

Abstract
Purpose of review
Hepatic bile acid synthesis is controlled, in part, by a complex enterohepatic feedback regulatory mechanism. In this review, we focus on the role of the intestinal FGF15/19 hormone in modulating bile acid levels, and additional metabolic effects on glucose metabolism, non-alcoholic liver disease (NAFLD), and liver regeneration. We also highlight the newly identified intestinal protein, Diet1, which is a modulator of FGF15/19 levels.
Recent findings
Low FGF19 levels are associated with bile acid diarrhea and NAFLD. In contrast, high FGF19 levels are associated with diabetes remission following Roux-en-Y gastric bypass surgery, suggesting new therapeutic approaches against type 2 diabetes. The effect of FGF15/19 on liver plasticity is a double-edged sword: whereas elevated FGF15/19 levels improve survival of mice after partial hepatectomy, FGF19 mitogenic activity is associated with liver carcinoma. Finally, a recent study has identified Diet1, an intestinal factor that influences FGF15/19 levels in mouse intestine and human enterocytes. Diet1 represents the first factor shown to influence FGF15/19 levels at a post-transcriptional level.
Summary
The biological effects of FGF15/19 make it an attractive target for treating metabolic dysregulation underlying conditions such as fatty liver and type 2 diabetes. Further elucidation of the role of Diet1 in FGF15/19 secretion may provide a control point for pharmacological modulation of FGF15/19 levels.
Keywords: enterohepatic circulation, fibroblast growth factor, NAFLD, gastric bypass
Introduction
Bile acids have been long known to be critical for efficient intestinal absorption of fats and lipid-soluble vitamins. The past dozen years have revealed a second life for bile acids as ligands for nuclear receptors that regulate bile acid synthesis, transport, and metabolism, and in metabolic processes such as glucose metabolism and energy expenditure. These roles necessitate precise regulation of bile acid levels. It is now established that bile acid homeostasis is achieved, in part, through communication from the small intestine to the liver to regulate hepatic bile acid synthesis. In response to bile acid reabsorption in the ileum, enterocytes secrete fibroblast growth factor 19 (FGF19; FGF15 in mouse) into the enterohepatic circulation. Uptake of FGF15/19 by hepatocyte receptors causes repression of bile acid synthesis, thereby coordinating bile acid synthesis in liver with reabsorption in intestine to maintain homeostasis. Herein, we describe recent findings related to the enterohepatic axis for bile acid regulation, with a focus on studies of FGF15/19 function in normal and pathophysiology, and the identification of Diet1, an intestinal protein that influences FGF15/19 production by enterocytes.
A very short history of bile
Bile has a prominent place in the history of Western medicine. In the ancient world, bile accounted for two of the mystical “four humours”: blood, phlegm, choler (yellow bile), and melancholer (black bile) [1]. Hippocrates, the father of medicine, taught that the humours must be maintained in a delicate balance, and that imbalance resulted in disease. In the 1660’s, the physician Jan Baptista van Helmont hypothesized that the emptying of choler (bile) into the intestine was important for digestion. In the 1800’s, cholesterol and cholic acid were identified as components of bile [2], but it remained until the 1940’s, for Konrad Bloch and colleagues to trace the incorporation of labeled cholesterol into newly synthesized bile acids in dogs [3]. The importance of bile acids in digestion in humans was established in the 1960’s [4].
Bile acids and the enterohepatic circulation
Work throughout the last several decades established the itinerary of bile acids during digestion [5]. Bile acids are synthesized in the liver and conjugated with the amino acids glycine or taurine in order to pass from the hepatocyte into the bile pool. Upon feeding, conjugated bile acids are secreted into the bile ducts and enter into the gastrointestinal tract. In the proximal small intestine, bile acid-containing micelles allow solubilization and absorption of cholesterol and fat-soluble vitamins. When bile acids reach the ileum, > 95% of bile acids are reabsorbed and return to the liver through the enterohepatic circulation. The small proportion of intestinal bile acids that are not absorbed in the ileum are excreted in the feces.
Bile acids have several metabolic roles. Approximately half of the dietary cholesterol consumed daily is converted to bile acids, and loss of fecal bile acids serves as a route for the elimination of cholesterol from the body [5]. The maintenance of normal bile acid levels is important to prevent bacterial overgrowth and mucosal injury in the intestine [6] or cholestasis in the liver. Bile acids are ligands for the nuclear receptors farnesoid X receptor (FXR), pregnane X receptor (PXR), vitamin D receptor, and the G protein-coupled receptor, TGR5 (reviewed in [7]). Bile acids activate these receptors to regulate gene expression in several metabolic tissues (including intestine, liver, brown adipose tissue, macrophages, and brain). Aberrations in bile acid metabolism are associated with conditions ranging from gallstones and bile acid diarrhea to common metabolic diseases such as type 2 diabetes and atherosclerosis (Fig. 1) [8–12]. Here, we will focus on recent developments related to the enterohepatic regulation of bile acid levels.
Figure 1.

Human diseases associated with altered bile acid metabolism include gallstones, hepatic cholestasis, and primary bile acid diarrhea, as well as common metabolic diseases such as type 2 diabetes and atherosclerosis.
The enterohepatic regulation of bile acid homeostasis
Bile acid biosynthesis from cholesterol occurs primarily in hepatocytes via two pathways—the classic (or neutral) and alternate (or acidic) pathways [13]. The first and rate-limiting step in the classic pathway is the hydroxylation of cholesterol at the 7α-position, catalyzed by cholesterol 7α-hydroxylase (CYP7A1). Cyp7a1 gene expression is stimulated by dietary cholesterol, and is repressed by bile acids. The alternate pathway utilizing oxysterols contributes 10% (mice) to 25% (humans) of total bile acid synthesis.
In liver, bile acid synthesis is partly under negative regulation by the FXR-SHP axis. Activation of FXR by bile acids induces expression of Shp (encoding the short heterodimeric partner, SHP), which represses expression of Cyp8b1 (encoding the enzyme responsible for cholic acid synthesis) and to a lower extent, Cyp7a1 [14, 15]. A fascinating aspect of bile acid regulation is the coordination between bile acid uptake in intestine and the control of bile acid synthesis in the liver (Fig. 2) [16–18]. In response to bile acid uptake at the apical surface of the enterocyte, FXR is activated and induces transcription of the gene encoding human FGF19 or its mouse ortholog, FGF15. FGF15/19 is secreted at the basolateral surface of the enterocyte into the enterohepatic circulation. In liver, FGF15/19 binds to the FGFR4 receptor and, through actions of the JNK and ERK signaling pathways, represses Cyp7a1 transcription [15, 19]. In addition, FGF15/19 promotes gallbladder filling by acting on the relaxation of the gallbladder smooth muscle cells [6]. This ensures that hepatic bile acid synthetic levels and secretion to the gallbladder are coupled to bile acid reabsorption in the ileum. This prevents overproduction of bile acids, which is energetically undesirable and may result in deleterious conditions such as bile acid diarrhea.
Figure 2.

Role of Diet1 and FGF15 in enterohepatic bile acid homeostasis. Bile acids are synthesized in the liver from cholesterol and are secreted into the gallbladder by BSEP (bile salt export pump). In liver, bile acids bind and activate the FXR (farnesoid X receptor) nuclear receptor, which induces expression of SHP (short heterodimer partner). In turn, SHP represses the expression of bile acid synthesis enzymes (including CYP7A1) and bile acid transporters NTCP (sodium-taurocholate cotransporting polypeptide) and OATP (organic anion-transporting polypeptide). In the distal small intestine, bile acids are actively reabsorbed by ASBT (apical sodium-dependent bile acid transporter), and can activate FXR. Bile acid excretion from enterocytes occurs through the organic solute transporter (OSTα/OSTβ heterodimer) at the basolateral membrane. FXR activation can induce expression of FGF15 and OST, and inhibit expression of ASBT (via SHP). Diet1 promotes FGF15 secretion, which is thought to travel through the enterohepatic circulation and signals through the FGF receptor FGFR4 and β-Klotho to exert effects on hepatic gene expression, including Cyp7a1 repression. In Diet1-deficient mice, enterohepatic regulation is altered, as indicated by red and green arrows: despite high bile acid levels in the intestine of Diet1-deficient mice, FGF15 expression and secretion are reduced, and hepatic Cyp7a1 expression and bile acid pool is increased.
Metabolic effects of FGF19
Recent studies in humans and non-human primates demonstrate the importance of FGF19 in the regulation of bile acid levels, and reveal additional metabolic effects of this hormone. In the first studies to examine bile acid and FGF19 levels in more than 400 individuals the general population, Gälman et al. reported 7-fold variations in serum FGF19 levels in healthy individuals, and demonstrated that FGF19 levels are inversely related to bile acid levels [20]. Bile acid levels were also positively correlated with serum triglyceride levels. Interestingly, women had lower bile acid and triglyceride levels than men. It will be interesting to determine if sex differences in FGF19 levels and/or bile acid production relate to the reduced prevalence of hypertriglyceridemia, and increased prevalence of gallstones, observed in women.
The presence of increased colonic bile acids causes primary bile acid diarrhea, which may affect approximately 1% of the European population [21]. It was previously shown that individuals with primary bile acid diarrhea have lower median levels of FGF19 than the general population [10]. This finding was extended in a recent prospective study in individuals assessed for 7-day retention of a radiolabeled bile acid (selenium homocholic acid taurine, SeHCAT). FGF19 levels were significantly lower in individuals classified with primary bile acid diarrhea compared to normal controls and to individuals with secondary bile acid diarrhea [22]. In agreement with these observations in humans, the inhibition of FGF19 action in cynomolgus monkeys with specific antibodies led to increased CYP7A1 expression, enhanced bile acid synthesis and efflux, reduced bile acid uptake, and diarrhea [23]. Since the SeHCAT retention test is not available in the United States and many other countries, it is notable that serum FGF19 levels appear to be a viable marker for the diagnosis of bile acid diarrhea [24].
In recent studies, the FGF19 levels in serum have been associated with several metabolic conditions beyond bile acid diarrhea. For example, reduced FGF19 levels have been observed in children, adolescents, and adults with non-alcoholic fatty liver disease (NAFLD) [25–27]. In adults, the FGF19 levels did not independently predict liver histology findings [26]. However, in children with NAFLD, FGF19 levels were inversely correlated with the progression to steatohepatitis and fibrosis [25]. Additionally, in adolescents with NAFLD, those with insulin resistance had more pronounced decreases in FGF19 levels than the insulin-sensitive group [27]. No assessment of bile acid levels was performed in these studies, but may be valuable in the future to elucidate the mechanistic association between FGF19 and NAFLD.
A link between bile acid metabolism and type 2 diabetes mellitus has been noted by numerous investigators, although the results have not always been consistent [11]. Notably, in diabetic patients that do not respond to the typical arsenal of anti-diabetic therapeutics (including insulin), treatment with bile acid sequestrants tends to improve glucose levels and whole-body insulin sensitivity [28, 29]. Recently, FGF19 levels have also been implicated in diabetes remission after Roux-en-Y gastric bypass surgery to promote weight loss and glycemic control. Substantial evidence indicates that gastric bypass increases both bile acid and FGF19 levels [30–34], with the FGF19 increase likely occurring secondary to elevated bile acids and FXR activation [35, 36]. Among individuals undergoing gastric bypass surgery, there is inter-individual variation in the normalization of diabetic phenotypes despite similar weight loss, and the underlying mechanisms are mysterious. A recent study detected larger increases in FGF19 levels from pre-operative concentrations in patients that experienced diabetes remission for 12 months following surgery than those who did not [30].
A better understanding of the mechanisms involved in glycemic improvement in gastric bypass could suggest novel treatments for type 2 diabetes. One hypothesis is that changes in bile flow resulting from the altered anatomy following bypass could be involved. It is likely that additional factors also change as a result of bypass, such as intestinal microbiota, which play a role in bile acid modification and alter the bile acid pool composition. Effects of FGF15/19 on glucose metabolism could also occur by direct signaling in target tissues. For example, FGF19 administration to mice stimulates hepatic glycogen synthesis (through activation of mitogen-activated protein kinase signaling) [37], and blocks gluconeogenesis (through inactivation of the cAMP regulatory element-binding protein transcription factor) [38]. These effects are insulin-independent. Studies in rodents also suggest that the central nervous system is a target for FGF19 action. Intracerebroventricular administration of FGF19 to rats caused acute reductions in food intake and body weight, and improved glucose tolerance [39]. Additionally, studies in the mouse characterized an insulin-independent effect of FGF19 on glucose disposal mediated through activity in the brain [40].
FGF19 may also be important during liver regeneration. After liver resection, hepatic bile acid levels must be controlled to prevent cholestasis. Studies in mice indicate that FGF15 deficiency leads to greater risk for liver injury and mortality following partial hepatectomy, and this was associated with elevated intrahepatic bile acid levels [41]. Importantly, FGF15 administration to mice after liver resection improved survival in both FGF15-deficient mice with partial hepatectomy and in wild-type mice after extensive liver resection [41]. These effects may be related to direct mitogenic activity of FGF15 on hepatocytes and biliary epithelial cells. However, alongside the apparent beneficial effects of FGF19 in liver regeneration, FGF19 mitogenic activity is associated with hepatic carcinoma. In recent studies, FGF19 mRNA and protein levels were positively correlated with larger tumor size and early recurrence of hepatocellular carcinomas [42], and negatively correlated with disease-free survival [43]. Furthermore, reduction of FGF19 or FGFR4 expression levels using siRNA in human hepatocellular carcinoma cell lines inhibited proliferation and enhanced apoptosis [43]. These findings suggest that FGF19 signaling is a potential therapeutic target in hepatocellular carcinoma.
The identification of Diet1 as a component of the enterohepatic signaling axis
As evidence builds for critical roles of FGF19/15 and bile acids in metabolism, there is impetus to identify the factors that regulate FGF15/19 levels and activity. Although substantial information exists regarding FGF15/19 transcriptional regulation and mechanism of action in liver, the factors governing the intracellular trafficking and secretion of FGF15/19 in enterocytes have not been addressed. The recent identification of Diet1 may shed some light on this process.
Several years ago, it was noted that the C57BL/6ByJ inbred mouse strain exhibits aberrant bile acid homeostasis. This year, the underlying genetic mutation was identified in a novel gene, Diet1. The C57BL/6ByJ (B6By) strain originated from the C57BL/6J strain, and the two substrains remain nearly genetically identical. However, a dramatic difference in lipid metabolism exists between the two strains: C57BL/6J mice are highly susceptible to diet-induced hyperlipidemia and atherosclerosis, whereas B6By mice are nearly resistant [44]. This difference could not be attributed to differences in food intake, dietary cholesterol absorption, or endogenous cholesterol synthesis [44, 45]. A clue to the underlying mechanism for maintenance of low cholesterol levels in B6By mice was the detection of enhanced bile acid excretion in the feces and urine [45]. Serum bile acid levels were also elevated, likely due to spillover of bile acids from the portal vein. Microarray analysis revealed elevated expression of Cyp7a1 and numerous other genes involved in bile acid synthesis and transport in liver, suggesting that low cholesterol levels result from enhanced conversion to bile acids followed by excretion [45]. However, the genetic lesion in B6By mice could not be attributed to known genes. The bile acid phenotype was therefore mapped to high resolution in a cross of nearly a thousand mice, leading to the identification of an underlying null mutation in the novel Diet1 gene [46].
Diet1 encodes a 236 kD protein with a modular structure containing nine copies of the MAM (meprin-A5-tyrosine phosphatase μ) domain, which alternate with nine copies of the low density lipoprotein receptor (LDLR) class A domain (Fig. 3). At the C-terminus of Diet1 is a predicted transmembrane spanning sequence, raising the possibility that the protein is membrane associated. The Diet1 protein sequence is 70% identical between mouse and human, and is evolutionarily conserved in mammals, amphibians, and fish [46]. The only other protein with substantial similarity is endotubin (also known as apical early endosomal glycoprotein and MAM domain-containing 4), which is about half the size of Diet1 (Fig. 3). The physiological function of endotubin is unknown, but studies in cultured cells have implicated it in trafficking of tight junction proteins in polarized epithelial cells [47]. According to publicly available gene expression databases, endotubin is ubiquitously expressed, whereas Diet1 is expressed almost exclusively in the small intestine in both mice and humans, with lower levels in kidney cortex [46].
Figure 3.
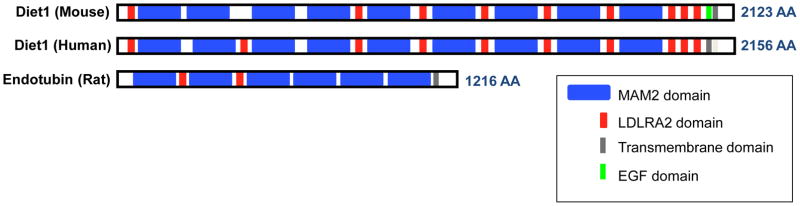
Predicted domain structure of the Diet1 protein. Diet1 is characterized by alternating MAM (meprin-A5-tyrosine phosphatase μ) and LDL receptor class A2 domains, with C-terminal EGF (epidermal growth factor) and transmembrane motifs. Endotubin is the most closely related protein structurally.
Diet1 has a role in modulating the levels of FGF15/19 (Fig. 2). In B6By mice, the loss of Diet1 activity leads to reduced FGF15 mRNA and protein levels in intestine, and (presumably compensatory) increases in liver genes involved in FGF15 and bile acid uptake, bile acid synthesis, and bile acid export [46]. In human enterocyte cell lines, Diet1 levels modulate the amount of FGF19 that accumulates in the culture medium: Diet1 expression promotes, and Diet1 knockdown reduces, secreted FGF19 protein levels [46]. Furthermore, Diet1 and FGF15 co-localize in vesicle-like structures in cultured enterocytes, suggesting that the two proteins have a functional interaction [46]. Together, these data suggest that Diet1 is a determinant of FGF15/19 production by enterocytes, with downstream effects on hepatic bile acid synthesis and cholesterol levels.
A comparison of Diet1–deficient mice with other models with perturbations in bile acid metabolism is consistent with a role for Diet1 in the FGF15/19–FGFR4–Cyp7a1 axis. Although the information available in the literature is incomplete, the comparisons in Table 1 demonstrate strong similarities between Diet1–deficient mice and genetically engineered models with aberrant bile acid synthesis or signaling from the small intestine to liver [14, 15, 48–58]. Diet1–deficient mice resemble all models listed in having enhanced Cyp7a1 mRNA expression, most of which also have increased bile acid pool size and enhanced fecal bile acid excretion. However, only a subset of models have reduced plasma cholesterol levels as observed in Diet1–deficient mice, raising the possibility that Diet1 has additional independent effects on cholesterol metabolism; it would be necessary to compare cholesterol levels across models under similar dietary conditions and genetic backgrounds to make firm conclusions.
Table 1.
Mouse models with genetic defects in enterohepatic bile acid homeostasis.
| Mouse model | BA pool | Fecal BA | Plasma BA | Plasma cholesterol | Ileal FGF15 | Ileal SHP | Liver Cyp7a1 | Liver SHP | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Diet1-deficient | ↑ | ↑ | ↑ | ↓ | ↓ | ↔ | ↑ | ↔ | [46] |
| FXRα KO, whole body | ↑ | ↔ | ↑ | ↑ | ↓ | NA | ↑ | ↓ | [14, 15, 52, 56] |
| FXRα Liver KO | ↑ | ↔ | ↔ | ↑ | ↔ or ↑ | ↔ | ↑ | ↔ | [14, 15, 53] |
| FXRα Intestine KO | ↑ | ↔ | ↔ | ↔ or ↓ | ↔ or ↓ | ↔ | ↔ or ↑ | ↔ | [14, 15, 53] |
| SHP KO | ↑ | ↑ | ↔ | ↔ | NA | Absent | ↑ | Absent | [15, 48] |
| Cyp8b1 KO | ↑ | ↑ | NA | NA | ↓ | ↓ | ↑ | ↓ or ↔ | [49, 54] |
| Cyp7a1 Tg | ↑ | ↑ | ↑ | ↓ | ↑ | ↑ | ↑ | ↑ | [53, 65] |
| βKlotho KO | ↑ | ↑ | NA | ↔ | ↑ | NA | ↑ | ↔ | [51, 57] |
| FGF15 KO | NA | ↑ | NA | NA | Absent | NA | ↑ | ↔ | [50, 55] |
| FGFR4 KO | ↑ | ↑ | NA | ↑ | ↔ | ↑ | ↓ or ↔ | [15, 50, 55, 58] |
Questions remain about the proportional contribution of the FGF15/19 signaling axis to determining bile acid levels [16], and multiple mechanisms may be at work in specific models. For example, it has been suggested that in CYP8B1–deficient mice, changes in bile acid composition (rather than simply bile acid levels) lead to diminished FXRα activation, and subsequent effects on bile acid homeostasis [49]. In addition, the role of FGF15/19 levels in portal blood in the regulation of hepatic Cyp7a1 expression remains unclear [16]. In a study performed in rats and rabbits, it was not possible to correlate FGF15/19 concentrations in the portal circulation with levels of ileal FGF15/19 mRNA or effects on hepatic gene expression [59]. With negative results, it is difficult to rule out that unknown parameters (such as the single time-point analyzed) did not allow optimal detection. Continued study is warranted, especially in light of strong evidence that the activation of FXRα–FGF15 in the intestine—but not liver—suppresses Cyp7a1 [14], and that signaling through FGFR4 is the predominant mechanism for this regulation [15].
Association of DIET1 locus with Alzheimer’s Disease
It remains to be determined whether genetic polymorphisms or rare mutations in human DIET1 influence bile acid levels and/or have a role in conditions such as bile acid diarrhea. Interestingly, a DNA polymorphism (D10S1423) that has been associated with late-onset Alzheimer’s Disease (AD) in three independent case-control samples is located within what is now known to be an intron of the DIET1 gene [60–62]. The association of D10S1423 with AD has further been confirmed in a prospective, longitudinal study performed over the course of 14 years [63]. These several studies indicate that a particular D10S1423 allele has a synergistic interaction with the well-characterized APOE E4 risk allele: carriers of the D10S1423 risk allele have an odds ratio for late-onset AD of 2.5; APOE E4 carriers have an odds ratio of 8.3; and carriers of both risk alleles have an odds ratio of 23.1 [64]. D10S1423 was originally identified in a predicted open reading frame, which is largely similar to DIET1, aside from errors in prediction of the intron/exon boundaries.
Future work is required to evaluate the potential role of DIET1 in AD. It has been reported that the predicted open-reading frame containing D10S1423 is expressed in human brain [64]. However, the mRNA species detected was only 1.2 kb in length and was not detected in intestine, whereas DIET1 mRNA is 7 kb in length and is prominently expressed in human small intestine, but not detectable in brain [46]. It is conceivable that a small, alternatively spliced form of DIET1 is expressed is brain, or perhaps an additional gene resides within the DIET1 locus and is expressed in brain. It is tempting to speculate that the role of Diet1 as a determinant of plasma cholesterol levels could be a link to the function of apolipoprotein E and lipid metabolism in the brain. Clearly, the elucidation of the relationship between DIET1, D10S1423, and late-onset AD merits further study.
Conclusion
Although bile acids have a long history in medicine and biomedical research, recent findings underscore new associations with prevalent diseases such as type 2 diabetes and non-alcoholic fatty liver disease. A critical area for understanding bile acid homeostasis is the further elucidation of the FGF15/19 enterohepatic signaling axis. Recent studies have shed light on the myriad roles of FGF15/19, and have revealed a novel potential regulator of FGF15/19 levels, Diet1. It should be noted that although evidence in human, primate, and mouse studies points to a key role for FGF15/19 in the maintenance of enterohepatic feedback regulation of bile acid synthesis, it remains possible that additional mechanisms contribute.
KEY POINTS.
Dysregulation of bile acid levels is associated with conditions ranging from gallstones and cholestasis to type 2 diabetes and metabolic syndrome.
The intestinally-derived hormone FGF15/19 signals to liver to regulate bile acid synthesis and maintain bile acid homeostasis.
FGF15/19 levels appear to influence non-alcoholic fatty liver disease, glycemic improvement following gastric bypass surgery, liver regeneration following partial hepatectomy, and liver carcinogenesis.
A novel intestinal protein, Diet1, has been identified as a regulator of FGF15/19 production by enterocytes, with effects on bile acid and cholesterol levels.
A genetic polymorphism in DIET1 is associated with late-onset Alzheimer’s disease, suggesting potential links between bile acid and cholesterol homeostasis and brain.
Acknowledgments
We gratefully acknowledge support from Public Health Service grants HL102661 and HL028481.
Footnotes
Conflicts of Interest
None.
References and Recommended Reading
- 1.Arikha N. Passions and Tempers: A History of the Humours. New York: Ecco; 2007. [Google Scholar]
- 2.Olson RE. Discovery of the lipoproteins, their role in fat transport, and their significance as risk factors. J Nutr. 1998;128:439S–443S. doi: 10.1093/jn/128.2.439S. [DOI] [PubMed] [Google Scholar]
- 3.Bloch K, Roittenberg D. The biological conversion of cholesterol to cholic acid. J Biol Chem. 1943;149:511–517. [Google Scholar]
- 4.Simmonds WJ, Hofman AF, Theodor E. Absorption of cholesterol from a micellar solution: intestinal perfusion studies in man. J Clin Invest. 1967;46:874–890. doi: 10.1172/JCI105587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russell DW. Fifty years of advances in bile acid synthesis and metabolism. J Lipid Res. 2009;50(Suppl):S120–5. doi: 10.1194/jlr.R800026-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi M, Moschetta A, Bookout AL, et al. Identification of a hormonal basis for gallbladder filling. Nat Med. 2006;12:1253–1255. doi: 10.1038/nm1501. [DOI] [PubMed] [Google Scholar]
- 7•.de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17:657–69. doi: 10.1016/j.cmet.2013.03.013. A comprehensive review of bile acids, their regulation, and roles in varied aspects of metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balistreri WF. Inherited disorders of bile Acid transport or synthesis. Gastroenterol Hepatol (N Y) 2007;3:343–5. [PMC free article] [PubMed] [Google Scholar]
- 9.Charach G, Grosskopf I, Rabinovich A, et al. The association of bile acid excretion and atherosclerotic coronary artery disease. Ther Adv Gastroenterol. 2011;4:95–101. doi: 10.1177/1756283X10388682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pattni S, Walters JR. Recent advances in the understanding of bile acid malabsorption. Br Med Bull. 2009;92:79–93. doi: 10.1093/bmb/ldp032. [DOI] [PubMed] [Google Scholar]
- 11.Prawitt J, Caron S, Staels B. Bile acid metabolism and the pathogenesis of type 2 diabetes. Curr Diab Rep. 2011;11:160–6. doi: 10.1007/s11892-011-0187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steiner C, Othman A, Saely CH, et al. Bile acid metabolites in serum: interindividual variation and associations with coronary heart disease, metabolic sydrome and diabetes mellitus. PLoS One. 2011;6:e25006. doi: 10.1371/journal.pone.0025006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russell DW, Setchell KD. Bile acid biosynthesis. Biochemistry. 1992;31:4737–49. doi: 10.1021/bi00135a001. [DOI] [PubMed] [Google Scholar]
- 14.Kim I, Ahn SH, Inagaki T, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48:2664–72. doi: 10.1194/jlr.M700330-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Kong B, Wang L, Chiang JY, et al. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 2012;56:1034–43. doi: 10.1002/hep.25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16•.Angelin B, Larsson TE, Rudling M. Circulating fibroblast growth factors as metabolic regulators--a critical appraisal. Cell Metab. 2012;16:693–705. doi: 10.1016/j.cmet.2012.11.001. Review of the family of circulating FGF hormones, with emphasis on unanswered questions and controversial findings. [DOI] [PubMed] [Google Scholar]
- 17.Jones SA. Physiology of FGF15/19. Adv Exp Med Biol. 2012;728:171–82. doi: 10.1007/978-1-4614-0887-1_11. [DOI] [PubMed] [Google Scholar]
- 18.Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 2012;26:312–24. doi: 10.1101/gad.184788.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song KH, Li T, Owlsey E, et al. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology. 2009;49:297–305. doi: 10.1002/hep.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galman C, Angelin B, Rudling M. Pronounced variation in bile acid synthesis in humans is related to gender, hypertriglyceridaemia and circulating levels of fibroblast growth factor 19. J Intern Med. 2011;270:580–8. doi: 10.1111/j.1365-2796.2011.02466.x. [DOI] [PubMed] [Google Scholar]
- 21.Wedlake L, A’Hern R, Russell D, et al. Systematic review: the prevalence of idiopathic bile acid malabsorption as diagnosed by SeHCAT scanning in patients with diarrhoea-predominant irritable bowel syndrome. Aliment Pharmacol Ther. 2009;30:707–17. doi: 10.1111/j.1365-2036.2009.04081.x. [DOI] [PubMed] [Google Scholar]
- 22••.Pattni SS, Brydon WG, Dew T, et al. Fibroblast growth factor 19 in patients with bile acid diarrhoea: a prospective comparison of FGF19 serum assay and SeHCAT retention. Aliment Pharmacol Ther. 2013;38:967–76. doi: 10.1111/apt.12466. Study demonstrating that serum FGF19 levels have good agreement with bile acid retention measurements to diagnose primary bile acid diarrhea, and to distinguish it from secondary bile acid diarrhea. [DOI] [PubMed] [Google Scholar]
- 23••.Pai R, French D, Ma N, et al. Antibody-mediated inhibition of fibroblast growth factor 19 results in increased bile acids synthesis and ileal malabsorption of bile acids in cynomolgus monkeys. Toxicol Sci. 2012;126:446–56. doi: 10.1093/toxsci/kfs011. Reduction in FGF19 using a specific antibody led to reduced FGF19 levels and increased CYP7A1 expression and bile acid excretion in a primate model. [DOI] [PubMed] [Google Scholar]
- 24.Pattni SS, Brydon WG, Dew T, Walters JR. Fibroblast Growth Factor 19 and 7alpha-Hydroxy-4-Cholesten-3-one in the Diagnosis of Patients With Possible Bile Acid Diarrhea. Clin Transl Gastroenterol. 2012;3:e18. doi: 10.1038/ctg.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Alisi A, Ceccarelli S, Panera N, et al. Association between Serum Atypical Fibroblast Growth Factors 21 and 19 and Pediatric Nonalcoholic Fatty Liver Disease. PLoS One. 2013;8:e67160. doi: 10.1371/journal.pone.0067160. FGF19 levels are inversely correlated with progression of non-alcoholic fatty liver disease to steatohepatitis and fibrosis in children. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eren F, Kurt R, Ermis F, et al. Preliminary evidence of a reduced serum level of fibroblast growth factor 19 in patients with biopsy-proven nonalcoholic fatty liver disease. Clin Biochem. 2012;45:655–8. doi: 10.1016/j.clinbiochem.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 27•.Wojcik M, Janus D, Dolezal-Oltarzewska K, et al. A decrease in fasting FGF19 levels is associated with the development of non-alcoholic fatty liver disease in obese adolescents. J Pediatr Endocrinol Metab. 2012;25:1089–93. doi: 10.1515/jpem-2012-0253. In adolescents with non-alcoholic fatty liver disease, decreases in FGF19 levels are associated with insulin resistance. [DOI] [PubMed] [Google Scholar]
- 28•.Beysen C, Murphy EJ, Deines K, et al. Effect of bile acid sequestrants on glucose metabolism, hepatic de novo lipogenesis, and cholesterol and bile acid kinetics in type 2 diabetes: a randomised controlled study. Diabetologia. 2012;55:432–42. doi: 10.1007/s00125-011-2382-3. Bile acid sequestrants improved tissue glucose metabolism and increased circulating incretins. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz SL, Lai YL, Xu J, et al. The effect of colesevelam hydrochloride on insulin sensitivity and secretion in patients with type 2 diabetes: a pilot study. Metab Syndr Relat Disord. 2010;8:179–88. doi: 10.1089/met.2009.0049. [DOI] [PubMed] [Google Scholar]
- 30•.Gerhard GS, Styer AM, Wood GC, et al. A role for fibroblast growth factor 19 and bile acids in diabetes remission after Roux-en-Y gastric bypass. Diabetes Care. 2013;36:1859–64. doi: 10.2337/dc12-2255. Pre- and post-operative data from subjects undergoing Roux-en-Y gastric bypass surgery implicate the FGF19-CYP7A1-bile acid pathway in remission of type 2 diabetes following surgery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jansen PL. Transient elevation of serum bile salts after partial hepatectomy is due to metabolic overload and not to cholestasis. J Hepatol. 2011;56:743–4. doi: 10.1016/j.jhep.2011.08.004. author reply 744–5. [DOI] [PubMed] [Google Scholar]
- 32.Kohli R, Kirby M, Setchell KD, et al. Intestinal adaptation after ileal interposition surgery increases bile acid recycling and protects against obesity-related comorbidities. Am J Physiol Gastrointest Liver Physiol. 2010;299:G652–60. doi: 10.1152/ajpgi.00221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patti ME, Houten SM, Bianco AC, et al. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity (Silver Spring) 2009;17:1671–7. doi: 10.1038/oby.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pournaras DJ, Glicksman C, Vincent RP, et al. The role of bile after Roux-en-Y gastric bypass in promoting weight loss and improving glycaemic control. Endocrinology. 2012;153:3613–9. doi: 10.1210/en.2011-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyata M, Hata T, Yamakawa H, et al. Involvement of multiple elements in FXR-mediated transcriptional activation of FGF19. J Steroid Biochem Mol Biol. 2012;132:41–7. doi: 10.1016/j.jsbmb.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 36•.Zhang JH, Nolan JD, Kennie SL, et al. Potent stimulation of fibroblast growth factor 19 expression in the human ileum by bile acids. Am J Physiol Gastrointest Liver Physiol. 2013;304:G940–8. doi: 10.1152/ajpgi.00398.2012. Studies using human ileal explants demonstrate the dramatic responsiveness ofFGF19 transcription to specific bile acid species. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kir S, Beddow SA, Samuel VT, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011;331:1621–1624. doi: 10.1126/science.1198363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Potthoff MJ, Boney-Montoya J, Choi M, et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway. Cell Metab. 2011;13:729–38. doi: 10.1016/j.cmet.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39••.Ryan KK, Kohli R, Gutierrez-Aguilar R, et al. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology. 2012;154:9–15. doi: 10.1210/en.2012-1891. Acute intracerebroventricular administration of FGF19 reduced 24-hour food intake and body weight in rats. The central nervous system may be a significant target of the beneficial effects of FGF19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40••.Morton GJ, Matsen ME, Bracy DP, et al. FGF19 action in the brain induces insulin-independent glucose lowering. J Clin Invest. 2013;123:4799–808. doi: 10.1172/JCI70710. FGF19 enhances insulin-independent glucose disposal in the brain of obese mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41•.Uriarte I, Fernandez-Barrena MG, Monte MJ, et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut. 2013;62:899–910. doi: 10.1136/gutjnl-2012-302945. Liver regeneration after partial hepatectomy in mice is enhanced by adenoviral expression of FGF15. [DOI] [PubMed] [Google Scholar]
- 42•.Hyeon J, Ahn S, Lee JJ, et al. Expression of fibroblast growth factor 19 is associated with recurrence and poor prognosis of hepatocellular carcinoma. Dig Dis Sci. 2013;58:1916–22. doi: 10.1007/s10620-013-2609-x. FGF19 protein levels in human hepatocellular carcinomas was a marker of larger tumor size and were correlated with early recurrence of tumors. [DOI] [PubMed] [Google Scholar]
- 43•.Miura S, Mitsuhashi N, Shimizu H, et al. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer. 2012;12:56. doi: 10.1186/1471-2407-12-56. FGF19 may contribute to human hepatocellular carcinoma progression through effects on cellular apoptosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mouzeyan A, Choi J, Allayee H, et al. A locus conferring resistance to diet-induced hypercholesterolemia and atherosclerosis on mouse chromosome 2. J Lipid Res. 2000;41:573–82. [PubMed] [Google Scholar]
- 45.Phan J, Pesaran T, Davis RC, Reue K. The Diet1 locus confers protection against hypercholesterolemia through enhanced bile acid metabolism. J Biol Chem. 2002;277:469–77. doi: 10.1074/jbc.M107107200. [DOI] [PubMed] [Google Scholar]
- 46••.Vergnes L, Lee JM, Chin RG, et al. Diet1 functions in the FGF15/19 enterohepatic signaling axis to modulate bile acid and lipid levels. Cell Metab. 2013;17:916–28. doi: 10.1016/j.cmet.2013.04.007. Identification of the Diet1 gene by positional cloning and characterization of its role in modulating FGF15/19 production in intestine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCarter SD, Johnson DL, Kitt KN, et al. Regulation of tight junction assembly and epithelial polarity by a resident protein of apical endosomes. Traffic. 2010;11:856–66. doi: 10.1111/j.1600-0854.2010.01052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anakk S, Watanabe M, Ochsner SA, et al. Combined deletion of Fxr and Shp in mice induces Cyp17a1 and results in juvenile onset cholestasis. J Clin Invest. 2011;121:86–95. doi: 10.1172/JCI42846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49•.Hu X, Bonde Y, Eggertsen G, Rudling M. Muricholic bile acids are potent regulators of bile acid synthesis via a positive feedback mechanism. J Intern Med. 2013 doi: 10.1111/joim.12140. Enhanced bile acid production in mice lacking CYP8B1 occurs through increased levels of bile acids with antagnoistic effects on FXRα. [DOI] [PubMed] [Google Scholar]
- 50.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–25. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 51.Ito S, Fujimori T, Furuya A, et al. Impaired negative feedback suppression of bile acid synthesis in mice lacking betaKlotho. J Clin Invest. 2005;115:2202–8. doi: 10.1172/JCI23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kok T, Hulzebos CV, Wolters H, et al. Enterohepatic circulation of bile salts in farnesoid X receptor-deficient mice: efficient intestinal bile salt absorption in the absence of ileal bile acid-binding protein. J Biol Chem. 2003;278:41930–7. doi: 10.1074/jbc.M306309200. [DOI] [PubMed] [Google Scholar]
- 53.Li T, Matozel M, Boehme S, et al. Overexpression of cholesterol 7alpha-hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology. 2011;53:996–1006. doi: 10.1002/hep.24107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li-Hawkins J, Gafvels M, Olin M, et al. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J Clin Invest. 2002;110:1191–200. doi: 10.1172/JCI16309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmidt DR, Holmstrom SR, Fon Tacer K, et al. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J Biol Chem. 2010;285:14486–94. doi: 10.1074/jbc.M110.116004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinal CJ, Tohkin M, Miyata M, et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–44. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 57.Tomiyama K, Maeda R, Urakawa I, et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc Natl Acad Sci U S A. 2010;107:1666–71. doi: 10.1073/pnas.0913986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu C, Wang F, Kan M, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275:15482–9. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 59.Miyake JH, Doung XD, Strauss W, et al. Increased production of apolipoprotein B-containing lipoproteins in the absence of hyperlipidemia in transgenic mice expressing cholesterol 7alpha-hydroxylase. J Biol Chem. 2001;276:23304–11. doi: 10.1074/jbc.M101853200. [DOI] [PubMed] [Google Scholar]
- 60•.Shang Q, Guo GL, Honda A, et al. FGF15/19 protein levels in the portal blood do not reflect changes in the ileal FGF15/19 or hepatic CYP7A1 mRNA levels. J Lipid Res. 2013;54:2606–14. doi: 10.1194/jlr.M034827. Study that seeks to clarify the role of FGF15/19 in portal blood in the regulation of hepatic CYP7A1 expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Majores M, Bagli M, Papassotiropoulos A, et al. Allelic association between the D10S1423 marker and Alzheimer’s disease in a German population. Neurosci Lett. 2000;289:224–6. doi: 10.1016/s0304-3940(00)01283-0. [DOI] [PubMed] [Google Scholar]
- 62.Zubenko GS, Hughes HB, 3rd, Stiffler JS. D10S1423 identifies a susceptibility locus for Alzheimer’s disease in a prospective, longitudinal, double-blind study of asymptomatic individuals. Mol Psychiatry. 2001;6:413–9. doi: 10.1038/sj.mp.4000900. [DOI] [PubMed] [Google Scholar]
- 63.Zubenko GS, Hughes HB, Stiffler JS, et al. A genome survey for novel Alzheimer disease risk loci: results at 10-cM resolution. Genomics. 1998;50:121–8. doi: 10.1006/geno.1998.5306. [DOI] [PubMed] [Google Scholar]
- 64.Zubenko GS, Hughes HB, 3rd, Zubenko WN. D10S1423 identifies a susceptibility locus for Alzheimer’s disease (AD7) in a prospective, longitudinal, double-blind study of asymptomatic individuals: results at 14 years. Am J Med Genet B Neuropsychiatr Genet. 2009;153B:359–64. doi: 10.1002/ajmg.b.31017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zubenko GS, Hughes HB., 3rd Predicted gene sequence C10orf112 is transcribed, exhibits tissue-specific expression, and may correspond to AD7. Genomics. 2009;93:376–82. doi: 10.1016/j.ygeno.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]