

Abstract
Nickel exposure is associated with changes in cellular energy metabolism which may contribute to its carcinogenic properties. Here, we demonstrate that nickel strongly represses mitochondrial fatty acid oxidation—the pathway by which fatty acids are catabolized for energy—in both primary human lung fibroblasts and mouse embryonic fibroblasts. At the concentrations used, nickel suppresses fatty acid oxidation without globally suppressing mitochondrial function as evidenced by increased glucose oxidation to CO2. Pre-treatment with L-carnitine, previously shown to prevent nickel-induced mitochondrial dysfunction in neuroblastoma cells, did not prevent the inhibition of fatty acid oxidation. The effect of nickel on fatty acid oxidation occurred only with prolonged exposure (>5 hr), suggesting that direct inhibition of the active sites of metabolic enzymes is not the mechanism of action. Nickel is a known hypoxia-mimetic that activates hypoxia inducible factor-1α (HIF1α). Nickel-induced inhibition of fatty acid oxidation was blunted in HIF1α knockout fibroblasts, implicating HIF1α as one contributor to the mechanism. Additionally, nickel down-regulated the protein levels of the key fatty acid oxidation enzyme very long-chain acyl-CoA dehydrogenase (VLCAD) in a dose-dependent fashion. In conclusion, inhibition of fatty acid oxidation by nickel, concurrent with increased glucose metabolism, represents a form of metabolic reprogramming that may contribute to nickel-induced carcinogenesis.
Keywords: nickel, mitochondria, fatty acid oxidation, lung fibroblast, hypoxia inducible factor-1α, very long-chain acyl-CoA dehydrogenase
1. Introduction
Nickel compounds represent an environmental threat to human health [1,2]. Nickel is widely used in industrial processes including electroplating, electroforming, and welding. The use of nickel is increasing due to its presence in stainless steel and particularly in nickel-cadmium rechargeable batteries. The lungs and skin are the major target organs for human nickel exposure [3]. Nickel concentrations in the air can reach high levels at industrial workplaces where it is used [4]. Additionally, nickel is present in cigarette smoke [5]. Inhalation exposure to nickel is associated with pulmonary inflammation, epithelial hyperplasia, fibrosis, asthma, and lung cancers [1,2,3]. The mechanisms of nickel-induced carcinogenesis are not clear, but may involve damage to mitochondria [6,7]. It has been proposed that stabilization of hypoxia inducible factor-1α (HIF1α) by nickel leads to hypoxia-like alterations in energy metabolism such as enhancement of glycolysis and repression of the TCA cycle [8,9]. In one study, the negative effects of nickel on mitochondrial function could be prevented with carnitine treatment [10]. The primary biological role for carnitine is to facilitate transport of fatty acids across the mitochondrial membrane [11]. However, the effects of nickel on mitochondrial fatty acid metabolism have not been studied. Here, we have used cell culture models to demonstrate that nickel suppresses mitochondrial fatty acid oxidation (FAO), an important mitochondrial energy metabolism pathway in the heart, muscle, liver, and lung [12,13].
2. Materials & Methods
Cell culture and treatments
Human lung fibroblasts (HLF) were isolated as outgrowths from explanted surplus transbronchial biopsy tissues obtained during routine follow-up bronchoscopy of lung transplant recipients as previously described in accordance with a protocol approved by the University of Pittsburgh Institutional Review Board [14]. Wild-type and HIF1α knockout MEFs were a gift of Dr. John LaPres. Nickel sulfate treatments were conducted as described, at the concentrations and times indicated in the text and figure legends. L-carnitine pre-treatments were conducted as described by He, et al [10].
Substrate oxidation
The oxidation of 14C-palmitate, 14C-glucose, and 14C-palmitoylcarnitine (Perkin Elmer) to 14CO2 and acid-soluble short-chain metabolites (ASM) was conducted in quadruplicate in 24-well sealed trapping plates as described [15]. Rates of metabolism were normalized to cellular protein content. After one hour of exposure to radiolabeled substrates, the wells were acidified by injection of perchloric acid, and then the plates were further incubated at 37°C for two hours to trap the 14CO2.
Western blotting
Western blotting was conducted as previously described [15]. Antibodies used were: anti-very long-chain acyl-CoA dehydrogenase (VLCAD) and anti-log-chain acyl-CoA dehydrogenase (LCAD) (1:1000; gifts of Dr. Jerry Vockley), anti-acetyllysine antibody (1:1000; Cell Signaling Technology, Danvers, MA), and a respiratory chain antibody cocktail (1:1000; Mitosciences, Eugene, OR). Staining of the membranes with Ponceau S was used to verify equal loading.
Acyl-CoA dehydrogenase activity assays
Acyl-CoA dehydrogenase activity was measured with palmitoyl-CoA (Sigma, St. Louis, MO) as substrate exactly as described [16].
3. Results & Discussion
Inhalation represents a major route of human exposure to nickel. Nickel has previously been shown to induce an inflammatory response in human primary lung fibroblasts (HLFs)[14]. In the present studies we tested the effects of nickel on mitochondrial energy metabolism in HLFs. First, HLFs were treated with 200 μM nickel for 48 hours and then assayed for FAO using 14C-palmitate. During the assay, 14C-palmitate is broken down to 14C-acetyl-CoA which then enters the TCA cycle and is ultimately oxidized completely to 14CO2. We measured both the rate of breakdown to 14C-acetyl-CoA/TCA cycle intermediates (acid soluble metabolites) and 14CO2. Both measures indicated that nickel treatment significantly inhibits FAO in cultured HLFs (Fig 1A,B).Nickel has been proposed to inhibit the TCA cycle and induce the Warburg effect, characterized by increased glycolysis concomitant with reduced mitochondrial function [9]. Inhibition of the TCA cycle could secondarily affect the FAO pathway, which is functionally integrated with both the TCA cycle and the electron transport chain [17]. To test this, we measured oxidation of uniformly labeled 14C-glucose to 14CO2 in nickel-treated HLFs. The result showed that in contrast to its inhibitory effect on FAO, nickel significantly promoted the rate of glucose oxidation to CO2 (Fig 1C). This indicates that in HLFs nickel selectively inhibits FAO, and that the inhibition is not due to dysfunction of the TCA cycle. Further, the inhibition of FAO by nickel is not an inherent property of HLFs, because FAO in normal wild-type mouse embryonic fibroblasts (MEFs) was also found to be sensitive to nickel inhibition in a dose-dependent fashion (Fig 1D).
Figure 1. Nickel (Ni) inhibits mitochondrial fatty acid oxidation.
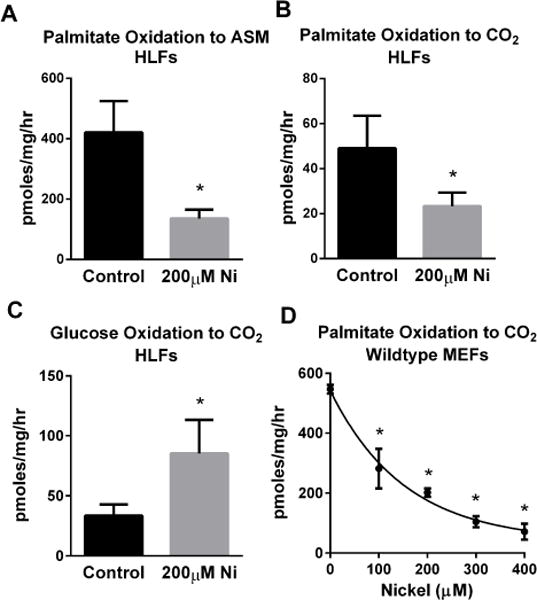
Human lung fibroblasts (HLFs) were treated with 200 μM nickel sulfate for 48 hours prior to measuring 14C-palmitate oxidation to (A) labeled short-chain acid-soluble metabolites (ASM) and (B) trapped 14CO2. (C) Glucose oxidation to 14CO2 is increased by 48 hours of nickel treatment. (D) The effect of nickel is not specific to HLFs, as palmitate oxidation was dose-dependently reduced by nickel sulfate in mouse embryonic fibroblasts (MEFs). All data are presented as the means and standard deviations of quadruplicate assays. *P <0.05 versus untreated control cells.
We next set out to determine the mechanism(s) by which nickel treatment might lead to selective inhibition of mitochondrial FAO. The rate-limiting step for FAO is carnitine-dependent transport across the mitochondrial membrane, mediated by the enzymes carnitine palmitoyltransferase-1 (CPT1) and carnitine palmitoyltransferase-2 (CPT2)[18]. He, et al [10] recently showed that pre-treatment of neuroblastoma cells with carnitine could prevent the cytotoxicity and mitochondrial dysfunction induced by high doses of nickel. Because of the known function of carnitine in the transport of fatty acids, we reasoned that nickel may reduce FAO by inhibiting CPT1 on the outer mitochondrial membrane. To explore this mechanism we measured the effects of nickel on the oxidation of 14C-palmitoylcarnitine, which can bypass CPT1 and directly traverse the mitochondrial membrane. MEFs were incubated with 400 μM nickel for 24 hr and then 14C-palmitoylcarnitine was added to the media. The rate of palmitoylcarnitine oxidation to CO2 was significantly inhibited by nickel (Fig 2A), excluding CPT1 as a direct nickel target. Furthermore, pre-treatment of MEFs with carnitine prior to the nickel treatment did not rescue FAO (Fig 2B), consistent with the idea that nickel does not inhibit FAO via effects on the carnitine-dependent transport steps.
Figure 2. Nickel (Ni) does not inhibit fatty acid oxidation (FAO) via direct effects on mitochondrial FAO enzyme activities.
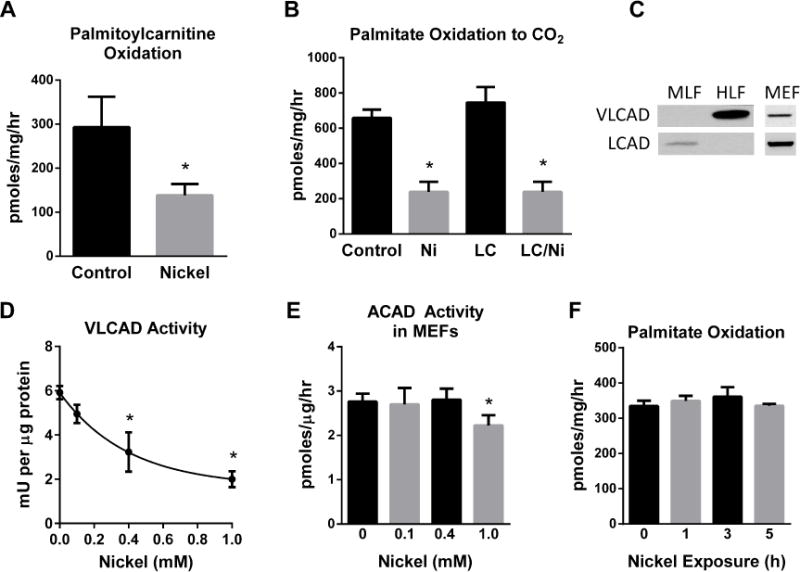
FAO is carnitine-dependent. (A) If nickel inhibited entry of fatty acids into mitochondria via the carnitine shuttle, then palmitoylcarnitine oxidation should not be inhibited by nickel, but the inhibition persists. (B) Administration of 1 mM L-carnitine (LC) with Ni does not relieve the inhibition of FAO. (C) VLCAD is the only acyl-CoA dehydrogenase expressed in human lung fibroblasts (HLF) while LCAD is expressed in murine lung fibroblasts (MLF). Mouse embryonic fibroblasts (MEF) express both acyl-CoA dehydrogenases. (D) Enzymatic activity of purified recombinant VLCAD is dose-dependently inhibited by Ni doses greater than 400 μM, but in (E) extracts of Ni-treated MEFs it takes ≥1 mM Ni to reduce the combined acyl-CoA dehydrogenase activity (LCAD + VLCAD). (F) Short-term treatment of MEFs with 400 μM Ni has no effect on FAO. All data are presented as the means and standard deviations of quadruplicate assays. *P <0.05 versus untreated control cells.
Once inside the mitochondria, long-chain fatty acids are chain-shortened in four enzymatic steps. The rate-limiting of the four steps is the first step, catalyzed by the acyl-CoA dehydrogenases [12,18]. Long-chain acyl-CoA dehydrogenase (LCAD) and very long-chain acyl-CoA dehydrogenase (VLCAD) are the major long-chain enzymes in the family, showing significant overlap in their utilization of long-chain substrates [18]. In humans, VLCAD predominates in most tissue types while LCAD predominates in rodent tissues [19]. HLFs were found to express VLCAD but not LCAD, while murine lung fibroblasts (MLFs) express only LCAD, and MEFs express both enzymes (Fig 2C). Due to the dominant role of VLCAD in HLFs, we tested whether nickel could directly interfere with its enzymatic activity. Incubation of purified recombinant VLCAD with increasing concentrations of nickel reduced its ability to dehydrogenate palmitoyl-CoA and pass electrons to its redox partner electron transferring flavoprotein (ETF)(Fig 2D). However, in extracts prepared from MEFs treated with nickel for 24 hr, there was no significant loss of acyl-CoA dehydrogenase activity toward palmitoyl-CoA at nickel concentrations less than 1 mM (Fig 2E), whereas the pathway is clearly inhibited at nickel concentrations <400 μM (see Fig 1D). This suggests that the primary mechanism by which nickel blocks FAO in an intact cell does not involve direct inhibition of acyl-CoA dehydrogenase enzymatic activity. Moreover, short-term treatment of MEFs with 400 μM nickel does not inhibit FAO (Fig 2F), indicating that nickel impinges on the FAO pathway through mechanisms that require prolonged exposure rather than through acute inhibition of metabolic enzymes.
We further explored mechanisms involving chronic nickel exposure, such as changes in gene expression or mitochondrial content. As an indicator of mitochondrial content, extracts from nickel-treated MEFS were western-blotted with an antibody cocktail recognizing several subunits of the respiratory chain. No significant effects of nickel were seen on the amounts of the respiratory chain subunits (Fig 3A, top panel). Protein acetylation is another mechanism known to inhibit mitochondrial FAO [20]. Nickel had no effect on the total amount of mitochondrial protein acetylation (Fig 3A, middle panel). Nickel treatment did, however, modestly reduce the abundance of VLCAD in nickel-treated MEFs in a dose-dependent fashion (Fig 3B). LCAD protein abundance was not affected (Fig 3B), which likely explains the fact that <400 μM of nickel does not cause a measureable loss in combined acyl-CoA dehydrogenase activity toward palmitoyl-CoA as measured in cell extracts (Fig 2E). The selective loss of VLCAD may play a bigger role in HLFs, which do not express LCAD. Nickel did indeed reduce expression of VLCAD in HLFs (Fig 3C).
Figure 3. Nickel reduces expression of VLCAD and the effect of nickel on FAO is partially abrogated in HIF1α knockout cells.
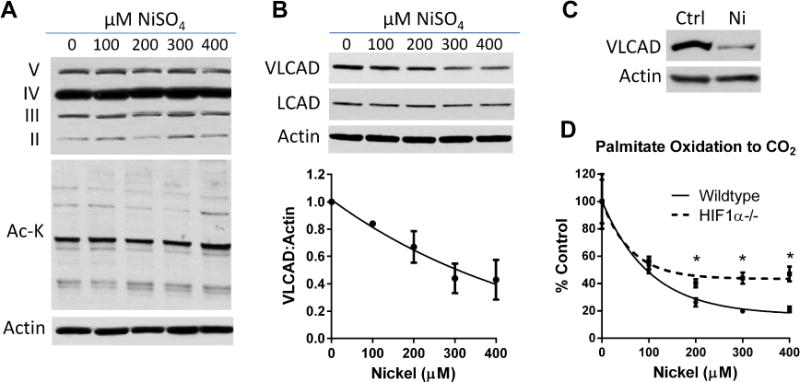
(A) The mechanism of nickel sulfate (NiSO4) on FAO does not involve changes to the abundance of the respiratory chain (top panel) or changes in mitochondrial protein lysine acetylation (Ac-K; middle panel). (B) Nickel dose-dependently reduces VLCAD, but not LCAD, protein levels in MEFs. Densitometry from repeated VLCAD blots was used to generate the line graph. (C) Nickel also reduces VLCAD protein abundance in human lung fibroblasts. (D) Wildtype and HIF1α knockout MEFs were treated with the indicated concentrations of nickel for 24 hrs and palmitate oxidation measured. The effect of nickel is significantly blunted at concentrations of nickel >200μM. *P <0.05, HIF1α−/− versus wildtype cells. Assays were performed in quadruplicate.
There is considerable literature implicating nickel as a hypoxia mimetic that activates hypoxia inducible factor-1α (HIF1α), with many downstream effects that ultimately result in metabolic reprogramming [14,21,22]. We sought to determine whether the effect of nickel on FAO was HIF1α-dependent by comparing the response to nickel between wildtype and HIF1α knockout MEFs. Palmitate oxidation was significantly reduced by nickel in both MEF cell lines, but the effect was attenuated in HIF1α knockout cells (Fig 3D). Thus, activation of HIF1α by nickel is partially responsible for the observed effect on FAO. Other, as yet unidentified mechanisms clearly contribute to the suppression of FAO by nickel.
In HLFs, the cytokine transforming growth factor-β (TGFβ) is a major driver of inflammatory, proliferative, and pro-fibrotic changes in cellular function [23]. TGFβ has been shown to down-regulate expression of nuclear-encoded mitochondrial proteins in A549 lung cancer cells [24]. We hypothesized that TGFβ would suppress FAO similar to nickel. HLFs were treated with 200 μM nickel, 2 ng/mL TGFβ, or both compounds for 48 hr. In contrast to nickel, TGFβ treatment significantly increased FAO (Fig 4A). The rate of FAO in cells treated simultaneously with nickel/TGFβ resembled that of cells receiving nickel alone. With regards to glucose oxidation, both nickel and TGFβ significantly increased the rate of glucose metabolism to CO2, but the effects were not synergistic (Fig 4B).
Figure 4. Effects of nickel and TGFβ on human lung fibroblast metabolism.
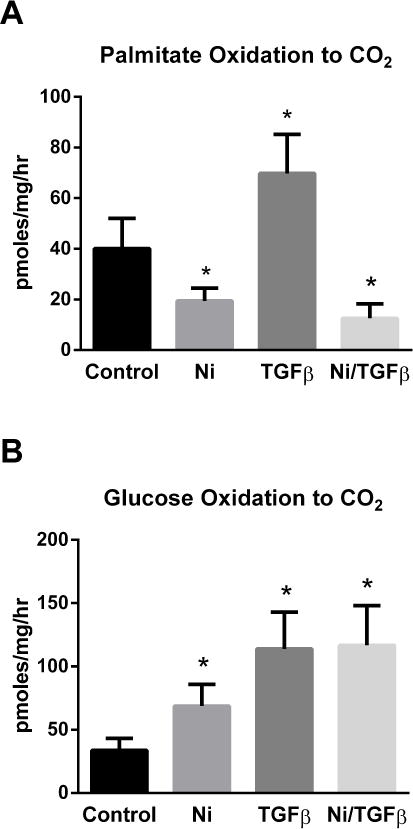
HLFs were grown to confluency and treated with 200 μM nickel, 2 ng/mL TGFβ, or both for 48 hours. (A) 14C-palmitate oxidation, and (B) 14C-glucose oxidation to 14CO2. All data are presented as the means and standard deviations of quadruplicate assays. *P <0.05 versus untreated control cells.
Tumorigenesis is characterized by promotion of anabolic processes which are required for enhanced cellular growth and proliferation, such as lipid synthesis. Lipid synthesis from glucose requires glycolyisis (glucose to pyruvate) as well as mitochondrial pyruvate oxidation to produce acetyl-CoA, which can then be exported back to the cytosol and used for anabolic pathways. Fatty acid synthesis and FAO cannot operate simultaneously. Malonyl-CoA, an intermediate in fatty acid synthesis, effectively inhibits CPT1 to prevent newly synthesized fatty acids from entering the mitochondria and being degraded [18]. The suppressive effects of nickel on mitochondrial FAO may contribute to its ability to drive tumor formation by consequently creating a permissive environment for fatty acid synthesis [25]. TGFβ, in contrast, drives both glucose metabolism and FAO (Fig 4). TGFβ can promote proliferation but the effect appears to be specific to fibroblasts, as proliferation of epithelial cells is potently inhibited by TGFβ [26]. While TGFβ has been shown to promote progression of advanced tumors, it shows suppressive effects with regards to tumor initiation [26]. The fact that nickel can override TGFβ and inhibit FAO when co-administered to HLFs suggests that nickel may also override the tumor suppressive benefits of TGFβ during cancer initiation. Future work is needed to determine the importance of metabolic reprogramming in nickel-associated cancers, and whether targeting the FAO pathway may be of therapeutic benefit in counteracting the carcinogenic effects of nickel.
Highlights.
Nickel is an environmental threat linked to lung inflammation, fibrosis, and cancer.
Nickel alters cellular metabolism by inhibiting fatty acid oxidation.
This effect is partially dependent upon hypoxia inducible factor-1α.
Nickel down-regulates expression of a key fatty acid oxidation enzyme.
Acknowledgments
This work was supported by National Institutes of Health grant DK090242 (E.S.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schaumloffel D. Nickel species: analysis and toxic effects. J Trace Elem Med Biol. 2012;26:1–6. doi: 10.1016/j.jtemb.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Zhao J, Shi X, Castranova V, Ding M. Occupational toxicology of nickel and nickel compounds. J Environ Pathol Toxicol Oncol. 2009;28:177–208. doi: 10.1615/jenvironpatholtoxicoloncol.v28.i3.10. [DOI] [PubMed] [Google Scholar]
- 3.Cameron KS, Buchner V, Tchounwou PB. Exploring the molecular mechanisms of nickel-induced genotoxicity and carcinogenicity: a literature review. Rev Environ Health. 2011;26:81–92. doi: 10.1515/reveh.2011.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pasanen K, Pukkala E, Turunen AW, Patama T, Jussila I, Makkonen S, Salonen RO, Verkasalo PK. Mortality among population with exposure to industrial air pollution containing nickel and other toxic metals. J Occup Environ Med. 2012;54:583–591. doi: 10.1097/JOM.0b013e3182492050. [DOI] [PubMed] [Google Scholar]
- 5.Caruso RV, O’Connor RJ, Stephens WE, Cummings KM, Fong GT. Toxic metal concentrations in cigarettes obtained from U.S. smokers in 2009: results from the International Tobacco Control (ITC) United States survey cohort. Int J Environ Res Public Health. 2014;11:202–217. doi: 10.3390/ijerph110100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu HC, Yang CY, Hung DZ, Su CC, Chen KL, Yen CC, Ho TJ, Su YC, Huang CF, Chen CH, Tsai LM, Chen YW. Nickel(II) induced JNK activation-regulated mitochondria-dependent apoptotic pathway leading to cultured rat pancreatic beta-cell death. Toxicology. 2011;289:103–111. doi: 10.1016/j.tox.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Forti E, Salovaara S, Cetin Y, Bulgheroni A, Tessadri R, Jennings P, Pfaller W, Prieto P. In vitro evaluation of the toxicity induced by nickel soluble and particulate forms in human airway epithelial cells. Toxicol In Vitro. 2011;25:454–461. doi: 10.1016/j.tiv.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 8.He M, Lu Y, Xu S, Mao L, Zhang L, Duan W, Liu C, Pi H, Zhang Y, Zhong M, Yu Z, Zhou Z. MiRNA-210 modulates a nickel-induced cellular energy metabolism shift by repressing the iron-sulfur cluster assembly proteins ISCU1/2 in Neuro-2a cells. Cell Death Dis. 2014;5:e1090. doi: 10.1038/cddis.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, Costa M. Effect of soluble nickel on cellular energy metabolism in A549 cells. Exp Biol Med (Maywood) 2006;231:1474–1480. doi: 10.1177/153537020623100905. [DOI] [PubMed] [Google Scholar]
- 10.He MD, Xu SC, Lu YH, Li L, Zhong M, Zhang YW, Wang Y, Li M, Yang J, Zhang GB, Yu ZP, Zhou Z. L-carnitine protects against nickel-induced neurotoxicity by maintaining mitochondrial function in Neuro-2a cells. Toxicol Appl Pharmacol. 2011;253:38–44. doi: 10.1016/j.taap.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Reuter SE, Evans AM. Carnitine and acylcarnitines: pharmacokinetic, pharmacological and clinical aspects. Clin Pharmacokinet. 2012;51:553–572. doi: 10.1007/BF03261931. [DOI] [PubMed] [Google Scholar]
- 12.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goetzman ES, Alcorn JF, Bharathi SS, Uppala R, McHugh KJ, Kosmider B, Chen R, Zuo YY, Beck ME, McKinney RW, Skilling H, Suhrie KR, Karunanidhi A, Yeasted R, Otsubo C, Ellis B, Tyurina YY, Kagan VE, Mallampalli RK, Vockley J. Long-chain Acyl-CoA dehydrogenase deficiency as a cause of pulmonary surfactant dysfunction. J Biol Chem. 2014 doi: 10.1074/jbc.M113.540260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brant KA, Fabisiak JP. Role of HIF1A and CREB1 in Synergistic Release of IL8 by PGE2 and Nickel in Lung Fibroblasts. Am J Respir Cell Mol Biol. 2013 doi: 10.1165/rcmb.2012-0297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Moura MB, Uppala R, Zhang Y, Van Houten B, Goetzman ES. Overexpression of mitochondrial sirtuins alters glycolysis and mitochondrial function in HEK293 cells. PLoS One. 2014;9:e106028. doi: 10.1371/journal.pone.0106028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goetzman ES. The regulation of acyl-CoA dehydrogenases in adipose tissue by rosiglitazone. Obesity (Silver Spring) 2009;17:196–198. doi: 10.1038/oby.2008.467. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Mohsen AW, Mihalik SJ, Goetzman ES, Vockley J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J Biol Chem. 2010;285:29834–29841. doi: 10.1074/jbc.M110.139493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartlett K, Eaton S. Mitochondrial beta-oxidation. Eur J Biochem. 2004;271:462–469. doi: 10.1046/j.1432-1033.2003.03947.x. [DOI] [PubMed] [Google Scholar]
- 19.Chegary M, Brinke H, Ruiter JP, Wijburg FA, Stoll MS, Minkler PE, van Weeghel M, Schulz H, Hoppel CL, Wanders RJ, Houten SM. Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim Biophys Acta. 2009;1791:806–815. doi: 10.1016/j.bbalip.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kessler R. Nickel ENMs activate HIF-1alpha, Environ Health Perspect. 2011;119:A512. doi: 10.1289/ehp.119-a512a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davidson TL, Chen H, Di Toro DM, D’Angelo G, Costa M. Soluble nickel inhibits HIF-prolylhydroxylases creating persistent hypoxic signaling in A549 cells. Mol Carcinog. 2006;45:479–489. doi: 10.1002/mc.20176. [DOI] [PubMed] [Google Scholar]
- 23.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 24.Sohn EJ, Kim J, Hwang Y, Im S, Moon Y, Kang DM. TGF-beta suppresses the expression of genes related to mitochondrial function in lung A549 cells. Cell Mol Biol (Noisy-le-grand) 2012;(Suppl. 58):OL1763–1767. [PubMed] [Google Scholar]
- 25.Mounier C, Bouraoui L, Rassart E. Lipogenesis in cancer progression (review) Int J Oncol. 2014;45:485–492. doi: 10.3892/ijo.2014.2441. [DOI] [PubMed] [Google Scholar]
- 26.Bachman KE, Park BH. Duel nature of TGF-beta signaling: tumor suppressor vs. tumor promoter. Curr Opin Oncol. 2005;17:49–54. doi: 10.1097/01.cco.0000143682.45316.ae. [DOI] [PubMed] [Google Scholar]