

Abstract
Sodium-chloride coupled neurotransmitter transporters achieve reuptake of their physiological substrate by exploiting the pre-existing sodium-gradient across the cellular membrane. This terminates the action of previously released substrate in the synaptic cleft. However, a change of the transmembrane ionic gradients or specific binding of some psychostimulant drugs to these proteins, like amphetamine and its derivatives, induce reverse operation of neurotransmitter:sodium symporters. This effect eventually leads to an increase in the synaptic concentration of non-exocytotically released neurotransmitters [and – in the case of the norepinephrine transporters, underlies the well-known indirect sympathomimetic activity]. While this action has long been appreciated, the underlying mechanistic details have been surprisingly difficult to understand. Some aspects can be resolved by incorporating insights into the oligomeric nature of transporters, into the nature of the accompanying ion fluxes, and changes in protein kinase activities.
Keywords: carrier-mediated release, clinical therapeutics, drug targets, neurotransmitter transporters, reverse operation, transporter-mediated release
Synaptic transmission is terminated by diffusion of neurotransmitter out of the synaptic cleft, by enzymatic degradation or by the action of neurotransmitter transporters (Iversen 1971). The latter re-accumulate the neurotransmitter into the presynaptic specialization by using the sodium gradient as the driving force (Rudnick and Clark 1993). This secondary-active transport process constitutes the most economical – and also, fastest means to retrieve released neurotransmitter from the neuronal vicinity. Among the transporters using electrochemical gradients to drive the accumulation of substrates, the neurotransmitter:sodium symporters (NSS; Saier et al. 2006) comprise the largest family (Nelson 1998). They include transporters for monoamines (i.e. dopamine, DAT, norepinephrine, NET, and serotonin, SERT, also abbreviated as 5-HTT), amino acids [i.e. GABA (GABA transporter), glycine, proline and taurine], as well as osmolytes (i.e. betaine and creatine) and the so-called ‘orphan’ transporters with currently unknown substrate specificity (Masson et al. 1999; Saier et al. 2006). The NSS family is of obvious medical relevance since a number of different disorders can be efficiently treated by inhibiting transporters: here, it is primarily mental disease like depression and inhibition of SERT and NET and epilepsy and inhibition of GABA-transporter-1 (GAT1) (Iversen 2000).
There is a wealth of data on ionic requirements, stoichiometry, regulation, physiological and pharmacological properties of the re-uptake process mediated by NSS (for review see Torres et al. 2003b). Moreover, there is general consensus that these transport proteins share a common structural motif of 12 hydrophobic transmembrane helices as indicated by hydropathy plot analysis (Amara and Kuhar 1993), studies employing antibodies (Bruss et al. 1995), electron microscopic immunocytochemistry (Nirenberg et al. 1997) and functional assays using thiol-reactive reagents (Chen and Reith 2000; Sen et al. 2005).
Currently, the interest in the transport process has been rekindled because several snapshots have become available that allow to re-examine previous educated guesses: the x-ray crystal structures of the bacterial transporter LeuTAa from Aquifex aeolicus has been solved in several conformations. LeuTAa is homologous to NSS transport proteins (Yamashita et al. 2005; Singh 2008; Singh et al. 2008). In spite of the low overall homology (some 20–25% residues in LeuTAa and mammalian NSS are identical), there is remarkable conservation around the binding site (i.e., more than 50% identity): this information clearly allows for making more educated guesses than those relying solely on site-directed mutagenesis. It is also gratifying to see that the crystallographic snapshots delineate a sequence of events (Singh 2008) that is consistent with the venerable alternating access model: some 40 years ago, Jardetzky (1966) conceptualized the conformational steps required for substrate translocation by inferring a minimum of two states during the transport cycle that differed by substrate accessibility: ‘open-to-out/closed-to-in’ (i.e. ‘outward-facing conformation’) or ‘closed-to-out/open-to-in’ (i.e. ‘inward-facing conformation’. Importantly, only one of the gates (extracellular or cytoplasmic) should be open at any given time point (otherwise the protein functions as a channel rather than a transporter). Furthermore, the hypothesis predicted a transition state in which the protein changes from outward- to inward-facing conformations via an occluded state. Here, both putative gates are closed. The various crystal structures (Singh 2008) support this scheme and thus have proven very fruitful in guiding attempts to elucidate the molecular mechanism of transport (Forrest et al. 2008; Shi et al. 2008).
It is currently well established that NSS are constitutive oligomers (Sitte and Freissmuth 2003; Sitte et al. 2004); this has been shown by numerous research groups by using a plethora of different biochemical methods (Jess et al. 1996; Kilic and Rudnick 2000; Hastrup et al. 2001) and microscopic techniques in living cells (Schmid et al. 2001; Sorkina et al. 2003; Just et al. 2004; Egana et al. 2009) including neurons expressing transporters in their natural environment (Egana et al. 2009; Fjorback et al. 2009). It is not clear, why transporters should adopt a quaternary structure: in fact, the transport process is apparently not affected in mutants that have been rendered monomeric (Scholze et al. 2002a). From a teleological perspective, it can be argued that oligomer formation has evolved to support efficient export of transporters from the endoplasmic reticulum (Farhan et al. 2006). Here, we will argue that the oligomeric nature of NSS does have an impact on the transport process because it is also essential for the action of amphetamine and its congeners on DAT, SERT and NET.
Plasmalemmal neurotransmitter transporters are necessary to support reverse transport
The discovery of reverse transport is intricately linked to the analysis of the pharmacological action of sympathomimetic amines: the original classification of sympathomimetic amines did not differentiate between a direct action of catechol compounds (that is now known to arise from a stimulation of the pertinent receptors) and the indirect action of non-catechol compounds (that is now appreciated to be contingent on reverse transport; Barger and Dale 1910). However, it became evident that cocaine potentiated the action of adrenaline (Fröhlich and Loewi 1910) but abolished the action of tyramine (Tainter and Chang 1927). Subsequent experiments documented that the effect of tyramine was contingent on the presence of endogenous noradrenaline because it was absent in denervated organs (Fleckenstein and Burn 1953) and in reserpine-treated animals (Burn and Rand 1958). These and related experiments, where sympathetic nerve endings in organ preparations were challenged with amphetamine or its congeners (Axelrod et al. 1961; Furchgott et al. 1963; Glowinski and Axelrod 1965; Ross and Renyi 1966), paved the way for a concept, in which release of noradrenaline (or dopamine) by reverse transport was the crucial step to understand their mode of action. This concept was also extensively verified in brain slices or synaptosomes (Gobbi et al. 1997). However, these paradigms share the common feature that they contain presynaptic nerve terminals with the complete machinery for synaptic vesicle exocytosis. The same is true for platelets and membranes prepared thereof, which contain SERT and the exocytotic machinery of platelets (Fishkes and Rudnick 1982; Rudnick and Wall 1992). Because of the simultaneous presence of vesicular and membrane transporters – and hence of different pools of substrates – it is difficult to dissect the contribution of each substrate pool to reverse transport under these conditions.
With the availability of the cDNA’s encoding individual NSS’s, it was possible to heterologously express a given monoamine transporter and to perform uptake and release experiments in cells that only contain the transporter of interest. This approach by definition eliminates the confounding effects of vesicular storage mechanisms and of the exocytotic release machinery: under these conditions – i.e., in cells heterologously expressing a given monoamine transporter – amphetamine and its derivatives induce reverse transport (Eshleman et al. 1994; Pifl et al. 1995, 1999; Wall et al. 1995; Pifl and Singer 1999). These observations and observations in DAT-knockout mice (Jones et al. 1998) provided a formal proof for the interpretation that monoamine transporters are the principle site of action of amphetamine and its congeners. While this statement may appear like a trivial truism, it is worth pointing out that, in vivo, the action of amphetamines and other indirectly acting sympathomimetic amines were abrogated in reserpine-treated animals (Burn and Rand 1958). Reserpine, however, targets the vesicular monoamine transporters-1 and -2 (VMAT1 and 2; Chaudhry et al. 2008; Schuldiner et al. 1993, 1995). However, it may be worth mentioning that genetic deletion of VMAT2 decreased the amount of amphetamine-induced release by 65% (Fon et al. 1997).
A unifying concept of counter-transport versus several modes of reverse transport
Importantly, not only amphetamines are capable of inducing reverse transport. In fact, all compounds recognized as substrates which are inwardly transported trigger reverse transport in NSS expressing cells (Eshleman et al. 1994; Pifl et al. 1995; Cinquanta et al. 1997; Sitte et al. 1998, 2001; Scholze et al. 2000; Gobbi et al. 2002, 2008; Willeit et al. 2008). We argue, though, that this is an experimental artefact: physiological substrates do not act as releasers under physiological conditions: in fact, this would lead to futile cycling – rather than efficient removal of neurotransmitter from the synaptic cleft – and there isn’t any evidence that this occurs in vivo (see below). In contrast, the biological actions of amphetamines and other naturally occurring amphetamine-like releasers (like ‘khat’, ‘ephedrine’, and ‘tyramine’; see also Sulzer et al. 2005) are contingent on transport reversal. In addition, if transmembrane ion gradients are changed, reverse transport is triggered: this is true for both, lowering the extracellular sodium concentration (Pifl et al. 1995, 1997; Pifl and Singer 1999) or raising the extracellular potassium concentration (Scholze et al. 2000, 2002a). Thus, it may be conceptually important to consider at least two different types of reverse transport: the first one is triggered upon binding of releasers to the transporter protein and the second one upon changes of the ion composition of the extra- or intracellular fluid. The latter situation differs from the former by the fact that releasers need not be present on the extracellular side. Obviously, in either case, a releasable pool of substrate must be accessible on the cytoplasmic side.
Several models have been put forth to account for and explain reverse transport; these were initially based on the ‘alternating access hypothesis’ that explained the inward transport of substrates from the extracellular side by a substrate-induced conformational change in a sodium- and chloride-dependent manner. Hence, based on this hypothesis, reverse transport was conceptualized as a revolving door, where outward transport of a given substrate located within the cell was driven by and coupled to the inward transport of another substrate residing on the extracellular side. The fact that both, forward and reverse transport is accomplished by the very same transporter molecule was inferred from the substrate selectivity and the blockage by specific inhibitors; e.g., cocaine blocked both substrate uptake and amphetamine-induced substrate efflux. This revolving door model was termed ‘facilitated exchange diffusion model’ (Widdas 1952; Stein 1967; Fischer and Cho 1979; Trendelenburg et al. 1987; Bönisch and Trendelenburg 1989).
In brief, the releaser (R) binds to the outward-facing conformation (To) of the transporter together with the co-substrates sodium and chloride to produce R-To. A change in conformation transports and releases the bound substrates instantaneously across the plasma membrane to the intracellular side. The unloading of the transported substrate on the intracellular side may be caused by the concentration gradient for the co-substrate(s): there is a steep concentration gradient for sodium (≥ 30-fold) and an additional electrical driving force between the extracellular and intracellular side. Given the low intracellular Na+ concentration, Na+ must be instantaneously released on the intracellular side resulting in a drop in the affinity for the substrate and rapid dissociation (i.e., cytoplasmic release) of the substrate-transporter complex. Subsequently, the inward-facing conformation of the transporter protein (Ti) is available for binding of intracellular substrate (S) (together with co-transported substrates to produce S-Ti). Reverse transport to the extracellular side of this substrate requires a reorientation step of the transporter protein. Here, the exchanged substrate is released by the transporter protein, which is then available for the next releaser-molecule to be taken up to the cytoplasmic side.
While the individual steps in this reaction cycle are readily delineated in the schematic rendering of Fig. 1, it does not allow to understand what keeps the transporter from futile cycling. Obviously, inward transport is the favored mode and outward transport of substrate is per se a rare event (because [Na+]i is low). If, however, the transporter supports influx of large amounts of substrate, reverse transport is more likely to occur (e.g., because [Na+]i builds up). Accordingly, EC50 for substrate-induced substrate release is higher than the KM for inward transport (Scholze et al. 2000). Similarly, Vmax for outward transport only reaches < 20% of Vmax for inward transport (Sitte et al. 2001, 2002).
Fig. 1.
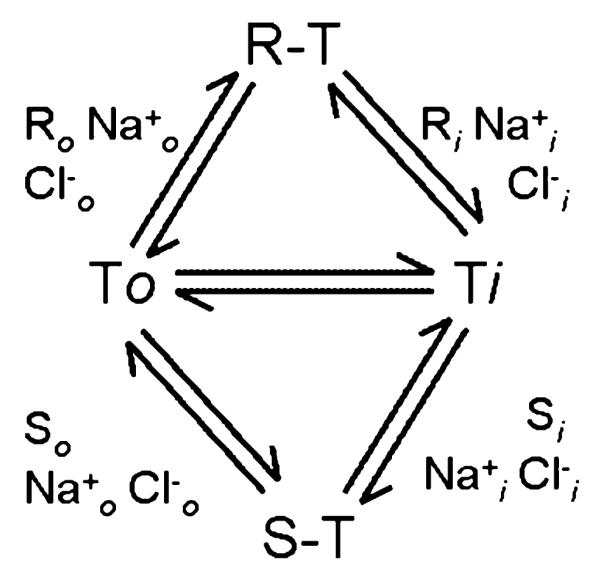
A kinetic scheme of substrate (S) and/or releaser (R) interaction with either the outward (o) or inward (i) facing transporter (T; adapted from Jones et al. 1999).
The scheme in Fig. 1 also postulates that the switch from outward-facing to inward-facing conformation enhances the availability of inward-facing transporters by any given substrate. This, in turn, enhances the probability of the binding and subsequent translocation of cytoplasmic substrates to the cell exterior. Fischer and Cho (1979) noted that a tight coupling between substrate uptake and reverse transport is to be expected: If uptake of the releasing amine is the first step in the chemical release process, then it must follow that the initial rate of release of [3H]dopamine induced by an IAS amine must be related to the initial rate of uptake of the releasing agent. This ‘facilitated exchange diffusion model’ was supported by numerous publications on reverse transport examined in different NSS family members (Hilgemann and Lu 1999; Lu and Hilgemann 1999a,b; Roux and Supplisson 2000; Wang et al. 2003).
As mentioned, appreciable transporter-mediated reverse transport is not expected to occur under basal conditions. In neurons (and platelets), the free cytosolic concentration of substrate is low because neurotransmitters are sequestered into synaptic vesicles by the action of vesicular transporters. This presumably limited the selective pressure, because there was no need to further optimize the system and preclude reverse transport completely. [dopamine]i has been estimated to about 2 μM (Ewing et al. 1992). This concentration, however, is too small to initiate a detectable reverse transport, because the KM for outward transport exceeds that for inward transport by three orders of magnitude (Sitte et al. 2001). Jones et al. (1999) measured reverse transport of dopamine by cyclic voltammetry in mouse striatal slices: a [dopamine]in elevation by VMAT inhibition, however, failed to induce DAT-mediated reverse transport. The detection limit for cyclic voltammetry is 25 nM; thus, the concentration did not apparently raise enough to allow for measurement of [dopamine]out. In this context, it may be noted that, once redistributed from the vesicle to the cytosol, DA is quickly metabolized by monoamine-oxidase (MAO), often accompanied by an increase in its metabolite 3,4-dihydroxyphenylacetic acid (DOPAC). However, reverse transport can be initiated by raising the intracellular dopamine concentration (in the absence of any further manipulation other than the co-injected Cl−): Sulzer et al. (1995) observed a clear increase in [dopamine]out after injecting 4 pL of 0.5 mM dopamine (i.e., 2 fmol) into the giant DA neuron of Planorbis corneus. It is difficult to interpret the phenomenon, because the dilution of dopamine cannot be estimated; however, under these conditions, reverse transport was specifically triggered because dopamine efflux was blocked by nomifensine (a transport inhibitor) (Sulzer et al. 1995). It is evident that this efflux did not require the presence of substrate on the extracellular side; it is also quite clear that the amount of co-injected Cl− did not suffice to change the ionic composition; apparently, no additional ions were co-injected. To the best of our knowledge, this is the only instance where raising dopamine alone – in the absence of an increase on intracellular Na+ – sufficed to support efflux. Mosharov et al. (2003) further characterized the mechanisms, by which amphetamine affects cytosolic dopamine levels by using intracellular patch electrochemistry in chromaffin cells: extracellular amphetamine (50 μM) was found to increase cytosolic dopamine levels 6-fold during the first 10–15 min, i.e., amphetamine can increase cytosolic substrate concentration also in the presence of vesicles. A follow-up study in neuronal cell bodies (Mosharov et al. 2009) showed decreased cytosolic dopamine levels on amphetamine exposure, presumably because there was no vesicular dopamine in neuronal cell bodies (in contrast to chromaffin cells); this decrease was blocked by cocaine or nomifensine, indicating that extracellular amphetamine caused release of cytosolic dopamine via reverse transport: the authors concluded that reverse transport was not induced by the elevated cytosolic dopamine alone, but required additional amphetamine-mediated effects on DAT.
As mentioned above, we proposed two conceptually separate triggers for favoring the efflux mode in the transport cycle outlined in Fig. 1: (i) Inward facing conformations are favored, if Na+ accumulates on the intracellular side. Similarly, a reduction in external Na+ eliminates the inward driving force and precludes the accumulation of outward facing conformations. The latter statement is supported by the following circumstantial evidence: inhibitors of NET, DAT and SERT (e.g., cocaine analogues, tricyclic anti-depressants) bind preferentially to the outward facing conformations (Singh 2008; Singh et al. 2008); high-affinity binding is strictly dependent on Na+ (Humphreys et al. 1994) and proceeds in an ordered fashion with Na+ binding prior to the inhibitor (Korkhov et al. 2006). Last but not least, currents associated with amphetamine translocation are also consistent with the interpretation that high Na+ drives the transporter into the outward conformation (Erreger et al. 2008). Manipulations of the Na+-gradient result in the predicted change in influx and efflux rates: Ouabain blocks the Na+/K+-ATPase and thereby enhances the [Na+]in; this has long been known to reduce substrate uptake and facilitate efflux (Bönisch 1986; Trendelenburg et al. 1987; Wolfel and Graefe 1992; Sitte et al. 1998; Jones et al. 1999) and this can readily be recapitulated upon heterologous expression, i.e. in the absence of vesicular transporters (Scholze et al. 2000; Sitte et al. 2000, 2002). Khoshbouei et al. (2003) directly correlated an increase in the cytoplasmic [Na+] and DAT-mediated dopamine efflux. Taken together, these results indicate that substrate efflux can be triggered by an increase in the intracellular [Na+] (see also below and Fig. 2). (ii) Amphetamines and other releasers must elicit a conformational change that drives reverse transport.
Fig. 2.

Schematic illustration of the oligomer-based counter-transport model (described in more detail in Seidel et al. 2005). The left side illustrates the effect of low concentrations of amphetamine (in this figure, pCA stands for para-chloroamphetamine); only a fraction of SERT moieties is occupied by pCA in the oligomeric complex (shown here for the sake of simplicity as a dimer). Occupancy by pCA precludes phosphorylation by PKC (Ramamoorthy and Blakely 1999). The other SERT moieties in the oligomeric complex that has not been occupied by pCA are subject to phosphorylation and thereby primed for outward transport of substrate. Right, at high pCA concentrations, all transporter subunits are occupied by pCA, which impairs the action of PKC and thus prevents the accumulation of inward-facing conformations: efflux of substrate is impaired.
Transport associated currents as a prerequisite for release
The second trigger that allows for efflux is the presence of a releasing substrate, the prototype being amphetamine and its congeners. However, it will be evident that in all instances amphetamine triggers a substantial movement of excess positive charges, i.e. Na+ influx. Na+ is a co-transported substrate in all NSS family members. In the alternating access model the concomitant translocation of substrates and co-transported ions is strictly stoichiometric. While the majority of the transporters of the NSS-family work in an electrogenic manner, SERT is thought to be an exception because it works in an electroneutral manner because of the counter-transport of K+ (Rudnick 1999): in theory, charge-flow must not necessarily be translated in measureable steady-state current; for SERT, the prediction would be that no net current can be measured. Theoretically, this movement of ions may be assessed in electrophysiological experiments by an increased noise upon transporter activity. Similar to the unexpected observations of SERT-mediated currents (Bruns et al. 1993; Mager et al. 1994), there was surprising excessive current measured in DAT-expressing cells (Sonders et al. 1997; Sitte et al. 1998). Hence, electrophysiological investigations have shown that plasma membrane neurotransmitter transporters can function in a way similar to ion channels (Sonders and Amara 1996). Still, the charge fluxes carried by NSS-members are an enigmatic property of the transporters, but they are central to understanding amphetamine-induced efflux.
Currents in transporters and their role in amphetamine-induced efflux
Excess ion flux is observed during substrate influx in many members of the NSS family; in addition, there is leak current, which can be detected under basal conditions and blocked by inhibitors (Mager et al. 1994, 1996; Galli et al. 1995, 1997, 1998). While the substrate-induced ion flow through neurotransmitter transporters is reminiscent of ligand-gated ion channels, it is worth noting that the currents of the transporters are smaller by orders magnitudes than those of ion channels and their conductance is presumably also much smaller.
It is a challenge to reconcile the view of the transporter as a ‘pump with a channel’ with the alternating access model, because the transport mechanism may be more complicated than predicted from the scheme in Fig. 1. The issue is succinctly summarized by Accardi and Miller (2004) who concluded their description of secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels in that ‘transporters and channels may be separated by an exceedingly fine line’. Segments in the structure of LeuTAa proteins can also be conceptually treated as gates that may open simultaneously and thus convert the transporter into a channel. However, there isn’t any evidence so far that LeuTAa can also generate a transport-associated current (see also below).
Small structural changes suffice to cross the ominous ‘line’ between a transporter and a channel, and this line is crossed in nature: a good example for such subtleties is the sodium-glucose transporter SGLT-3: a mere glucose transporter in pigs, but a glucose-activated Na+-channel in man and functional as a glucose sensor (Diez-Sampedro et al. 2003). Conversely, both bacterial (Accardi and Miller 2004) and mammalian chloride channels are actually H+/Cl−-antiporters (Scheel et al. 2005). Similarly, the mammalian SERT has significantly lower conducting properties than its homolog from Drosophila melanogaster: in flies, the currents carry 5- to 10-times more charged moieties/transport cycle (Galli et al. 1997; Petersen and DeFelice 1999). In theory, a channel like-mode can be envisaged by assuming that the transporter moves excess Na+ (and Cl−) because the upper (external) gate opens faster than the substrate is released at the internal vestibule. In this instance, the transporter would still be considered to operate in a transport mode albeit a leaky one. However, a true channel-like mode was observed for the substrate translocation in SERT of Drosophila melanogaster: at high concentrations, substrate and ions move through the translocation pathway in random order (Petersen and DeFelice 1999) consistent with a ‘single file model’ (Lester et al. 1996; Su et al. 1996) that is indicative of diffusion through a pore. Ramsey and DeFelice (2002) suggested that the formation of a pore might rely on oligomerization of the SERT at the plasma membrane; however, further evidence in support of this notion is still lacking.
Uncoupled conductance can only be explained by the formation of a pore within the transporter molecule. The difficulty in interpreting the data at the current stage also arises from the fact that – in spite of the presence of candidate gates at the external and internal vestibule – the crystal structure of LeuTAa does not reveal a cavity consistent with the random movement of charge through the molecule (Singh 2008).
Regardless of these conceptual difficulties, currents are important to understand the actions of amphetamines: binding of amphetamine to the transporter appears to favour a channel mode, i.e., a conducting pore may be stabilized and support efflux: brief spike-like elevations of extracellular dopamine can be observed by amperometry in DAT-expressing cells. These spikes were detected only after application of amphetamine but not of dopamine (Kahlig et al. 2005). The spikes are consistent with fast, burst-like release events of neurotransmitter through a channel. Amperometry is limited in its sensitivity (and the amount of released detectable substrate), thus, it is difficult to discern the relative proportion of dopamine that is released by bursts and by counter-transport. Kahlig et al. (2005) estimated a ratio of 9 : 1 in favour of continuous efflux; this estimate suggests that amphetamine does allow the transporter to cycle efficiently through outward and inward facing conformations and that the channel-like mode is a comparably rare event.
Originally, reverse transport was defined as a carrier-mediated, Ca2+-independent non-exocytotic release of neurotransmitter (Levi and Raiteri 1993). This led to the implicit assumption that the membrane potential was irrelevant for amphetamine-induced efflux. However, monoamine substrates are transported as charged species (Berfield et al. 1999). Hence, a negative membrane potential provides an additional driving force for influx. Similarly reverse transport also depends on the membrane potential (Khoshbouei et al. 2003) and depolarization adds some additional driving force for counter-transport. It is worth noting that the action of amphetamine on DAT per se depolarizes the cell (Carvelli et al. 2004; Meinild et al. 2004) and that Na+ is the principal charge carrier involved in this process (Sitte et al. 1998). Similarly, experiments in SERT-expressing Xenopus laevis oocytes also support Na+ as the charge carrier (Quick et al. 2004). The uncoupled conductance is postulated to supply enough Na+ to trigger reverse transport: based on the scheme in Fig. 1, the local accumulation of intracellular Na+ is predicted to favour the accumulation of transporters in the inward facing conformation and thus render the transporter more prone to support substrate efflux; binding of substrate to the inward-facing conformation of the transporter becomes a more likely event as [Na+]in rises. The transporter will return to the outward-facing conformation in a substrate-loaded state.
The physiological role of the uncoupled currents in neurotransmitter transporters remains enigmatic. Moreover, the channel within the DAT is also permeable for anions (usually chloride; Ingram et al. 2002; Meinild et al. 2004) which can be decisive in shifting the membrane potential: depending on the actual membrane potential and the reversal potential for Cl−, the net effect can either result in hyperpolarisation or depolarization of the cell membrane, (Ingram et al. 2002; Sulzer and Galli 2003; Meinild et al. 2004). Thus, it is conceivable that neurotransmitter transport modulates the excitability of neurons (Falkenburger et al. 2001; Ingram et al. 2002) and, in addition, that reverse transport takes place if cells are overloaded with Na+, e.g. by depolarization via glutamate receptors (Falkenburger et al. 2001) or by hypoxia/anoxia (Schömig et al. 1987).
Taken together, the observations show that all transporter substrates induce a transport-associated current. This current, however, only rarely suffices to trigger substrate efflux. However, the pharmacological actions of amphetamines are contingent on reverse transport. Reverse transport by monoamine transporters has also been discovered as a target during evolution: long before man has elaborated the synthesis of amphetamine, plants have acquired the ability to produce related alkaloids such as ephedrine and cathinon to fend off grazers and insects.
What are the differences between physiological substrates and amphetamines that finally lead to their dissimilar properties? The apparent affinity in substrate and amphetamine uptake is very similar in the low micromolar range. But the number of transported substrate molecules differs. Admittedly, it is difficult to measure uptake of amphetamines precisely owing to their notorious lipophilic nature (which causes rapid back-diffusion; Bonisch 1984; Zaczek et al. 1991). Thus, there are only a few systematic investigations in this area: HEK293 cells stably expressing the human DAT differed in substrate uptake rates (dopamine, tyramine and the two isomers of amphetamine) by more than a factor of 20 (Sitte et al. 1998). In contrast, superfusion experiments performed in parallel sister cultures revealed that d-amphetamine induced RT with the highest potency and efficacy. It is worth pointing out that the released substrate was MPP+, which is metabolically stable (Langston et al. 1984; Javitch et al. 1985); this obviates any confounding effects because of the action of amphetamines on MAO-A or MAO-B. Furthermore, patch-clamp experiments carried out in parallel showed that the rank order of potency for the induction of inwardly directed current was comparable to that obtained in superfusion experiments (Sitte et al. 1998). Importantly, when currents were consecutively assessed in the same DAT-expressing cells (Sitte et al. 1998), amphetamine induced a 25% higher inwardly-directed uncoupled conductance compared to dopamine. Hence, the propensity of amphetamines to induce larger currents suggests that they are more effective in triggering the channel mode in neurotransmitter transporters. This may account for the differences between amphetamines and physiological substrates.
There is only one observation that is inconsistent with the conjecture that transport-associated currents are essential to drive the transporter into an efflux mode: this is the action of syntaxin 1a on DAT mediated efflux. Syntaxin 1a is a SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) that supports fusion of neurotransmitter vesicles with the target membrane (i.e. the presynaptic membrane). Syntaxin 1A also directly interacts with neurotransmitter transporters (Dipace et al. 2007) and inhibits excess currents through SERT (Quick et al. 2004) and GAT1 (Wang et al. 2003). Surprisingly DAT-mediated efflux, however, is enhanced upon co expression of syntaxin 1a (Binda et al. 2008). These data are difficult to interpret because (i) current is a prerequisite for transporter-mediated efflux (see above) and (ii) the apparent disparity to the experiments performed by Wang et al. (2003) where the presence of syntaxin 1a clearly reduces the efflux of GABA via GAT1. It would be crucial to see, if this action can be recapitulated in neuronal cells that had been treated with for instance clostridial toxins (of C. tetani and C. botulinum) to elicit the predicted opposite effect.
Multiple targets of amphetamine and its congeners within the presynaptic specialization
As outlined earlier, the monoamine transporters DAT, NET and SERT are the primary site of action of amphetamine and its congeners, [e.g., 3,4-methylene-dioxy-met-amphetamine (MDMA), ‘ecstasy’], methamphetamine [‘ice’] or amphetamine [‘speed’] (Seiden et al. 1993; Sulzer et al. 2005). Individual compounds differ by their affinity to the three transporters but the mechanism of action is the same. In the absence of monoamine transporters (or upon inhibition of the transporters), the compounds do not elicit their biological actions. However, amphetamines have additional targets that contribute to their mechanism of action: (i) inhibition of VMAT1 and VMAT2 and (ii) MAO-A, MAO-B. Under physiological conditions, there are two processes that keep the cytosolic concentration of monoamine neurotransmitters low in the presynaptic specialization: (i) the reserpine-sensitive, proton-driven VMAT1 and VMAT2 retrieves neurotransmitters into the synaptic vesicles (Liu et al. 1992). (ii) Monoamines that escape the action of VMAT are rapidly degraded by the action of mitochondrial MAO-A and MAO-B and to a lesser extent by catechol O-methyltransferase (Seiden et al. 1993). Amphetamines inhibit MAO and are substrates for VMAT. The concerted action on all targets can be summarized as follows: (i) charged amphetamines are recognized by DAT, NET and/or SERT as substrates and enter the cells. More amphetamine molecules will be in their protonated form, because the intracellular pH is lower than the extracellular pH. Nevertheless, the remaining unprotonated amphetamine molecules will diffuse back into the synaptic cleft through the membrane and be quickly available for another round of transporter-mediated uptake (Rudnick 2002). Hence, one molecule of amphetamine can, in principle, cycle repeatedly between the interior of the cell and the extracellular space; this cycle is thought to underlie the accumulation of Na+, Cl− and H+ in the cytoplasm (Rudnick 1999). (ii) Amphetamines interfere with vesicular uptake of monoamines by competing for VMAT and, upon accumulation in the vesicles, by their buffering action, which dissipates the pH gradient. This has lead Sulzer and colleagues to hypothesize a succinct model, termed the ‘weak base hypothesis’ (Sulzer et al. 1993, 1995): here, the amphetamine action leads to the elevation of cytosolic substrate which is then available for reverse transport. (iii) Amphetamines also inhibit the degradation of monoamines by MAO (Robinson 1985). Accordingly, these actions explain why amphetamines readily induce reverse transport while natural substrates do not.
Shortcomings of the present hypotheses (weak base/facilitated exchange diffusion): futile cycling, phosphorylation and Zn2+
There are several observations that the currently held models fail to explain:
-
(i)
The facilitated exchange diffusion hypothesis suffers from one conceptual shortcoming. If amphetamine resides in the inner vestibule of the transporter and has raised intracellular Na+ by multiple rounds of inward transport, why should it be released and replaced by the physiological substrate (which is not yet in the vestibule), rather than not per se be subject to reverse transport? In other words, there must be a mechanism that prevents futile cycling of amphetamines in the transporter. This conceptual problem can be solved in two ways: (a) amphetamines have a negligible affinity for the internal vestibule such that they cannot be subject to reverse transport in the presence of (competing) substrate on the cytosolic side. (b) The actions of amphetamines are contingent on the oligomeric nature of the transporters (see below).
-
(ii)
The cogent framework outlined above serves to integrate several actions of amphetamines in the presynaptic terminal and explains how substrates become available for outward transport in the nerve terminal. However, any model on the action of amphetamines must account for the requirement of protein phosphorylation: inhibition of protein kinase C (PKC) or of calmodulin-dependent kinase-2 blunts the action of amphetamines but does not eliminate substrate-induced efflux (i.e., exchange diffusion) (Kantor and Gnegy 1998; Kantor et al. 2001; Gnegy 2003; Seidel et al. 2005; Fog et al. 2006). These observations can also be recapitulated upon genetic ablation of PKCβ (Chen et al. 2009).
-
(iii)
Any model on the action of amphetamines must explain why amphetamines can, in principle, give rise to bell-shaped concentration-response curves. This can readily be seen in superfusion experiments, where the concentrations can be varied appropriately over a large range (Seidel et al. 2005).
-
(iv)
Last but not least, the observations obtained with Zn2+ are difficult to reconcile with a model that treats efflux and influx as coupled processes (see above for the quotation of Fischer and Cho, which can be distilled into ‘the faster in, the faster out’): Zn2+ binds to an endogenous allosteric site in the DAT (Norregaard et al. 1998). The residues for the coordination of Zn2+-binding are His189, Glu377 and Glu396 (Norregaard et al. 1998; Loland et al. 1999). Upon binding to this tridentate coordination sphere, Zn2+ inhibits substrate uptake by DAT (Norregaard et al. 1998; Scholze et al. 2002b). This non-competitive effect on uptake indirectly suggests that Zn2+, by binding to the transporter, constrains relative movements between ECL2, TM7 and TM8 critical for the inward translocation process (Loland et al. 1999). However, Zn2+ stimulates amphetamine-induced efflux (Scholze et al. 2002b; Pifl et al. 2004). The asymmetrical effect is obvious, the Zn2+-induced inhibition of uptake is accompanied by an increase in efflux and this may be accounted for by the fact that Zn2+ promotes an additional ion current (Meinild et al. 2004; Pifl et al. 2009). Regardless of the underlying mechanistic details, the observations stress two pertinent aspects: influx and efflux do not necessarily occur via the same route and enhanced inward currents favor outward transport.
Amphetamine-induced activation of protein kinases
Originally, reverse transport was defined as a carrier-mediated, Ca2+-independent non-exocytotic release of neurotransmitter (Levi and Raiteri 1993). However, Gnegy et al. (2004) showed that removal of internal Ca2+ by the membrane-impermeant Ca2+-chelator BAPTA reduces amphetamine-induced dopamine efflux in cells expressing DAT. This requirement for low calcium concentrations can be rationalized by the fact that the action of amphetamines is – in part – contingent on PKC-mediated (Kantor and Gnegy 1998) and Ca2+/calmodulin-dependent phosphorylation (Kantor et al. 1999; Fog et al. 2006). This was first observed for DAT but subsequently confirmed for NET (Kantor et al. 2001) and SERT (Seidel et al. 2005). It is at present not clear, how amphetamines cause activation of PKC (Giambalvo 1992a,b) and of Ca2+/calmodulin-dependent protein kinase, but the stimulation is dependent on intracellular Ca2+ (Giambalvo 2003). The pleiotropic actions of kinases may allow for the recruitment of many different mechanisms that afford transport reversal, e.g. phosphorylation-dependent inhibition of Na+/K+-ATPase (Kazanietz et al. 2001) which would directly favour intracellular accumulation of Na+. Initially, activators of PKC such as 12-O-tetra-decanoyl phorbol-13-acetate, diacylglycerol and arachidonic acid, have been shown to enhance both DAT-mediated efflux of dopamine (Pozzan et al. 1984; Davis and Patrick 1990; L’hirondel et al. 1995). However, there is circumstantial evidence for a direct action of PKC-isoforms on transporters: truncation of the 22 N-terminal residues of DAT, eliminates the ability of DAT to undergo PKC-dependent phosphorylation (Granas et al. 2003) and suppresses reverse transport induced by amphetamine (Khoshbouei et al. 2004). The relevant PKC isoform has been identified as PKC beta(II), interacting with DAT in rat striatum (Johnson et al. 2005); however, there is no formal proof that this PKC isoform actually phosphorylates DAT. Foster et al. (2002) detected DAT phosphorylation after stimulation of PKC-isoforms with phorbol myristate (which activates typical and novel isoforms, e.g. PKCs α, β, γ, δ, ε). The N-terminus of human DAT can be phosphorylated by PKCα (Fog et al. 2006), and this is also observed with the N terminus of rat-DAT (Gorentla et al. 2009). However, PKCα does not associate with DAT (Johnson et al. 2005). Hence, the role of the different PKC isoforms has not yet been clarified. Nevertheless, amphetamine-induced reverse transport is blunted after mutational replacement of the five serine residues in this segment of DAT by alanine (Khoshbouei et al. 2004). From these mutagenesis experiments, the authors concluded that amino terminal phosphorylation by PKC shifts the DAT from a ‘reluctant’ to a ‘willing’ state for efflux (Robertson et al. 2009). This was further underscored by mutation of the same five serine residues to aspartate and thereby restoring the efflux property of the transporter (Khoshbouei et al. 2004). It must be conceded, though, that there is no direct evidence for phosphorylation of these sites.
There are many discrepancies between heterologous expression systems and endogenously expressed transporters and these are evident when examining the actions of protein kinases on transporters: Sorkina et al. (2006) and Eriksen et al. (2009) did neither find any appreciable effect of PKC by activation with phorbol myristate nor by inhibition with staurosporine or GF109203X on dopamine uptake by dopaminergic neurons. However, inhibition of uptake (presumably by down-regulation of surface transporters) had been described many times for heterologous expression systems (see Foster et al. 2006) and this has also been recapitulated in rat striatal synaptosomes (Vaughan et al. 1997; Chi and Reith 2003; Hoover et al. 2007). Hence, to date it is unclear if PKC exerts a direct effect on transporter activity.
Protein kinase C’s are not the only intracellular kinases postulated to phosphorylate neurotransmitter transporters. As mentioned above, calmodulin-dependent kinase-2α phosphorylates DAT (but not NET or SERT) and the mechanistic details are understood: first, the enzyme engages the carboxyl terminus of DAT and to subsequently phosphorylates the amino terminus (Fog et al. 2006). However, it is unclear to date if the two kinases exert their effects independently from each other or in strictly hierarchical order. Furthermore, other kinases (e.g., Moron et al. 2003; Zhu et al. 2005; Ramamoorthy et al. 2007) impinge on neurotransmitter transporters: this causes changes in trafficking (Moron et al. 2003) and/or velocity of substrate influx (Zhu et al. 2005). It is not clear if these kinases also affect amphetamine-induced outflow. However, it may be worth considering this possibility: amphetamines are used to treat attention-deficit hyperactivity disorder (ADHD; Mazei-Robinson and Blakely 2006). The effectiveness of amphetamine is counter-intuitive, because amphetamines elicit psycho stimulant effects (an action which is sought in their illicit consumption). However, a (rare) mutation in the dopamine transporter allows for rationalizing the paradoxical effect of amphetamines in ADHD: a substitution of alanine559 by valine (found in an affected family) results in a transporter which is targeted to the cell surface and mediates dopamine uptake in a manner indistinguishable form the wild type. However, DAT-A559V has augmented substrate basal efflux, which is blocked by amphetamine (and methylphenidate) (Mazei-Robison et al. 2008). It is conceivable that in other individuals affected by ADHD, it is not the transporter that is mutated but that the defect is in a regulatory pathway which switches the transporter into a mode analogous to that adopted by DAT-A559V.
An oligomer-based counter-transport model as a unifying concept
Neurotransmitter transporters are oligomers (Kilic and Rudnick 2000; Schmid et al. 2001; Kocabas et al. 2003; Sorkina et al. 2003; Torres et al. 2003a). Oligomerization has been recognized as an important prerequisite for exit from the endoplasmic reticulum (Farhan et al. 2004, 2007, 2008). It has furthermore been shown that – throughout the secretory pathway from the endoplasmic reticulum to the plasma membrane – DAT is an oligomer and this is also true for the endocytosed transporter (Sorkina et al. 2003). For endoplasmic reticulum export, oligomeric assembly has been proposed to facilitate efficient recruitment of the transporter into endoplasmic reticulum exit sites by the assembling coat protein complex II (COPII)-coat (Farhan et al. 2006). This may also be true for other cargo receptors (e.g., ARF-GAP1; Reiterer et al. 2008) and proteins that regulate trafficking through the secretory pathway (e.g. GTRAP3-18, Liu et al. 2008; Ruggiero et al. 2008; Maier et al. 2009). In addition, we posit that oligomerization is also relevant to understand the mechanism of action of amphetamines. We refer to this model as oligomer-based counter-transport model, in which the releaser is taken up by one moiety and transporter-mediated substrate efflux is achieved by the neighbouring moiety. There are several arguments that support this concept:
-
(i)
It is evident that a single transporter moiety can hardly allow for permeation of amphetamine and simultaneously support the efflux of substrate. It is much more plausible to assume that amphetamine triggers the channel mode in an adjacent transporter moiety within the oligomeric complex. If this were the case, amphetamine ought to induce maximum substrate release at a concentration where it occupies about 50% of the available transporters. A further increase of amphetamine is predicted to cause a decline in substrate release, because progressive occupation of transporters with inward moving amphetamine reduces the number of those available for outward transport of substrate resulting in a bell-shaped concentration-response curve. Bell-shaped concentration-response curves have actually been observed with amphetamine and several times reported (Ross and Kelder 1977; Langeloh and Trendelenburg 1987; Pifl et al. 1999; Seidel et al. 2005).
-
(ii)
The role of an oligomeric arrangement has been tested by using a concatemer composed of SERT and the GAT1. In this SERT-GAT1 concatemer, the SERT-selective amphetamines employed (pCA; MDMA) elicited [3H]GABA efflux (Seidel et al. 2005). This effect demonstrates that – in the concatemer – GAT1 can sense the action of amphetamines on the adjacent SERT. It is likely that the signal that is sensed is the Na+ influx in the vicinity of the inner vestibule. This finding is most parsimoniously explained by the oligomer-based counter-transport model (see Fig. 2). It is impossible to formally rule out other mechanisms, e.g. amphetamine-induced changes in second messenger-triggered cascades or alterations of the membrane potential propagated through ion channels. But, based on our current understanding of the biological actions of amphetamine, these alternative possibilities cannot readily account for driving the GAT1-moiety of the concatemer into the reverse-transport conformation.
-
(iii)
It is worth noting that pCA-induced efflux in this concatemer is still sensitive to PKC-inhibition. This observation indicates that accumulation of internal Na+ per se does not suffice to explain amphetamine-induced efflux and again stresses the importance of amphetamine-induced protein kinase activation (see above).
It is not clear, how individual subunits communicate within the oligomer. A testable model, however, can be developed by examining the recently solved structure of the bacterial betaine transporter BetP (Ressl et al. 2009). BetP is only distantly related to neurotransmitter transporters; however, the pseudo symmetrical fold is conserved. The topology is somewhat different from LeuTAa, the bacterial homolog of neurotransmitter transporters. In LeuTAa where the pseudo symmetric fold comprises transmembrane helices TM1–TM10 (with symmetrical positions held by TM1 and TM6, TM2 and TM7 etc.), while TM11 and TM12 make up the crystallographic dimer interface (Yamashita et al. 2005; Singh 2008) and a candidate interaction site in neurotransmitter transporters (Just et al. 2004). In BetP, the trimer interface is comprised by the two N-terminally located transmembrane helices TM1 and TM2 and the pseudo symmetrical fold is comprised by the TM3–TM12. BetP is an osmolyte transporter and senses (hyper)osmotic stress by the ensuing change in the intracellular K+ concentration. Interestingly, the stress signal is communicated via the C-terminus to a partner within the trimer to trigger inward transport. Based on the topology, a speculative model can be developed that posits that in the monoamine transporters the Na+-triggered signal is communicated from one transporter moiety to the adjacent moiety by the N-terminus which is topologically equivalent to the C-terminus in BetP. Efficient communication requires a priming action of protein kinase-dependent phosphorylation of the N-terminus. This model is consistent with the observation that the N-terminus of DAT contains the phosphorylation sites essential for amphetamine-induced substrate efflux (Khoshbouei et al. 2004; Fog et al. 2006). It is also consistent with the intermolecular distances of N- and C-termini (Just et al. 2004): the distance between C-termini within oligomers is smaller than that of N-termini. Last but not least, the C-terminus of one BetP monomeric moiety communicates via a network of ionic bonds with charged residues in loop 2 and loop 8 of the adjacent monomer (Ressl et al. 2009). In GAT1, the N-terminus apparently binds to the intracellular loop 4 (equivalent to loop 8 in BetP) via interaction of charged residues (Hansra et al. 2004). It is obvious that phosphorylation of the N-terminus may have a substantial effect on these ionic interactions. Thus, while the model is highly speculative, it can account for most findings and – most importantly – it provides a starting point to locate the latch by which for amphetamine-induced substrate efflux is triggered (Fig. 3). It is obvious that at this stage, we cannot discriminate between the sequence of events that follow the conformational change induced by binding of amphetamine, i.e. whether the N-terminal latch is simply triggered by binding of amphetamine, whether it requires ‘channel mode’ and/or the accompanying Na+ influx or amphetamine-induced protein kinase activation.
Fig. 3.

Hypothetical representation of a possible cooperative interplay within a transporter oligomer (for the sake of simplicity, the transporter oligomer is shown as a dimer only). The amino-termini drawn with 11 helical turns suffice to reach the centre of the putative oligomeric partner to locate the latch by which for amphetamine-induced substrate efflux is triggered. The scheme is based on the published coordinates of LeuTAa (Yamashita et al. 2005) and the artwork created by using UCSF Chimera (http://www.cgl.ucsf.edu/chimera; Pettersen et al. 2004).
In an oligomer-based counter-transport model, the substrate is taken up by one moiety and transporter-mediated release is achieved by the neighbouring moiety (Fig. 2). However, from a pharmacological perspective, the occupancy of the single transporter moieties in an oligomeric complex by substrate must reduce the probable efflux; in other words, the more substrate available at the cell exterior, the fewer transporters can be available for efflux – hence, amphetamine-induced SERT efflux ought to subside when the amphetamine concentrations rises above a certain level; a biphasic concentration response curve again reflected these predictions (Seidel et al. 2005).
Most importantly, we hypothesized that carrier-mediated efflux relies on the spatial proximity within a transporter oligomer. To verify this conjecture, we examined the amphetamine effects on GABA-preloaded SERT-GAT1-expressing cells. A tiagabine sensitive, concentration dependent GABA efflux was triggered by amphetamine application. This finding confirmed the oligomer-based counter-transport model. Most importantly, increasing the doses of amphetamine to induce efflux did not result in a bell-shaped concentration-response curve but reached saturation: this was to be expected since GAT1 is resistant against amphetamine and therefore, the amphetamine uptake was not able to prevent GAT1 phosphorylation needed for efflux.
How to test the oligomer-based counter-transport model
Our model predicts that monomeric transporter molecules cannot support amphetamine induced efflux. The model cannot readily be tested, because monomeric transporters are not exported (Scholze et al. 2002a). However, the export defect may be remedied by linking an engineered FK506-binding domain that can be reversibly dimerized/oligomerized by using a pharmacologically inert analogue of tacrolimus.
Truncation of the N-terminus is known to abolish amphetamine-induced efflux. It is difficult though to differentiate between the loss of (potential) protein phosphorylation sites and the absence of the putative amphetamine-operated latch. An alternative approach is to tether the N-terminus and thus to restrict its mobility.
This strategy can be further developed by creating various SERT concatemers (SERT is the molecule of choice because concatemers of DAT and/or NET are not readily inserted into the plasma membrane): if the first moiety (of SERT) is truncated at its N-terminus, the concatemer is predicted to be amphetamine-resistant. The approach with SERT concatemers may also be combined with engineered Zn2+-binding sites: if the first SERT moiety carries the Zn2+ binding site, the oligomer-based counter-transport model predicts that Zn2+ ought to depress amphetamine-induced release. Conversely, if the engineered Zn2+-binding site is in the second moiety, the amphetamine action will be enhanced by Zn2+. The Zn2+-experiment has the advantage that it provides a handle to model the propagation of conformational changes through the protein by providing a fixed geometrical constraint for molecular dynamics studies.
As mentioned earlier, the actions of amphetamines are thought to be in part mediated via protein kinases. These observations must be accounted for by mechanistic hypotheses on the action of amphetamines. The oligomer-based counter-transport model is not superior to the facilitated exchange diffusion model when rationalizing the effects of the protein kinases. The facilitated exchange diffusion model and the oligomer-based counter-transport model do also not differ in their ability to accommodate the peculiar actions of Zn2+.
Concluding remarks
Monoaminergic neurotransmitter transporters are the prime target of the amphetamines; they comprise a large variety of widely abused, psychotropic substances including d-amphetamine (= speed), methamphetamine (= ice) and methylene-dioximethamphetamine (MDMA; = ecstasy). Amphetamines were originally marketed as appetite suppressants (before they were withdrawn on a worldwide basis because of their addictive potential and because some compounds caused pulmonary hypertension). At present, the amphetamine derivative methylphenidate is used in hyperactive children; this compound acts in a manner similar to cocaine because it blocks uptake and also inhibits transporter-mediated current (Sonders et al. 1997). All commonly used and abused amphetamine-like compounds induce carrier-mediated efflux (Levi and Raiteri 1993), with the notable exception of methylphenidate. Regardless of the underlying molecular mechanism, all amphetamine-like compounds elevate synaptic monoamine concentrations. Hence, a prolonged stimulation of postsynaptic receptors ensues. Amphetamines differ in their affinities for monoamine transporters: for instance, d-amphetamine predominantly targets DAT while MDMA acts mainly on SERT (Seiden et al. 1993; Green et al. 2003). Many insights have been generated that shed some light on the mechanism of action of amphetamines. However, the mechanistic basis for their effect has remained enigmatic at the molecular level. The strength of the oligomer-based counter-transport model is that it attempts to integrate different observations: (i) the quaternary structure of the monoamine transporters, (ii) the unique action of compounds that act as releasers in vivo, (iii) the electrophysiological properties of transporters, and (iv) the putative regulation by intracellular kinases. By proposing the N-terminus as a latch, it also provides for testable hypotheses. Like any hypothetical model, it may not withstand the test of experimental scrutiny.
Acknowledgements
The author’s work is supported of the Austrian Science fund/FWF, a stand-alone project P18706 (to HHS) and a program project grant SFB35 (to HHS and MF). The authors wish to express their gratefulness to all the members of their laboratories that participated in the tedious part of the work: generation of data to fuel hypotheses. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
Abbreviations used
- ADHD
attention-deficit hyperactivity disorder
- DAT
dopamine transporter
- GAT1
GABA-transporter-1
- MAO
monoamine-oxidase
- MDMA
3,4-methylene-dioxy-met-amphetamine
- NET
norepinephrine transporter
- NSS
neurotransmitter:sodium symporters
- PKC
protein kinase C
- SERT
serotonin transporter
- VMAT
vesicular monoamine transporter
References
- Accardi A, Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 2004;427:803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- Amara SG, Kuhar MJ. Neurotransmitter transporters: recent progress. Annu. Rev. Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- Axelrod J, Whitby LG, Hertting G. Effect of psychotropic drugs on the uptake of 3H-Norepinephrine by tissues. Science. 1961;133:383–384. doi: 10.1126/science.133.3450.383. [DOI] [PubMed] [Google Scholar]
- Barger G, Dale HH. Chemical structure and sympathomimetic action of amines. J. Physiol. 1910;41:19–59. doi: 10.1113/jphysiol.1910.sp001392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berfield JL, Wang LC, Reith ME. Which form of dopamine is the substrate for the human dopamine transporter: the cationic or the uncharged species? J. Biol. Chem. 1999;274:4876–4882. doi: 10.1074/jbc.274.8.4876. [DOI] [PubMed] [Google Scholar]
- Binda F, Dipace C, Bowton E, et al. Syntaxin 1A interaction with the dopamine transporter promotes amphetamine-induced dopamine efflux. Mol. Pharmacol. 2008;74:1101–1108. doi: 10.1124/mol.108.048447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonisch H. The transport of (+)-amphetamine by the neuronal noradrenaline carrier. Naunyn Schmiedebergs Arch. Pharmacol. 1984;327:267–272. doi: 10.1007/BF00506235. [DOI] [PubMed] [Google Scholar]
- Bönisch H. The role of co-transported sodium in the effect of indirectly acting sympathomimetic amines. Naunyn Schmiedebergs Arch. Pharmacol. 1986;332:135–141. doi: 10.1007/BF00511403. [DOI] [PubMed] [Google Scholar]
- Bönisch H, Trendelenburg U. The mechanism of action of indirectly acting sympathomimetic amines. In: Trendelenburg U, Weiner N, editors. Handbook of Experimental Pharmacology: Catecholamines. Springer; Berlin, Hamburg, New York: 1989. pp. 247–277. [Google Scholar]
- Bruns D, Engert F, Lux H-D. A fast activating presynaptic reuptake current during serotonergic transmisson in identified neurons of Hirudo. Neuron. 1993;10:559–572. doi: 10.1016/0896-6273(93)90159-o. [DOI] [PubMed] [Google Scholar]
- Bruss M, Hammermann R, Brimijoin S, Bonisch H. Anti-peptide antibodies confirm the topology of the human norepinephrine transporter. J. Biol. Chem. 1995;270:9197–9201. doi: 10.1074/jbc.270.16.9197. [DOI] [PubMed] [Google Scholar]
- Burn JH, Rand MJ. The action of sympathomimetic amines in animals treated with reserpine. J. Physiol. 1958;144:314–336. doi: 10.1113/jphysiol.1958.sp006104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvelli L, McDonald PW, Blakely RD, DeFelice LJ. Dopamine transporters depolarize neurons by a channel mechanism. Proc. Natl Acad. Sci. USA. 2004;101:16046–16051. doi: 10.1073/pnas.0403299101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Boulland JL, Jenstad M, Bredahl MK, Edwards RH. Pharmacology of neurotransmitter transport into secretory vesicles. Handb. Exp. Pharmacol. 2008;184:77–106. doi: 10.1007/978-3-540-74805-2_4. [DOI] [PubMed] [Google Scholar]
- Chen N, Reith ME. Structure and function of the dopamine transporter. Eur. J. Pharmacol. 2000;405:329–339. doi: 10.1016/s0014-2999(00)00563-x. [DOI] [PubMed] [Google Scholar]
- Chen R, Furman CA, Zhang M, Kim MN, Gereau RW, Leitges M, Gnegy ME. Protein kinase Cbeta is a critical regulator of dopamine transporter trafficking and regulates the behavioral response to amphetamine in mice. J. Pharmacol. Exp. Ther. 2009;328:912–920. doi: 10.1124/jpet.108.147959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi L, Reith ME. Substrate-induced trafficking of the dopamine transporter in heterologously expressing cells and in rat striatal synaptosomal preparations. J. Pharmacol. Exp. Ther. 2003;307:729–736. doi: 10.1124/jpet.103.055095. [DOI] [PubMed] [Google Scholar]
- Cinquanta M, Ratovitski T, Crespi D, Gobbi M, Mennini T, Simantov R. Carrier-mediated serotonin release induced by d-fenfluramine: studies with human neuroblastoma cells transfected with a rat serotonin transporter. Neuropharmacology. 1997;36:803–809. doi: 10.1016/s0028-3908(97)00064-6. [DOI] [PubMed] [Google Scholar]
- Davis ME, Patrick RL. Diacylglycerol-induced stimulation of neurotransmitter release from rat brain striatal synaptosomes. J. Neurochem. 1990;54:662–668. doi: 10.1111/j.1471-4159.1990.tb01922.x. [DOI] [PubMed] [Google Scholar]
- Diez-Sampedro A, Hirayama BA, Osswald C, Gorboulev V, Baumgarten K, Volk C, Wright EM, Koepsell H. A glucose sensor hiding in a family of transporters. Proc. Natl Acad. Sci. USA. 2003;100:11753–11758. doi: 10.1073/pnas.1733027100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipace C, Sung U, Binda F, Blakely RD, Galli A. Amphetamine induces a calcium/calmodulin-dependent protein kinase II-dependent reduction in norepinephrine transporter surface expression linked to changes in syntaxin 1A/transporter complexes. Mol. Pharmacol. 2007;71:230–239. doi: 10.1124/mol.106.026690. [DOI] [PubMed] [Google Scholar]
- Egana LA, Cuevas RA, Baust TB, et al. Physical and functional interaction between the dopamine transporter and the synaptic vesicle protein synaptogyrin-3. J. Neurosci. 2009;29:4592–4604. doi: 10.1523/JNEUROSCI.4559-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen J, Rasmussen SG, Rasmussen TN, Vaegter CB, Cha JH, Zou MF, Newman AH, Gether U. Visualization of dopamine transporter trafficking in live neurons by use of fluorescent cocaine analogs. J. Neurosci. 2009;29:6794–6808. doi: 10.1523/JNEUROSCI.4177-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger K, Grewer C, Javitch JA, Galli A. Currents in response to rapid concentration jumps of amphetamine uncover novel aspects of human dopamine transporter function. J. Neurosci. 2008;28:976–989. doi: 10.1523/JNEUROSCI.2796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshleman AJ, Henningsen RA, Neve KA, Janowsky A. Release of dopamine via the human transporter. Mol. Pharmacol. 1994;45:312–316. [PubMed] [Google Scholar]
- Ewing AG, Stein TS, Lau YY. Analytical chemistry in microenvironments: single nerve cells. Accts. Chem. Res. 1992;25:440–447. [Google Scholar]
- Falkenburger BH, Barstow KL, Mintz IM. Dendrodendritic inhibition through reversal of dopamine transport. Science. 2001;293:2465–2470. doi: 10.1126/science.1060645. [DOI] [PubMed] [Google Scholar]
- Farhan H, Korkhov VM, Paulitschke V, Dorostkar MM, Scholze P, Kudlacek O, Freissmuth M, Sitte HH. Two discontinuous segments in the carboxy terminus are required for membrane targeting of the rat GABA transporter-1 (GAT1) J. Biol. Chem. 2004;279:28553–28563. doi: 10.1074/jbc.M307325200. [DOI] [PubMed] [Google Scholar]
- Farhan H, Freissmuth M, Sitte HH. Oligomerization of neurotransmitter transporters: a ticket from the endoplasmic reticulum to the plasma membrane. Handb. Exp. Pharmacol. 2006;175:233–249. doi: 10.1007/3-540-29784-7_12. [DOI] [PubMed] [Google Scholar]
- Farhan H, Reiterer V, Korkhov VM, Schmid JA, Freissmuth M, Sitte HH. Concentrative export from the endoplasmic reticulum of the gamma-aminobutyric acid transporter 1 requires binding to SEC24D. J. Biol. Chem. 2007;282:7679–7689. doi: 10.1074/jbc.M609720200. [DOI] [PubMed] [Google Scholar]
- Farhan H, Reiterer V, Kriz A, Hauri HP, Pavelka M, Sitte HH, Freissmuth M. Signal-dependent export of GABA transporter 1 from the ER-Golgi intermediate compartment is specified by a C-terminal motif. J. Cell Sci. 2008;121:753–761. doi: 10.1242/jcs.017681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer JF, Cho AK. Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J. Pharmacol. Exp. Ther. 1979;208:203–209. [PubMed] [Google Scholar]
- Fishkes H, Rudnick G. Bioenergetics of serotonin transport by membrane vesicles derived from platelet dense granules. J. Biol. Chem. 1982;257:5671–5677. [PubMed] [Google Scholar]
- Fjorback AW, Pla P, Muller HK, Wiborg O, Saudou F, Nyengaard JR. Serotonin transporter oligomerization documented in RN46A cells and neurons by sensitized acceptor emission FRET and fluorescence lifetime imaging microscopy. Biochem. Biophys. Res. Commun. 2009;380:724–728. doi: 10.1016/j.bbrc.2009.01.128. [DOI] [PubMed] [Google Scholar]
- Fleckenstein A, Burn JH. The effect of denervation on the action of sympathomimetic amines on the nictitating membrane. Br. J Pharmacol. Chemother. 1953;8:69–78. doi: 10.1111/j.1476-5381.1953.tb00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fog JU, Khoshbouei H, Holy M, et al. Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron. 2006;51:417–429. doi: 10.1016/j.neuron.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Fon EA, Pothos EN, Sun BC, Killeen N, Sulzer D, Edwards RH. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proc. Natl Acad. Sci. USA. 2008;105:10338–10343. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JD, Pananusorn B, Vaughan RA. Dopamine transporters are phosphorylated on N-terminal serines in rat striatum. J. Biol. Chem. 2002;277:25178–25186. doi: 10.1074/jbc.M200294200. [DOI] [PubMed] [Google Scholar]
- Foster JD, Cervinski MA, Gorentla BK, Vaughan RA. Regulation of the dopamine transporter by phosphorylation. Handb. Exp. Pharmacol. 2006;175:197–214. doi: 10.1007/3-540-29784-7_10. [DOI] [PubMed] [Google Scholar]
- Fröhlich A, Loewi O. Über eine Steigerung der Adrenalin-empfindlichkeit durch Cocain. (On the increase in the adrenalin sensitivity induced by cocaine) Arch. exp. Pathol. Pharmakol. 1910;62:159–169. [Google Scholar]
- Furchgott RF, Kirpekar SM, Rieker M, Schwab A. Actions and interactions of norepinephrine, tyramine and cocaine on aortic strips of rabbit and left atria of guinea pig and cat. J. Pharmacol. Exp. Ther. 1963;142:39–58. [PubMed] [Google Scholar]
- Galli A, DeFelice LJ, Duke BJ, Moore KR, Blakely RD. Sodium-dependent norepinephrine-induced currents in norepinephrine-transporter-transfected HEK-293 cells blocked by cocaine and antidepressants. J. Exp. Biol. 1995;198:2197–2212. doi: 10.1242/jeb.198.10.2197. [DOI] [PubMed] [Google Scholar]
- Galli A, Petersen CI, deBlaquiere M, Blakely RD, DeFelice LJ. Drosophila serotonin transporters have voltage-dependent uptake coupled to a serotonin-gated ion channel. J. Neurosci. 1997;17:3401–3411. doi: 10.1523/JNEUROSCI.17-10-03401.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli A, Blakely RD, DeFelice LJ. Patch-clamp and amperometric recordings from norepinephrine transporters: channel activity and voltage-dependent uptake. Proc. Natl Acad. Sci. USA. 1998;95:13260–13265. doi: 10.1073/pnas.95.22.13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambalvo CT. Protein kinase C and dopamine transport–1. Effects of amphetamine in vivo. Neuropharmacology. 1992a;31:1201–1210. doi: 10.1016/0028-3908(92)90048-t. [DOI] [PubMed] [Google Scholar]
- Giambalvo CT. Protein kinase C and dopamine transport—2. Effects of amphetamine in vitro. Neuropharmacology. 1992b;31:1211–1222. doi: 10.1016/0028-3908(92)90049-u. [DOI] [PubMed] [Google Scholar]
- Giambalvo CT. Differential effects of amphetamine transport vs. dopamine reverse transport on particulate PKC activity in striatal synaptoneurosomes. Synapse. 2003;49:125–133. doi: 10.1002/syn.10223. [DOI] [PubMed] [Google Scholar]
- Glowinski J, Axelrod J. Effect of drugs on the uptake, release, and metabolism of H3-norepinephrine in the rat brain. J. Pharmacol. Exp. Ther. 1965;149:43–49. [PubMed] [Google Scholar]
- Gnegy ME. The effect of phosphorylation on amphetamine-mediated outward transport. Eur. J. Pharmacol. 2003;479:83–91. doi: 10.1016/j.ejphar.2003.08.059. [DOI] [PubMed] [Google Scholar]
- Gnegy ME, Khoshbouei H, Berg KA, Javitch JA, Clarke WP, Zhang M, Galli A. Intracellular Ca2+ regulates amphetamine-induced dopamine efflux and currents mediated by the human dopamine transporter. Mol. Pharmacol. 2004;66:137–143. doi: 10.1124/mol.66.1.137. [DOI] [PubMed] [Google Scholar]
- Gobbi M, Mennini T, Garattini S. Mechanism of neurotransmitter release induced by amphetamine derivatives: pharmacological and toxicological aspects. Curr. Top. Pharmacol. 1997;3:217–227. [Google Scholar]
- Gobbi M, Moia M, Pirona L, Ceglia I, Reyes-Parada M, Scorza C, Mennini T. p-Methylthioamphetamine and 1-(m-chlorophenyl)piperazine, two non-neurotoxic 5-HT releasers in vivo, differ from neurotoxic amphetamine derivatives in their mode of action at 5-HT nerve endings in vitro. J. Neurochem. 2002;82:1435–1443. doi: 10.1046/j.1471-4159.2002.01073.x. [DOI] [PubMed] [Google Scholar]
- Gobbi M, Funicello M, Gerstbrein K, et al. N,N-dimethylthioamphetamine and methyl-thioamphetamine, two non-neurotoxic substrates of 5-HT transporters, have scant in vitro efficacy for the induction of transporter-mediated 5-HT release and currents. J. Neurochem. 2008;105:1770–1780. doi: 10.1111/j.1471-4159.2008.05272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorentla BK, Moritz AE, Foster JD, Vaughan RA. Proline-directed phosphorylation of the dopamine transporter N-terminal domain. Biochemistry. 2009;48:1067–1076. doi: 10.1021/bi801696n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granas C, Ferrer J, Loland CJ, Javitch JA, Gether U. N-terminal truncation of the dopamine transporter abolishes phorbol ester- and substance P receptor-stimulated phosphorylation without impairing transporter internalization. J. Biol. Chem. 2003;278:4990–5000. doi: 10.1074/jbc.M205058200. [DOI] [PubMed] [Google Scholar]
- Green AR, Mechan AO, Elliott JM, O’Shea E, Colado MI. The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”) Pharmacol. Rev. 2003;55:463–508. doi: 10.1124/pr.55.3.3. [DOI] [PubMed] [Google Scholar]
- Hansra N, Arya S, Quick MW. Intracellular domains of a rat brain GABA transporter that govern transport. J. Neurosci. 2004;24:4082–4087. doi: 10.1523/JNEUROSCI.0664-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastrup H, Karlin A, Javitch JA. Symmetrical dimer of the human dopamine transporter revealed by cross- linking Cys-306 at the extracellular end of the sixth transmembrane segment. Proc. Natl Acad. Sci. USA. 2001;98:10055–10060. doi: 10.1073/pnas.181344298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann DW, Lu CC. GAT1 (GABA:Na+:Cl−) co-transport function. Database reconstruction with an alternating access model. J. Gen. Physiol. 1999;114:459–475. doi: 10.1085/jgp.114.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Everett CV, Sorkin A, Zahniser NR. Rapid regulation of dopamine transporters by tyrosine kinases in rat neuronal preparations. J. Neurochem. 2007;101:1258–1271. doi: 10.1111/j.1471-4159.2007.04522.x. [DOI] [PubMed] [Google Scholar]
- Humphreys CJ, Wall SC, Rudnick G. Ligand binding to the serotonin transporter: equilibria, kinetics, and ion dependence. Biochemistry. 1994;33:9118–9125. doi: 10.1021/bi00197a014. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Prasad BM, Amara SG. Dopamine transporter-mediated conductances increase excitability of midbrain dopamine neurons. Nat. Neurosci. 2002;5:971–978. doi: 10.1038/nn920. [DOI] [PubMed] [Google Scholar]
- Iversen LL. Role of transmitter uptake mechanisms in synaptic neurotransmission. Br. J. Pharmacol. 1971;41:571–591. doi: 10.1111/j.1476-5381.1971.tb07066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen L. Neurotransmitter transporters: fruitful targets for CNS drug discovery. Mol. Psychiatry. 2000;5:357–362. doi: 10.1038/sj.mp.4000728. [DOI] [PubMed] [Google Scholar]
- Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl Acad. Sci. USA. 1985;82:2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jess U, Betz H, Schloss P. The membrane-bound rat serotonin transporter, SERT1, is an oligomeric protein. FEBS Lett. 1996;394:44–46. doi: 10.1016/0014-5793(96)00916-7. [DOI] [PubMed] [Google Scholar]
- Johnson LA, Guptaroy B, Lund D, Shamban S, Gnegy ME. Regulation of amphetamine-stimulated dopamine efflux by protein kinase C beta. J. Biol. Chem. 2005;280:10914–10919. doi: 10.1074/jbc.M413887200. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J. Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Joseph JD, Barak LS, Caron MG, Wightman RM. Dopamine neuronal transport kinetics and effects of amphetamine. J. Neurochem. 1999;73:2406–2414. doi: 10.1046/j.1471-4159.1999.0732406.x. [DOI] [PubMed] [Google Scholar]
- Just H, Sitte HH, Schmid JA, Freissmuth M, Kudlacek O. Identification of an additional interaction domain in trans-membrane domains 11 and 12 that supports oligomer formation in the human serotonin transporter. J. Biol. Chem. 2004;279:6650–6657. doi: 10.1074/jbc.M306092200. [DOI] [PubMed] [Google Scholar]
- Kahlig KM, Binda F, Khoshbouei H, Blakely RD, McMahon DG, Javitch JA, Galli A. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proc. Natl Acad. Sci. USA. 2005;102:3495–3500. doi: 10.1073/pnas.0407737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor L, Gnegy ME. Protein kinase C inhibitors block amphetamine-mediated dopamine release in rat striatal slices. J. Pharmacol. Exp. Ther. 1998;284:592–598. [PubMed] [Google Scholar]
- Kantor L, Hewlett GH, Gnegy ME. Enhanced amphetamine- and K+-mediated dopamine release in rat striatum after repeated amphetamine: differential requirements for Ca2+- and calmodulin-dependent phosphorylation and synaptic vesicles. J. Neurosci. 1999;19:3801–3808. doi: 10.1523/JNEUROSCI.19-10-03801.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor L, Hewlett GH, Park YH, Richardson-Burns SM, Mellon MJ, Gnegy ME. Protein kinase C and intracellular calcium are required for amphetamine-mediated dopamine release via the norepinephrine transporter in undifferentiated PC12 cells. J. Pharmacol. Exp. Ther. 2001;297:1016–1024. [PubMed] [Google Scholar]
- Kazanietz MG, Caloca MJ, Aizman O, Nowicki S. Phosphorylation of the catalytic subunit of rat renal Na+, K+-ATPase by classical PKC isoforms. Arch. Biochem. Biophys. 2001;388:74–80. doi: 10.1006/abbi.2000.2264. [DOI] [PubMed] [Google Scholar]
- Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A. Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J. Biol. Chem. 2003;278:12070–12077. doi: 10.1074/jbc.M212815200. [DOI] [PubMed] [Google Scholar]
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A, Javitch JA. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2004;2:E78. doi: 10.1371/journal.pbio.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic F, Rudnick G. Oligomerization of serotonin tranporter and its functional consequences. Proc. Natl Acad. Sci. USA. 2000;97:3106–3111. doi: 10.1073/pnas.060408997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocabas AM, Rudnick G, Kilic F. Functional consequences of homo- but not hetero-oligomerization between transporters for the biogenic amine neurotransmitters. J. Neurochem. 2003;85:1513–1520. doi: 10.1046/j.1471-4159.2003.01793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkhov VM, Holy M, Freissmuth M, Sitte HH. The conserved glutamate (Glu136) in transmembrane domain 2 of the serotonin transporter is required for the conformational switch in the transport cycle. J. Biol. Chem. 2006;281:13439–13448. doi: 10.1074/jbc.M511382200. [DOI] [PubMed] [Google Scholar]
- L’hirondel M, Cheramy A, Godeheu G, Glowinski J. Effects of arachidonic acid on dopamine synthesis, spontaneous release, and uptake in striatal synaptosomes from the rat. J. Neurochem. 1995;64:1406–1409. doi: 10.1046/j.1471-4159.1995.64031406.x. [DOI] [PubMed] [Google Scholar]
- Langeloh A, Trendelenburg U. The mechanism of the 3H-noradrenaline releasing effect of various substrates of uptake1: role of monoamine oxidase and of vesicularly stored 3H-noradrenaline. Naunyn Schmiedebergs Arch. Pharmacol. 1987;336:611–620. doi: 10.1007/BF00165751. [DOI] [PubMed] [Google Scholar]
- Langston JW, Irwin I, Langston EB, Forno LS. 1-Methyl-4-phenylpyridinium ion (MPP+): identification of a metabolite of MPTP, a toxin selective to the substantia nigra. Neurosci. Lett. 1984;48:87–92. doi: 10.1016/0304-3940(84)90293-3. [DOI] [PubMed] [Google Scholar]
- Lester HA, Cao Y, Mager S. Listening to neurotransmitter transporters. Neuron. 1996;17:807–810. doi: 10.1016/s0896-6273(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Levi G, Raiteri M. Carrier-mediated release of neurotransmitters. Trends Neurosci. 1993;16:415–419. doi: 10.1016/0166-2236(93)90010-j. [DOI] [PubMed] [Google Scholar]
- Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenberg D, Brecha N, Edwards RH. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell. 1992;70:539–551. doi: 10.1016/0092-8674(92)90425-c. [DOI] [PubMed] [Google Scholar]
- Liu Y, Vidensky S, Ruggiero AM, Maier S, Sitte HH, Rothstein JD. Reticulon RTN2B regulates trafficking and function of neuronal glutamate transporter EAAC1. J. Biol. Chem. 2008;283:6561–6571. doi: 10.1074/jbc.M708096200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loland CJ, Norregaard L, Gether U. Defining proximity relationships in the tertiary structure of the dopamine transporter. Identification of a conserved glutamic acid as a third coordinate in the endogenous Zn(2+)-binding site. J. Biol. Chem. 1999;274:36928–36934. doi: 10.1074/jbc.274.52.36928. [DOI] [PubMed] [Google Scholar]
- Lu CC, Hilgemann DW. GAT1 (GABA:Na+:Cl−) cotransport function. Kinetic studies in giant Xenopus oocyte membrane patches. J. Gen. Physiol. 1999a;114:445–457. doi: 10.1085/jgp.114.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CC, Hilgemann DW. GAT1 (GABA:Na+:Cl−) cotransport function. Steady state studies in giant Xenopus oocyte membrane patches. J. Gen. Physiol. 1999b;114:429–444. doi: 10.1085/jgp.114.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager S, Min C, Henry DJ, Chavkin C, Hoffman BJ, Davidson N, Lester HA. Conducting states of a mammalian serotonin transporter. Neuron. 1994;12:845–859. doi: 10.1016/0896-6273(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Mager S, Kleinberger DN, Keshet GI, Davidson N, Kanner BI, Lester HA. Ion binding and permeation at the GABA transporter GAT1. J. Neurosci. 1996;16:5405–5414. doi: 10.1523/JNEUROSCI.16-17-05405.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier S, Reiterer V, Ruggiero AM, Rothstein JD, Thomas S, Dahm R, Sitte HH, Farhan H. GTRAP3-18 serves as a negative regulator of Rab1 in protein transport and neuronal differentiation. J. Cell Mol. Med. 2009;13:114–124. doi: 10.1111/j.1582-4934.2008.00303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson J, Sagne C, Hamon M, el Mestikawy S. Neurotransmitter transporters in the central nervous system. Pharmacol. Rev. 1999;51:439–464. [PubMed] [Google Scholar]
- Mazei-Robinson MS, Blakely RD. ADHD and the dopamine transporter: are there reasons to pay attention? Handb. Exp. Pharmacol. 2006;175:373–415. doi: 10.1007/3-540-29784-7_17. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Bowton E, Holy M, Schmudermaier M, Freissmuth M, Sitte HH, Galli A, Blakely RD. Anomalous dopamine release associated with a human dopamine transporter coding variant. J. Neurosci. 2008;28:7040–7046. doi: 10.1523/JNEUROSCI.0473-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinild AK, Sitte HH, Gether U. Zinc potentiates an uncoupled anion conductance associated with the dopamine transporter. J. Biol. Chem. 2004;279:49671–49679. doi: 10.1074/jbc.M407660200. [DOI] [PubMed] [Google Scholar]
- Moron JA, Zakharova I, Ferrer JV, et al. Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transport capacity. J. Neurosci. 2003;23:8480–8488. doi: 10.1523/JNEUROSCI.23-24-08480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Gong LW, Khanna B, Sulzer D, Lindau M. Intracellular patch electrochemistry: regulation of cytosolic catecholamines in chromaffin cells. J. Neurosci. 2003;23:5835–5845. doi: 10.1523/JNEUROSCI.23-13-05835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson N. The family of Na+/Cl− neurotransmitter transporters. J. Neurochem. 1998;71:1785–1803. doi: 10.1046/j.1471-4159.1998.71051785.x. [DOI] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM. Immunogold localization of the dopamine transporter: an ultrastructural study of the rat ventral tegmental area. J. Neurosci. 1997;17:5255–5262. doi: 10.1523/JNEUROSCI.17-14-05255.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norregaard L, Frederiksen D, Nielsen EO, Gether U. Delineation of an endogenous zinc-binding site in the human dopamine transporter. EMBO J. 1998;17:4266–4273. doi: 10.1093/emboj/17.15.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CI, DeFelice LJ. Ionic interactions in the Drosophila serotonin transporter identify it as a serotonin channel. Nat. Neurosci. 1999;2:605–610. doi: 10.1038/10158. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pifl C, Singer EA. Ion dependence of carrier-mediated release in dopamine or norepinephrine transporter-transfected cells questions the hypothesis of facilitated exchange diffusion. Mol. Pharmacol. 1999;56:1047–1054. doi: 10.1124/mol.56.5.1047. [DOI] [PubMed] [Google Scholar]
- Pifl C, Drobny H, Reither H, Hornykiewicz O, Singer EA. Mechanism of the dopamine-releasing actions of amphetamine and cocaine: plasmalemmal dopamine transporter versus vesicular monoamine transporter. Mol. Pharmacol. 1995;47:368–373. [PubMed] [Google Scholar]
- Pifl C, Agneter E, Drobny H, Reither H, Singer EA. Induction by low Na+ or Cl− of cocaine sensitive carrier-mediated efflux of amines from cells transfected with the cloned human catecholamine transporters. Br. J. Pharmacol. 1997;121:205–212. doi: 10.1038/sj.bjp.0701137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pifl C, Agneter E, Drobny H, Sitte HH, Singer EA. Amphetamine reverses or blocks the operation of the human noradrenaline transporter depending on its concentration: superfusion studies on transfected cells. Neuropharmacology. 1999;38:157–165. doi: 10.1016/s0028-3908(98)00155-5. [DOI] [PubMed] [Google Scholar]
- Pifl C, Rebernik P, Kattinger A, Reither H. Zn2+ modulates currents generated by the dopamine transporter: parallel effects on amphetamine-induced charge transfer and release. Neuropharmacology. 2004;46:223–231. doi: 10.1016/j.neuropharm.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Pifl C, Wolf A, Rebernik P, Reither H, Berger ML. Zinc regulates the dopamine transporter in a membrane potential and chloride dependent manner. Neuropharmacology. 2009;56:531–540. doi: 10.1016/j.neuropharm.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Pozzan T, Gatti G, Dozio N, Vicentini LM, Meldolesi J. Ca2+-dependent and -independent release of neurotransmitters from PC12 cells: a role for protein kinase C activation? J. Cell Biol. 1984;99:628–638. doi: 10.1083/jcb.99.2.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Hu J, Wang D, Zhang HY. Regulation of a gamma-aminobutyric acid transporter by reciprocal tyrosine and serine phosphorylation. J. Biol. Chem. 2004;279:15961–15967. doi: 10.1074/jbc.M306924200. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Blakely RD. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 1999;285:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Samuvel DJ, Buck ER, Rudnick G, Jayanthi LD. Phosphorylation of threonine residue 276 is required for acute regulation of serotonin transporter by cyclic GMP. J. Biol. Chem. 2007;282:11639–11647. doi: 10.1074/jbc.M611353200. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, DeFelice LJ. Serotonin transporter function and pharmacology are sensitive to expression level: evidence for an endogenous regulatory factor. J. Biol. Chem. 2002;277:14475–14482. doi: 10.1074/jbc.M110783200. [DOI] [PubMed] [Google Scholar]
- Reiterer V, Maier S, Sitte HH, Kriz A, Ruegg MA, Hauri HP, Freissmuth M, Farhan H. Sec24- and ARFGAP1-dependent trafficking of GABA transporter-1 is a prerequisite for correct axonal targeting. J. Neurosci. 2008;28:12453–12464. doi: 10.1523/JNEUROSCI.3451-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressl S, Terwisscha van Scheltinga AC, Vonrhein C, Ott V, Ziegler C. Molecular basis of transport and regulation in the Na(+)/betaine symporter BetP. Nature. 2009;458:47–52. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]
- Robertson SD, Matthies HJ, Galli A. A closer look at amphetamine-induced reverse transport and trafficking of the dopa-mine and norepinephrine transporters. Mol. Neurobiol. 2009;39:73–80. doi: 10.1007/s12035-009-8053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JB. Stereoselectivity and isoenzyme selectivity of monoamine oxidase inhibitors. Enantiomers of amphetamine, N-methylamphetamine and deprenyl. Biochem. Pharmacol. 1985;34:4105–4108. doi: 10.1016/0006-2952(85)90201-1. [DOI] [PubMed] [Google Scholar]
- Ross SB, Kelder D. Efflux of 5-hydroxytryptamine from synaptosomes of rat cerebral cortex. Acta Physiol. Scand. 1977;99:27–36. doi: 10.1111/j.1748-1716.1977.tb10348.x. [DOI] [PubMed] [Google Scholar]
- Ross SB, Renyi AL. Uptake of some tritiated sympathomimetic amines by mouse brain cortex slices in vitro. Acta Pharmacol. Toxicol. (Copenh) 1966;24:297–309. doi: 10.1111/j.1600-0773.1966.tb00392.x. [DOI] [PubMed] [Google Scholar]
- Roux MJ, Supplisson S. Neuronal and glial glycine transporters have different stoichiometries. Neuron. 2000;25:373–383. doi: 10.1016/s0896-6273(00)80901-0. [DOI] [PubMed] [Google Scholar]
- Rudnick G. Ion-coupled neurotransmitter transport: thermodynamic vs. kinetic determinations of stoichiometry. Methods Enzymol. 1999;296:233–247. doi: 10.1016/s0076-6879(98)96018-9. [DOI] [PubMed] [Google Scholar]
- Rudnick G. Mechanism of biogenic amine neurotransmitter transporters. In: Reith MEA, editor. Neurotransmitter Transporters: Structure, Function, and Regulation. Humana Press Inc; Totowa, NJ: 2002. pp. 25–52. [Google Scholar]
- Rudnick G, Clark J. From synapse to vesicle: the reuptake and storage of biogenic amine neurotransmitters. Biochim. Biophys. Acta. 1993;1144:249–263. doi: 10.1016/0005-2728(93)90109-s. [DOI] [PubMed] [Google Scholar]
- Rudnick G, Wall SC. p-Chloroamphetamine induces serotonin release through serotonin transporters. Biochemistry. 1992;31:6710–6718. doi: 10.1021/bi00144a010. [DOI] [PubMed] [Google Scholar]
- Ruggiero AM, Liu Y, Vidensky S, Maier S, Jung E, Farhan H, Robinson MB, Sitte HH, Rothstein JD. The endoplasmic reticulum exit of glutamate transporter is regulated by the inducible mammalian Yip6b/GTRAP3-18 protein. J. Biol. Chem. 2008;283:6175–6183. doi: 10.1074/jbc.M701008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH, Jr, Tran CV, Barabote RD. TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006;34:D181–D186. doi: 10.1093/nar/gkj001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- Schmid JA, Scholze P, Kudlacek O, Freissmuth M, Singer EA, Sitte HH. Oligomerization of the human serotonin trans porter and of the rat GABA transporter 1 visualized by fluorescence resonance energy transfer microscopy in living cells. J. Biol. Chem. 2001;276:3805–3810. doi: 10.1074/jbc.M007357200. [DOI] [PubMed] [Google Scholar]
- Scholze P, Zwach J, Kattinger A, Pifl C, Singer EA, Sitte HH. Transporter-mediated release: a superfusion study on human embryonic kidney cells stably expressing the human serotonin transporter. J. Pharmacol. Exp. Ther. 2000;293:870–878. [PubMed] [Google Scholar]
- Scholze P, Freissmuth M, Sitte HH. Mutations within an intramembrane leucine heptad repeat disrupt oligomer formation of the rat GABA transporter 1. J. Biol. Chem. 2002a;277:43682–43690. doi: 10.1074/jbc.M205602200. [DOI] [PubMed] [Google Scholar]
- Scholze P, Norregaard L, Singer EA, Freissmuth M, Gether U, Sitte HH. The role of zinc ions in reverse transport mediated by monoamine transporters. J. Biol. Chem. 2002b;277:21505–21513. doi: 10.1074/jbc.M112265200. [DOI] [PubMed] [Google Scholar]
- Schömig A, Fischer S, Kurz T, Richardt G, Schömig E. Nonexocytotic release of endogenous noradrenaline in the ischemic and anoxic rat heart: mechanism and metabolic requirements. Circ. Res. 1987;60:194–205. doi: 10.1161/01.res.60.2.194. [DOI] [PubMed] [Google Scholar]
- Schuldiner S, Steiner MS, Yelin R, Wall SC, Rudnick G. Amphetamine derivatives interact with both plasma membrane and secretory vesicle biogenic amine transporters. Mol. Pharmacol. 1993;44:1227–1231. [PubMed] [Google Scholar]
- Schuldiner S, Shirvan A, Linial M. Vesicular neurotransmitter transporters: from bacteria to humans. Physiol. Rev. 1995;75:369–392. doi: 10.1152/physrev.1995.75.2.369. [DOI] [PubMed] [Google Scholar]
- Seidel S, Singer EA, Just H, et al. Amphetamines take two to tango: an oligomer-based counter-transport model of neurotransmitter transport explores the amphetamine action. Mol. Pharmacol. 2005;67:140–151. doi: 10.1124/mol.67.1.. [DOI] [PubMed] [Google Scholar]
- Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu. Rev. Pharmacol. Toxicol. 1993;33:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- Sen N, Shi L, Beuming T, Weinstein H, Javitch JA. A pincer-like configuration of TM2 in the human dopamine transporter is responsible for indirect effects on cocaine binding. Neuropharmacology. 2005;49:780–790. doi: 10.1016/j.neuropharm.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter–inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol. Cell. 2008;30:667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK. LeuT: A prokaryotic stepping stone on the way to a eukaryotic neurotransmitter transporter structure. Channels (Austin.) 2008;2:380–389. doi: 10.4161/chan.2.5.6904. [DOI] [PubMed] [Google Scholar]
- Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitte HH, Freissmuth M. Oligomer formation by Na+-Cl−-coupled neurotransmitter transporters. Eur. J. Pharmacol. 2003;479:229–236. doi: 10.1016/j.ejphar.2003.08.072. [DOI] [PubMed] [Google Scholar]
- Sitte HH, Huck S, Reither H, Boehm S, Singer EA, Pifl C. Carrier-mediated release, transport rates, and charge transfer induced by amphetamine, tyramine, and dopamine in mammalian cells transfected with the human dopamine transporter. J. Neurochem. 1998;71:1289–1297. doi: 10.1046/j.1471-4159.1998.71031289.x. [DOI] [PubMed] [Google Scholar]
- Sitte HH, Scholze P, Schloss P, Pifl C, Singer EA. Characterization of carrier-mediated release in human embryonic kidney 293 cells stably expressing the rat serotonin transporter: a superfusion study. J. Neurochem. 2000;74:1317–1324. doi: 10.1046/j.1471-4159.2000.741317.x. [DOI] [PubMed] [Google Scholar]
- Sitte HH, Hiptmair B, Zwach J, Pifl C, Singer EA, Scholze P. Quantitative analysis of inward and outward transport rates in cells stably expressing the cloned human serotonin transporter: inconsistencies with the hypothesis of facilitated exchange diffusion. Mol. Pharmacol. 2001;59:1129–1137. doi: 10.1124/mol.59.5.1129. [DOI] [PubMed] [Google Scholar]
- Sitte HH, Singer EA, Scholze P. Bi-directional transport of GABA in human embryonic kidney (HEK-293) cells stably expressing the rat GABA transporter GAT-1. Br. J. Pharmacol. 2002;135:93–102. doi: 10.1038/sj.bjp.0704446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitte HH, Farhan H, Javitch JA. Sodium-dependent neurotransmitter transporters: oligomerization as a determinant of transporter function and trafficking. Mol. Interv. 2004;4:38–47. doi: 10.1124/mi.4.1.38. [DOI] [PubMed] [Google Scholar]
- Sonders MS, Amara SG. Channels in transporters. Curr. Opin. Neurobiol. 1996;6:294–302. doi: 10.1016/s0959-4388(96)80111-5. [DOI] [PubMed] [Google Scholar]
- Sonders MS, Zhu SJ, Zahniser NR, Kavanaugh MP, Amara SG. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J. Neurosci. 1997;17:960–974. doi: 10.1523/JNEUROSCI.17-03-00960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina T, Doolen S, Galperin E, Zahniser NR, Sorkin A. Oligomerization of dopamine transporters visualized in living cells by FRET microscopy. J. Biol. Chem. 2003;278:28274–28283. doi: 10.1074/jbc.M210652200. [DOI] [PubMed] [Google Scholar]
- Sorkina T, Miranda M, Dionne KR, Hoover BR, Zahniser NR, Sorkin A. RNA interference screen reveals an essential role of Nedd4-2 in dopamine transporter ubiquitination and endocytosis. J. Neurosci. 2006;26:8195–8205. doi: 10.1523/JNEUROSCI.1301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein WD. The Movement of Molecules Across Cell Membranes. Academic Press; New York: 1967. [Google Scholar]
- Su A, Mager S, Mayo SL, Lester HA. A multi-substrate single-file model for ion-coupled transporters. Biophys. J. 1996;70:762–777. doi: 10.1016/S0006-3495(96)79616-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Galli A. Dopamine transport currents are promoted from curiosity to physiology. Trends Neurosci. 2003;26:173–176. doi: 10.1016/S0166-2236(03)00063-8. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Maidment NT, Rayport S. Amphetamine and other weak bases act to promote reverse transport of dopamine in ventral midbrain neurons. J. Neurochem. 1993;60:527–535. doi: 10.1111/j.1471-4159.1993.tb03181.x. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J. Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog. Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Tainter ML, Chang DK. The antagonism of sympathetic and adrenaline content of the spleen, kidney, and salivary glands in the sheep. J. Pharmacol. Exp. Ther. 1927;30:193–207. [Google Scholar]
- Torres GE, Carneiro A, Seamans K, Fiorentini C, Sweeney A, Yao WD, Caron MG. Oligomerization and trafficking of the human dopamine transporter. Mutational analysis identifies critical domains important for the functional expression of the transporter. J. Biol. Chem. 2003a;278:2731–2739. doi: 10.1074/jbc.M201926200. [DOI] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat. Rev. Neurosci. 2003b;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- Trendelenburg U, Langeloh A, Bönisch H. Mechanism of action of indirectly acting sympathomimetic amines. Blood Vessels. 1987;24:261–270. doi: 10.1159/000158702. [DOI] [PubMed] [Google Scholar]
- Vaughan RA, Huff RA, Uhl GR, Kuhar MJ. Protein kinase C-mediated phosphorylation and functional regulation of dopamine transporters in striatal synaptosomes. J. Biol. Chem. 1997;272:15541–15546. doi: 10.1074/jbc.272.24.15541. [DOI] [PubMed] [Google Scholar]
- Wall SC, Gu H, Rudnick G. Biogenic amine flux mediated by cloned transporters stably expressed in cultured cell lines: amphetamine specificity for inhibition and efflux. Mol. Pharmacol. 1995;47:544–550. [PubMed] [Google Scholar]
- Wang D, Deken SL, Whitworth TL, Quick MW. Syntaxin 1A inhibits GABA flux, efflux, and exchange mediated by the rat brain GABA transporter GAT1. Mol. Pharmacol. 2003;64:905–913. doi: 10.1124/mol.64.4.905. [DOI] [PubMed] [Google Scholar]
- Widdas WF. Inability of diffusion to account for placental glucose transfer in the sheep and consideration of the kinetics of a possible carrier transfer. J. Physiol. 1952;118:23–39. doi: 10.1113/jphysiol.1952.sp004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willeit M, Sitte HH, Thierry N, et al. Enhanced serotonin transporter function during depression in seasonal affective disorder. Neuropsychopharmacology. 2008;33:1503–1513. doi: 10.1038/sj.npp.1301560. [DOI] [PubMed] [Google Scholar]
- Wolfel R, Graefe KH. Evidence for various tryptamines and related compounds acting as substrates of the platelet 5-hydroxytryptamine transporter. Naunyn Schmiedebergs Arch. Pharmacol. 1992;345:129–136. doi: 10.1007/BF00165727. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Zaczek R, Culp S, De SE. Interactions of [3H]amphetamine with rat brain synaptosomes. II. Active transport. J. Pharmacol. Exp. Ther. 1991;257:830–835. [PubMed] [Google Scholar]
- Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA, Blakely RD. p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J. Biol. Chem. 2005;280:15649–15658. doi: 10.1074/jbc.M410858200. [DOI] [PubMed] [Google Scholar]