

SUMMARY
Age-related frailty may be due to decreased skeletal muscle regeneration. The role of TGF-β molecules myostatin and GDF11 in regeneration is unclear. Recent studies showed an age-related decrease in GDF11 and that GDF11 treatment improves muscle regeneration, which were contrary to prior studies. We now show that these recent claims are not reproducible and the reagents previously used to detect GDF11 are not GDF11 specific. We develop a GDF11-specific immunoassay and show a trend toward increased GDF11 levels in sera of aged rats and humans. GDF11 mRNA increases in rat muscle with age. Mechanistically, GDF11 and myostatin both induce SMAD2/3 phosphorylation, inhibit myoblast differentiation, and regulate identical downstream signaling. GDF11 significantly inhibited muscle regeneration and decreased satellite cell expansion in mice. Given early data in humans showing a trend for an age-related increase, GDF11 could be a target for pharmacologic blockade to treat age-related sarcopenia.
Graphical Abstract
INTRODUCTION
Aging is associated with a decrease in the mass and function of skeletal muscle, termed “sarcopenia.” Sarcopenia contributes to the overall frailty that is observed in the elderly. The decrease in muscle mass and strength observed has been associated with disability and is unfortunately a reliable predictor of the loss of independence, and even mortality—independent of other risk factors (Roubenoff and Hughes, 2000). The molecular mechanisms that are causal for sarcopenia are thought to be multi-factorial (Egerman and Glass, 2014). An unbiased profiling study in rats demonstrated that a decline in mitochondrial pathways and neuromuscular junction competence were among the primary perturbations correlated with sarcopenia (Ibebunjo et al., 2013).
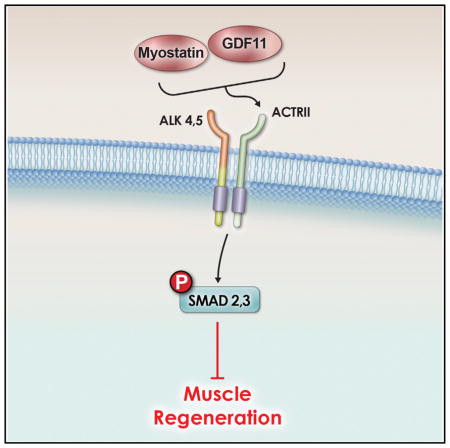
Other studies have implicated a role for TGF-β family members in aging and frailty. For example, TGF-β itself has been associated with the fibrosis seen in older tissue and as an inhibitor of muscle differentiation (Beggs et al., 2004; Carlson et al., 2009; Massagué et al., 1986). Myostatin, also called growth differentiation factor 8 (GDF8), has also been demonstrated to be an inhibitor of muscle differentiation (McPherron et al., 1997a; Sartori et al., 2009; Trendelenburg et al., 2009). It can also induce atrophy on post-differentiated myotubes (Sartori et al., 2009; Trendelenburg et al., 2009). Myostatin null mice (McPherron et al., 1997a) and cattle demonstrate a doubling in muscle mass (Kambadur et al., 1997; McPherron and Lee, 1997b). This increase in muscularity upon the loss of myostatin has been demonstrated in multiple animals and even in humans (Lee, 2010). It has also been shown that other TGF-β family molecules, distinct from myostatin, play a role in modulating skeletal muscle size, since myostatin−/− mice that are mated with mice that are transgenic for follistatin (TGfollistatin), which is capable of inhibiting not only myostatin but also its close relative GDF11 and other TGF-β molecules, such as the activins (Hill et al., 2002; Schneyer et al., 2008; McPherron et al., 2009), resulted in an even greater increase in muscle size (Lee et al., 2010). Myostatin induces cellular signaling by binding either of the type II activin receptors (IIa or IIb), which then allows for activation of type I receptors ALK4 or ALK5 (Tsuchida et al., 2008). The binding of myostatin to these receptor complexes results in the phosphorylation and activation of the transcription factors SMAD2 and SMAD3, which translocate to the nucleus upon phosphorylation (Rebbapragada et al., 2003).
GDF11 is highly related to myostatin, sharing 90% homology in the mature active regions of both proteins (Nakashima et al., 1999). As in the case of other TGF-β family members, myostatin and GDF11 precursor proteins are proteolytically processed to form biologically active carboxy-terminal dimers. They both have similar signaling pathways; both bind activin type II receptors and activate the intracellular mediator SMAD 2/3 pathway (Lee and McPherron, 2001; Oh et al., 2002; Sartori et al., 2009; Trendelenburg et al., 2009; Tsuchida et al., 2008). While myostatin is predominantly expressed in developing and adult skeletal muscle (Bass et al., 1999; Gonzalez-Cadavid et al., 1998; Ji et al., 1998; Kambadur et al., 1997), GDF11 expression is quite different (Nakashima et al., 1999). GDF11−/− mice display homeotic transformations of the axial skeleton, without an obvious effect on skeletal muscle (McPherron et al., 1999), although it is a negative regulator of myogenesis and chondrogenesis in the developing chick (Gamer et al., 2001). While the phenotypes caused by myostatin versus GDF11 deletion appear to be nonoverlapping, the high sequence identity and similarity in signaling mechanisms suggest the two molecules may be functionally redundant and that the different phenotypes are due to differences in sites of expression.
A couple of recent manuscripts reported that GDF11 has distinct effects in comparison to myostatin. In one case, it was reported that GDF11 decreased cardiac-related muscle hypertrophy (Loffredo et al., 2013); in this study it was also shown that GDF11 levels decreased as a function of age in mice, and therefore it was suggested that replenishment of GDF11 would help block cardiac muscle hypertrophy. Later, in a distinct study, another group showed that GDF11 had positive effects on aged satellite cells (SCs) and an improvement in skeletal muscle regeneration when GDF11 was administered to older mice (Sinha et al., 2014). These more recent findings seemed surprising given that the very closely related myostatin is a direct inhibitor of muscle differentiation (Ríos et al., 2002; Trendelenburg et al., 2009; Sartori et al., 2009). Therefore, given the claimed dramatically positive effects of GDF11 on skeletal muscle, in contrast to the demonstrated negative effects induced by myostatin, it seemed important to study the role of GDF11 in particular on skeletal muscle both in vitro and in vivo.
RESULTS
Prior Reagents Used to Detect GDF11 Are Not GDF11 Specific
Previous reports had identified GDF11, through both proteomic and western blot analyses, as a circulating factor in mice whose serum levels decrease with age (Loffredo et al., 2013; Sinha et al., 2014). In the Loffredo study, the SOMAmer technology was used to make an assessment of GDF11 levels. We first sought to test the specificity of the GDF11 SOMAmer used in the prior study by determining whether it might cross-react with myostatin. This seemed possible given the 90% sequence identity between the two proteins in their mature active form. In a direct binding test of GDF11 and myostatin, we observed that the GDF11 SOMAmer does indeed bind both proteins (Figure 1A, apparent KD for GDF11: 6.6 ± 1.1 nM; for myostatin: 11.8 ± 1.1 nM), while a chemically related control SOMAmer binds neither protein (data not shown). Since the SOMAmer that was previously used to demonstrate a decrease of GDF11 levels in age actually could not distinguish between myostatin and GDF11, we next tried the antibody that was used to demonstrate that GDF11 declines with age (Loffredo et al., 2013; Sinha et al., 2014). This antibody was also first tested for its specificity. By western blot analysis, the antibody was found to recognize both recombinant myostatin and GDF11 to a similar degree, indicating cross-reactivity and a lack of preferential binding to GDF11 (Figure 1B). Importantly, under the reducing conditions used in this study, both the mature dimer (~25 kDa) and reduced monomer (~12.5 kDa) of recombinant myostatin and GDF11 were observed. In addition, some higher molecular weight material was observed, consistent with aggregated GDF11 or myostatin, since the molecular weight of the higher bands are multiples of the monomer.
Figure 1. Prior Reagents Used to Measure GDF11 Are Not Specific, but Show that the Combination of GDF11 and Myostatin Increases with Age; Specific Methods show GDF11 Levels Increase with Age.

(A) Affinity of GDF11 SOMAmer for recombinant GDF11 and myostatin. Binding of the GDF11 SOMAmer to GDF11 (shown in blue) and myostatin (shown in red) proteins as measured by dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA). Data represent means ± SD from three technical replicates.
(B) Western blot analysis to determine specificity of Abcam antibody to GDF11 versus myostatin (GDF8). An anti-GDF11 antibody from Abcam was tested for specificity using a concentration gradient of recombinant GDF11 and myostatin, ranging from 6.25 ng to 100 ng, and was found to cross-react with myostatin. Even though this is a denaturing gel, bands consistent with dimer and even high molecular forms consistent with aggregates of the recombinant material are evident.
(C) Western analysis on sera from young and old mice. Sera samples from four different young animals (4 months old; 1, 2, 3, 4) and four different old animals (23 months old; 5, 6, 7, 8) were tested by western analysis for myostatin/GDF11 levels (top). Coomasie staining (bottom) demonstrates equivalent loading of each lane. Lane with ladder is indicated. The dimer band was not fully denatured to monomer. There was an increase in GDF11/myostatin dimer levels in the sera from older animals in comparison to young animals. Densitometry of monomer + dimer is provided on the right, indicating an overall increase in myostatin/GDF11 levels in the mouse sera (*p < 0.05).
(D) GDF11 and myostatin mRNA content in in skeletal muscles of Sprague-Dawley male rats aged 6, 12, 18, 21, and 24 months (data derived from the RNA-seq analyses). RNA-seq analysis demonstrates that GDF11 expression increases as a function of age (comparing mRNA obtained from muscles from 6-, 12-, 18-, 21-, and 24-month-old rats). In contrast, myostatin (MSTN) expression decreases with age in rats. The y axis is the standardized expression level, with mean of 0 and standard deviation of 1. GOF, goodness of fit to a sigmoidal curve; FC, fold change between 24 m and 6 m.
(E) GDF11 protein levels in sera from young and old rats determined by immunoassay. GDF11 protein content in serum from young (6 months) or old (24 months) rats was measured by immunoassay. Old rats had higher levels of GDF11 compared with young. Data are mean ± SEM (p = 0.0534, Student’s t test).
(F) GDF11 protein levels in sera from young and older humans determined by immunoassay. GDF11 protein content was measured in serum samples from nine older (aged >60 years, males, shown in red) or ten young (aged 20–30 years, males, shown in blue). The median GDF11 concentration in serum of older humans was higher than in younger humans, but this did not reach statistical significance. Serum samples from three young and one old subject had GDF11 below a detection limit (less than 0.274 ng/ml), shown with a dotted horizontal line.
The Combination of Myostatin and GDF11 Increases in Mouse Sera
Nevertheless, reasoning that we could at least confirm whether the combination of myostatin and GDF11 declined with age, as had been previously reported by detection with this antibody, serum samples from young and old mice were analyzed by western blot to determine the relative abundance of myostatin/GDF11 in mouse blood. Although the level of monomer decreased with age, as had been previously reported, the active dimer was also observed, and its level actually significantly increased (Figure 1C and Figure S1A). Both the monomer band and the dimer band co-migrated with recombinant GDF11 (Figure 1C and Figure S1A). We were assured that we were able to detect actual GDF11 in the serum by injecting young mice with recombinant GDF11; with the antibody, we could then observe an increase in the dimer band, corresponding to an overall increase in serum GDF11 levels (Figure S1B). A Coomassie stained gel is also provided to demonstrate equivalent protein loading (75 μg total protein/lane) (Figure 1C) and a Ponceau stain is provided in a second experiment, repeating these results (Figure S1A). Overall, it appears that the total levels of myostatin/GDF11 actually increase with age, contradicting the prior reports (Loffredo et al., 2013; Sinha et al., 2014).
A GDF11-Specific Method Demonstrates a Trend of GDF11 Increasing with Age in Rat and Human Sera
In order to specifically detect the levels of GDF11 in serum samples, an immunoassay was established that was validated to be specific for GDF11 (Figures S2A and S2B). This immunoassay did not detect myostatin (Figures S2A and S2B). The validation work further established that the immunoassay could detect endogenous GDF11 in human sera (Figure S2C, bar graph on left, blue bars) and that it was actually measuring GDF11, since recombinant GDF11 when spiked into human sera was detected (Figure S2C, bar graph on left, red bars). Furthermore, when sera were diluted, GDF11 was recovered in proportion to that dilution, demonstrating that the assay can quantitatively measure GDF11 within a range, even when it is diluted (Figure S2C, graph on right). With this assay, we could not detect endogenous GDF11 in either young or old mice (data not shown), since the levels were below the sensitivity of detection for this immunoassay. We next tried to detect GDF11 in sera from other species. We measured the blood serum concentration of GDF11 in both young and old rats (6 months versus 24 months) and humans (20–30 years versus 60+ years). We found that there was a nearly significant increase of GDF11 (~1.4 fold increase, p = 0.0534) in serum from older rats compared to the younger rats (Figure 1E). A similar trend toward an increase in median levels was observed comparing serum from humans over the age of 60 in comparison to sera from humans between 20 and 30 years old (Figure 1F).
GDF11 Expression Increases with Age in Rat Skeletal Muscle
To use a distinct method to detect GDF11 specifically, we performed RNA-seq on skeletal muscle from the rats, using 6-, 12-, 18-, 21-, and 24-month-old animals (Ibebunjo et al., 2013), spanning the lifespan of the animal. This study demonstrated that GDF11 expression increased dramatically as a function of age, while surprisingly, myostatin expression was shown to decrease with age (Figure 1D). These findings were confirmed by qPCR in a distinct cohort (Figure S1C).
GDF11 and Myostatin Similarly Induce SMAD, p38, and ERK Phosphorylation
To rationalize whether GDF11 could have distinct effects in comparison to myostatin, we decided to compare the two molecules for their ability to induce signaling and cellular effects in vitro. Human skeletal muscle-derived cells (hSkMDCs) were used, since human cells were shown to respond to TGF-β family members at physiological concentrations in a prior study (Trendelenburg et al., 2009). Differentiated hSkMDC myotubes were serum-starved and then treated with multiple doses of either recombinant protein. Myostatin and GDF11 treatment both resulted in increases in SMAD2 and SMAD3 phosphorylation, and these increases occurred in a concentration-dependent manner (Figure 2A). Myostatin and GDF11 activated SMAD2/3 transcription factor function, as indicated by dose-dependent increases on CAGA-luc SMAD reporter activity (Figure 2B). These data demonstrate that myostatin and GDF11 can signal through SMAD2/3 activation in skeletal muscle and other cells. Since myostatin is also able to signal via the mitogen-activated protein kinase (MAPK) pathways (ERK1/2, JNK, p38) (Philip et al., 2005; Yang et al., 2006), GDF11 and myostatin were compared for their ability to activate p38 and ERK. Indeed, treatment with myostatin and GDF11 also increased phosphorylation of p38 and ERK in C2C12 cells, indicating that both GDF11 and myostatin induce signaling via these pathways to a similar degree (Figure S3).
Figure 2. GDF11 and Myostatin Signal through Identical Pathways in Skeletal Muscle.

(A) Western blot analysis to determine myostatin versus GDF11 activation of downstream signaling. Human myotubes were stimulated with vehicle (UNT), as a negative control, or with increasing doses of myostatin or GDF11 (10, 30, 100, 300 ng/ml). Both proteins stimulated SMAD2 and SMAD3 phosphorylation (pSMAD2 and pSMAD3) in a dose-dependent manner.
(B) SMAD2/3 reporter assay. A CAGA-luc reporter gene assay treated with either myostatin (GDF8) or GDF11 was used to assess recombinant protein activity. Data are expressed as chemiluminescence units, relative to untreated, and shown as means ± SEM. Both myostatin and GDF11 were shown to be active.
(C) Human myoblast differentiation assay. Myoblasts were differentiated into myotubes without any treatment (−) as a negative control, TNF-α (30ng/ml) as a positive control, and two concentrations of myostatin (GDF8) (10 ng/ml and 300 ng/ml) and GDF11 (10 ng/ml and 300 ng/ml). Biological triplicates are shown for each treatment, with myotubes identified using anti-MyHC antibody staining. Both myostatin and GDF11 can block myoblast differentiation. Differentiation was quantified in the bar graph by evaluating the percentage of nuclei within myotubes that were positively identified using anti-MyHC antibody staining. Differences between groups were analyzed using one-way ANOVA (compared to UNT group). Data are means ± SEM. Values were considered statistically significant at p < 0.05 (*).
GDF11 and Myostatin Both Inhibit Myoblast Differentiation
We next sought to compare the phenotypic effects of myostatin and GDF11 in hSkMDCs, in a muscle cell culture system from human adult donor cells, to see if they might have divergent effects on differentiation. This seemed to be relevant since it was claimed that GDF11 improved regeneration in older mice—therefore, one might expect to see an improvement in myoblast differentiation brought on by GDF11 treatment. This was especially of interest since it was previously reported that in fetal donor cells the TGF-β family actually blocks skeletal muscle differentiation—specifically, myostatin, GDF11, TGF-β1, and activin A have all been shown to block myogenic differentiation with varying degrees of potency (Gamer et al., 2001; Ríos et al., 2002; Trendelenburg et al., 2009). Therefore, human primary myoblasts were differentiated for 3 days in either the presence or absence of recombinant myostatin or GDF11 (10 ng/ml or 300 ng/ml). Cell cultures were fixed and immunostained with antibodies against myosin heavy chain (MyHC) (and DAPI to visualize nuclei) to assess myotube formation (Figure 2C). Treatment with either recombinant myostatin or GDF11 led to a decrease in the number of myotubes (Figure 2C). Inhibition of differentiation was observed at both doses for GDF11, while only a high dose of myostatin resulted in a similar effect. The decrease was quantified by determining the percentage of nuclei that were positive for MyHC. Both myostatin and GDF11 significantly decreased the percentage of MyHC-positive fibers (Figure 2C). Therefore GDF11, like myostatin, is a direct inhibitor of skeletal muscle differentiation.
GDF11 and Myostatin Induce Almost Identical Gene Changes
Even though we demonstrated similar direct signaling of GDF11 and myostatin and that this correlated to inhibitory effects by both molecules on the differentiation of human skeletal myoblasts, it was still formally possible that GDF11 might have some unique activity on muscle, distinct from its activation of ActRII/ALK/SMAD signaling. We thought the most unbiased way to approach this possibility was to perform a microarray study on myotubes with and without stimulation with myostatin and GDF11 and to then ask if there were any differences in gene activation. Any unique effect of GDF11 in comparison to myostatin should be captured by seeing a differential downstream modulation of gene expression. Human primary muscle cells were stimulated for 24 hr with 300 ng/ml of myostatin, or GDF11, or buffer alone (as a negative control), and the log fold change in gene expression was examined. The effect on gene expression induced by GDF11 in comparison to myostatin was essentially identical (Figures 3A and 3B). Therefore, we could not detect any unique effect of GDF11 on skeletal muscle cells in comparison to myostatin; both seem to behave identically—and their effects are inhibitory.
Figure 3. Microarray Analysis of hSkMDCs Treated with GDF11 or Myostatin Demonstrate that GDF11 and Myostatin Induce Almost Identical Expression Changes.

(A) The log fold-change (FC) versus control samples for samples (n = 4 biological replicates per group) stimulated for 24 hr with 300 ng/ml myostatin or GDF11 (x and y axes, respectively). Data points are colored by absolute FC difference, with darker points representing larger differences. Reference lines are included for log FC differences of 1/−1 (dashed) and 0 (solid).
(B) Gene expression was generated for hSkMDCs treated with GDF11 or myostatin. There were 243 genes (356 probe sets) regulated by either GDF11 or MSTN. Intensities are shown for vehicle and the two treatments (GDF11 and myostatin; 24 hr at 300 ng/ml). Blue, low expression; red, high expression; gray, median expression. Genes are regulated similarly by GDF11 and MSTN.
GDF11 Inhibits Muscle Regeneration
We next sought to corroborate claims that GDF11 administration improved regenerative capacity of skeletal muscle in aged mice (Sinha et al., 2014). Therefore, 23-month-old C57BL/6 mice were treated with GDF11 (0.1 mg/kg) or Vehicle for 28 days to attempt to “rejuvenate the stem cell pool” prior to receiving a cardiotoxin (CTX) injection into the tibialis anterior muscle. Treatments were continued for an additional 7 days after CTX was administered, and then animals were necropsied. In this initial experiment, the animals were sacrificed at 7 days post-CTX treatment because in the prior study reporting positive effects on regeneration, this 7-day time point was used (Sinha et al., 2014). In our study, no differences were observed in regenerative capacity of skeletal muscle of aged mice treated with GDF11 or Vehicle at the 7-day time point (Figure S4). In addition, the number of Pax7+ cells between control and GDF11-treated aged mice was not different (Figures S5A and S5B). This argues against the “rejuvenating” potential of GDF11 on the aged muscle stem cell pool in vivo.
Next we wanted to determine whether higher systemic levels of GDF11 could positively impact muscle regeneration in settings where GDF11 was lower, which in our study was in younger mice. Therefore, we administered a 3-fold-higher dose of GDF11 (0.3 mg/kg) than what was used in the prior experiment. Sixteen-week-old C57BL/6 mice were administered GDF11 or Vehicle for 3 days prior to receiving a CTX injection into the tibialis anterior muscle, and GDF11 was administered daily throughout the study. Mice were necropsied 14 days following CTX injections. Classically, in this sort of approach, injured fibers are scored by looking for central nuclei, which is a marker of regeneration. When muscle is first formed, the nuclei are in the center of the fiber, and then over the next 50 days (in mice), they migrate to the periphery; therefore, if one observes a nucleus in the center of the fiber, it is a sign that the muscle fiber has recently formed and is actively regenerating. However, in this study, we noticed that in the GDF11-treated muscle, there were fibers that were so small that one could not even appreciate the location of the nuclei—this is a sign of absolutely nascent myofibers. Therefore, we decided to present the fiber distributions both by the usual way, where central nuclei could be definitively determined, and by simply presenting all of the fibers within the area of injury. In regenerating muscle, GDF11 treatment was associated with a greater frequency of small fibers with centralized nuclei (CN), as indicated by a moderate leftward shift in frequency distribution (Figure 4A). This, however, did not result in a significant change in total mean fiber cross-sectional area (Figure 4B). When we narrowed our focus to the smaller fibers with clear central nuclei (≤600 μm2), to see if there was a shift in this population, we observed a statistically significant reduction in the mean area with GDF11 treatment (p = 0.0045; Figure 4C).
Figure 4. GDF11 Delays Regeneration of Tibialis Anterior Muscle following Cardiotoxin Injury.

(A–D) In regenerating fibers positive for centralized nuclei, GDF11 treatment induced (A) a leftward shift in fiber cross-sectional area frequency distribution, but no change in (B) total mean fiber area; however, (C) mean fiber area of fibers with cross-sectional areas ≤600 μm2 was significantly reduced with GDF11 treatment (p = 0.0045). In GDF11-treated mice, regenerating muscles had (D) a greater frequency of smaller-size fibers (positive and undetectable for centralized nuclei). (E and F) Total mean fiber area (E) and mean area of fibers ≤ 600 μm2 (F) was significantly decreased with GDF11 treatment (p = 0.028 and p = 0.014, respectively).
(G) Representative images of regenerating tibialis anterior muscles stained with an anti-laminin antibody (green) and Hoechst (blue) show regions of much smaller fibers that are indicative of delayed regeneration (yellow dashed box) with GDF11 compared to Vehicle treatment.
(H) Representative images of H&E-stained tissues showing region of delayed regeneration with GDF11 treatment. *p < 0.05, **p < 0.01. CN, centralized nuclei. Scale bar, 100 μm.
Next, we expanded our analysis to include all fibers within the area of injury (i.e., fibers that were both positive for CN and those where we could not determine nuclear localization). Interestingly, we observed a striking leftward shift in the GDF11-treated animals (Figure 4D), consistent with the idea that regeneration had been delayed, since there were so many more tiny myofibers. Consequently, mean area was significantly reduced in total fibers (p = 0.028; Figure 4E) and in fibers ≤ 600 μm2 (p = 0.014; Figure 4F) with GDF11 treatment. Histological analysis of tissue sections stained with anti-laminin antibodies and Hoechst showed that muscles from GDF11-treated mice contained numerous pockets of small densely packed fibers that were positive or undetectable for CN (yellow dashed box; Figure 4G), suggesting delayed regeneration. These pockets with small fibers were also evident on H&E-stained muscle sections (Figure 4H). Therefore, in contrast to what had been shown in the previous study (Sinha et al., 2014), higher systemic levels of GDF11 are associated with impaired regeneration, as indicated by a greater number of very small myofibers in the GDF11-treated muscles.
GDF11 Decreased the Growth of Adult and Aged SCs
Finally, we sought to examine the impact of GDF11 on growth and lineage progression of SCs from adult and aged muscle, since an improvement in regenerative capacity could be due to a positive effect on SCs. SCs (Lin−, Inta7+, VCAM+, and PI+) isolated by flow cytometry were cultured in high-serum conditions (20% FBS) in 96-well plates (200 cells per well) in GDF11 or vehicle for 3 days. SC cultures were stained for Pax7, MyoD, and Myogenin (Myog), which are markers of muscle differentiation, and counted using automated software analysis (Figures 5A–5C). Quantification of cell number based on DAPI shows that treatment with GDF11 decreased the growth of adult and aged SC cultures in a dose-dependent manner (Figures 5B and 5C). Analysis of myogenic fate markers did not reveal any significant changes in their expression, suggesting that GDF11 does not impact cell fate of SCs (Figures S5C and S5D). Therefore, GDF11 limits the expansion of adult and aged SCs and their progeny.
Figure 5. GDF11 Limits Satellite Cell Expansion in Adult and Aged Mice.

(A) Representative Pax7 (magenta) and MyoD (green) immunostaining of adult SCs after 3 days in culture treated with GDF11 (50 ng/ml) or vehicle control. DAPI marks myonuclei (white).
(B and C) Histograms show the total number of (B) adult and (C) aged myogenic cells per well after 3 days of GDF11 (15 ng/ml, low or 50 ng/ml, high) or vehicle control treatment in culture (n = 1,645–2,185 cells, performed in triplicate).
(D) Single muscle fibers from control (top) and GDF11-treated (bottom) 3-day cultures show Pax7+ (red) and myogenin (Myog)+ (green) cells. DAPI highlights myonuclei (blue). Arrowheads show representative cells.
(E and F) Data are represented as mean ± SD. Cell growth assays were statistically analyzed by Kruskal-Wallis nonparametric test with Dunn’s post hoc test (*p < 0.05). Student’s t test was performed on single-fiber data followed by a Mann-Whitney U post hoc test (**p < 0.01).
To analyze the growth of SCs in their niche, single muscle fibers were isolated from adult mice and cultured for 3 days in the absence or presence of GDF11 (Figure 5D). Quantification based on Pax7 and Myog immunohistochemistry reveals a significant decline in myogenic cell numbers in GDF11-treated compared to vehicle-treated fibers (Figure 5E). In contrast, lineage commitment was not impacted (Figure 5F). Therefore, GDF11 limits the expansion of adult SCs resident in their niche. This result is inconsistent with a restorative role of GDF11. Rather, treatment with exogenous GDF11 promotes an age-related SC phenotype.
DISCUSSION
Aging is often associated with the onset of frailty, caused by the associated loss of skeletal muscle known as sarcopenia, and a decrease in the ability of SCs to respond to injury by helping regenerate the muscle. One way to approach treating aging-related conditions might be to search for secreted molecules that are regulated with age and then ask whether returning them to levels more commonly seen in younger animals might be beneficial.
That is why it seemed to make sense that in prior studies just such an approach was taken (Loffredo et al., 2013; Sinha et al., 2014). The combination of these two reports demonstrated that the myostatin-related factor GDF11 decreased with age and that treating older mice with GDF11 improved muscle regeneration (Sinha et al., 2014), a process that is impaired in elderly animals, including humans. However, the beneficial finding of GDF11 treatment on skeletal muscle was quite surprising, since GDF11 is closely related structurally to myostatin and myostatin has been well published for its ability to inhibit muscle differentiation and to induce muscle atrophy.
The present study contradicts every aspect of the prior studies as they relate to skeletal muscle. We demonstrate first that GDF11 cannot be shown to decrease in mouse sera; we found that the reagents used in the prior studies are not GDF11 specific, since they pick up myostatin as well, but use of the antibody shows an increase rather than a decrease in GDF11/myostatin levels if you include both GDF11/myostatin bands that the antibody identifies, which includes the active dimer. The upper band was not presented in the prior studies (Loffredo et al., 2013).
Fortunately, we were able to develop a new immunoassay method to specifically measure GDF11 in blood; the validation work for this immunoassay is presented in this study, in the Supplemental Information, where we show that GDF11 and not myostatin can be detected in a quantitative manner, including in serum. Using this immunoassay, we show that GDF11 can be specifically measured in the sera of rats and humans. There is a strong trend for GDF11 increasing in the blood serum of rats, and there is a dramatic increase in GDF11 mRNA in rat skeletal muscle, which undergoes sarcopenia when they age (Ibebunjo et al., 2013). In humans, we found just a trend toward an increase in blood serum, which at the very least shows that GDF11 does not seem to decline in humans as a function of age.
Consistent with the demonstration in the present study that GDF11 increases with age, in a prior study we demonstrated that the downstream myostatin/GDF11 signaling pathway, characterized by SMAD3 phosphorylation, is also elevated in the aged rat (Trendelenburg et al., 2012). Given the combination of these findings, the implication would be that it would be deleterious to add even more GDF11 to an aged animal, as one would do by giving GDF11 as a treatment, since this pathway is inhibitory to muscle regeneration. Our current study validates this concern, since supplementation of GDF11 was found to dramatically inhibit muscle regeneration.
Since there is a trend of GDF11 increasing in sera from humans as a function of age, that finding introduces the question as to whether GDF11 blockade might be helpful to treat age-associated human muscle pathology. What was striking was that there were a couple of aged individuals with especially high levels of GDF11. So perhaps a detection assay would be appropriate in humans, and if they were found to have very high levels of GDF11, and this was coincident with muscle pathology, then those individuals might be candidates for either GDF11-specific blockade or for a more general blockade of the GDF11, myostatin, and activin receptor, ActRII (Lach-Trifilieff et al., 2014).
EXPERIMENTAL PROCEDURES
DELFIA Immunoassay for SOMAmer Binding
SOMAmers against GDF11 and NR3C1 (negative control) were made synthetically (Integrated DNA Technologies) with a photocleavable biotin moiety at the 5′ end and benzyl-modified dUTP nucleotides as previously described (Gold et al., 2010). GDF11 and myostatin (R&D Systems, #1958-GD and #788-G8, respectively) were coated onto DELFIA (dissociation-enhanced lanthanide fluorescent immunoassay) microtitration plates (Perkin Elmer, #1244-550) at 2 μg/ml in 100 mM carbonate buffer (pH 9.6) overnight and washed thoroughly with PBS containing 0.05% Tween 20. SOMAmers were diluted to 10 μM in 5 mM HEPES (pH 7.5), heated to 85°C for 5 min and cooled to room temperature for 15 min, then further diluted in assay buffer (40 mM HEPES, 100 mM NaCl, 5 mM KCl, 5 mM MgCl2, 1 mM EDTA, 0.05% Tween 20, 0.5 μM dextran sulfate, 1% BSA) and added to the protein-coated plates for 30 min. Plates were then washed and incubated with Eu-labeled streptavidin (Perkin Elmer, #1244-360) for 45 min, followed by DELFIA enhancement solution (Perkin Elmer, #1244-105) for 15 min, and read on an EnVision 2103 multilabel reader according to manufacturer recommendations. Data were fit using a three-parameter nonlinear regression curve fit with a fixed Hill slope in GraphPad Prism v. 6.04.
Cell Culture and Protein Treatment
hSkMDCs (adult donor; Cook Myosite) were cultured in growth medium consisting of MyoTonic basal medium (Cook Myosite) supplemented with 20% FBS (Hyclone) and MyoTonic serum-free growth supplement (Cook Myosite). Differentiation was initiated 24 hr after seeding (day 0) by changing to serum-free differentiation medium consisting of MyoTonic differentiation medium (Cook Myosite) containing 2% horse serum (Hyclone) and 1% FBS and altering the atmospheric conditions from 5% to 7.5% CO2. Cells were switched back to growth medium on day 4 for 48–72 hr before returning to differentiation medium prior to treatments of myotubes. To assess effects on hSkMDC differentiation, compounds were added at day 0 at the onset of differentiation, and cells were differentiated into myotubes for 72 hr. To assess effects on mature hSkMDC myotubes, compounds were added either on day 6 for an additional 24 hr or on day 7. For signaling experiments, hSkMDC myotubes were serum starved for 3–4 hr and treated for 30 min prior to harvesting. HEK293T cells stably transfected with pGL3-CAGA12-luc were grown in culture medium containing 10% FBS at 5% CO2 and subsequently seeded in serum-reduced, phenol red-free assay medium (2% FBS) for 24 hr at 7.5% CO2. Cells were stimulated with compounds for another 24 hr prior to analysis. Murine C2C12 myoblasts (American Type Culture Collection [ATCC]) were maintained in defined media. Myoblasts were fused into myotubes at confluence by shifting from growth medium (DMEM/10% fetal bovine serum) to differentiation medium (DMEM/2% horse serum) and altering the atmospheric conditions from 5% to 7.5% CO2. Cells were differentiated into myotubes for 96 hr prior to treatment on day 4 myotubes. C2C12 myotubes were serum starved for 4 hr and treated for 30 min for signaling experiments.
Biochemicals and Antibodies
Myostatin (or GDF8), GDF11, and TNF-α were from R&D Systems. Stock solutions were prepared as per the manufacturer’s instructions, either in PBS with 0.1% BSA (TNF-α) or in 4 mM HCl supplemented with 0.1% bovine serum albumin (BSA; supplemented only for in vitro experiments) (myostatin and GDF11). For immunostaining, primary antibody against MyHC (clone A4.1025) was from Upstate (#05-716); secondary antibody (Alexa Fluor 488 F [AB]) was from Invitrogen (#A11017). For western blot analysis, antibody against GDF11 was from Abcam (#ab124721); antibody against phospho-Smad2 (Ser465/467) (pSmad2) was from Thermo Scientific (#MA5-15122l); antibodies against phospho-Smad3 (Ser423/425) and Smad2/3 were from Cell Signaling (#9520S and #3102, respectively); antibodies against phospho-p38 and p38 were from Cell Signaling (#9211S and 9212S, respectively); antibodies against phospho-Erk1/2 and Erk1/2 were from Cell Signaling (#9101L and 9102L, respectively); antibody against GAPDH was from Genetex (#GTX627408). As secondary antibodies, the following were used: goat anti-rabbit IgG horseradish peroxidase (HRP) and goat anti-mouse IgG HRP from Cell Signaling (#7074S and #7076S, respectively). For the GDF11 immunoassay, standard 96-well MULTI-ARRAY plates and 4× MSD (Meso Scale Discovery) Read Buffer with Surfactant were acquired from MSD. Phosphate-buffered saline (PBS; pH 7.0), PBS with 0.05% v/v Tween 20 (PBST), PBS with 3% BSA, and PBS with 1% BSA were prepared in-house. The mouse anti-human GDF11 (clone 743833) was purchased from R&D Systems. Ruthenium-labeled mouse anti-human GDF11 (clone 743833) was prepared in-house per the manufacturer’s recommendations.
Western Blot Analysis
HSkMDCs and C2C12 cells were homogenized in RIPA lysis buffer (Boston BioProducts) supplemented with protease and phosphatase inhibitors (Roche). Homogenates were centrifuged for 15 min at 4°C (13,000 rpm) and supernatants were collected. Protein quantification was performed using bi-cinchoninic acid (BCA) kit (Pierce). Samples were diluted in SDS-PAGE sample buffer (6× reducing buffer, Boston BioProducts) and denatured for 5 min at 95°C. Equal or equivalent amounts of protein (75 μg for all serum samples; 4 μl of plasma from single mouse per time point, Figure S1B) were loaded per lane of 4%–12% Bis-Tris NuPAGE gels, electrophoresed, and then transferred onto PVDF membranes using a wet transfer technique. Ponceau S stain was applied to PVDF membranes to ensure even transfer, or a second gel was run in parallel for Coomassie staining. Membranes were blocked in Tris-buffered saline (TBS) with 0.05% Tween 20 (TBST) with 5% (wt/vol) nonfat milk powder for 1 hr. Primary antibodies were incubated overnight in TBST with 5% BSA, and secondary antibodies were incubated in TBST with 5% nonfat milk. Immunoreactivity was detected by Amersham ECL western blotting detection reagents (GE Healthcare), Peirce ECL Plus western blotting substrate, or SuperSignal West Femto Maximum Sensitivity Substrate and exposed to film.
GDF11-Specific Assay
High Bind 96-well MULTI-ARRAY plates were coated with 50 μl/well of 2 μg/ml mouse anti-human GDF11 (clone 743833) in PBS overnight at 4°C. After washing with PBST, 300 μl of PBS with 3% BSA was added to each well and incubated with shaking at room temperature for 1–2 hr, followed by washing with PBST. An eight-point calibration curve was prepared using 3-fold dilutions, starting with a prepared sample of 100 ng/ml recombinant human GDF11 in PBS with 1% BSA. PBS with 1% BSA was used as a blank. Samples were diluted 1:2 in PBS with 1% BSA. Fifty microliters of the calibration curve dilutions or samples was added to duplicate wells and incubated with shaking for 3 hr at room temperature. Following a PBST wash, 50 μl of ruthenium-labeled mouse anti-human GDF11 (clone 743833) diluted to 4 μg/ml in PBS with 1% BSA was added to each well and allowed to incubate in the dark with shaking for 2 hr at room temperature. After another PBST wash step and addition of 150 μl/well 2× MSD Read Buffer with Surfactant (4× MSD Reader Buffer with Surfactant diluted 1:2 with distilled water), the plates were read on the SECTOR Imager 6000 plate reader. Sample concentrations were determined using a four-parameter logistic equation and 1/Y2 weighting.
Regeneration Model In Vivo
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the Novartis Institutes for Biomedical Research.
See Supplemental Experimental Procedures for additional procedures.
Supplementary Material
Highlights.
GDF11 inhibits rather than helps muscle regeneration
A GDF11-specific immunoassay shows a trend to GDF11 levels increasing in human and rat sera
GDF11 blockade may be an appropriate treatment for muscle disease
Acknowledgments
Thank you to Jiang Zhu for additional bioinformatics analysis, to Hua Li for help in processing tissues, and to Bo Guo for help with the SOMAmer binding assay. We thank the Muscle Diseases Group and the Aging Pathways Group at the Novartis Institutes for Biomedical Research (NIBR) for their enthusiastic support, along with the rest of the NIBR community. The portion of the work done by A.S.B. was supported by NIH grants R01 AR060868 and R01 AR061002. All authors except for H.N.N. and A.S.B. are employees of Novartis, and some are also stockholders of Novartis.
Footnotes
ACCESSION NUMBERS
The accession number for the data reported in this paper is GEO: GSE67326.
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2015.05.010.
H.N.N. and A.S.B. declare no conflicts.
References
- Bass J, Oldham J, Sharma M, Kambadur R. Growth factors controlling muscle development. Domest Anim Endocrinol. 1999;17:191–197. doi: 10.1016/s0739-7240(99)00036-3. [DOI] [PubMed] [Google Scholar]
- Beggs ML, Nagarajan R, Taylor-Jones JM, Nolen G, Macnicol M, Peterson CA. Alterations in the TGFbeta signaling pathway in myogenic progenitors with age. Aging Cell. 2004;3:353–361. doi: 10.1111/j.1474-9728.2004.00135.x. [DOI] [PubMed] [Google Scholar]
- Carlson ME, Conboy MJ, Hsu M, Barchas L, Jeong J, Agrawal A, Mikels AJ, Agrawal S, Schaffer DV, Conboy IM. Relative roles of TGF-beta1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell. 2009;8:676–689. doi: 10.1111/j.1474-9726.2009.00517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerman MA, Glass DJ. Signaling pathways controlling skeletal muscle mass. Crit Rev Biochem Mol Biol. 2014;49:59–68. doi: 10.3109/10409238.2013.857291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamer LW, Cox KA, Small C, Rosen V. Gdf11 is a negative regulator of chondrogenesis and myogenesis in the developing chick limb. Dev Biol. 2001;229:407–420. doi: 10.1006/dbio.2000.9981. [DOI] [PubMed] [Google Scholar]
- Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE. 2010;5:e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M, et al. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci USA. 1998;95:14938–14943. doi: 10.1073/pnas.95.25.14938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JJ, Davies MV, Pearson AA, Wang JH, Hewick RM, Wolfman NM, Qiu Y. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem. 2002;277:40735–40741. doi: 10.1074/jbc.M206379200. [DOI] [PubMed] [Google Scholar]
- Ibebunjo C, Chick JM, Kendall T, Eash JK, Li C, Zhang Y, Vickers C, Wu Z, Clarke BA, Shi J, et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol Cell Biol. 2013;33:194–212. doi: 10.1128/MCB.01036-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji S, Losinski RL, Cornelius SG, Frank GR, Willis GM, Gerrard DE, Depreux FF, Spurlock ME. Myostatin expression in porcine tissues: tissue specificity and developmental and postnatal regulation. Am J Physiol. 1998;275:R1265–R1273. doi: 10.1152/ajpregu.1998.275.4.R1265. [DOI] [PubMed] [Google Scholar]
- Kambadur R, Sharma M, Smith TP, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 1997;7:910–916. doi: 10.1101/gr.7.9.910. [DOI] [PubMed] [Google Scholar]
- Lach-Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, Brachat S, Rivet H, Koelbing C, Morvan F, et al. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014;34:606–618. doi: 10.1128/MCB.01307-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ. Extracellular Regulation of Myostatin: A Molecular Rheostat for Muscle Mass. Immunol Endocr Metab Agents Med Chem. 2010;10:183–194. doi: 10.2174/187152210793663748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Lee YS, Zimmers TA, Soleimani A, Matzuk MM, Tsuchida K, Cohn RD, Barton ER. Regulation of muscle mass by follistatin and activins. Mol Endocrinol. 2010;24:1998–2008. doi: 10.1210/me.2010-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P, Sinha M, Dall’Osso C, Khong D, Shadrach JL, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153:828–839. doi: 10.1016/j.cell.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, Cheifetz S, Endo T, Nadal-Ginard B. Type beta transforming growth factor is an inhibitor of myogenic differentiation. Proc Natl Acad Sci USA. 1986;83:8206–8210. doi: 10.1073/pnas.83.21.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA. 1997b;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997a;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat Genet. 1999;22:260–264. doi: 10.1038/10320. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Huynh TV, Lee SJ. Redundancy of myostatin and growth/differentiation factor 11 function. BMC Dev Biol. 2009;9:24. doi: 10.1186/1471-213X-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima M, Toyono T, Akamine A, Joyner A. Expression of growth/differentiation factor 11, a new member of the BMP/TGFbeta superfamily during mouse embryogenesis. Mech Dev. 1999;80:185–189. doi: 10.1016/s0925-4773(98)00205-6. [DOI] [PubMed] [Google Scholar]
- Oh SP, Yeo CY, Lee Y, Schrewe H, Whitman M, Li E. Activin type IIA and IIB receptors mediate Gdf11 signaling in axial vertebral patterning. Genes Dev. 2002;16:2749–2754. doi: 10.1101/gad.1021802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip B, Lu Z, Gao Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell Signal. 2005;17:365–375. doi: 10.1016/j.cellsig.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor β-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23:7230–7242. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos R, Carneiro I, Arce VM, Devesa J. Myostatin is an inhibitor of myogenic differentiation. Am J Physiol Cell Physiol. 2002;282:C993–C999. doi: 10.1152/ajpcell.00372.2001. [DOI] [PubMed] [Google Scholar]
- Roubenoff R, Hughes VA. Sarcopenia: current concepts. J Gerontol A Biol Sci Med Sci. 2000;55:M716–M724. doi: 10.1093/gerona/55.12.m716. [DOI] [PubMed] [Google Scholar]
- Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, Sandri M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol. 2009;296:C1248–C1257. doi: 10.1152/ajpcell.00104.2009. [DOI] [PubMed] [Google Scholar]
- Schneyer AL, Sidis Y, Gulati A, Sun JL, Keutmann H, Krasney PA. Differential antagonism of activin, myostatin and growth and differentiation factor 11 by wild-type and mutant follistatin. Endocrinology. 2008;149:4589–4595. doi: 10.1210/en.2008-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, Miller C, Regalado SG, Loffredo FS, Pancoast JR, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344:649–652. doi: 10.1126/science.1251152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–C1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Jacobi C, Feige JN, Glass DJ. TAK-1/p38/nNFκB signaling inhibits myoblast differentiation by increasing levels of Activin A. Skelet Muscle. 2012;2:3. doi: 10.1186/2044-5040-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida K, Nakatani M, Uezumi A, Murakami T, Cui X. Signal transduction pathway through activin receptors as a therapeutic target of musculoskeletal diseases and cancer. Endocr J. 2008;55:11–21. doi: 10.1507/endocrj.kr-110. [DOI] [PubMed] [Google Scholar]
- Yang W, Chen Y, Zhang Y, Wang X, Yang N, Zhu D. Extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase pathway is involved in myostatin-regulated differentiation repression. Cancer Res. 2006;66:1320–1326. doi: 10.1158/0008-5472.CAN-05-3060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.