

Abstract
The positive transcription elongation factor b (P-TEFb) plays a pivotal role in productive elongation of nascent RNA molecules by RNA polymerase II. Core active P-TEFb is composed of CDK9 and cyclin T. In addition, mammalian cell extracts contain an inactive P-TEFb complex composed of four components, CDK9, cyclin T, the 7SK snRNA and the MAQ1/HEXIM1 protein. We now report an in vitro reconstitution of 7SK-dependent HEXIM1 association to purified P-TEFb and subsequent CDK9 inhibition. Yeast three-hybrid tests and gel-shift assays indicated that HEXIM1 binds 7SK snRNA directly and a 7SK snRNA-recognition motif was identified in the central part of HEXIM1 (amino acids (aa) 152–155). Data from yeast two-hybrid and pull-down assay on GST fusion proteins converge to a direct binding of P-TEFb to the HEXIM1 C-terminal domain (aa 181–359). Consistently, point mutations in an evolutionarily conserved motif (aa 202–205) were found to suppress P-TEFb binding and inhibition without affecting 7SK recognition. We propose that the RNA-binding domain of HEXIM1 mediates its association with 7SK and that P-TEFb then enters the complex through association with HEXIM1.
Keywords: HEXIM1, MAQ1, P-TEFb, RNA polymerase II, transcription
Introduction
The positive transcription elongation factor b (P-TEFb) plays a pivotal role in productive elongation of nascent RNA molecules by RNA polymerase II (RNAPII) (Price, 2000). Transcription of pre-mRNAs is blocked shortly after initiation by the DRB sensitivity-inducing factor (DSIF) assisted by the negative elongation factor (NELF) (Yamaguchi et al, 1999). Release from this block involves phosphorylation of the carboxyl-terminal domain (CTD) of RNAPII and phosphorylation of the Spt5 subunit of DSIF by P-TEFb (Wada et al, 1998; Ping and Rana, 2001; Shim et al, 2002).
Numerous cellular proteins have been reported to interact with P-TEFb but two P-TEFb complexes predominate in extracts from HeLa cells (Nguyen et al, 2001; Michels et al, 2003; Shore et al, 2003). The core active P-TEFb complex comprises two subunits, cyclin-dependent kinase-9 (CDK9) and cyclin T1, T2 or K. The ‘large' inactive P-TEFb complex comprises CDK9, cyclin T1 or T2, the 7SK small nuclear RNA (Nguyen et al, 2001; Yang et al, 2001) and the MAQ1/HEXIM1 protein (Michels et al, 2003; Yik et al, 2003; Chen et al, 2004). Human MAQ1 (‘ménage à quatre') (Michels et al, 2003) is identical to HEXIM1 (HMBA-inducible protein 1), which is induced in smooth muscle cells following exposure to hexamethylene bis acetamide (HMBA) (Ouchida et al, 2003). Furthermore, cardiac lineage protein-1 (Clp-1), the chicken and mouse ortholog of HEXIM1, accumulates in cardiac muscle cells during early development (Huang et al, 2002). HEXIM1 inhibits the expression of cotransfected genes (Ouchida et al, 2003; Yik et al, 2003).
Active and inactive P-TEFb complexes are in rapid equilibrium. Cardiac hypertrophy inducers have been reported as physiological triggers of P-TEFb/7SK dissociation in cardiomyocytes (Sano et al, 2002). A transcriptional arrest triggers dissociation of 7SK and HEXIM1 from CDK9 and cyclin T1 (or T2) and subsequent accumulation of kinase active P-TEFb (Nguyen et al, 2001; Yang et al, 2001; Michels et al, 2003). 7SK RNA and HEXIM1 thus form a kinase inhibitor that contributes to the regulation of gene transcription. HEXIM1 purified from HeLa cells was shown to inhibit P-TEFb in vitro (Yik et al, 2003). However, the individual contribution of HEXIM1 and 7SK RNA to the inhibition of P-TEFb kinase activity has remained unclear.
To unravel the mechanism of P-TEFb inhibition, we performed a yeast three-hybrid study as well as electrophoretic mobility shift assays (EMSAs), which indicated that HEXIM1 binds 7SK RNA. We next generated a series of point mutations in HEXIM1 that impaired P-TEFb or 7SK binding. The association of core P-TEFb with HEXIM1 and 7SK RNA and the repression of kinase activity were reconstituted in vitro in a defined system.
Results
HEXIM1/7SK interaction is observed in yeast three-hybrid systems
To investigate the structural determinants of 7SK binding, a yeast three-hybrid system was used (Zhang et al, 2000a). The LexA DNA-binding domain was fused to the MS2 coat protein. The RNA to be tested was fused to the 3′ end of MS2-binding RNA sequences. Constitutive binding of the LexA-MS2 hybrid protein to the MS2 RNA recruits the hybrid RNA upstream regulatory sequences of both HIS3 and LacZ integrated genes. The third hybrid consisted of a fusion of the Gal4 activation domain to the protein to be investigated. Interaction of the target hybrid RNA with the Gal4 protein fusion results in HIS3 and LacZ gene transcription.
As a positive control, yeast cells were transformed with an MS2-TAR RNA fusion and Tat fused to the Gal4 activation domain (Zhang et al, 2000a; Fraldi et al, 2001). A robust growth was observed on selective media lacking histidine (not shown) and a strong β-galactosidase activity was detected (Figure 1A). Next, the 7SK RNA was fused to the MS2-binding RNA sequences and the Gal4 activation domain was fused to the P-TEFb protein subunits (CDK9, cyclin T1 or HEXIM1). When cells were transformed with Gal4-HEXIM1 and the entire 7SK sequence fused to MS2, a small but reproducible increase in β-galactosidase activity was observed, suggesting a weak interaction between 7SK and HEXIM1 (Figure 1A). However, RNA–protein interactions are impaired when MS2 RNA is fused to RNA fragments longer than 150–200 nucleotides (Zhang et al, 2000a). Therefore, the 7SK was split into two fragments, 7SK(1–175) and 7SK(169–331). Transformation of yeast with the MS2-7SK(1–175) fusion and HEXIM1 resulted in a robust growth on media lacking histidine and led to a β-galactosidase activity as high as that observed with the TAR/Tat positive control. In contrast, neither the inverted sequence KS7(175–1), the 7SK(169–331) fragment nor TAR RNA resulted in a significant β-galactosidase activity. No significant β-galactosidase activity was detected when testing either the entire 7SK, 7SK(1–175) or 7SK(169–331) against CDK9 or cyclin T1. Notwithstanding the limitation of our three-hybrid protocol that did not allow the use of full-length 7SK RNA for efficient interactions, these results demonstrated a strong specific interaction of HEXIM1 with the first 175 nucleotides of 7SK RNA.
Figure 1.
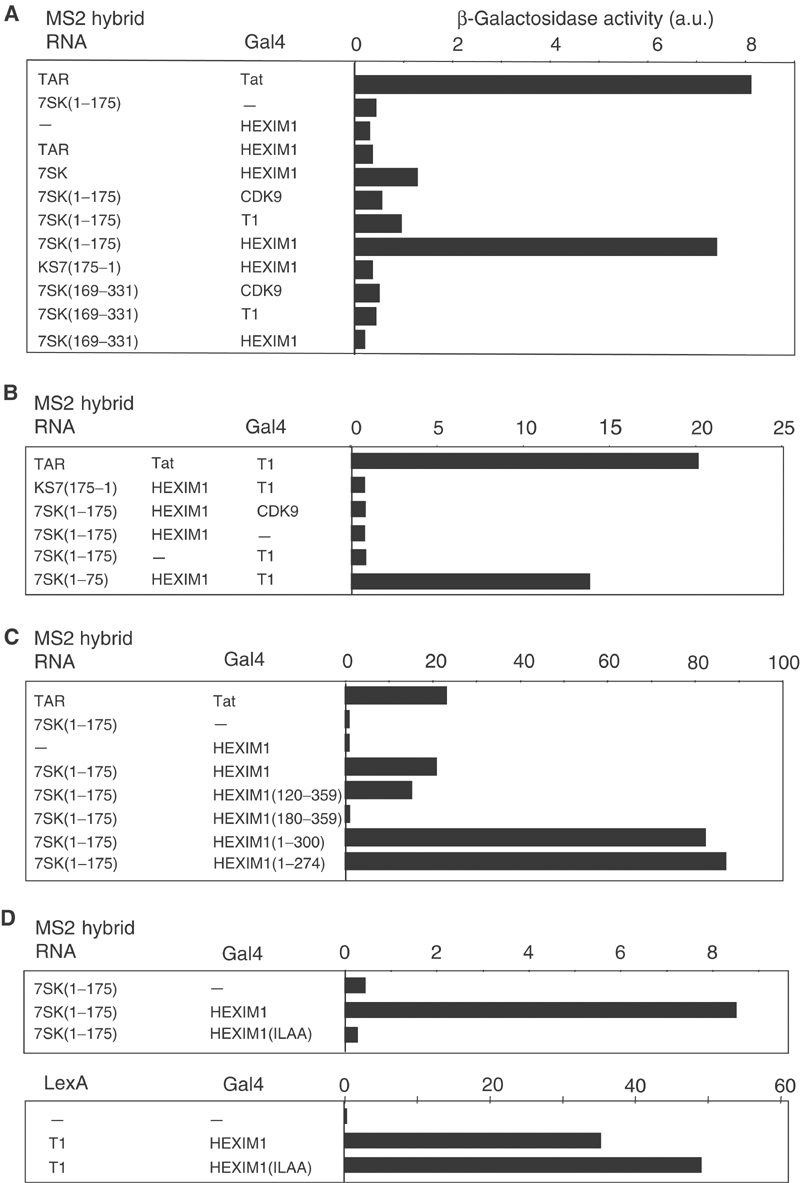
7SK snRNA interacts with HEXIM1 in yeast three-hybrid assays. β-Galactosidase activities in yeast three-hybrid (A, C, D), modified three-hybrid (B) and two-hybrid (D) assays. Proteins fused to the Gal4 activation or LexA DNA-binding domains and RNA fused to the MS2-binding sites are indicated in the left panels. Pools of more than 10 transformed yeast colonies grown on selective medium were assayed for β-galactosidase activities and quantified in arbitrary units (a.u.). Interaction between HIV TAR RNA and Tat was used as a positive control. (A) Three-hybrid assay testing truncated 7SK RNAs for interaction with full-length CDK9, cyclin T1 or HEXIM1. (B) Modified three-hybrid testing the capacity of HEXIM1 to link 7SK RNA to CDK9 or cyclin T1. The additional genuine protein is indicated in the central column. (C) Three-hybrid assay testing 7SK(1–175) for interaction with HEXIM1 deletion mutants. (D) Three-hybrid and two-hybrid assays testing 7SK(1–175) and cyclin T1 respectively for interaction with HEXIM1(ILAA).
Cyclin T1 had previously been shown to interact with the C-terminus of HEXIM1 (amino acids (aa) 181–359) (Michels et al, 2003). Hence, as both 7SK and cyclin T1 bind to HEXIM1, and since 7SK does not bind efficiently to cyclin T1, we speculated that HEXIM1 might link 7SK to cyclin T1 as the HIV Tat protein bridges the TAR RNA to cyclin T1 (Bieniasz et al, 1998; Fraldi et al, 2001). To test this hypothesis, a modified three-hybrid system was set up. As before, transformation with MS2-7SK(1–175) and Gal4-cyclin T1 hybrids did not result in significant β-galactosidase activity (Figure 1B). However, addition of a fourth plasmid driving the expression of the genuine HEXIM1 protein promoted a strong β-galactosidase activity. This interaction was specific as expression of HEXIM1 had no effect when transformed with 7SK and Gal4-CDK9, or with antisense MS2-KS7(175–1) and Gal4-cyclin T1. These findings indicated that a cyclin T1/HEXIM1/7SK complex could be reconstituted in a heterologous yeast system, and suggested that HEXIM1 might link 7SK to the cyclin T1 subunit of P-TEFb in vivo.
The KHRR motif in the basic domain of HEXIM1 is involved in 7SK binding
To map the HEXIM1 region required for interaction with 7SK, yeast cells were transformed with the MS2-7SK(1–175) hybrid and various Gal4-HEXIM1 deletion mutants. Both (1–274) and (1–300) C-terminal deletion mutants showed a stronger binding to 7SK compared to full-length HEXIM1, suggesting that HEXIM1 sequences C-terminal to aa 300 interfere with 7SK binding (Figure 1C). The (120–359) N-terminal deletion mutant interacted with 7SK like the entire HEXIM1(1–359). In contrast, the (181–359) mutant did not show significant β-galactosidase activity. Thus, sequences between aa 120 and 180 might be involved in HEXIM1/7SK interaction.
The HEXIM1 sequence between aa 120 and 180 contains a stretch of basic amino-acid residues (Michels et al, 2003). The KHRR amino-acid motif (aa 152–155) is the longest motif that has been conserved throughout evolution from flies to mammals in this region. When the KHRR sequence was replaced by ILAA, the mutant failed to interact with 7SK(1–175) in the three-hybrid assay (Figure 1D). Hence, the KHRR motif located in the basic domain of the central part of HEXIM1 is critical for the binding of 7SK to HEXIM1.
7SK RNA enhances the binding of P-TEFb to recombinant HEXIM1 in vitro
To investigate the association of HEXIM1 with P-TEFb in vitro, recombinant glutathione-S-adenosyl transferase (GST)-HEXIM1 fusion proteins were bound to glutathione beads and used to pull down P-TEFb from a HeLa cell extract. To eliminate endogenous P-TEFb/HEXIM1/7SK complexes in the extract, HeLa cells were treated with actinomycin D prior to lysis (Michels et al, 2003). When no RNA was added, GST-HEXIM1 beads bound a very low amount of cyclin T1 and CDK9 (Figure 2A, left, lanes 1 and 7). However, addition of T7-transcribed 7SK RNA markedly increased the binding of both CDK9 and cyclin T1 in a 7SK concentration-dependent manner (lanes 2–6). 7SK concentrations as low as 4 nM were efficient. In contrast, addition of an irrelevant RNA such as U2 snRNA at concentrations as high as 160 nM had no visible effect (Figure 2A, lanes 8–12). A subsequent RNase treatment completely released P-TEFb bound first to HEXIM1 in the presence of 7SK (Figure 2A, right). Thus, P-TEFb binding to HEXIM1 relies specifically on the presence of 7SK RNA.
Figure 2.
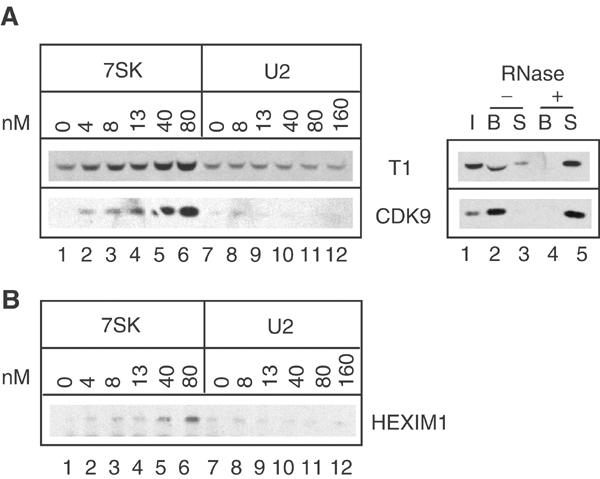
In vitro binding of HEXIM1 to P-TEFb requires 7SK snRNA. Proteins were detected by Western blot with anti-CDK9, anti-cyclin T1 or anti-HEXIM1 antibodies. (A) Left: Glutathione beads coated with GST-HEXIM1(1–359) were incubated with cell extracts in the presence of increasing concentrations of 7SK (0–80 nM) or U2 RNA (0–160 nM). Right: RNase A was added (+) or not (−) to HEXIM1/7SK/P-TEFb complexes preformed on the beads. Inputs (I), supernatants (S) and beads (B) were probed for cyclin T1 and CDK9. (B) An extract of actinomycin-treated HeLa cells was used to immunoprecipitate P-TEFb with anti-cyclin T1, which was incubated with purified His10-HEXIM1 and increasing concentrations of 7SK (nM) or U2 (nM) RNA.
To strengthen this observation, a reconstitution procedure involving the binding of purified recombinant histidine-tagged HEXIM1 protein to affinity-purified core P-TEFb was investigated. P-TEFb was immunoprecipitated from HeLa cell extracts with anti-cyclin T1. After thorough washes, the P-TEFb-coated beads were incubated with purified histidine-tagged HEXIM1. Binding of recombinant HEXIM1 to P-TEFb was hardly detectable (Figure 2B, lane 1). However, addition of T7-transcribed 7SK markedly increased the binding (lanes 2–6). In contrast, U2 snRNA had no effect (lanes 8–12). RNA concentrations used in this experiment were in the same range as in the previous GST pull-down assay.
Thus, binding of recombinant HEXIM1 to P-TEFb requires 7SK RNA and suggests that four components, namely CDK9, cyclin T, HEXIM1 and 7SK snRNA, are sufficient to form stable P-TEFb/HEXIM1/7SK complexes in vitro.
The KHRR motif is involved in assembling P-TEFb/HEXIM1/7SK complexes in vitro and in vivo
To test the importance of the KHRR motif in the P-TEFb/HEXIM1/7SK complex formation in vitro, the HEXIM1(ILAA) mutant was fused to GST and used in a pull-down assay with HeLa cell extracts. The cyclin T1/CDK9 binding was markedly reduced, and in contrast to wild-type (WT) HEXIM1 addition of 7SK did not stimulate cyclin T1/CDK9 binding to ILAA (Figure 3A). The ILAA mutation did not affect the cyclin T1/HEXIM1 interaction in a yeast two-hybrid assay (Figure 1D).
Figure 3.

The KHRR motif is involved in in vitro and in vivo formation of the P-TEFb/HEXIM1/7SK RNA complex. (A) Pull-down assay of GST, GST-HEXIM1 WT (WT) and GST-HEXIM1(ILAA) with (+) or without (−) addition of 7SK RNA (80 nM). GST (fusion) proteins bound to glutathione beads were probed with anti-GST. (B) HeLa cells transiently transfected with Flag-HEXIM1(WT), Flag-HEXIM1(ILAA), Flag-HEXIM1(150–359) or Flag-HEXIM1(156–359) were processed for immunofluorescence with anti-Flag antibodies. Nuclei were stained with DAPI. (C) HeLa cells transfected with Flag-HEXIM1(WT), Flag-HEXIM1(ILAA), Flag-HEXIM1(150–359) or Flag-HEXIM1(156–359) or an empty vector (control) were treated (+) or not (−) with actinomycin D (ActD), lysed and immunoprecipitated with anti-Flag antibodies. Proteins in the extracts (inputs) or immunoprecipitated (beads) were probed with anti-Flag, anti-cyclin T1 and anti-CDK9 antibodies. 7SK RNA was detected by Northern blot.
To substantiate the role of the KHRR sequence in P-TEFb/HEXIM1/7SK complex formation in vivo, a Flag-tagged ILAA and truncated protein were expressed in HeLa cells. Neither the ILAA mutation nor truncation of the 155 N-terminal amino acids interfered with nuclear localization of the transfected FlagHEXIM1 proteins (Figure 3B). Hence, the KHRR sequence is distinct from the nuclear localization signal (NLS) (aa 159–181) (Michels et al, 2003; Ouchida et al, 2003). CDK9, cyclin T1 and 7SK were immunoprecipitated from HeLa cells expressing the WT FlagHEXIM1 protein (Figure 3C, lane 3). This co-immunoprecipitation was suppressed by actinomycin D treatment prior to lysis (lane 4). Co-immunoprecipitation of 7SK RNA, cyclin T1 or CDK9 with the ILAA protein was markedly reduced compared to the WT protein, although Flag-tagged protein levels were similar (lanes 3–6). The remaining immunoprecipitated 7SK, cyclin T1 and CDK9 disappeared following actinomycin treatment (lane 6). Truncation of the 155 N-terminal amino acids removed the KHRR motif and likewise abolished HEXIM1 interaction with 7SK and P-TEFb in vivo (lanes 9 and 10). In contrast, a HEXIM1 protein with a truncation of the 149 N-terminal amino acids (still containing the KHRR motif) behaved like the WT protein (lanes 7 and 8). It is therefore concluded that the 149 N-terminal amino acids of HEXIM1, which diverge in non-mammalian vertebrate sequences (Michels et al, 2003), are not involved in P-TEFb and 7SK binding. These experiments indicate that the KHRR motif plays an important role in P-TEFb/HEXIM1/7SK complex formation in vitro and in vivo.
Binding of the HEXIM1 C-terminal domain to P-TEFb does not require 7SK RNA
A yeast two-hybrid assay suggested a direct interaction between the C-terminal domain of HEXIM1 and cyclin T1 (Michels et al, 2003). To extend this finding, we investigated HEXIM1 regions required to bind P-TEFb in the GST pull-down assay. The binding of P-TEFb was detected by Western blot with CDK9 antibodies. P-TEFb from a HeLa cell extract was not retained by the (1–274) and (1–300) C-terminal deletion mutants even after addition of 7SK (Figure 4A, lanes 9–12). In contrast, deletion of the N-terminal 120 aa had no effect. HEXIM1(120–359) bound P-TEFb but only after addition of 7SK (lanes 5 and 6) like HEXIM1(WT). A larger N-terminal deletion, up to 180 aa, exhibited constitutive binding of P-TEFb in the absence of 7SK (lane 7). Addition of 7SK to (181–359) did not further increase P-TEFb binding (lanes 7 and 8). Thus, as previously suggested by the two-hybrid assays, the C-terminal domain of HEXIM1 is involved in P-TEFb binding (Michels et al, 2003). The GST pull-down data also suggest that a region between aa 120 and 180 prevents this binding unless 7SK RNA is present.
Figure 4.
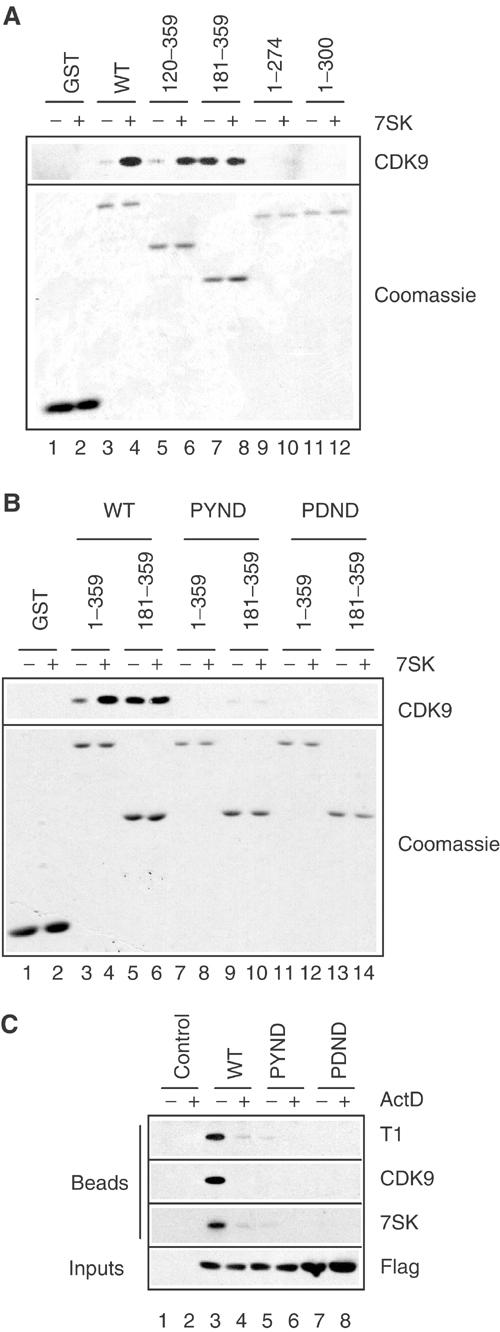
Involvement of the PYNT motif in HEXIM1 C-terminal domain in in vivo and in vitro binding to P-TEFb. (A) Glutathione beads coated with GST-HEXIM1 full-length (WT) or truncated proteins were incubated with cell extracts with (+) or without (−) 80 nM 7SK RNA. GST-HEXIM1 proteins bound to the beads were detected by Coomassie blue staining. (B) Full-length (1–359) or truncated (181–359) GST-HEXIM proteins with or without the PYND or PDND mutation were tested for binding to P-TEFb in the presence (+) and absence (−) of 80 nM 7SK. (C) Cells transfected with empty vector (control) or Flag-HEXIM1 with or without the PYND or PDND mutations were treated (+) or not (−) with actinomycin D (ActD), lysed and immunoprecipitated with anti-Flag. Beads were probed for cyclin T1, CDK9 and 7SK. Extracts were probed with anti-Flag (inputs).
The PYNT motif in the C-terminal domain of HEXIM1 is involved in P-TEFb binding
To gain further insight into this picture, we looked for point mutants in the HEXIM1 C-terminal domain that would be impaired for P-TEFb binding. The PYNT motif (aa 202–205) in the C-terminal domain was targeted because it has been conserved throughout evolution from insects to mammals (Michels et al, 2003). The mutant proteins were assayed in the GST pull-down assay. As seen in Figure 4A, P-TEFb binding to full-length (1–359)WT increased in the presence of 7SK (Figure 4B, lanes 3 and 4). When threonine 205 was replaced by aspartate (PYND) or after subsequent replacement of tyrosine 203 by aspartate in the double mutant (PDND), P-TEFb binding became undetectable (lanes 7, 8 and 11,12). As shown before, binding of P-TEFb to the truncated (181–359)WT was constitutive and was not affected by 7SK (lane 5 and 6). However, P-TEFb binding to the truncated PYND mutant (lanes 9 and 10) was very weak and below detection with the PDND double mutant (lanes 11 and 12). This finding underlines the specificity of P-TEFb binding to the truncated WT protein. These findings point to a possible contribution of tyrosine 203 and to the critical role of threonine 205 in P-TEFb binding, irrespective of 7SK function.
The formation of P-TEFb/HEXIM1/7SK RNA complex was assayed next in HeLa cells transiently transfected with FlagHEXIM1 proteins. All mutant proteins were nuclear and indistinguishable from the WT (not shown). CDK9, cyclin T1 and 7SK RNA co-immunoprecipitated with HEXIM1, but the association was suppressed by actinomycin D treatment prior to lysis (Figure 4C, lanes 3 and 4). The PYND point mutant was impaired in binding either P-TEFb or 7SK RNA in vivo (lanes 5 and 6). The PDND double mutant was completely deficient (lanes 7 and 8). As the 7SK-binding properties of these mutant proteins are similar to WT (see Electrophoretic mobility shift assay), these findings are consistent with a direct interaction between the HEXIM1 C-terminal domain and P-TEFb.
Interaction of HEXIM1 with P-TEFb represses CDK9 kinase activity
P-TEFb/HEXIM1/7SK complexes from cell extracts have a highly reduced kinase activity compared to core P-TEFb complexes (Nguyen et al, 2001; Yang et al, 2001). A kinase assay was performed following a GST-HEXIM1 pull-down of P-TEFb from HeLa cell extracts. The kinase activities used as substrate the CTD4 peptide consisting of four consensus repeats of the RNAP II CTD. HEXIM1(WT) bound little CDK9 protein in the absence of 7SK (Figure 5A, lanes 5–8) and a weak kinase activity was detected (Figure 5B and C, closed squares). However, although addition of 7SK strongly increased the amount of CDK9 protein retained on the beads (Figure 5A, lanes 9–12), the kinase activity increased marginally (Figure 5B and C, closed circles). Addition of RNase to the assay releases P-TEFb from GST-HEXIM1 beads (Figure 2A, right) and, indeed, a strong increase in kinase activity was observed (Figure 5C, open circles). Thus, although the amount of CDK9 molecules was the same, the kinase activity increased upon dissociation from HEXIM1/7SK. The activity level then matched that detected with equivalent amounts of P-TEFb immunoprecipitated (IP) from the same extracts using anti-cyclin T1 antibodies (Figure 5A, lanes 1–4, and Figure 5C, closed diamonds).
Figure 5.
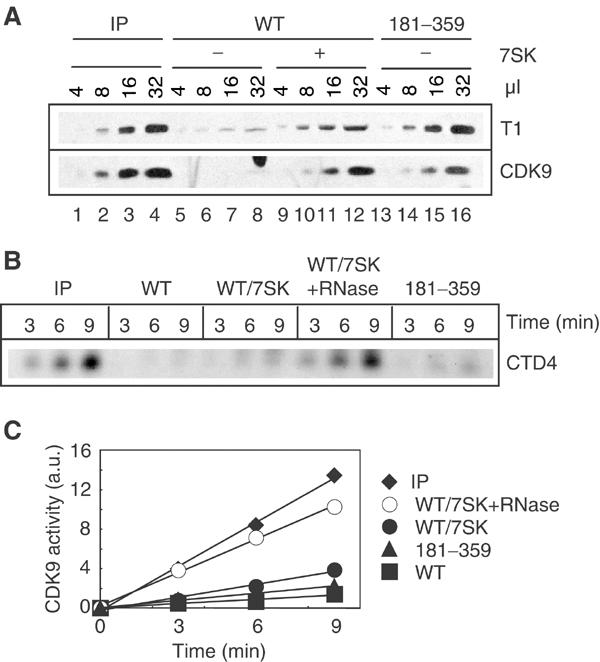
Binding of P-TEFb to GST-HEXIM1 represses its kinase activity. P-TEFb from a cell extract was retained on glutathione beads coated with WT (WT) or truncated (181–359) GST-HEXIM1 with (−) or without (+) 80 nM 7SK. As a control, P-TEFb was immunoprecipitated from a cell extract with anti-cyclin T1 (IP). (A) P-TEFb retained on beads was analyzed using anti-cyclin T1 and CDK9 antibodies. For an accurate comparison, increasing amounts of material (μl) were loaded on the gels. (B) The reactions were analyzed by SDS–PAGE using autoradiography. (C) 32P incorporation into the CTD4 peptide was quantified in arbitrary units (a.u.) and plotted versus time (min). This experiment was performed three times with similar results. A typical experiment is shown.
As the HEXIM1 C-terminal fragment (181–359) associates with the P-TEFb complex in the absence of 7SK, it was questioned whether this binding would be sufficient to inhibit CDK9. Levels of CDK9 retained by the (181–359) deletion mutant in the absence of RNA were equivalent to those retained by HEXIM1(WT) in the presence of 7SK (Figure 5A, compare lanes 13–16 to lanes 9–12). Again, only a weak kinase activity was detected (Figure 5C, closed triangles). These data suggest that binding of the HEXIM1 C-terminal domain to P-TEFb is sufficient to repress CDK9 kinase activity and that 7SK is not essential for this inhibition.
The kinase assays on GST pull-downs implied a comparison between kinase activity of P-TEFb bound to beads or released into the assay buffer. This experiment was therefore repeated following a different procedure. P-TEFb was immunoprecipitated first with anti-cyclin T1 antibodies. Then, synthetic 7SK and carboxyl-terminal His-tagged HEXIM1 were added to reconstitute a P-TEFb/HEXIM1/7SK complex on protein A beads. The kinase assay was performed on the washed beads (Figure 6). Addition of HEXIM1 or 7SK had no significant effect. In contrast, a much weaker kinase activity was observed when both HEXIM1 and 7SK were present in the reconstitution step. Subsequent addition of RNase to the assay restored the kinase activity. Thus, in vitro binding of P-TEFb to HEXIM1 fusion proteins in the presence of 7SK RNA represses CDK9 activity.
Figure 6.

Binding of HEXIM1 to P-TEFb represses its kinase activity. P-TEFb immunoprecipitated with anti-cyclin T1 was incubated with buffer (control), or recombinant histidine-tagged HEXIM1, or T7-transcribed 7SK RNA (80 nM), or a combination of both for 1 h at 21°C. Kinase activity was assayed on the beads as in Figure 5. When indicated, RNase A was added to the kinase assay. This experiment was performed twice with similar results. A typical experiment is shown.
Functional analysis using a defined reconstitution system
To extend results obtained using immunoprecipitation methods, we set up more defined kinase assays using P-TEFb purified from baculovirus-infected insect cells. Increasing amounts of histidine-tagged HEXIM1 and T7-transcribed 7SK were incubated with P-TEFb and DSIF as substrate. Incorporation of 32P into the large subunit of DSIF (Spt5) by P-TEFb containing either cyclin T1 or T2a was inhibited to between 10 and 30% (Figure 7A). Although there was some difference in the extent of inhibition, the results indicate that both forms of P-TEFb can be inhibited. To examine the effect of HEXIM1 and 7SK individually, similar reactions were carried out using P-TEFb containing cyclin T2a except that RNAPII was used as substrate. Increasing amounts of HEXIM1 up to 5 pmol per reaction had no significant effect on phosphorylation of the large subunit of RNAPII (Figure 7B, closed circles). 7SK inhibited P-TEFb slightly (open circles), but the level of inhibition varied during the four experimental trials carried out. In contrast, addition of an equal molar mixture of HEXIM1 and 7SK to P-TEFb had a very strong and highly reproducible inhibitory effect (closed diamonds). Significant inhibition occurred at 0.05 and 0.1 pmol of both HEXIM1 and 7SK, and was more than 50% complete at 0.5 pmol. Because the amount of P-TEFb in the reactions is around 0.2 pmol, it is possible that a 1:1:1 complex is forming.
Figure 7.

Analysis of P-TEFb inhibition by 7SK and HEXIM1 in a defined system. Kinase assays were performed with recombinant P-TEFb and HEXIM1 proteins, and T7-transcribed 7SK RNA. The reactions were analyzed by SDS–PAGE using autoradiography (inset) and quantified. (A) Using DSIF as substrate, the kinase reactions contained increasing amounts of a mixture of HEXIM1 and 7SK (8:1 molar ratio) and P-TEFb (containing cyclin T1 or T2a). (B) Using RNAPII as substrate, kinase assays were performed with cyclin T2a-containing P-TEFb and the indicated amounts of HEXIM1 and/or 7SK. When HEXIM1 and 7SK are both present, they are in an equal molar ratio with the indicated amounts of each individually. (C) Using RNAPII as substrate, kinase assays with cyclin T2a-containing P-TEFb and the indicated HEXIM1 proteins and/or 7SK were performed. HEXIM1 WT and mutant proteins (and 7SK) were present at either 5 or 2 pmol per reaction.
We next wanted to compare the ability of the HEXIM1 mutants to inhibit P-TEFb in the defined system. Reactions contained P-TEFb, HEXIM1 or HEXIM1 mutant proteins, and 7SK as indicated. In two otherwise identical experiments, HEXIM1 proteins and 7SK were held at either 5 or 2 pmol each (Figure 7C). None of the HEXIM1 proteins were able to inhibit P-TEFb in the absence of 7SK. Quantification of the 32P incorporation showed that WT HEXIM1 inhibited phosphorylation of RNAPII the most. Mutants that eliminated (181–359) or disrupted (ILAA) the RNA-binding domain of HEXIM1 had dramatically less inhibitory activity. The PYNT mutants had progressively less inhibitory activity, with PDNT showing the most, PYND less and PDND only slight inhibition. Overall, these functional assays with a defined reconstitution of proteins and RNA indicate that P-TEFb is inhibited by HEXIM1 in the presence of 7SK, and that both the RNA-binding region and the PYNT region of HEXIM1 are important for inhibitory function.
P-TEFb/HEXIM1/7SK complex formation assayed by electrophoretic mobility shift
To begin to examine the formation of the inhibitory complex containing P-TEFb, HEXIM1 and 7SK, we developed an EMSA using labelled, T7-transcribed 7SK RNA and recombinant HEXIM1 and P-TEFb. To block nonspecific interactions, especially those seen with purified IgGs, 1 μg of poly(I)·poly(C) was added to each reaction containing 0.5 ng of labelled 7SK RNA. When HEXIM1 was titrated into reactions, two complexes (1 and 2a) were formed (Figure 8A, lanes 2–6). P-TEFb was unable to bind to 7SK, but when HEXIM1 and P-TEFb were added together a new set of complexes (2b and 3) formed (lane 8). Inclusion of affinity-purified, anti-HEXIM1 antibody had no effect on 7SK RNA, but caused a further shift of complexes 1, 2a and 3 (lane 12). Affinity-purified, anti-CDK9 antibody gave a slight nonspecific complex between 1 and 2a with 7SK or 7SK and HEXIM1, but the 1 and 2a complexes were unaffected (lanes 13 and 14). When P-TEFb was included in the reactions, the nonspecific antibody shift was suppressed, presumably due to interaction between P-TEFb and the antibody, and complex 3 was supershifted (lane 16). These results demonstrate that complexes 1 and 2a contain only 7SK and HEXIM1, and that complex 3 contains both HEXIM1 and P-TEFb.
Figure 8.

EMSA analysis of HEXIM1, P-TEFb and 7SK interactions. HEXIM1 alone or when combined with P-TEFb shifts radioactive 7SK RNA. (A) Reactions contained indicated amounts of WT HEXIM1, about 1.5 pmol of P-TEFb, 150 ng of anti-CDK9 or affinity-purified anti-HEXIM1. (B) Indicated amounts of cold 7SK were added either during (D, lanes 2–7) or after (A, lanes 8–13) the formation of the complexes. Radioactive 7SK/HEXIM1 complexes were allowed to form first, and cold 7SK was added next with P-TEFb (asterisk, lanes 14–16). (C) Comparison between WT (WT) HEXIM1 or mutant proteins. Reactions contained 0.5 pmol of WT (WT) HEXIM1 or mutant proteins, about 1.5 pmol of P-TEFb. (D) DTT (5 mM) was added where indicated (lanes 13–17). Reactions contained 0.5 pmol of WT (WT) HEXIM1 or mutant proteins, about 1.5 pmol of P-TEFb, 150 ng of anti-CDK9 and 5 mM DTT as indicated (lanes 13–17).
To examine the specificity and stability of the interactions detected by the EMSA, competition experiments were designed. Increasing amounts of cold 7SK were mixed with the labelled 7SK and then incubated with HEXIM1 or HEXIM1 and P-TEFb. The HEXIM1 complexes (1 and 2a) and the HEXIM1/P-TEFb complex (3) were all competed in a dose-dependent manner (Figure 8B, lanes 2–7). Specificity of the interaction is demonstrated by the effective competition of complex formation by low amounts (50–250 ng) of 7SK in the presence of much higher amounts of poly(I)·poly(C) (1 μg). When the complexes were formed first and then incubated with cold 7SK, they remained intact, indicating that the interaction of HEXIM1 with the RNA is stable (lanes 8–13). Because, under the conditions used, HEXIM1 interacts directly with the RNA and P-TEFb does not, we examined the ability of P-TEFb to enter a preformed HEXIM1 complex. P-TEFb was able to enter the complex even in the presence of increasing amounts of 7SK (lanes 14–16). Overall, these results indicate that HEXIM1 interacts specifically with 7SK, and that P-TEFb then interacts primarily with HEXIM1 to enter into the complex.
We next examined the properties of the mutant proteins by EMSA. As expected, the 181–359 truncation mutant and the ILAA mutant demonstrated no affinity for 7SK (Figure 8C, lanes 3–6). Compared to WT, the PYNT mutants were able to bind 7SK equally well. However, the PYNT mutants displayed a graded effect on the formation of complex 3 with P-TEFb (lanes 7–12). The PYNT mutants were able to form complex 2b with about equal efficiency. However, formation of complex 3 was decreased (PDNT and PYND) or abrogated (PDND). An additional EMSA with a CDK9 antibody supershift verified that WT HEXIM1, but not the PDND mutant, allowed the inclusion of P-TEFb in complex 3 (Figure 8D, lanes 2–7). Our results suggested that in all EMSAs both 2a and 2b complexes contained only HEXIM1, and that the 2b complex only appeared upon addition of P-TEFb. Because the P-TEFb fraction contained dithiothreitol (DTT), we tested the effect of DTT directly. Compared to standard conditions, inclusion of 5 mM DTT favored the formation of complex 2b by HEXIM1 (WT or PDND) instead of complexes 1 and 2a (Figure 8D, lanes 9 and 14). Results from nondenaturing protein gels suggest that DTT causes a conformational change in HEXIM1 (data not shown), which might affect the nature of its association with 7SK. The presence of DTT did not alter the exclusion of P-TEFb from the PDND/7SK complex (lane 15). Overall, the EMSA data indicate that formation of a P-TEFb/HEXIM1/7SK complex determines kinase inhibition.
Discussion
Two major P-TEFb-containing complexes are found in human cells. One is active and restricted to CDK9 and a cyclin T, the other is inactive and contains HEXIM1 and 7SK RNA in addition to P-TEFb (Nguyen et al, 2001; Yang et al, 2001; Michels et al, 2003; Yik et al, 2003). HEXIM1 and 7SK RNA are the only components that have been identified so far. We now show that reconstitution of inactive P-TEFb associated with HEXIM1 and 7SK RNA can be recapitulated in vitro using purified recombinant components (P-TEFb (CDK9/cyclin T1 or T2), HEXIM1 and 7SK RNA). Three-hybrid tests and EMSA indicated that HEXIM1 binds specifically 7SK RNA. Activation of P-TEFb binding to HEXIM1 by 7SK RNA was observed by EMSA and pull-down by GST-HEXIM1 fusion proteins. P-TEFb kinase activity could be inhibited in vitro using a highly defined system using recombinant proteins in the presence of synthetic 7SK RNA.
Definition of P-TEFb-binding and regulatory domains in HEXIM1
Efficient pull-down of P-TEFb on GST-HEXIM1(181–359) did not require 7SK RNA. This observation was consistent with previous yeast two-hybrid data indicating that the cyclin T1 or T2 N-terminal ‘cyclin' domain directly interacts with a region located between aa 181 and 300 in the C-terminus of HEXIM1 (Michels et al, 2003). In additional support, point mutations in the PYNT motif (aa 202–205) had dramatic consequences on HEXIM1 binding to P-TEFb both in vitro and in vivo. The GST pull-down experiments also define a regulatory domain located between aa 120 and 181, which limits the interaction of HEXIM1 with P-TEFb in the absence of 7SK (Figure 9A). This domain contains a stretch of basic amino-acid residues (aa 150–163) overlapping the NLS (Michels et al, 2003; Ouchida et al, 2003). Truncation of the 150 N-terminal amino acids had no consequences for P-TEFb/HEXIM1/7SK formation in vivo and in the GST pull-down (not shown). Hence, we propose to narrow down the N-terminal boundary of the regulatory domain to 150 aa.
Figure 9.
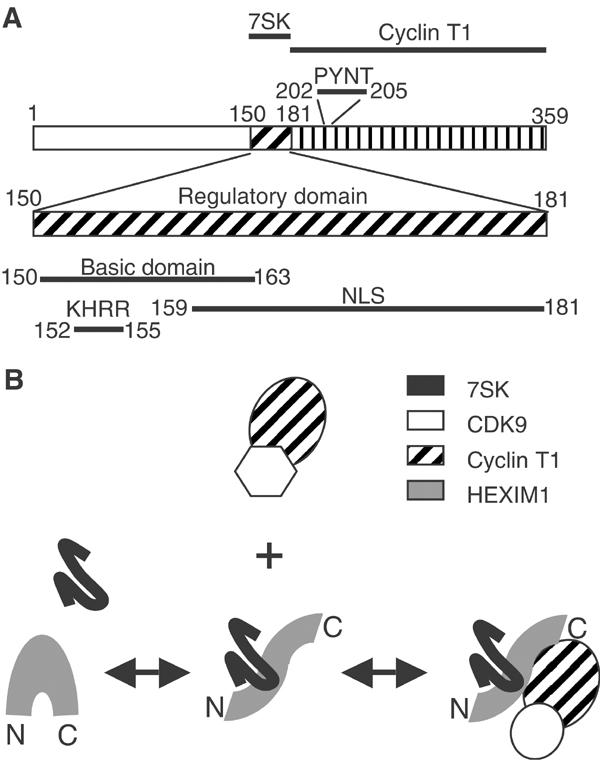
(A) HEXIM1 functional domains. Positions of amino acids are indicated. (B) Model: HEXIM1 binds 7SK RNA first and subsequently associates and inactivates P-TEFb. These processes are regulated in vivo.
7SK RNA binding to HEXIM1 precedes P-TEFb binding to HEXIM1
7SK RNA recognition is found to involve a stretch of four basic amino acids, KHRR (aa 152–155), in the regulatory domain of HEXIM1 and distinct from the NLS. Replacement of these residues by the ILAA sequence impaired the HEXIM1/7SK interaction in vivo and in vitro. Furthermore, truncation of the 156 N-terminal amino acids abolished the formation of P-TEFb/HEXIM1/7SK complexes in vivo, whereas truncation of the 150 N-terminal amino acids had no visible effects. It has been reported that purified 7SK RNA binds directly to immunoprecipitated P-TEFb (Yik et al, 2003; Chen et al, 2004). However, neither three-hybrid tests nor EMSA assays confirmed this possibility, and the modified three-hybrid assay suggests that HEXIM1 links 7SK RNA to the cyclin T1 subunit of P-TEFb.
The EMSA competition data indicate that 7SK association to HEXIM1 is very stable and precedes the formation of P-TEFb/HEXIM1/7SK complexes. We propose that the direct interaction between 7SK and HEXIM1 is a prerequisite for stable formation of the P-TEFb/HEXIM1/7SK complex (Figure 9B). 7SK RNA binds to HEXIM1 regulatory domain and promotes the binding of the C-terminal domain to cyclin T1 or T2. Binding of RNA molecules might influence protein folding as recently reported for the prion protein (Deleault et al, 2003). Further studies will be required to distinguish a remodelling of the HEXIM1 C-terminal domain tertiary structure from the removal of a steric hindrance masking the surfaces contacting cyclin T.
HEXIM1 binding to 7SK RNA in vitro does not require P-TEFb; it is spontaneous and does not require energy. In contrast, in vivo, the presence of the two partners within a cell is insufficient for their interaction. A significant proportion of cellular 7SK RNA is not bound to HEXIM1, although the latter is in large excess (Michels et al, 2003; Yik et al, 2003). Furthermore, an arrest in transcription results in almost complete HEXIM1/7SK complex disruption without changes in HEXIM1 or 7SK RNA levels (VT Nguyen, AA Michels and O Bensaude, unpublished). The HEXIM1/7SK interaction is a regulated process in vivo.
HEXIM1/7SK RNA, a new type of CDK inhibitor
The two-hybrid assays (Michels et al, 2003), the GST pull-down of P-TEFb with truncated HEXIM1 proteins in the absence of 7SK RNA and the deleterious effect of mutations in the PYNT motif converge to a critical direct interaction between the HEXIM1 C-terminal domain and the N-terminal domain of cyclin T1 or T2. The regulatory domain of HEXIM1 would prevent this association in the absence of 7SK RNA. A kinase assay following GST pull-down suggested that binding of the HEXIM1 C-terminal domain might be sufficient to inhibit P-TEFb kinase activity independently of 7SK RNA. It should be mentioned, however, that binding and inhibition of P-TEFb by a truncated His-tagged HEXIM1 could not be detected.
A variety of different mechanisms are known to regulate the activity of CDKs (Pavletich, 1999). Cyclin binding to the kinase subunit is a critical step that induces a conformational change in the catalytic site. Phosphorylation of CDKs by kinases such as the CDK-activating kinase (CAK) induces an additional conformational change. Indeed, phosphorylation of CDK9 is required for its activity and has been proposed to be required to assemble the P-TEFb/HEXIM1/7SK complex (Chen et al, 2004). Binding of a third protein partner to the CDK/cyclin pair adds a supplementary level of regulation. Proteins of the INK4 family prevent cyclin D binding to CDK4 or CDK6 and disrupt the catalytic pocket of Cdk6 by distorting the ATP-binding site (Jeffrey et al, 2000). Proteins of the Cip family bind both the kinase and the cyclin in Cdk2–cyclin A complexes and block the ATP-binding site inside the catalytic pocket (Pavletich, 1999). Unlike INK4 proteins, HEXIM1 binding would not prevent cyclin T binding to CDK9 but might affect the catalytic site. It is the first CDK inhibitor shown to be controlled by an RNA.
The HIV Tat/TAR ribonucleic complex as a viral antagonist of HEXIM1/7SK
Numerous proteins involved in transcription have been reported to associate with P-TEFb. Of particular interest, transcription driven by the long terminal repeat (LTR) promoter of the human immunodeficiency virus (HIV) involves the formation of a quaternary complex between P-TEFb, the viral transactivator protein Tat and the TAR element, an RNA loop at the 5′ end of the transcript (Garber et al, 1998; Peng et al, 1998). A highly specific interaction between Tat and human cyclin T1 is involved. Cyclin T1 binding to Tat enhances the affinity of TAR to Tat (Wei et al, 1998) and, conversely, TAR binding stabilizes the Tat/cyclin T1 interaction (Zhang et al, 2000b). TAR binding requires a stretch of basic amino acids, the arginine-rich motif (ARM) in Tat. Here we show that 7SK binding to P-TEFb in vivo involves a stretch of basic residues in a central evolutionarily conserved sequence of HEXIM1, which evokes the Tat ARM. TAR binding to P-TEFb in vitro also requires the Tat/TAR recognition motif (TRM) from aa 250 to 262 in human cyclin T1 (Garber et al, 1998). However, in HeLa cells, HEXIM1/7SK bound efficiently to an N-terminal-tagged cyclin T1(1–254) with a truncated TRM (Michels et al, 2003). The Tat/TAR RNA pair differs from the HEXIM1/7SK RNA pair in many aspects. Firstly, HEXIM1 interacts with cyclin T1 and T2 indistinctly, whereas Tat only binds to human cyclin T1. Secondly, Tat/TAR binding stimulates P-TEFb kinase activity (Zhou et al, 2000), whereas HEXIM1/7SK binding is inhibitory (Nguyen et al, 2001; Yang et al, 2001).
A limited number of RNAs have been reported to regulate the activity of proteins. Most of the known examples concern regulation of gene expression. For instance, the 6S RNA stabilizes the association of the core RNA polymerase with the sigma 70 subunit (Montzka Wassarman and Storz, 2000). U1 snRNA associates with and stimulates the activity of TFIIH transcriptional initiation factor (Kwek et al, 2002). Binding of double-stranded RNA to PKR activates its kinase activity and leads to repression of protein synthesis (Clemens, 1997). P-TEFb appears as a unique case of a protein kinase regulated by specific ribonucleic protein complexes.
Materials and methods
Yeast three-hybrid and modified three-hybrid assays
L40 coat yeast cells were cotransformed with plasmids derived from pIIIA/MS2-1 and plasmids derived from pACTII (Zhang et al, 2000a). pIIIA/MS2-derived plasmids carry the URA3 gene and pACTII-derived plasmids carry the LEU2 gene. Transformed cells were selected on media lacking uracil and leucine. For modified three-hybrid assays, L40 coat cells were transformed with plasmids derived from pIIIA/MS2-1, plasmids derived from pACTII and pRS313 derivatives driving or not the expression of genuine HEXIM1. Transformants were selected on media lacking uracil, histidine and leucine. Quantitative determination of β-galactosidase activity was performed as described previously (Fraldi et al, 2001).
Plasmids
GST was fused to the N-terminus of the various HEXIM1 mutant proteins in pGEX4T-2 (Pharmacia). N-terminal Flag-tagged HEXIM1 proteins were derived from pAdRSV-FlagMAQ1 (Michels et al, 2003). The WT plasmids were mutagenized by PCR to generate the ILAA, PDNT (Y203D), PYND (T205D) and PDND (Y203D, T205D) mutant proteins. C-terminal His6-tagged HEXIM1 was cloned in pET21a and pET21d (Novagen). The Gal4 activation domain was fused to HEXIM1 in pACTII (Clontech). The 7SK sequence was fused to the 3′ end of two MS2 RNA-binding sites after PCR amplification from pEMBL-7SK (generous gift of Shona Murphy) and cloned into the SmaI site of pIIIA/MS2-1 (Zhang et al, 2000a) to generate the pIIIA/MS2-7SK plasmid. To generate pIIIA/MS2-7SK(1–175), the pIIIA/MS2-7SK plasmid was digested by SphI/AgeI and subcloned into the SphI/XmaI sites of pIIIA/MS2-1. The 7SK(169–331) PCR fragment was cloned into the SmaI site of pIIIA/MS2-1 to generate pIIIA/MS2-7SK(169–331). HEXIM1 was inserted in pRS313 (Stone and Reed, 1990) to obtain pRS313-MAQ1. pACTII-CycT1, pACTII-Tat, pIIIA/TAR-MS2 and pRS313-Tat plasmids have been described previously (Fraldi et al, 2001). Synthetic 7SK and U2 RNAs were generated by T7 polymerase transcription (Promega) from plasmids pH7SK and pHU2 (generous gift of Tamàs Kiss) linearized by XbaI.
HeLa cell lysate preparation
HeLa cells (strain MRL2) were cultured, transfected and treated with actinomycin D as described before (Michels et al, 2003). Cells were lysed in ice-chilled buffer A (10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 200 mM NaCl, 0.2 mM EDTA), supplemented with 1 mM DTT, 40 U ml−1 of RNasin (Promega), protease inhibitor cocktail (P-8340; Sigma) and 0.5% Nonidet P-40. Lysates were vortexed and incubated for 20 min on ice and clarified by sequential centrifugations for 5 min each at 500 g and 9000 g at 4°C. The final supernatant was used in all experiments.
HEXIM1 protein purification
His-tagged HEXIM1 proteins used for kinase assays and EMSA with recombinant P-TEFb were expressed in Escherichia coli BL21 (DE3). After induction with IPTG and overnight growth at 18°C, cells were sonicated in PBS with 1% Triton X-100, 0.1% saturated solution of PMSF in EtOH, 5 mM imidazole and cleared by spinning for 55 min at 200 000 g. The lysate was brought to 750 mM NaCl and then added to 2 ml of Ni-NTA resin (Qiagen) that had been pre-equilibrated with 750 mM NaCl lysis buffer. After a 1 h incubation at 4°C, the flow through was collected, and the resin was washed with 10 volumes of 10 mM Tris (pH 7.8), 0.5 M NaCl, 1% Triton X-100, 0.1% PMSF, 10 mM imidazole and 10 volumes of wash buffer B (10 mM Tris (pH 7.8), 100 mM NaCl, 1% Triton X-100, 0.1% PMSF, 10 mM imidazole). The column was eluted with 5 ml of wash buffer B containing 250 mM imidazole. The protein was then loaded onto a 1 ml Mono Q column, which was eluted with a linear gradient from 100 to 600 mM HGKEDP (25 mM HEPES (pH 7.6), 15% glycerol, 100–600 mM KCl, 0.1 mM EDTA, 1 mM DTT, 1 mM PMSF). The fractions were analyzed by SDS–PAGE and fractions containing HEXIM1 (about 98% pure) were stored at −80°C.
Electrophoretic mobility shift assay
Reactions (12 μl final volume) were carried out in 25 mM HEPES (pH 7.6), 15% glycerol, 60 mM KCl, 0.1 mM EDTA, 0.01% NP-40 and 1 μg BSA, and contained 500 pg labelled 7SK RNA as well as 1 μg poly(I)·poly(C) (Amersham) as nonspecific competitor RNA. Recombinant P-TEFb comprised CDK9 and cyclin T2a. For the standard reaction, the 7SK (cold and/or hot) was added last, at t=0. Variations in the order of addition, where they occurred, are discussed in the text and figure legends. Reactions were incubated for 20 min at room temperature. For supershift lanes, 150 ng of anti-CDK9 IgG (sc-484, Santa Cruz) or affinity-purified anti-HEXIM1 IgG was included during the entire reaction. Complexes were separated on 4% Tris/glycine gel. Gel and buffer were precooled to 4°C. The gel was prerun for 10 min, and samples were loaded and run for 1.5 h at 6 W, after which the gel was dried and subjected to autoradiography.
Antibodies
Antibodies used were rabbit anti-cyclin T1 (H-245, Santa Cruz), goat anti-cyclin T1 (T-18, Santa Cruz), anti-CDK9 mouse monoclonal (D-7, Santa Cruz), rabbit anti-CDK9 (C-20, Santa Cruz), rabbit anti-HEXIM1 (C4) (Michels et al, 2003), anti-Flag mouse monoclonal (M2, Sigma) and anti-GST HRP-conjugated protein (Sigma). Primary antibodies were detected with HRP-labelled (Promega) or Cy3-conjugated (Amersham) antibodies. Anti-Flag M2 affinity gel (Sigma) was used for immunoprecipitations. Immunofluorescence was performed as described previously (Michels et al, 2003).
Kinase assays on immunoprecipitates and GST pull-downs
Reconstituted P-TEFb/HEXIM1 complexes on 20 μl of glutathione beads or protein A sepharose beads were suspended and stirred at 21°C in 80 μl of [γ-32P]ATP (0.1 μCi μl−1, 100 μM), (YSPTSPS)4 peptide (0.1 μg ml−1) (CTD4) in buffer A supplemented with DTT (1 mM) and RNasin (40 U ml−1). Aliquots (20 μl) of the slurry were pipetted at 3 min intervals and mixed with SDS–PAGE loading buffer to stop the reaction. The phosphorylated peptide migrated as a 19 kDa protein on a 15% SDS–polyacrylamide gel. Incorporation of [32P] into CTD4 was quantified on a FujiBAS 3000 Imager analysis.
Kinase assays using recombinant P-TEFb
Kinase reactions (20 μl) were carried out at 34 mM KCl, 20 mM HEPES (pH 7.6), 7 mM MgCl2 and 1 μg BSA per reaction. Cold ATP was at 30 μM, and each reaction had 1.3 μCi of [γ-32P]ATP (Amersham). P-TEFb (CDK9/cyclin T1 or T2a) was expressed in baculovirus (Peng et al, 1998). The substrate was either Drosophila RNAPII (Marshall et al, 1996) or DSIF (Renner et al, 2001). All reaction components excluding the ATP were preincubated together for 10 min at 23°C. 7SK cDNA was amplified by RT–PCR and cloned in pUC19 such that transcription by T7 polymerase resulted in 7SK RNA containing one extra G at the 5′ end. 7SK RNA was added to reactions last, after heating it to 75°C for 5 min and cooling on ice. After preincubation, ATP was added and reactions were allowed to proceed for 20 min at 30°C, then terminated by addition of one-fourth volume of SDS–PAGE loading buffer. Gels were dried, subjected to autoradiography and quantified on a Packard Instant Imager.
Note added in proof
Consistent with our findings, JH Yik and co-workers report that the basic domain of HEXIM1 is required for 7SK snRNA-mediated inactivation of P-TEFb (Molecular and Cellular Biology, in press).
Acknowledgments
This work was supported by grants from Ligue Nationale Contre le Cancer, ANRS, ARC, ACI Biologie Moléculaire et Structurale (OB), Sidaction (AAM), EMBO short-term fellowship (AF), National Research Program on AIDS, AIRC (LL) and NIH GM35500 (DHP). We thank Jeff Cooper and Daisuke Mayuzumi for purification of His-tagged proteins, Sarah Byers for affinity purification of HEXIM1 antibody and Damon Shutt for a 7SK construct. We thank Dr Qiang Zhou for communication of results prior to publication.
References
- Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR (1998) Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J 17: 7056–7065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Yang Z, Zhou Q (2004) Phosphorylated P-TEFb is tagged for inhibition through association with 7SK snRNA. J Biol Chem 279: 4153–4160 [DOI] [PubMed] [Google Scholar]
- Clemens MJ (1997) PKR—a protein kinase regulated by double-stranded RNA. Int J Biochem Cell Biol 29: 945–949 [DOI] [PubMed] [Google Scholar]
- Deleault NR, Lucassen RW, Supattapone S (2003) RNA molecules stimulate prion protein conversion. Nature 425: 717–720 [DOI] [PubMed] [Google Scholar]
- Fraldi A, Licciardo P, Majello B, Giordano A, Lania L (2001) Distinct regions of cyclinT1 are required for binding to CDK9 and for recruitment to the HIV-1 Tat/TAR complex. J Cell Biochem 36: 247–253 [DOI] [PubMed] [Google Scholar]
- Garber ME, Wei P, KewalRamani VN, Mayall TP, Herrmann CH, Rice AP, Littman DR, Jones KA (1998) The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev 12: 3512–3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Wagner M, Siddiqui MA (2002) Structure, expression, and functional characterization of the mouse CLP-1 gene. Gene 292: 245–259 [DOI] [PubMed] [Google Scholar]
- Jeffrey PD, Tong L, Pavletich NP (2000) Structural basis of inhibition of CDK–cyclin complexes by INK4 inhibitors. Genes Dev 14: 3115–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwek KY, Murphy S, Furger A, Thomas B, O'Gorman W, Kimura H, Proudfoot NJ, Akoulitchev A (2002) U1 snRNA associates with TFIIH and regulates transcriptional initiation. Nat Struct Biol 9: 800–805 [DOI] [PubMed] [Google Scholar]
- Marshall NF, Peng J, Xie Z, Price DH (1996) Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem 271: 27176–27183 [DOI] [PubMed] [Google Scholar]
- Michels AA, Nguyen VT, Fraldi A, Labas V, Edwards M, Bonnet F, Lania L, Bensaude O (2003) MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol Cell Biol 23: 4859–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montzka Wassarman K, Storz G (2000) 6S RNA regulates E. coli RNA polymerase activity. Cell 101: 613–623 [DOI] [PubMed] [Google Scholar]
- Nguyen VT, Kiss T, Michels AA, Bensaude O (2001) 7SK snRNA binds to and inhibits the activity of Cdk9/cyclin T complexes. Nature 414: 322–325 [DOI] [PubMed] [Google Scholar]
- Ouchida R, Kusuhara M, Shimizu N, Hisada T, Makino Y, Morimoto C, Handa H, Ohsuzu F, Tanaka H (2003) Suppression of NF-kappaB-dependent gene expression by a hexamethylene bisacetamide-inducible protein HEXIM1 in human vascular smooth muscle cells. Genes Cells 8: 95–107 [DOI] [PubMed] [Google Scholar]
- Pavletich NP (1999) Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol 287: 821–828 [DOI] [PubMed] [Google Scholar]
- Peng J, Zhu Y, Milton JT, Price DH (1998) Identification of multiple cyclin subunits of human P-TEFb. Genes Dev 12: 755–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping YH, Rana TM (2001) DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 Tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J Biol Chem 276: 12951–12958 [DOI] [PubMed] [Google Scholar]
- Price DH (2000) P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol 20: 2629–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner DB, Yamaguchi Y, Wada T, Handa H, Price DH (2001) A highly purified RNA polymerase II elongation control system. J Biol Chem 276: 42601–42609 [DOI] [PubMed] [Google Scholar]
- Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, Michael LH, DeMayo FJ, Schneider MD (2002) Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med 8: 1310–1317 [DOI] [PubMed] [Google Scholar]
- Shim EY, Walker AK, Shi Y, Blackwell TK (2002) CDK-9/cyclin T (P-TEFb) is required in two postinitiation pathways for transcription in the C. elegans embryo. Genes Dev 16: 2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore SM, Byers SA, Maury W, Price DH (2003) Identification of a novel isoform of Cdk9. Gene 307: 175–182 [DOI] [PubMed] [Google Scholar]
- Stone DE, Reed SI (1990) G protein mutations that alter the pheromone response in Saccharomyces cerevisiae. Mol Cell Biol 10: 4439–4446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Takagi T, Yamaguchi Y, Watanabe D, Handa H (1998) Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J 17: 7395–7403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang S-M, Fischer WH, Jones KA (1998) A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92: 451–462 [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, Hasegawa J, Handa H (1999) NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 97: 41–51 [DOI] [PubMed] [Google Scholar]
- Yang Z, Zhu Q, Luo K, Zhou Q (2001) The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414: 317–322 [DOI] [PubMed] [Google Scholar]
- Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q (2003) Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell 12: 971–982 [DOI] [PubMed] [Google Scholar]
- Zhang B, Kraemer B, SenGupta D, Fields S, Wickens M (2000a) Yeast three-hybrid system to detect and analyze RNA–protein interactions. Methods Enzymol 318: 399–419 [DOI] [PubMed] [Google Scholar]
- Zhang J, Tamilarasu N, Hwang S, Garber ME, Huq I, Jones KA, Rana TM (2000b) HIV-1 TAR RNA enhances the interaction between Tat and cyclin T1. J Biol Chem 275: 34314–34319 [DOI] [PubMed] [Google Scholar]
- Zhou M, Halanski MA, Radonovich MF, Kashanchi F, Peng J, Price DH, Brady JN (2000) Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Mol Cell Biol 20: 5077–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]