated that a Rev-deficient HIV mutant that contained one copy of the CTE replicated poorly (Bray et al, 1994). In contrast, the presence of four copies of the CTE led to very efficient Gag production, processing and budding (lane 6).
Figure 3.
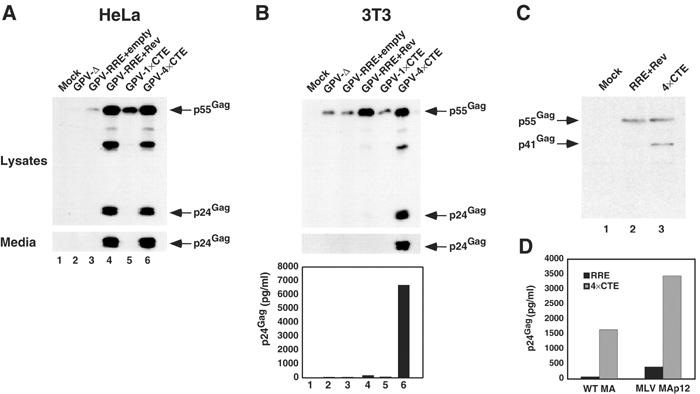
HIV Gag processing and budding are restored in murine cells by 4 × CTE/NXF1-mediated RNA export. (A, B) The 4 × CTE confers efficient budding in HeLa (A) and 3T3 (B) cells. Cells were transfected with the indicated GPV constructs and Gag proteins analyzed as in Figure 2. The 3T3 samples were examined further by ELISA (B, lower panel). (C) 3H myristylate labeling of Gag in 3T3 cells transfected with GPV-RRE+pcRev (lane 2) or GPV-4 × CTE (lane 3); lane 1 represents mock-transfected cells. Gag proteins were immunoprecipitated from cell lysates, resolved by SDS–PAGE and visualized by autoradiography. (D) The 4 × CTE enhances budding of the HIV Gag-Pol with the MLV MA membrane targeting domain. 3T3 cells transfected with GPV vectors based on wild-type NL4-3 or NL4-3 with the HIV MA domain replaced by the MLV MAp12 domains. Filtered media from the tranfections were analyzed by p24Gag ELISA.
Analysis of cell lysates from the transfected 3T3 cultures revealed that the general trends of relative Gag expression reflected those seen in HeLa cells. The patterns of particle budding were, however, quite different. Gag processing and budding were inefficient in rodent cells following RRE/Rev-mediated expression despite significant levels of Gag production (Figure 3B, lane 4). Strikingly, Gag processing and particle production were very efficient when expression was regulated by the 4 × CTE. By ELISA, the levels of HIV p24Gag in the 3T3 culture supernatants were as much as 40-fold higher for 4 × CTE samples compared to RRE/Rev samples (Figure 3B bottom panel, compare lane 4 to 6). Of note, Vif was not necessary for this phenotype as constructs encoding just Gag and the protease (PR) region of Pol gave similar results (data not shown).
As Gag must be cotranslationally myristoylated at its amino-terminus for it to be targeted to the plasma membrane (Freed, 1998), we 3H myristate labeled 3T3 cells transfected with the GPV-RRE (plus Rev) or GPV-4 × CTE constructs and evaluated the status of Gag by immunoprecipitation (Figure 3C). Intracellular Gag expressed from both constructs had similar 3H myristate incorporation, and, as expected, Gag in the GPV-4 × CTE transfected cells was cleaved into the p41Gag processing intermediate comprising p17Gag and p24Gag. Therefore, even though myristoylated Gag is expressed from mRNAs exported via Crm1- or NXF1-dependent pathways, only mRNA that utilizes the 4 × CTE/NXF1 pathway has the potential to generate efficiently Gag protein that is functional for budding in murine cells.
As it had been previously shown that substitution of the HIV MA region with the analogous region of MLV MA (MHIV) enhances HIV budding in murine cells (Chen et al, 2001; Reed et al, 2002), we tested whether utilization of the 4 × CTE/NXF1 nuclear export pathway could also enhance Gag budding in this context. Wild type and MHIV GPV-RRE and GPV-4 × CTE constructs were made, transfected into 3T3 cells, and p24Gag levels in the media then measured by ELISA (Figure 3D). While the MHIV Gag budded six-fold better than wild-type HIV Gag in the context of the RRE, Gag from the MHIV GPV-4 × CTE budded a further eight-fold better than Gag from the MHIV GPV-RRE. Thus, changing an unspliced RNA's nuclear export pathway from RRE/Rev/Crm1 to 4 × CTE/NXF1 can enhance budding in murine cells in the context of two different MA-encoded membrane targeting signals.
To eliminate the possibility that differences in intracellular Gag levels were responsible for differences in budding phenotypes (i.e., low Gag expression resulting in inefficient budding), we performed a titration assay (Figure 4A). Transfections of 3T3 cells with 2, 0.6 or 0.2 μg of the GPV-4 × CTE plasmid were performed using an empty vector to maintain DNA concentration, and compared to cells transfected with 2 μg of the RRE/GPV vector (plus Rev). Even when the intracellular levels of Gag expressed using the 4 × CTE were considerably lower than those expressed with the RRE/Rev vectors, budding remained much more efficient for the 4 × CTE construct (e.g., compare lanes 2 and 4). 3T3 cells transfected with GPV-RRE or GPV-4 × CTE also showed no differences in the splicing of viral RNAs as determined by Northern blot analysis of cytoplasmic RNA samples (Figure 4B). Therefore, the increased level of budding seen in 3T3 cells transfected with the GPV-4 × CTE construct is not due to changes in intracellular Gag expression levels or aberrant splicing.
Figure 4.

The 4 × CTE RNA export element does not enhance budding in murine cells by increasing intracellular Gag concentration or by altering splicing. (A) The induction of Gag budding from 3T3 cells transfected with GPV-4 × CTE (lanes 1–3) versus GPV-RRE+pcRev (lane 4) is not due to variations in intracellular Gag concentration. Cells were transfected either with a titration of GPV-4 × CTE (100%=2 μg pGPV-4 × CTE+1 μg pcDNA3.1, total DNA levels were maintained by the addition of pcDNA3.1) or 2 μg of GPV-RRE+1 μg pcRev, and the Gag proteins were analyzed as in Figure 1A. (B) The RRE and 4 × CTE RNA export elements do not lead to differences in splicing. Cytoplasmic RNA was isolated from 3T3 cells transfected with GPV-RRE or GPV-4 × CTE and analyzed by Northern blot with a probe specific for the gag gene. The difference in transcript sizes is due to the 441 bp difference in the length of the RRE compared to the 4 × CTE.
As discussed earlier, the inefficient budding phenotype of Gag expressed using the RRE/Rev system in murine cells can be attributed to a lack of membrane targeting. To demonstrate more directly that Gag membrane trafficking in murine cells was restored by the addition of 4 × CTE, membrane flotation assays were performed using lysates of transfected 3T3 cells (Figure 5A). Whereas only minor amounts of the Gag produced from the GPV-RRE vector were detected in the plasma membrane fractions, Gag expressed using the 4 × CTE construct efficiently trafficked to the plasma membrane with substantial amounts of both p55Gag and p24Gag present in fractions 3 and 4. As expected, Gag expressed in HeLa cells from RRE- or 4 × CTE-containing RNAs displayed similar profiles in the flotation assay to that of the GPV-4 × CTE sample in 3T3 cells (data not shown).
Figure 5.

4 × CTE/NXF1-mediated nuclear export promotes the association of Gag with the plasma membrane. (A) RNA export elements modulate Gag's ability to associate with membranes using a membrane flotation assay. 3T3 cells were transfected with GPV-RRE+pcRev (left panels) or GPV-4 × CTE (right panels) and the resulting postnuclear supernatants were analyzed using a membrane flotation assay. Following fractionation, all samples were examined for Gag content by immunoprecipitation/immunoblot and for transferrin receptor (TR) by immunoblot. The smaller TR bands (marked with *) are degradation products, but as the antibody used is a monoclonal to the cytoplasmic tail, these are additive with the full-length 95 kDa protein. Fractions 3 and 4 contain membrane-associated proteins and, potentially, demembranated immature viral cores. (B) Indirect immunofluorescence shows that RNA export elements modulate Gag's ability to be targeted to the plasma membrane. Subconfluent mouse 3T3 cells were seeded on glass coverslips and transfected with GPV-RRE (panels (i) and (ii)) or GPV-4 × CTE (panels (iii) and (iv)). At 20 h post-transfection, the cells were fixed and stained with rabbit polyclonal anti-MA antibody.
Indirect immunofluorescence analysis of Gag in 3T3 cells transfected with the GPV-RRE or the GPV-4 × CTE constructs showed that Gag translated from RNA containing the 4 × CTE is targeted specifically to the plasma membrane. In particular, Gag expressed using the GPV-RRE vector was dispersed throughout the cytosol (Figure 5B, panels (i) and (ii)) while Gag expressed from 4 × CTE-containing transcripts was detected predominantly in close proximity to the plasma membrane (Figure 5B, panels (iii) and (iv)). Indeed, this staining pattern was also seen in HeLa cells transfected with either construct (data not shown). Thus, the identity of the RNA nuclear export element can determine whether or not the HIV p55Gag polyprotein is trafficked to the plasma membrane in murine cells.
Finally, it remained plausible that the 4 × CTE and RRE export elements could influence Gag fate by acting as a 3′ UTR in the cytoplasm, rather than through events initiated in the nucleus. To eliminate this possibility, recombinant vaccinia viruses that expressed GPV-Δ, GPV-RRE and GPV-4 × CTE were generated. Because vaccinia virus replicates in the cytosol (Moss, 2001), the influence of regulatory elements that act in the nucleus should be lost. When 3T3 cells (Figure 6) or HeLa cells (data not shown) were infected with vvGPV-Δ, vvGPV-RRE or vvGPV-4 × CTE, the levels of p24Gag released into the culture supernatants (as well as the levels of intracellular p55Gag, data not shown) were similarly high. Therefore, the export elements can only regulate HIV budding when expressed in the nucleus.
Figure 6.

The 4 × CTE does not enhance Gag budding in murine cells when the RNA is directly expressed in the cytoplasm. 3T3 cells were infected with vaccinia viruses expressing GPV-Δ, GPV-RRE or GPV-4 × CTE. At 48 h after infection, lysates and media were harvested and the Gag proteins analyzed as in Figure 2. The relative levels of p24Gag in the media represent the mean of three independent experiments normalized to the vvGPV-Δ samples (mean value of 1.81 μg/ml) with error bars representing standard deviation. All three viruses expressed similar amounts of intracellular Gag (not shown).
Discussion
The results described in this paper indicate that HIV unspliced RNA export and Gag trafficking to the plasma membrane are linked. By simply changing the RNA export element from the RRE to 4 × CTE, we can restore Gag assembly and budding in murine cells (Figures 3 and 5). To explain how a pretranslational event, RNA export, could modulate a post-translational event, membrane trafficking, we hypothesize that HIV RNA is ‘marked' at (or by) nuclear export such that the cytosolic fate of the encoded Gag is predetermined. Based on our findings, both the RRE/Rev/Crm1 and 4 × CTE/NXF1 nuclear export pathways successfully ‘mark' unspliced gag-pol mRNA in human cells and promote proper assembly. However, in murine cells, ‘marking' through the action of RRE/Rev/Crm1 is defective and HIV assembly is inhibited. Possibilities for the ‘mark' include the structure of the mRNA itself or proteins that comprise the mRNP; these could be added or removed as the export complex is formed, as it transits the NPC, or as it is remodeled in the cytoplasm upon the completion of export. Because experiments using mouse–human cell fusions have demonstrated that the assembly-permissive human cell phenotype is dominant (Trono and Baltimore, 1990; Bieniasz and Cullen, 2000; Mariani et al, 2001), it is most likely that murine cells lack a critical cellular factor (or express a defective version thereof) necessary for RNA marking in the context of the Rev/Crm1 export machinery. In contrast, 4 × CTE/NXF1-mediated export results in productive marking of the gag-pol RNA mRNP in both murine and human cells. Our results show that the nuclear history of an RNA, mediated by the mode of RNA export, can regulate a protein's activity after translation.
We posit that differential cytosolic RNA localization, that is, where mRNA is localized to a specific region(s) of a cell or embryo (Palacios and Johnston, 2001; Kloc et al, 2002), could explain why 4 × CTE/NXF1-mediated RNA export rescues HIV Gag assembly and virion release in murine cells. Due to the viscosity of the cytosol, only RNAs that are shorter than 1.6 kb can freely diffuse and as much as 30% of the cytosolic mRNAs may be actively targeted to cytoskeletal polysomes (Jansen, 1999). Because a large RNA such as the ∼9 kb HIV genome is unlikely to diffuse randomly, we suggest that murine cells fail to mediate proper cytosolic RNA localization following export via the RRE/Rev/Crm1 pathway. The 4 × CTE/NXF1 nuclear export pathway results in no such defect as the RNA would be properly localized and translated Gag would then enter a trafficking pathway that culminates in efficient virus assembly. In human cells, RNA localization would be functional for unspliced RNA exported by both pathways. This model raises several interesting questions. First, as a protein ‘mark' on the RNA would likely mediate the cytosolic localization, what are the differences in RNP composition between HIV genomes using each nuclear export pathway? Second, what are the cis-acting determinants that may regulate RNP composition, and, if the CTE itself is important, is there an important RNA regulation step for its natural host virus, M-PMV? Third, where is the HIV RNA localized for translation (as opposed to packaging) under assembly-permissive versus -nonpermissive conditions?
We do not know why Gag translated from RNA exported via RRE/Rev/Crm1 is not targeted to the plasma membrane of murine cells. We believe that Gag expressed this way should be competent for routing to the plasma membrane as it is myristoylated and no other post-translational modifications are known to be required for membrane trafficking. Based on the fact that multiple Rev/Crm1-independent constructs generate Gag protein that efficiently buds in murine cells, the cellular factors necessary for Gag trafficking to the plasma membrane appear to be functional (Figures 2, 3 and 6). Recently, it has been shown for MLV and M-PMV that Gag is trafficked to the plasma membrane in the presence of the viral envelope glycoproteins using Rab11, transferrin-positive vesicles from the recycling endosome (Basyuk et al, 2003; Sfakianos and Hunter, 2003). HIV Gag has also been shown to localize to multivesicular bodies and endosomes in multiple cell types (Goff et al, 2003; Nydegger et al, 2003; Pelchen-Matthews et al, 2003; Sherer et al, 2003; von Schwedler et al, 2003; Ono and Freed, 2004), although mechanistically how this mediates Gag trafficking to the plasma membrane is unclear. If HIV gag-pol mRNA is indeed actively localized to a cytosolic compartment or microdomain, this could facilitate access of both the RNA and encoded proteins to important cellular assembly cofactors. Some potential consequences for nascent Gag molecules being exposed to appropriate spatially restricted cofactors include interactions with factors involved in trafficking to the plasma membrane or with chaperones such as Hsp70, TRiC and HP68 (Hong et al, 2001; Gurer et al, 2002; Zimmerman et al, 2002). As Gag oligomerization has been shown to be necessary for myristate exposure (Tang et al, 2004), chaperones could regulate an early cytosolic oligomerization step.
The notion of a ‘viral assembly factory' for HIV that is established by active RNA localization is not without precedent. Other viruses such as hepatitis C virus, herpesviruses and poxviruses have been shown to assemble in viral factories where the structural proteins and cellular factors necessary for assembly are concentrated (Sanchez et al, 2000; Heath et al, 2001; Egger et al, 2002; Shi et al, 2003). Recently, it has been shown that M-PMV assembles in a pericentriolar compartment and that trafficking to this compartment is necessary for correct capsid assembly and the production of infectious virus (Sfakianos et al, 2003). M-PMV Gag is trafficking to this region cotranslationally by a dominant protein signal within the MA region (Rhee and Hunter, 1990; Choi et al, 1999). Since an analogous signal does not appear to be present in HIV Gag, it is plausible that the targeting to its assembly compartment could be mediated by RNA localization rather than by protein localization.
Genome RNA marking as a means of regulating critical cytosolic events may be a common feature in the life cycles of multiple retroviruses. Perturbation of the RNA export element of avian leukosis virus (ALV) has been associated with budding and genome packaging defects, as well as with reductions in cytoplasmic RNA levels (Boris-Lawrie et al, 2001). Also, the ALV CTE is nonfunctional in mammalian (as opposed to avian) cells, leading to assembly defects as well as reduced genome RNA export (Nasioulas et al, 1995; Ogert et al, 1996). Reminiscent of the data presented herein, altering the ALV genome RNA export pathway by the addition of HIV RRE (and Rev) conferred efficient viral budding in mammalian cells (Nasioulas et al, 1995).
As an obligate intracellular parasite, HIV exploits cellular machinery to achieve productive infection and replication. Therefore, understanding the cellular factors necessary for the regulation of HIV genomic RNA function may aid in the general understanding of cellular gene regulation. For example, we liken these findings in the HIV and ALV systems to some previous studies of U snRNA biogenesis and fate (Ohno et al, 2002). In particular, when the pathway of U1 snRNA export was manipulated from normal Crm1 dependence to NXF1 dependence, the exported RNA, while comprising the identical RNA sequence, was incapable of being reimported into the nucleus (as the U1 snRNP). Our results using HIV further support the concept that RNA nuclear export and associated processes can regulate RNA and protein activity in the cytoplasm.
Finally, the demonstration that efficient HIV assembly and budding is rescued in murine cells by the addition of 4 × CTE in cis may have important implications for the development of small animal models of HIV replication and pathogenesis. As noted earlier, even though a number of essential replication cofactors have been identified in humans, their collective introduction into murine cells has, thus far, failed to render such cells permissive to productive HIV replication (Garber et al, 1998; Bieniasz and Cullen, 2000; Mariani et al, 2000). We propose that using an alternative RNA export element will now help to overcome the barrier to virion assembly.
Materials and methods
Cell culture and plasmids
HeLa and 3T3 cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1% Penn-Strep. The Rev-independent (pSYNGP, codon-optimized) Gag-Pol vectors have been described (Kotsopoulou et al, 2000). The GPV vectors were constructed by first inserting the RRE (HIV-1IIIB isolate nucleotides 7708–8058) or the 1 × CTE from M-PMV (nucleotides 8007–8176) as XhoI/XbaI fragments into pcDNA3.1 (Invitrogen); the multimeric 4 × CTE (Wodrich et al, 2000) was inserted as an EcoRI/NotI fragment. A SacI/EcoRI fragment of HIV-1YU2, HIVNL4-3 or HIVNL4-3 with the MLV MAp12 (Chen et al, 2001) that encompassed the Gag-Pol-Vif region (nucleotides 678–5741 for HIV-1YU2, 487–5743 for HIVNL4-3) was then inserted between the HindIII and EcoRI sites of these vectors as well as of pcDNA3.1. The HIV-1YU2–based vectors were used for all experiments except that reported in Figure 3D. The HIV Rev expression vector pcREV contains the Rev cDNA cloned into pcDNA3.
Budding assays
Gag expression vectors were transfected into HeLa or 3T3 monolayers using FuGENE 6 (Roche) according to the manufacturer's directions and the medium was changed at ∼24 h. At 40 h post-transfection, the culture supernatants were removed, filtered through 0.45 μm filters and adjusted to 1 × RIPA buffer (10 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate) for immunoprecipitation: corresponding cell pellets were harvested and lysed in 1 × RIPA buffer. All samples were incubated with a polyclonal CA-specific antiserum raised in rabbits (Simon et al, 1997), and the immunoprecipitates were subjected to immunoblot analysis using a murine CA-specific monoclonal antibody (Gaddis et al, 2003), an HRP-conjugated secondary antibody and enhanced chemiluminescence. Supernatant p24Gag levels were measured by ELISA (Perkin Elmer) according to the manufacturer's directions.
Membrane flotation assays
Analyses of Gag interactions with membranes were performed similar to that described by Ono and Freed (1999). Briefly, transfected HeLa or 3T3 cells were harvested at ∼40 h, and postnuclear supernatants (PNSs) were generated by sonicating the cells and pelleting nuclei at 1000 g. PNSs were adjusted to 70% sucrose (w/v) in 2.5 ml, and overlaid with 6 ml of 65% (w/v) and 2.5 ml of 10% (w/v) sucrose in a 14 × 89 mm tube (Beckman) to create a discontinuous gradient between the 10 and 65% sucrose fractions. Gradients were centrifuged at 120 000 g for 18 h and eleven 1 ml fractions were collected. Each fraction was adjusted to 1 × RIPA, and either analyzed for Gag content by immunoprecipitation followed by immunoblot (as described above) or directly by immunoblot using a transferrin receptor (TR)-specific antibody raised in mice (Zymed), an HRP-conjugated secondary antibody and enhanced chemiluminescence. Fractions 3 and 4 contain the membrane-bound Gag and any potential demembranated immature viral cores.
Immunofluorescence
Subconfluent 3T3 cell monolayers were seeded onto coverslips (Cover Glass, 23 mm, BDH) in six-well plates and transfected using FuGENE 6. Cells were fixed 20 h post-transfection with 4% paraformaldehyde (PFA) in 250 mM HEPES pH 7.4 for 10 min on ice. This was followed by further fixation with 8% PFA in 250 mM HEPES pH 7.4 for 50 min at RT. Cells were then washed, incubated with 50 mM ammonium chloride in PBS (to quench the unreacted PFA), permeabilized with 0.2% Triton X-100 in PBS and incubated with blocking solution (10% FBS in PBS) for 1 h at RT. Cells were stained with rabbit polyclonal anti-serum UP595 (raised against HIV MA) at a 1:1000 dilution for 1 h, and bound antibody was detected with a goat anti-rabbit IgG conjugated to tetramethyl rhodamine isothiocyanate (TRITC). The stained cells were then washed and mounted onto Citifluor AF-1 mounting medium containing 1 μg/ml of 4′, 6-diamin-2-phenylin-dol-dihydrochloride (DAPI) for DNA staining. Images were collected with a laser confocal scanning microscope (DM IRE2, Leica) and processed with the LCS (version 2.02) software (Leica) and Adobe Photoshop (version 6.0). Each image represents a single horizontal optical slice (0.1–0.25 μM) taken through the estimated centre of the sample.
3H myristylate labeling
Metabolic labeling was performed similar to that described by Bijlmakers and Marsh (2000). At 40 h post-transfection, cells were preincubated in labeling media (DMEM, 5% serum, 5 mM sodium pyruvate and 4 × nonessential amino acids) for 1 h. In all, 0.56 mCi/ml [9,10(n)-3H]myristic acid (Amersham) was then added in DMSO, and the cultures were maintained for a further 6 h. The cells were washed with PBS, lysed in RIPA buffer and the Gag proteins were immunoprecipitated with a CA-specific polyclonal antiserum raised in rabbits (Simon et al, 1997). Samples were resolved by SDS–PAGE and visualized by autoradiography.
Northern blot
Cytoplasmic RNA was isolated from transfected 3T3 cells using the Quigen RNeasy kit according to the manufacturer's directions. A Northern blot was performed as previously described (Sheehy et al, 2002) using a probe for the gag gene (bp 1083–1710 of the YU2 provirus).
Vaccinia virus cloning and infections
Gag expression cassettes were cloned into the vaccinia recombination plasmid pSC65 to create pSCGPV-Δ, pSCGPV-RRE and pSCGPV4 × CTE. Recombinant vaccinia viruses were generated as described in Current Protocols in Molecular Biology. Briefly, recombination plasmids were transfected into HEK293T cells that had been infected with the WR strain of vaccinia at an MOI of 0.05. At 48 h post-transfection, the cells were harvested, freeze thawed to release recombinant viruses, and the recombinants isolated through three rounds of plaque purification using HuTK-143B cells in the presence of BrdU. Stocks of vvGPV-Δ, vvGPV-RRE or vvGPV4 × CTE were expanded using HeLaS3 cells, and titers determined on BSC-1 cells. Budding assays were performed by infecting 90% confluent monolayers of HeLa or 3T3 cells at an MOI of 10 in DMEM, 2.5% FCS for 60 min, washing and examining cell supernatants and lysates by ELISA or immunoprecipitation/immunoblot as above.
Acknowledgments
We thank members of the Malim lab for critically reading this manuscript. Kyriacos Mitrophanous, Hans-Georg Krausslich, Bryan Cullen, Benjamin Chen, David Rekosh and Marie-Louise Hammarskjold kindly provided reagents. We also thank Marie-Jose Bijlmakers for advice on myristylate labeling. This work was supported by grants from the National Institutes of Health (AI46942) and the Medical Research Council. CMS was supported in part by a National Institutes of Health Training Grant (AI07325). MHM is an Elizabeth Glaser Scientist supported by the Elizabeth Glaser Pediatric AIDS Foundation.
References
- Atchison RE, Gosling J, Monteclaro FS, Franci C, Digilio L, Charo IF, Goldsmith MA (1996) Multiple extracellular elements of CCR5 and HIV-1 entry: dissociation from response to chemokines. Science 274: 1924–1926 [DOI] [PubMed] [Google Scholar]
- Basyuk E, Galli T, Mougel M, Blanchard JM, Sitbon M, Bertrand E (2003) Retroviral genomic RNAs are transported to the plasma membrane by endosomal vesicles. Dev Cell 5: 161–174 [DOI] [PubMed] [Google Scholar]
- Bieniasz PD, Cullen BR (2000) Multiple blocks to human immunodeficiency virus type 1 replication in rodent cells. J Virol 74: 9868–9877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijlmakers MJ, Marsh M (2000) Hsp90 is essential for the synthesis and subsequent membrane association, but not the maintenance, of the Src-kinase p56(lck). Mol Biol Cell 11: 1585–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boris-Lawrie K, Roberts TM, Hull S (2001) Retroviral RNA elements integrate components of post-transcriptional gene expression. Life Sci 69: 2697–2709 [DOI] [PubMed] [Google Scholar]
- Bray M, Prasad S, Dubay JW, Hunter E, Jeang KT, Rekosh D, Hammarskjold ML (1994) A small element from the Mason-Pfizer monkey virus genome makes human immunodeficiency virus type 1 expression and replication Rev-independent. Proc Natl Acad Sci USA 91: 1256–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BK, Rousso I, Shim S, Kim PS (2001) Efficient assembly of an HIV-1/MLV Gag-chimeric virus in murine cells. Proc Natl Acad Sci USA 98: 15239–15244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi G, Park S, Choi B, Hong S, Lee J, Hunter E, Rhee SS (1999) Identification of a cytoplasmic targeting/retention signal in a retroviral Gag polyprotein. J Virol 73: 5431–5437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR (2003) Nuclear RNA export. J Cell Sci 116: 587–597 [DOI] [PubMed] [Google Scholar]
- Daneholt B (2001) Assembly and transport of a premessenger RNP particle. Proc Natl Acad Sci USA 98: 7012–7017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G, Kim VN, Kataoka N (2002) Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 3: 195–205 [DOI] [PubMed] [Google Scholar]
- Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K (2002) Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76: 5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE, Berger EA (1996) HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272: 872–877 [DOI] [PubMed] [Google Scholar]
- Freed EO (1998) HIV-1 gag proteins: diverse functions in the virus life cycle. Virology 251: 1–15 [DOI] [PubMed] [Google Scholar]
- Gaddis NC, Chertova E, Sheehy AM, Henderson LE, Malim MH (2003) Comprehensive investigation of the molecular defect in vif-deficient human immunodeficiency virus type 1 virions. J Virol 77: 5810–5820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallouzi IE, Steitz JA (2001) Delineation of mRNA export pathways by the use of cell-permeable peptides. Science 294: 1895–1901 [DOI] [PubMed] [Google Scholar]
- Garber ME, Wei P, KewalRamani VN, Mayall TP, Herrmann CH, Rice AP, Littman DR, Jones KA (1998) The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev 12: 3512–3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI (2001) Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107: 55–65 [DOI] [PubMed] [Google Scholar]
- Goff A, Ehrlich LS, Cohen SN, Carter CA (2003) Tsg101 control of human immunodeficiency virus type 1 Gag trafficking and release. J Virol 77: 9173–9182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruter P, Tabernero C, von Kobbe C, Schmitt C, Saavedra C, Bachi A, Wilm M, Felber BK, Izaurralde E (1998) TAP, the human homolog of Mex67p, mediates CTE-dependent RNA export from the nucleus. Mol Cell 1: 649–659 [DOI] [PubMed] [Google Scholar]
- Gurer C, Cimarelli A, Luban J (2002) Specific incorporation of heat shock protein 70 family members into primate lentiviral virions. J Virol 76: 4666–4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachet O, Ephrussi A (2004) Splicing of oskar RNA in the nucleus is coupled to its cytoplasmic localization. Nature 428: 959–963 [DOI] [PubMed] [Google Scholar]
- Heath CM, Windsor M, Wileman T (2001) Aggresomes resemble sites specialized for virus assembly. J Cell Biol 153: 449–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold A, Klymenko T, Izaurralde E (2001) NXF1/p15 heterodimers are essential for mRNA nuclear export in Drosophila. RNA 7: 1768–1780 [PMC free article] [PubMed] [Google Scholar]
- Herold A, Teixeira L, Izaurralde E (2003) Genome-wide analysis of nuclear mRNA export pathways in Drosophila. EMBO J 22: 2472–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieronymus H, Silver PA (2003) Genome-wide analysis of RNA–protein interactions illustrates specificity of the mRNA export machinery. Nat Genet 33: 155–161 [DOI] [PubMed] [Google Scholar]
- Hong S, Choi G, Park S, Chung AS, Hunter E, Rhee SS (2001) Type D retrovirus Gag polyprotein interacts with the cytosolic chaperonin TRiC. J Virol 75: 2526–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RP (1999) RNA-cytoskeletal associations. FASEB J 13: 455–466 [PubMed] [Google Scholar]
- Kloc M, Zearfoss NR, Etkin LD (2002) Mechanisms of subcellular mRNA localization. Cell 108: 533–544 [DOI] [PubMed] [Google Scholar]
- Koito A, Shigekane H, Matsushita S (2003) Ability of small animal cells to support the postintegration phase of human immunodeficiency virus type-1 replication. Virology 305: 181–191 [DOI] [PubMed] [Google Scholar]
- Kotsopoulou E, Kim VN, Kingsman AJ, Kingsman SM, Mitrophanous KA (2000) A Rev-independent human immunodeficiency virus type 1 (HIV-1)-based vector that exploits a codon-optimized HIV-1 gag-pol gene. J Virol 74: 4839–4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei EP, Silver PA (2002) Protein and RNA export from the nucleus. Dev Cell 2: 261–272 [DOI] [PubMed] [Google Scholar]
- Maddon PJ, Dalgleish AG, McDougal JS, Clapham PR, Weiss RA, Axel R (1986) The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell 47: 333–348 [DOI] [PubMed] [Google Scholar]
- Malim MH, McCarn DF, Tiley LS, Cullen BR (1991) Mutational definition of the human immunodeficiency virus type 1 Rev activation domain. J Virol 65: 4248–4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5: 89–99 [DOI] [PubMed] [Google Scholar]
- Mariani R, Rasala BA, Rutter G, Wiegers K, Brandt SM, Krausslich HG, Landau NR (2001) Mouse–human heterokaryons support efficient human immunodeficiency virus type 1 assembly. J Virol 75: 3141–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani R, Rutter G, Harris ME, Hope TJ, Krausslich HG, Landau NR (2000) A block to human immunodeficiency virus type 1 assembly in murine cells. J Virol 74: 3859–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Yarovoy A, Perez-Caballero D, Bieniasz PD (2003) Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci USA 100: 12414–12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B (2001) Poxviridae: the viruses and their replication. In Fields Virology, Knipe DH, PM (ed) Vol. 2, pp 2849–2884. Philadelphia: Lippincott Williams & Wilkins [Google Scholar]
- Nasioulas G, Hughes SH, Felber BK, Whitcomb JM (1995) Production of avian leukosis virus particles in mammalian cells can be mediated by the interaction of the human immunodeficiency virus protein Rev and the Rev-responsive element. Proc Natl Acad Sci USA 92: 11940–11944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Le Hir H, Moore MJ (2004) Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev 18: 210–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nydegger S, Foti M, Derdowski A, Spearman P, Thali M (2003) HIV-1 egress is gated through late endosomal membranes. Traffic 4: 902–910 [DOI] [PubMed] [Google Scholar]
- Ogert RA, Lee LH, Beemon KL (1996) Avian retroviral RNA element promotes unspliced RNA accumulation in the cytoplasm. J Virol 70: 3834–3843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Segref A, Kuersten S, Mattaj IW (2002) Identity elements used in export of mRNAs. Mol Cell 9: 659–671 [DOI] [PubMed] [Google Scholar]
- Ono A, Freed EO (1999) Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J Virol 73: 4136–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO (2004) Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J Virol 78: 1552–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios IM, Gatfield D, St Johnston D, Izaurralde E (2004) An eIF4AIII-containing complex required for mRNA localization and nonsense-mediated mRNA decay. Nature 427: 753–757 [DOI] [PubMed] [Google Scholar]
- Palacios IM, Johnston DS (2001) Getting the message across: the intracellular localization of mRNAs in higher eukaryotes. Annu Rev Cell Dev Biol 17: 569–614 [DOI] [PubMed] [Google Scholar]
- Pelchen-Matthews A, Kramer B, Marsh M (2003) Infectious HIV-1 assembles in late endosomes in primary macrophages. J Cell Biol 162: 443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard VW, Malim MH (1998) The HIV-1 Rev protein. Annu Rev Microbiol 52: 491–532 [DOI] [PubMed] [Google Scholar]
- Reed M, Mariani R, Sheppard L, Pekrun K, Landau NR, Soong NW (2002) Chimeric human immunodeficiency virus type 1 containing murine leukemia virus matrix assembles in murine cells. J Virol 76: 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed R, Hurt E (2002) A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell 108: 523–531 [DOI] [PubMed] [Google Scholar]
- Rhee SS, Hunter E (1990) A single amino acid substitution within the matrix protein of a type D retrovirus converts its morphogenesis to that of a type C retrovirus. Cell 63: 77–86 [DOI] [PubMed] [Google Scholar]
- Sanchez V, Greis KD, Sztul E, Britt WJ (2000) Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J Virol 74: 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segref A, Sharma K, Doye V, Hellwig A, Huber J, Luhrmann R, Hurt E (1997) Mex67p, a novel factor for nuclear mRNA export, binds to both poly(A)+ RNA and nuclear pores. EMBO J 16: 3256–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfakianos JN, Hunter E (2003) M-PMV capsid transport is mediated by Env/Gag interactions at the pericentriolar recycling endosome. Traffic 4: 671–680 [DOI] [PubMed] [Google Scholar]
- Sfakianos JN, LaCasse RA, Hunter E (2003) The M-PMV cytoplasmic targeting-retention signal directs nascent Gag polypeptides to a pericentriolar region of the cell. Traffic 4: 660–670 [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418: 646–650 [DOI] [PubMed] [Google Scholar]
- Sherer NM, Lehmann MJ, Jimenez-Soto LF, Ingmundson A, Horner SM, Cicchetti G, Allen PG, Pypaert M, Cunningham JM, Mothes W (2003) Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4: 785–801 [DOI] [PubMed] [Google Scholar]
- Shi ST, Lee KJ, Aizaki H, Hwang SB, Lai MM (2003) Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J Virol 77: 4160–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JH, Fouchier RA, Southerling TE, Guerra CB, Grant CK, Malim MH (1997) The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J Virol 71: 5259–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, Gottlinger HG (2003) AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114: 689–699 [DOI] [PubMed] [Google Scholar]
- Stutz F, Izaurralde E (2003) The interplay of nuclear mRNP assembly, mRNA surveillance and export. Trends Cell Biol 13: 319–327 [DOI] [PubMed] [Google Scholar]
- Tan W, Zolotukhin AS, Bear J, Patenaude DJ, Felber BK (2000) The mRNA export in Caenorhabditis elegans is mediated by Ce-NXF-1, an ortholog of human TAP/NXF and Saccharomyces cerevisiae Mex67p. RNA 6: 1762–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF (2004) Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci USA 101: 517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trono D, Baltimore D (1990) A human cell factor is essential for HIV-1 Rev action. EMBO J 9: 4155–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VerPlank L, Bouamr F, LaGrassa TJ, Agresta B, Kikonyogo A, Leis J, Carter CA (2001) Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc Natl Acad Sci USA 98: 7724–7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, Scott A, Krausslich HG, Kaplan J, Morham SG, Sundquist WI (2003) The protein network of HIV budding. Cell 114: 701–713 [DOI] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang SM, Fischer WH, Jones KA (1998) A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92: 451–462 [DOI] [PubMed] [Google Scholar]
- Wilkie GS, Zimyanin V, Kirby R, Korey C, Francis-Lang H, Van Vactor D, Davis I (2001) Small bristles, the Drosophila ortholog of NXF-1, is essential for mRNA export throughout development. RNA 7: 1781–1792 [PMC free article] [PubMed] [Google Scholar]
- Wodrich H, Schambach A, Krausslich HG (2000) Multiple copies of the Mason-Pfizer monkey virus constitutive RNA transport element lead to enhanced HIV-1 Gag expression in a context-dependent manner. Nucleic Acids Res 28: 901–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YH, Yu HF, Peterlin BM (2003) Human p32 protein relieves a post-transcriptional block to HIV replication in murine cells. Nat Cell Biol 5: 611–618 [DOI] [PubMed] [Google Scholar]
- Zimmerman C, Klein KC, Kiser PK, Singh AR, Firestein BL, Riba SC, Lingappa JR (2002) Identification of a host protein essential for assembly of immature HIV-1 capsids. Nature 415: 88–92 [DOI] [PubMed] [Google Scholar]