

Abstract
Protein Phosphatase 2A (PP2A) is a tumor suppressor whose function is lost in many cancers. An emerging, though counterintuitive, therapeutic approach is inhibition of PP2A to drive damaged cells through the cell cycle, sensitizing them to radiation therapy. We investigated the effects of PP2A inhibition on U251 glioblastoma cells following radiation treatment in vitro and in a xenograft mouse model in vivo. Radiation therapy alone augmented PP2A activity, though this was significantly attenuated with combination LB100 treatment. LB100 treatment yielded a radiation dose enhancement factor of 1.45 and increased the rate of post-radiation mitotic catastrophe at 72 and 96 hours. Glioblastoma cells treated with combination LB100 and radiation therapy maintained increased γ-H2AX expression at 24 hours, diminishing cellular repair of radiation-induced DNA double-strand breaks. Combination therapy significantly enhanced tumor growth delay and mouse survival and decreased p53 expression 3.68-fold, compared to radiation therapy alone. LB100 treatment effectively inhibited PP2A activity and enhanced U251 glioblastoma radiosensitivity in vitro and in vivo. Combination treatment with LB100 and radiation significantly delayed tumor growth, prolonging survival. The mechanism of radiosensitization appears to be related to increased mitotic catastrophe, decreased capacity for repair of DNA double-strand breaks, and diminished p53 DNA damage response pathway activity.
Keywords: protein phosphatase 2, glioblastoma, drug evaluation, preclinical, protein phosphatase 2A, radiosensitization
1. Introduction
Glioblastoma multiforme is the most common malignant primary brain tumor in adults and has limited treatment options. Despite modest improvements in the multimodality therapy of malignant gliomas, the overall prognosis remains poor with median survival rates of approximately 14 months and few long-term survivors (1-3). Radiation therapy has been the mainstay of treatment along with systemic chemotherapy (2). Further advances are needed to combat the evasion and highly resistant nature of glioblastoma to radiation and chemotherapy.
The protein phosphatase 2A (PP2A) family of serine-threonine phosphatases has been well described to play a role in many human cancers. PP2A is a tumor suppressor, and its function can be lost by inactivating mutations of structural subunits or up-regulation of cellular PP2A inhibitors (4-7). PP2A is a negative regulator of the cell cycle and inhibition of PP2A leads to abrogation of cell cycle checkpoints and acceleration of the cell cycle (8-10). The majority of conventional oncologic therapies are cytotoxic or cytostatic and target rapidly proliferating cells. A potential consequence of this approach however is that quiescent tumor cell populations remain relatively resistant to these therapies. Quiescent tumor cells are reported to represent a treatment resistant subpopulation that is frequently responsible for therapeutic failure and tumor recurrence (11-13). A therapy which preferentially targets these subpopulations by accelerating the cell cycle affords a novel strategy for sensitization to ionizing radiation. Combining a DNA damaging treatment such as radiation with PP2A inhibition could enhance the sensitivity of cancer cells to these treatments and could convert quiescent and resistant tumor cells to a more sensitive phenotype. Inhibition of PP2A represents a counterintuitive but emerging paradigm of driving damaged cells through the cell cycle, inducing cell death. Quiescence in tumor cells is regulated via the nuclear co-repressor (NCOR) amongst other signaling molecules and transcription factors that are targets of PP2A (14).
LB100 is a hydrophilic small molecule inhibitor of PP2A. Previous studies have shown that administration of LB100 to tumor cells results in phosphorylation of Akt-1 and Plk-1, bypassing the G2/M checkpoint and inappropriately accelerating cells to enter mitosis, leading to mitotic cell death (15). The aim of this current study was to evaluate the effects of radiation therapy on PP2A activity and the radiation modifying effects of PP2A inhibition with LB100 on glioblastoma cells in vivo and in vitro.
2. Materials and Methods
2.1 LB100
The drug, LB100, was designed and provided by Lixte Biotechnology Holdings, Inc. (East Setauket, NY) (15). The structure of LB100 is shown the supplementary material (Supplementary Fig. S1). LB100 was reconstituted in saline (1 μM), and was stored at -80°C.
2.2 Cell lines and radiation treatment
U251 and U87 glioblastoma cells and H1915 non-small cell lung cancer cells were obtained from the National Cancer Institute (Frederick, MD). All three cell lines were validated on July 19, 2010 by Research Animal Diagnostic Laboratory (Columbia, MO) through analysis of 9 different short tandem repeat markers (amelogenin, CSF1PO, D13S317, D16S539, D5S818, D7S820, TH01, TPOX, and vWA). Cells were grown in DMEM (Invitrogen, Grand Island, NY) with 10% fetal bovine serum, maintained at 37°C in 5% CO2. Cultures were irradiated at a dose rate of 2.28 Gy/min by a Pantak X-ray source.
2.3 In Vitro PP2A activity assay
Cultured tumor cells were plated in 175-cm3 flasks. When the cells were 80% confluent, the media was replaced with media containing different concentrations of LB100 (2.5 μM) or an equivalent volume of vehicle 3 hours prior to 5 Gy or sham radiation. After 1 hour, the cells were washed 3 times in a 0.9% normal saline solution. Tissue protein extraction reagent (T-PER) (Pierce Biotechnology, Rockford, IL) solution was added to the cells, and cells were prepared for protein extraction. Lysates from each treatment group containing 300-μg protein were assayed by using a Malachite Green Phosphatase assay specific for serine/threonine phosphatase activity (Ser/Thr phosphatase assay kit 1; Millipore, Billerica, MA).
2.4 In Vivo PP2A activity assay
Nude mice bearing U251 subcutaneous xenografts (methods described below) were treated with LB100 (1.5 mg/kg), radiation (4 Gy), or combination of LB100 and radiation. Mice were treated with LB100 or vehicle control 3 hours before radiation. Animals were sacrificed 3 hours following treatment and tumors were excised for measurement of PP2A activity, assayed in the same conditions as above.
2.5 γ-H2AX ELISA
Cells were seeded in a 96-well plate for 6 hours followed by drug treatment (2 and 5 μM LB100) and irradiated 4 hours later (5 Gy) and assayed after 24 hours. A commercially available cellular histone-H2AX phosphorylation ELISA was used following manufacturer's protocol. A monoclonal antibody against Phospho-Histone H2AX (S139) was added for 1 hour at room temperature. Cells were washed and then anti-mouse IgG conjugated to HRP was added for 1 hour. HRP substrate was added for 15 minutes followed by stop solution. Assay was read at 450 nm on a spectrophotometric microplate reader.
2.6 Clonogenic assay
Single-cell suspensions and cells were seeded into six-well tissue culture plates. Cells were allowed to attach 6 hours followed by drug treatment (2.5 μM LB100) and irradiated (5 Gy) 4 hours later with drug removed after 24 hours. Twelve days after seeding, colonies were stained with crystal violet and the number of colonies containing at least 50 cells was determined. The surviving fractions were calculated and survival curves generated after normalizing for cytotoxicity from LB100 treatment alone.
2.7 Cell cycle analysis
Evaluation of cell cycle and G2-checkpoint integrity was performed by flow cytometry. Cells were exposed to LB100 (2.5 μM) for 4 hours prior to administration of 5 Gy or sham radiation. Cells were trypsinized, fixed and stained per manufacturer's instructions with Cell Cycle Reagent and analyzed on an EasyCyte Plus flow cytometer (Guava Technologies, Hayward, CA). G2-checkpoint integrity was evaluated as previously reported (16, 17) utilizing rabbit polyclonal antibody against phospho-H3 histone (Millipore) followed by staining with goat anti-rabbit-FITC conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA) to distinguish mitotic cells.
2.8 Apoptosis assay
Apoptotic fraction was evaluated by flow cytometry using the Guava Nexin assay (Guava Technologies, Hayward, CA). Cells were exposed LB100 (2.5 μM) for 4 hours prior to administration of 5 Gy or sham radiation. Cells were trypsinized and stained per manufacturer's instructions with Nexin Reagent to assess annexin-V conjugated to phycoerithrin as a marker of cells in early apoptosis and 7-AAD as an indicator of late apoptosis (Guava Technologies). Analysis was performed on an EasyCyte Plus flow cytometer (Guava Technologies).
2.9 γ-H2AX assay
Immunofluorescent cytochemical staining for γ-H2AX foci was performed. Cells were grown in chamber slides and exposed LB100 (2.5 μM) for 4 hours prior to administration of 5 Gy or sham radiation. Cells were fixed with 2% paraformaldehyde, washed with PBS, permeabilized with 1% Triton X-100, washed again with PBS and blocked with 1% BSA. Mouse anti-γ-H2AX antibody (Millipore) was added at 1:500 and incubated overnight at 4°C. Cells were washed with 1% BSA and goat anti-mouse-FITC antibody (Jackson ImmunoResearch) was added at 1:100 and incubated 1 hr at room temperature. Nuclei were counterstained with DAPI (Sigma, St. Louis, MO). Cover slips were mounted with VectaShield anti-fade solution (Vector Labs, Burlingame, CA) and slides examined on a Leica DMRXA fluorescent microscope (Leica Microsystems). γ-H2AX foci were quantitated in 50 cells per condition.
2.10 Mitotic catastrophe
The presence of fragmented nuclei was used to define cells undergoing mitotic catastrophe. Cells were grown on chamber slides under identical treatment conditions as above. At 24, 48, 72, and 96 hours after radiation, cells were fixed with methanol, blocked with 1% BSA, and stained overnight at 4°C with mouse anti-α-tubulin antibody (Sigma) followed by staining with goat anti-mouse-Texas Red antibody (Jackson ImmunoResearch) 2 hours at room temperature. Nuclei were counterstained with DAPI (Sigma). Coverslips were mounted with VectaShield anti-fade solution (Vector Labs) and visualized on a Leica DMRXA fluorescent microscope (Leica Microsystems). Cells were manually counted with the presence of nuclei fragmented with ≥2 lobes as the criteria defining cells undergoing mitotic catastrophe. For each condition, 100 cells were scored.
2.11 Subcutaneous xenograft model
Six to eight week old, female, athymic NCr nu/nu, nude mice weighing approximately 20 grams (NCI Animal Production Program, Frederick, MD) were used for all in vivo studies. Animals were fed animal chow and water ad libitum, maintained on a 12-hour light/12-hour dark cycle. Mice were injected subcutaneously in the right flank with U251 (1 × 106) cells. When tumors reached ≈ 172 mm3 ([L × W2] × π/2), animals were randomized into groups: untreated controls, LB100, radiation, and combination LB100 and radiation. LB100 and radiation therapy was administered daily Monday to Friday for 3 weeks (15 treatments). LB100 dose administered was 1.5 mg/kg and radiation dose was 4 Gy per fraction. Treatments proceeded for 3 weeks to a total radiation dose of 60 Gy. In the combination treatment group, radiation of tumors took place 3 hours after treatment with LB100. Animals were restrained in lead jigs custom made by the Radiation Biology Branch of the National Cancer Institute. Tumors were measured three times per week; animals were euthanized when tumors reached ≥ 1800 mm3. Survival was assessed by the Kaplan-Meier method with the day of injection assigned as day zero and a log-rank test used to compare groups. All animal studies were conducted in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Animals and approved by the Animal Care and Use Committee of the National Cancer Institute.
2.12 Protein extraction and western blotting
Cell pellets were lysed in RIPA (Thermo Fischer Scientific, Waltham, MA) with Halt Protease Inhibitor (Thermo Fischer Scientific), sonicated, and centrifuged. Protein quantity was determined in the supernatant by Bio-Rad Protein Assay. Equal amount of proteins were denatured at 95 °C for 5-min in protein loading buffer, and loaded on a NuPAGE 4–12% Bis-Tris gel (Invitrogen Life Technologies) and transferred to PVDF membranes (Invitrogen Life Technologies). Membranes were blocked in 5% dried skim milk in PBST and blotted with primary antibody. Primary antibodies were as follows: Caspase-3 (1:1000, Cell Signaling, Danvers, MA).
2.13 Quantitative real-time PCR
Total RNA was extracted using RNeasy mini kit (Qiagen, Venlo, Netherlands) and cDNA was synthesized using SuperScript III First-Strand Synthesis SuperMix (Invitrogen Life Technologies). RT-PCR was performed using Eco RT PCR System (Illumina, San Diego, CA) and SYBR Green Master Mix (Applied Biosystems, Foster City, CA).
2.14 Statistical analysis
In vitro studies were subject to three independent experiments. Data is presented as mean ± SE. A two-sided Student's t test was used to compare sample means with a p value of <0.05 considered significant.
3. Results
3.1 Radiation increases PP2A activity in glioblastoma cells while LB100 attenuates PP2A activity alone and following radiation
PP2A has been shown to play a role in the ATM/ATR mediated activation of the G2/M cell cycle checkpoint following radiation-induced DNA damage (9, 18). LB100 is a small molecule competitive inhibitor of PP2A. PP2A activity was measured in U251 cells after 2 μM LB100 treatment, 5 Gy radiation, and LB100 followed by 5 Gy radiation in vitro. Three and six hours after radiation alone, U251 cells showed 194% and 200% of the PP2A activity in comparison to control cells (p<0.001) (Fig. 1A). Notably, 24 hours after radiation PP2A levels fell to 80% compared to controls but had returned to baseline at 48 hours (Supplementary Fig. S2). In contrast, treatment with 2 μM LB100 reduced PP2A activity to 61% of control cells after both three and six hours of LB100 exposure. The addition of 5 Gy radiation to LB100-treated cells resulted in PP2A activity at 3 hours and 6 hours after radiation treatment of 91% and 112% compared to control cells, respectively (Fig. 1A). LB100 effectively prevented the post-radiation increase in PP2A activity although levels were higher than LB100 treatment alone.
Figure 1. PP2A activity increases after radiation and is inhibited by LB100 in vivo and in vitro.
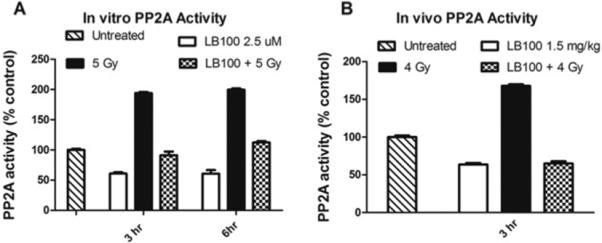
(A) PP2A activity is expressed as a percentage of control activity based on a phosphatase assay. In vitro PP2A activity in U251 cells is shown, measured at 3 hours and 6 hours after LB100 treatment, radiation (5 Gy), or combined LB100 and radiation treatment. (B) In vivo PP2A activity is shown in U251 xenografts, measured at three hours after treatment with LB100 (2.5 mg/kg), radiation (4 Gy), or combined treatment.
To assess the effect of LB100 on in vivo, nude mice bearing U251 subcutaneous xenografts were treated with LB100, radiation, or combination of LB100 and radiation. LB100 treatment resulted in a decrease of PP2A activity to 64% of baseline (p<0.05) (Fig. 1B). PP2A activity increased 1.67 fold after 4 Gy radiation, consistent with the radiation-induced increase in PP2A activity seen previously in vitro. However, tumors treated with combination LB100 and 4 Gy showed a decrease of PP2A activity to 65% of baseline, indicating potent suppression of the radiation-induced increase in PP2A activity.
3.2 LB100 sensitizes glioblastoma cells to the effects of radiation
To screen LB100 as a potential radiation sensitizer in several cell lines, we screened U251 glioblastoma, U87 glioblastoma, and H1915 non-small cell lung cancer (NSCLC) cells. An ELISA for γ-H2AX was performed 24 hours after radiation, as PP2A levels at this time point were no longer elevated (Supplementary Fig. S2). With 2 μM LB100 treatment, γ-H2AX expression was increased 147% in U87, 146% in U251, and 124% in H1915, compared to expression in untreated controls. Likewise, with 5 μM LB100 treatment, γ-H2AX expression was increased 136% in U87, 196% in U251, and 135% in H1915 cells, compared to untreated controls. These data indicated that LB100 treatment resulted in retention of γ-H2AX foci at 24 hours after radiation in several cell lines (data not shown). For subsequent mechanistic experiments, U251 cells were used.
To assess the effects of LB100 treatment on DNA damage and repair, γ-H2AX expression was assessed by immunofluorescence in U251 cells as a measure of DNA double strand breaks at 1, 6 and 24 hours (Fig. 2A and B). LB100 alone caused no significant change in γ-H2AX levels. At 1 and 6 hours, cells treated with radiation or the combination of LB100 and radiation demonstrated similar significant elevations in γ-H2AX levels compared to control or LB100 alone treated cells (p<0.001). At 24 hours, γ-H2AX level had returned to near baseline in the cells treated with radiation alone; however, γ-H2AX levels were significantly higher in the cells treated with combination LB100 and radiation than cells treated with vehicle control (p=0.009), LB100 (p=0.002), or radiation alone (p=0.001). This finding at 24 hours confirmed that LB100 diminished the cellular repair of radiation induced DNA double strand breaks.
Figure 2. LB100 enhances radiation sensitivity.
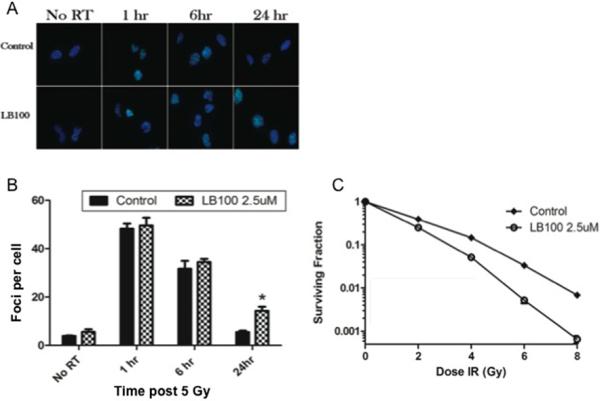
(A) Immunocytochemical staining to detect γ-H2AX foci was performed. Cells were plated in 4 well chamber slides, allowed to attach (6 hours) and then incubated in LB100 (2.5 μM) for 4 hours prior to radiation (5 Gy). Foci were counted in at least 50 cells per experiment. (B) Quantitative assessment of γ-H2AX foci per cell at 1 hour, 6 hours, and 24 hours after radiation is shown. Data presented are the mean ± SE from at least three independent experiments. *, P < 0.05 (comparing both radiation alone and LB100 alone treated cells to the combination treatment). (C) The effects of LB100 on radiosensitivity of U251 are demonstrated by clonogenic assay. Cells were seeded as a single-cell suspension with a specified number of cells. After allowing cells time to attach (6 hours), 2.5 μM LB100 was added and the plates were radiated 4 hours later and drug removed after 24 hours. Twelve days after seeding, survival curves were generated after normalizing for the cytotoxicity generated by LB100 alone. Plating efficiency was 0.31 and treatment with LB100 yielded a surviving fraction of 0.68. The addition of LB100 to radiation resulted in a dose enhancement factor of 1.45 at a surviving fraction of 0.10.
To confirm our findings, we performed immunofluorescence of rad51 foci in U251 cells to detect homologous recombinational repair of double strand breaks in DNA-damaged cells. Compared to controls, 5 Gy radiation alone increased rad51 foci formation two-fold. Moreover, the addition of LB100 to radiation further elevated rad51 foci formation to levels 3.5 times that of the controls (Supplementary Fig. S3).
To determine the effects of LB100 on the radiosensitivity of U251 cells, a clonogenic assay was performed. Plating efficiency was 0.31 and treatment of U251 cells with 2.5 μM LB100 yielded a surviving fraction of 0.68 (an appropriate degree of cytotoxicity for evaluation in combination with radiation). Cells were then radiated 4 hours following LB100 treatment with drug removal at 24 hours. LB100 treatment resulted in a dose enhancement factor of 1.45 at a surviving fraction of 0.10 (Fig. 2C).
3.3 LB100 increases the proportion of cells within the M phase of the cell cycle
PP2A activity has been previously shown to be necessary for G2/M arrest in some cancer cell lines (9, 19-21). Flow cytometry performed 24 hours after exposure of U251 cells to 2.5 μM LB100 showed no significant difference in the distribution of cells in G0/G1, S, G2, or M phase. However, cells treated with combination LB100 and radiation had a significantly higher proportion of cells in M phase than control cells (Fig. 3A) and exhibited a higher mitotic index measured by H3-pS10 staining (Fig. 3B). This suggests that the radiation sensitization induced by LB100 is not due to drug-induced alterations in cell cycle distribution at the time of radiation; however, that combination therapy results in the accumulation of an increased proportion of cells in M-phase after radiation.
Figure 3. LB100 and radiation increase mitotic index and induce mitotic catastrophe.
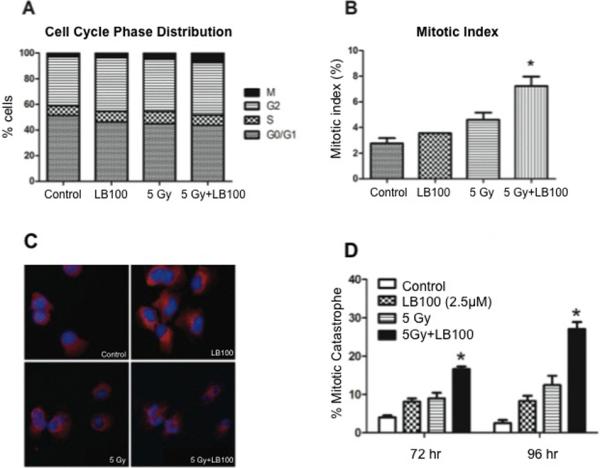
(A) The percentage of cells in G0/G1, S, G2 and M phase of the cell cycle are depicted before and at 24 hours after radiation (5 Gy), with and without pre-exposure (for 4 hours) to LB100 treatment (2.5 μM). There was no difference in distribution of cells in G0/G1, S, or G2, but an increased number of cells in M phase (B) was seen after radiation and LB100 treatment. (C) U251 cells growing in chamber slides were exposed to LB100 (2.5 μM) for 4 hours, radiated (5 Gy), and after radiation were subjected to immunocytochemical analysis of mitotic catastrophe. Nuclear fragmentation (defined as the presence of two or more distinct lobes within a single cell) was evaluated in at least 150 cells per treatment per experiment. (D) Quantitative assessment of percentage of cells in mitotic catastrophe is shown. *, P < 0.05 (comparing cells in the combination group compared to either drug or radiation alone groups at the same time point).
3.4 LB100 does not alter cellular apoptosis though augments mitotic catastrophe
To determine if induction of apoptosis was contributing to radiosensitization in vitro, we measured apoptosis by flow cytometry 24 hours after treatment. Less than 4% of cells in all treatment groups were apoptotic, with no difference seen between untreated controls and cells treated with 2.5 μM LB100, 5 Gy, or the combination of 5 Gy and 2.5 μM LB100 (data not shown). Previous work has suggested that LB100 treatment may promote nuclear changes associated with mitotic catastrophe (15). The number of cells in mitotic catastrophe was significantly greater in cells treated with combination LB100 and radiation than cells receiving radiation alone at 72 and 96 hours (p=0.0083 and 0.0041, respectively) (Fig. 3C and D).
3.5 LB100 enhances tumor growth delay after radiation
To evaluate the benefit of combination treatment in vivo, mice bearing U251 subcutaneous xenografts were randomized into four groups: vehicle, LB100 (1.5 mg/kg), radiation alone, and combination LB100 and radiation. Treatments were administered daily Monday-Friday for three weeks for a total of 15 treatments (60 Gy cumulative radiation dose). The time for tumor to grow from 172 mm3 to 1,000 mm3 (a 5-fold increase in tumor size) increased from 5.7 ± 1.3 days for vehicle-treated mice to 11.7 ± 1.3 days for LB100-treated mice (p=0.01) (Fig. 4A). Irradiated mice reached 1,000 mm3 in 65.0 ± 4.4 days (p<0.001). The time for tumors to reach 1,000 mm3 in mice treated with combination radiation and LB100 was 100-150 days in 40% of the mice and 60% of the mice did not have tumors regrow during the evaluation period of 200 days. Survival time analysis showed significant differences between all groups (p=0.01 for vehicle vs. LB100 and p<0.001 between all other groups) (Fig. 4B). These data demonstrate synergy in the combination of LB100 with radiation therapy and that combination treatment significantly enhanced tumor growth delay and survival compared to radiation alone.
Figure 4. LB100 enhances in vivo tumor growth delay from radiation.
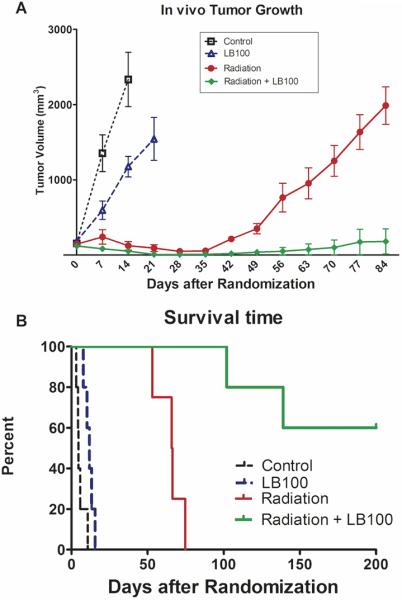
(A) Mice were implanted with 1×106 U251 cells on the lateral aspect of the rear leg. Tumors were randomized at 172 mm3 into four groups and treated with vehicle control, LB100 alone (1.5 mg/kg by intraperitoneal injection), radiotherapy (4 Gy/fraction × 15 treatments over 3 weeks), or combination treatment with LB100 and radiotherapy. Tumors were measured three times per week and followed until they reached at least 1,500 mm3. Volumes were calculated using the formula (L × W2) × π/2. (B) Percent survival is shown, expressed in number of days after treatment randomization.
3.6 LB100 attenuates p53-induced cell cycle arrest and enhances p53-independent apoptosis following radiation
Combination LB100 and radiation enhanced G2/M phase transition and mitotic catastrophe. To gain further insight into the mechanisms underlying these effects we examined the expression of p53 in vivo. PP2A subunit B56γ forms a complex with p53 following DNA damage, leading to Thr55 dephosphorylation of p53 and subsequent inhibition of cell cycle transition and proliferation through p53 transcriptional activation and p21 synthesis (22). Real-time quantitative PCR of U251 xenograft tissues demonstrated a 3.68-fold decrease in p53 expression in radiated mice with LB100 treatment compared to those receiving radiation alone (p53: one-way ANOVA F(3,8) = 148.0, p < 0.0001) (Fig. 5A). In addition, western blot analysis revealed a significant increase in the expression of cleaved caspase-3 in U251 xenograft tissues in radiated mice with LB100 treatment compared to control and LB100 or radiation alone (cleaved caspase-3: one-way ANOVA F(3,8) = 28.17, p < 0.001) (Fig. 5B). These results indicate that combination LB100 and radiation may inhibit B56γ (PP2A)-p53 interaction and diminish p53 DNA damage response pathway, driving cell proliferation and enhancing p53-independent apoptosis in vivo.
Figure 5. Combination LB100 and radiation attenuates p53 expression and induces p53-independent apoptosis.

(A) Quantitative real-time PCR data are shown with expression of factors being conveyed as fold-induction relative to control sample expression (fold-induction=1). (B) Western blot analysis for caspase-3 is shown, demonstrating significantly increased expression of cleaved caspase-3 with combined LB100 and radiation compared to radiation alone. Data are presented as the mean ± SEM (n=3). *, P ≤ 0.05.
4. Discussion
This study demonstrated LB100, a small molecule inhibitor of PP2A, enhances radiosensitivity of human glioblastoma cells in vitro and in vivo. The potential for PP2A inhibition to be an effective strategy for radiosensitization has been suggested previously, (9, 23) and radiosensitization by LB100 has been shown in pancreatic cancer and nasopharyngeal carcinoma (24, 25). In this study, we have demonstrated that glioblastoma may also be susceptible to radiosensitization after LB100 exposure. Our findings suggest that apoptosis was not an important acute mechanism of radiosensitization of U251 cells by LB100 in vitro. However, in vivo results demonstrated that combination LB100 and radiation suppressed p53 transcriptional activation and induced p53-independent apoptosis at 3-4 weeks following initial treatment. The discrepancy between these results was most likely due to temporal differences in tissue assessment. Nonetheless, it appears that the role of PP2A in apoptosis is actually quite complex due to the appreciated diversity of PP2A as it displays a mixture of pro-apoptotic and anti-apoptotic functions (26).
We further explored the effects of LB100 on cell cycle. LB100 exposure accumulated an increased percentage of cells in mitosis 24 hours after radiation treatment, which may have occurred due to several possibilities. One possibility is that PP2A inhibition resulted in abrogation of the G2/M checkpoint, allowing more cells to enter mitosis after radiation. However, our data suggested that 24 hours after 5 Gy treatment, there was minimal residual G2 arrest. An alternative possibility was that PP2A inhibition with LB100 prevented cells from exiting mitosis. There is accumulating evidence that PP2A activity in association with subunit B55 is necessary for mitotic exit in mammalian cells and that disruption of this process through PP2A inhibition may result in mitotic catastrophe (27, 28). Likewise, our data demonstrated induction of mitotic catastrophe as well as an increased mitotic fraction after combination therapy. However, it is unclear whether mitotic catastrophe occurred secondary to or was the cause of this increased mitotic index.
We found no initial increase in radiation induced DNA damage based on γ-H2AX levels. However, at 24 hours after radiation, there was increased γ-H2AX foci retention as well as rad51 foci formation, suggesting LB100 inhibits homologous recombination repair of radiation induced DNA damage in U251 cells. Extension of the in vitro data to an in vivo model confirmed that LB100 could effectively inhibit PP2A and prevent radiation induced increases in PP2A activity. With daily treatment, LB100 caused minor delay in tumor growth. This finding was not unexpected based on the mechanism of action of the drug, which would suggest that it likely requires concurrent administration of a DNA damaging treatment for efficacy. We have previously demonstrated the necessity of additional genotoxic combination therapy in the study of chemosensitization in glioblastoma (15). In our in vivo experiments, we found that animals treated with 60 Gy of fractionated radiation initially experienced significant tumor growth delay, but after 4-5 weeks, exhibited tumor recurrence. This finding was expected as regression, remission, and early recurrence is a common feature of clinical glioblastoma after radiation treatment. Notably, however, in the combination treatment arm, no tumors were observed until 3-4 weeks after treatment and furthermore, 60% of the mice failed to demonstrate macroscopic tumor formation after 6 months.
These data provide support for further evaluation of LB100 as a radiation sensitizer and a basis for additional preclinical exploration of the radiosensitizing properties of LB100 and other PP2A inhibitors. Further investigations to understand the specific molecular mechanisms leading to radiosensitization in other tumor cell lines are warranted.
Supplementary Material
Acknowledgements
The authors wish to thank Lixte Biotechnology Holdings, Inc. (East Setauket, NY) for generously providing the PPA2 inhibitor, LB100, for this study. All co-authors were supported by the Intramural Research Program at the National Institute of Neurological Disorders and Stroke and National Cancer Institute at the National Institutes of Health (NIH). K. Huntoon, J.M. Frerich, M.J. Feldman, and D. Ye were additionally supported by the NIH Medical Research Scholars Program, a public–private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from Pfizer, Inc., the Doris Duke Charitable Foundation, Alexandria Real Estate Equities, Inc., and Mr. and Mrs. Joel S. Marcus and the Howard Hughes Medical Research Institute, as well as other private donors. For a complete list, please visit http://fnih.org/work/education-training-0/medical-research-scholars-program.
Footnotes
The authors declare no conflicts of interest.
References
- 1.Stupp R, Dietrich PY, Ostermann Kraljevic S, Pica A, Maillard I, Maeder P, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002;20:1375–82. doi: 10.1200/JCO.2002.20.5.1375. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Gorlia T, Stupp R, Brandes AA, Rampling RR, Fumoleau P, Dittrich C, et al. New prognostic factors and calculators for outcome prediction in patients with recurrent glioblastoma: A pooled analysis of EORTC Brain Tumour Group phase I and II clinical trials. Eur J Cancer. 2012 doi: 10.1016/j.ejca.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Ruediger R, Ruiz J, Walter G. Human cancer-associated mutations in the Aalpha subunit of protein phosphatase 2A increase lung cancer incidence in Aalpha knock-in and knockout mice. Molecular and cellular biology. 2011;31:3832–44. doi: 10.1128/MCB.05744-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cristobal I, Garcia-Orti L, Cirauqui C, Alonso MM, Calasanz MJ, Odero MD. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia. 2011;25:606–14. doi: 10.1038/leu.2010.294. [DOI] [PubMed] [Google Scholar]
- 6.Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, et al. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130:51–62. doi: 10.1016/j.cell.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 7.Mumby M. PP2A: unveiling a reluctant tumor suppressor. Cell. 2007;130:21–4. doi: 10.1016/j.cell.2007.06.034. [DOI] [PubMed] [Google Scholar]
- 8.Krasinska L, Domingo-Sananes MR, Kapuy O, Parisis N, Harker B, Moorhead G, et al. Protein phosphatase 2A controls the order and dynamics of cell-cycle transitions. Mol Cell. 2011;44:437–50. doi: 10.1016/j.molcel.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Yan Y, Cao PT, Greer PM, Nagengast ES, Kolb RH, Mumby MC, et al. Protein phosphatase 2A has an essential role in the activation of gamma-irradiation-induced G2/M checkpoint response. Oncogene. 2010;29:4317–29. doi: 10.1038/onc.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan LY, Amon A. The protein phosphatase 2A functions in the spindle position checkpoint by regulating the checkpoint kinase Kin4. Genes Dev. 2009;23:1639–49. doi: 10.1101/gad.1804609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das S, Srikanth M, Kessler JA. Cancer stem cells and glioma. Nat Clin Pract Neurol. 2008;4:427–35. doi: 10.1038/ncpneuro0862. [DOI] [PubMed] [Google Scholar]
- 12.Moore N, Lyle S. Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J Oncol. 2011:1–11. doi: 10.1155/2011/396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roth S, Fodde R. Quiescent stem cells in intestinal homeostasis and cancer. Cell Commun Adhes. 2011;18:33–44. doi: 10.3109/15419061.2011.615422. [DOI] [PubMed] [Google Scholar]
- 14.Lu J, Zhuang Z, Song DK, Mehta GU, Ikejiri B, Mushlin H, et al. The effect of a PP2A inhibitor on the nuclear receptor corepressor pathway in glioma. J Neurosurg. 2009;113:225–33. doi: 10.3171/2009.11.JNS091272. [DOI] [PubMed] [Google Scholar]
- 15.Lu J, Kovach JS, Johnson F, Chiang J, Hodes R, Lonser R, et al. Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11697–702. doi: 10.1073/pnas.0905930106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu B, Kim ST, Lim DS, Kastan MB. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol. 2002;22:1049–59. doi: 10.1128/MCB.22.4.1049-1059.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu B, Kastan MB. Analyzing cell cycle checkpoints after ionizing radiation. Methods Mol Biol. 2004;281:283–92. doi: 10.1385/1-59259-811-0:283. [DOI] [PubMed] [Google Scholar]
- 18.Petersen P, Chou DM, You Z, Hunter T, Walter JC, Walter G. Protein phosphatase 2A antagonizes ATM and ATR in a Cdk2- and Cdc7-independent DNA damage checkpoint. Molecular and cellular biology. 2006;26:1997–2011. doi: 10.1128/MCB.26.5.1997-2011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Molecular cell. 2005;20:801–9. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Li G, Elder RT, Qin K, Park HU, Liang D, Zhao RY. Phosphatase type 2A-dependent and -independent pathways for ATR phosphorylation of Chk1. The Journal of biological chemistry. 2007;282:7287–98. doi: 10.1074/jbc.M607951200. [DOI] [PubMed] [Google Scholar]
- 21.Jang YJ, Ji JH, Choi YC, Ryu CJ, Ko SY. Regulation of Polo-like kinase 1 by DNA damage in mitosis. Inhibition of mitotic PLK-1 by protein phosphatase 2A. The Journal of biological chemistry. 2007;282:2473–82. doi: 10.1074/jbc.M605480200. [DOI] [PubMed] [Google Scholar]
- 22.Shouse GP, Nobumori Y, Liu X. A B56gamma mutation in lung cancer disrupts the p53-dependent tumor-suppressor function of protein phosphatase 2A. Oncogene. 2010;29:3933–41. doi: 10.1038/onc.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price WA, Stobbe CC, Park SJ, Chapman JD. Radiosensitization of tumour cells by cantharidin and some analogues. International journal of radiation biology. 2004;80:269–79. doi: 10.1080/09553000410001679785. [DOI] [PubMed] [Google Scholar]
- 24.Wei D, Parsels LA, Karnak D, Davis MA, Parsels JD, Marsh AC, et al. Inhibition of protein phosphatase 2A radiosensitizes pancreatic cancers by modulating CDC25C/CDK1 and homologous recombination repair. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:4422–32. doi: 10.1158/1078-0432.CCR-13-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lv P, Wang Y, Ma J, Wang Z, Li JL, Hong CS, et al. Inhibition of protein phosphatase 2A with a small molecule LB100 radiosensitizes nasopharyngeal carcinoma xenografts by inducing mitotic catastrophe and blocking DNA damage repair. Oncotarget. 2014;5:7512–24. doi: 10.18632/oncotarget.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janssens V, Rebollo A. The role and therapeutic potential of Ser/Thr phosphatase PP2A in apoptotic signalling networks in human cancer cells. Curr Mol Med. 2012;12:268–87. doi: 10.2174/156652412799218930. [DOI] [PubMed] [Google Scholar]
- 27.Wurzenberger C, Gerlich DW. Phosphatases: providing safe passage through mitotic exit. Nature reviews Molecular cell biology. 2011;12:469–82. doi: 10.1038/nrm3149. [DOI] [PubMed] [Google Scholar]
- 28.Manchado E, Guillamot M, de Carcer G, Eguren M, Trickey M, Garcia-Higuera I, et al. Targeting mitotic exit leads to tumor regression in vivo: Modulation by Cdk1, Mastl, and the PP2A/B55alpha,delta phosphatase. Cancer cell. 2010;18:641–54. doi: 10.1016/j.ccr.2010.10.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.