

Abstract
Glucocorticoids (GCs) are known to induce apoptosis of leukemia cells via gene regulatory changes affecting key pro-and anti-apoptotic genes. Three genes previously implicated in GC-evoked apoptosis in the CEM human T-cell leukemia model, RCAN1, E4BP4 and BIM, were studied in a panel of human lymphoid and myeloid leukemia cell lines. Of the two RCAN1 transcripts, the synthetic GC Dexamethasone (Dex) selectively upregulates RCAN1-1, but not RCAN1-4, in GC-susceptible Sup-B15, RS4;11, Kasumi-1 cells but not in GC-resistant Sup T1 and Loucy cells. E4BP4 and BIM regulation correlated with that of RCAN1-1. A putative GRE and four EBPREs were identified within 1500bp upstream from the transcription start site of RCAN1-1. GC-refractory CEM C1-15 cells sensitized to GC-evoked apoptosis by ectopic E4BP4 expression, CEM C1-15mE#3, showed restored RCAN1-1 upregulation, suggesting that RCAN1-1 is a downstream target of E4BP4. A model for coordinated regulation of RCAN1-1, E4BP4 and BIM, and their role in GC-evoked apoptosis is proposed.
Keywords: glucocorticoids, leukemia, apoptosis, RCAN1, E4BP4, BIM
INTRODUCTION
Glucocorticoids (GCs) are used widely in chemotherapy either as a primary cytotoxic drug targeting cancer cells (as in acute leukemia, lymphoma and multiple myeloma) or to reduce inflammation, prevent allergic reactions or reduce the side effects of chemotherapy [1, 2]. The pharmacological actions of GC therapy include, among other effects, the suppression of the immune response and loss of the lymphocyte population via apoptosis. Relapse of leukemia after GC therapy is often associated with development of resistant cells that no longer respond to GC-evoked apoptosis [3]. The primary action of GCs is mediated through their interactions with the GR, a transcription factor from the nuclear receptor family, which modulates up or down regulation of genes containing GC Responsive Elements (GRE) or through interactions with other transcription factors, coactivators and corepressors [2]. We [4, 5] and others [6, 7] have analyzed changes in gene expression profiles induced by GCs in an effort to identify candidate genes modulating GC-evoked apoptosis of leukemic lymphoid cells. We have identified a panel of genes, including RCAN1, E4BP4 and Bcl2L11 (BIM) as significantly upregulated in CEM-C7-14 cells which are susceptible to GC-evoked apoptosis, but not in CEM-C1-15 cells, which are refractory to GC-evoked apoptosis [4, 5, 8, 9].
Bim is a BH3-only proapoptotic member of the Bcl-2 family, and its upregulation plays a vital role in apoptosis in response to various stimuli, including GCs, in normal cells as well as in lymphoid and solid tumors [6, 10]. Bim is a direct activator of apoptosis because it can interact with and inhibit the anti-apoptotic members of the Bcl-2 family, as well as promote oligomerization of pro-apoptotic Bak and Bax to open up the mitochondrial apoptosis-induced channel (MAC) [10, 11]. E4BP4 (or NFIL3) is a bZIP transcription factor with a role in anti-inflammatory response, circadian oscillation, apoptosis regulation, and immune cell development [12, 13]. E4BP4 is homologous to the C. elegans pro-apoptotic cell death specification gene ces-2 [14], which facilitates the downstream upregulation of the pro-apoptotic gene egl-1, an ortholog of human BIM. In the CEM cell culture model of human leukemia, we have demonstrated that GC-dependent upregulation of E4BP4 facilitates BIM upregulation and subsequent apoptosis [9].
The RCAN1 gene is located on chromosome 21q22.12, near the minimal critical region implicated in the Down Syndrome phenotype. The gene consists of six exons, and codes for two major protein isoforms, each with a unique first exon, exon 1 (RCAN1-1) or exon 4 (RCAN1-4) and shared exons 5-7 [15]. Additionally, exon 1 has two translation start codons, corresponding to a long and short variant: RCAN1-1L and RCAN1-1S. All RCAN1 isoforms share a highly conserved central region, the FLISPP motif, a potential target for serine phosphorylation, and a C-terminal calcineurin binding motif (PKIIQT), through which it inhibits the calcineurin phosphatase (PP3C) activity [16]. RCAN1-1 expression is upregulated in response to oxidant- stress while Ca2+-induced stress causes RCAN1-4 upregulation [16]. Increased abundance of both isoforms has been reported in Down syndrome, and may contribute to the phenotype of cardiac and immune dysfunction [16, 17]. Down syndrome patients are also susceptible to early onset Alzheimer’s disease, and chronic RCAN1 expression has been shown to promote formation of neurofibrillary tangles and amyloid beta plaques [17, 18]. Transient expression of RCAN1 has been shown to mediate the adaptation to and protection from oxidative and calcium-induced stress [18]. RCAN1 has been shown to be protective in Huntington disease, primarily because of its ability to inhibit calcineurin activity and prevent dephosphorylation of the mutated huntingtin protein, reducing its toxicity [19]. We have previously reported GC-induced upregulation of RCAN1-1, but not RCAN1-4 in CEM cells [8], and have demonstrated that RCAN1-1 binds to and inhibits calcineurin PP3C activity.
In studies reported here, we extended our investigations to a panel of lymphoid and myeloid leukemia cell lines, and demonstrate a correlation between sensitivity to GC-evoked apoptosis and upregulation of RCAN1-1, E4BP4 and BIM. We have identified a novel GC response element (GRE), and potential E4BP4 response elements (EBPRE) within the RCAN1-1 promoter, in addition to previously reported ones [20]. Using a previously characterized [9] mouse E4BP4 expressing CEM C1-15 cell line (CEM C1-15mE#3), we demonstrate that E4BP4 regulates RCAN1-1 expression. We present a model by which E4BP4, BIM and RCAN1-1 coordinately regulate GC-evoked apoptosis in lymphoid cells.
MATERIALS AND METHODS
Leukemic Cell Lines
Sup-B15, RS4;11, Kasumi-1, Sup T1, and Loucy cell lines were obtained from the American Type Culture Collection. Sup-B15 is a human B cell ALL line with a t(9;22) translocation (Philadelphia chromosome). RS4;11 is a human acute leukemia cell line with a t(4;11) chromosomal rearrangement exhibiting B-cell and myeloid lineage. The Kasumi-1 cell line has a t(8;21) translocation representing human AML. The Sup T1 line is a human T lymphoblastic leukemia cell line expressing multiple T- cell markers including CD1a, CD3, CD4, CD5, CD7 and CD8. Loucy cells are human T-cell ALL cells bearing a t(16;20) translocation. CEM C7-14 and CEM C1-15 cells, are derived from CCRF-CEM cells, and are sensitive and resistant, respectively, to GC-evoked apoptosis, and are generously donated by Dr. E. Brad Thompson (UTMB, Galveston). CEM C1-15mE#3 cells are obtained by stable transfection of mouse E4BP4 in CEM C1-15 cells, rendering them sensitive to GC-evoked apoptosis [9], in correlation with BIM upregulation.
Cell Culture
RS4;11, Sup T1, Kasumi-1, Loucy and CEM cells were cultured in RPMI 1640 medium supplemented with 5% (CEM) 10% (RS4;11, Sup T1, Loucy) or 20% (Kasumi-1) FBS. Sup-B15 cells were cultured in Isocove’s modified DMEM with 4mM L-glutamine, 1.5g/L sodium bicarbonate, 0.05mM 2-mercaptoethanol and 20% FBS. All cell lines were maintained at 37°C in a humidified 5% CO2 incubator in log phase between 3×105 cells/ml and 3×106 cells/ml.
Reagents
Dexamethasone (Dex) was purchased from EMD Biosciences (Madison, WI). TRIzol reagent was from Invitrogen Life Technologies (La Jolla, CA). Reagents for reverse transcription, endpoint PCR and Real-time qPCR (RT-qPCR), including M-MLV reverse transcriptase, oligo(dT)15 primer, RNasin®Ribonuclease inhibitor, dNTP mix, and Taq DNA polymerase were purchased from Promega Life Sciences (Madison, WI). SYBR®JumpStartTMTaqReadyMix (Cat #4438) was from Sigma-Aldrich (St. Louis, MO). Other reagent grade chemicals were purchased from Fisher Scientific (Pittsburgh, PA) or Sigma-Aldrich.
Assessment of Cell viability
Cells were plated at a density of 1–5 × 105 cells/ml and treated for 96h with 0.1% ethanol or Dex at a final concentration of 10nM, 100nM, 1μM, or 10μM. Aliquots were removed at 24 h intervals for cell counts. Viable cells were counted by the trypan blue dye exclusion method using a Hemocytometer.
Apoptosis Assay
To confirm that cell death occurred via apoptosis, we employed CF™594 Annexin V and Hoechst 33342 dyes from Biotium (Hayward, CA). Cells were treated with ethanol or 1μM Dex for 33h, washed and incubated for 30 min with CF™594 Annexin V (detects exposed phospatidylserine) and Hoechst 33342 (stains nuclei), and observed using Texas Red (ex/em 593/614) and Hoechst (ex/em 350/461) filters on an Accu-Scope 3025 epifluorescence microscope equipped with a ProgRes MF camera (JENOPTIK, Germany), and the 2.7 ProRes software.
RNA Extraction and Reverse Transcription
All cells were treated at a density of 5 × 105 cells/ml for 24h with either 1μM Dex or 0.1% ethanol. RNA was extracted from approximately 2.5 × 107 cells using TRIzol reagent. For reverse transcription, 7μg of total RNA was incubated for 3h at 42°C in the presence of 0.5μg of oligo(dT)15, 1μl (~200U) of M-MLV reverse transcriptase, 0.5mM dNTP mix, and 100U of RNase inhibitor.
Endpoint and Real-Time Quantitative PCR (RT-PCR) Analysis
End-point PCR was performed for 25 cycles on a GeneAmp® PCR System 9700 (Applied Biosystems, Foster City, CA) using 1μl of the reverse transcription product, 500nM each of the forward and reverse primers (Table 1), 250μM of dNTP mix, 1.5mM MgCl2, and Taq DNA polymerase at 25 U/μl. Amplicons were resolved on a 1% Agarose gel. For RT-qPCR, 1μl of the reverse transcription product was mixed with SYBR® Green JumpStart Taq Ready mix and the appropriate primers (Table 1) in a final volume of 25μl, and run on an Applied Biosystems 730 real-time PCR instrument. Data were analyzed using the freeware program LinRegPCR, to determine efficiency (E) and cycle threshold (CT) values. Relative expression levels and fold induction were quantitated using the Pfaffl method: (E) CTtarget(control-sample)/(E) CTreference(control-sample), where β-actin was the reference gene.
Table 1.
Primers used for PCR
| Transcript | Forward Primer | Reverse Primer | Product Size |
|---|---|---|---|
| RCAN1-1 | 5′-ACCATCGCCTGTCACCTGGA-3′ | 5′-GGTGATGTCCTTGTCATACGTCCT-3′ | 96bp |
| RCAN1-4 | 5′-CTCCCTGATTGCCTGTGTGG-3′ | 5′-TTCCTCTTCTTCCTCCTTCT-3′ | 484bp |
| E4BP4 | 5′ATGGGGAATTCTTTCTCTGG3′ | 5′CTTTGATCCGGAGCTTGTGT3′ | 250bp |
| BIM | 5′CAGATATGCGCCCAGAGATA3′ | 5′ACCAGGCGGACAATGTAAC3′ | 163bp |
| β-actin | 5′AGTCCTCTCCCAAGTCCACA3′ | 5′CACGAAGGCTCATCATTCAA3′ | 130bp |
RESULTS AND DISCUSSION
In the T-lymphoblastic leukemia model of CEM cells, we have previously reported a correlation between GC-evoked apoptosis and upregulation of E4BP4 and BIM, as well as upregulation of RCAN1-1 but not RCAN1-4. We have also demonstrated that E4BP4 at least partially modulated BIM expression. To determine whether genetic pathways for GC sensitivity were comparable in different types of leukemias, we evaluated a panel of well characterized human T- B- and myeloid leukemia cell lines for their sensitivity and dose dependence to Dex, and correlated this with the expression of the two transcripts of RCAN1 as well as E4BP4 and BIM.
Susceptibility of leukemic cell lines to Dex-evoked death
Sup-B15 cells were responsive to Dex in a dose-dependent manner, with 10nM Dex reducing viable cell numbers to 32% of ethanol treated controls by Day 2 and 1μM Dex causing 100% loss of cell viability by day 4. RS4;11 cells were also susceptible to Dex-induced death, with 62% viable cells by day 2 in the 10nM Dex treated group compared to ethanol treated controls, and greater than 95% loss of cell viability at day 4 with 1μM Dex. Kasumi-1 cells were responsive to Dex-mediated cell death, with 10nM Dex reducing viable cells to 65% of ethanol treated controls, and 1μM Dex causing 100% loss of cell viability by day 4. Sup-T1 and. Loucy cells were found to be resistant to Dex-evoked cell death, with Sup-T1 cells having virtually identical growth profiles in ethanol or Dex up to a concentration of 10μM. Loucy cells showed a 50% decrease in viable cells by day 1, but subsequently recovered to exhibit a parallel growth profile to ethanol treated control cells.
Cell death occurs via apoptosis
To confirm that the Dex-induced cell death occurred via apoptosis, cells treated with 1μM Dex for 33h were evaluated for phosphatidylserine exposure to the outer leaflet of the plasma membrane using CF 594 Annexin V. Hoechst 33342 was used to stain the nuclei. Stained cells were observed under an epifluorescence microscope, using appropriate filters, as described in the methods section. Representative images documenting Dex-induced apoptosis of RS4;11 cells are shown in Figure 1, bottom right.
Figure 1. Sensitivity of Leukemia Cell lines to Dex.
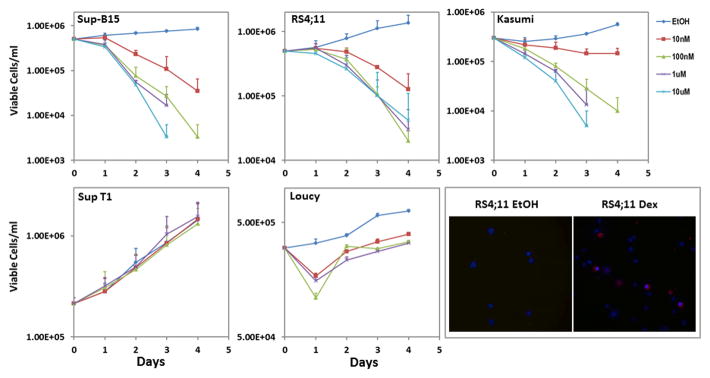
Sup B-15, RS4;11, Kasumi-1, Sup-T1 and Loucy cells were plated at a density of 1 to 5 × 105 cells/ml and treated for 4 days with either ethanol or 10nM, 100nM, 1μM or 10μM Dex. Cells were incubated at 37°C in a humidified 5% CO2 incubator and trypan blue excluding viable cells were counted at 24 hour intervals. Each point represents a mean ± SD of three independent experiments conducted in duplicates. Lower right panel shows representative epifluorescence images of ethanol and Dex treated RS4;11 cells stained for 30 min with CF 594 Annexin V and Hoechst 33342, to detect apoptosis. Cells with phosphatidylserine membrane eversion show a red rim.
Dex-mediated upregulation of RCAN1-1 correlates with apoptosis
RNA extracted from cells treated with ethanol or 1μM Dex was subjected to reverse transcription and RT-qPCR as described in the methods section. The data (Figure 2A) show that RCAN1-1 was induced in cells that are susceptible to apoptosis, but cells that were resistant to Dex treatment showed little to no change in expression of RCAN1-1. The data also show that RCAN1-4 was either not induced or minimally affected by Dex treatment in all cell lines (Figure 2A). Sup-B15 cells showed an 8.8 fold (±2.7) induction of RCAN1-1, and a modest upregulation of RCAN1-4 (2.5 ±1.5 fold). RCAN1-1 was upregulated 5.5 fold (±0.3) by Dex in RS4;11 cells, while RCAN1-4 expression was increased 2.7 fold (±1.3). Expression of RCAN1-1 in Kasumi-1 cells was increased by 4.5 fold (±0.3) upon Dex treatment while RCAN1-4 was induced by 2.2 fold (±0.4). The inductin of RCAN1-4 by Dex could be a consequence of Dex-evoked increase in intracellular Ca2+ levels in correlation with apoptosis. Dex treatment had little to no effect on the Loucy and Sup T1 cell lines. RCAN1-1 expression decreased by 0.7 fold (±0.03) in Sup T1 cells, and was 1.2 fold (±0.5) in Loucy cells in response to Dex. RCAN1-4 expression after Dex treatment was essentially unaffected in both Sup T1 and Loucy cells.
Figure 2. Dex-mediated upregulation of RCAN1-1, E4BP4and BIM in leukemic cells correlates with their ability to undergo apoptosis.
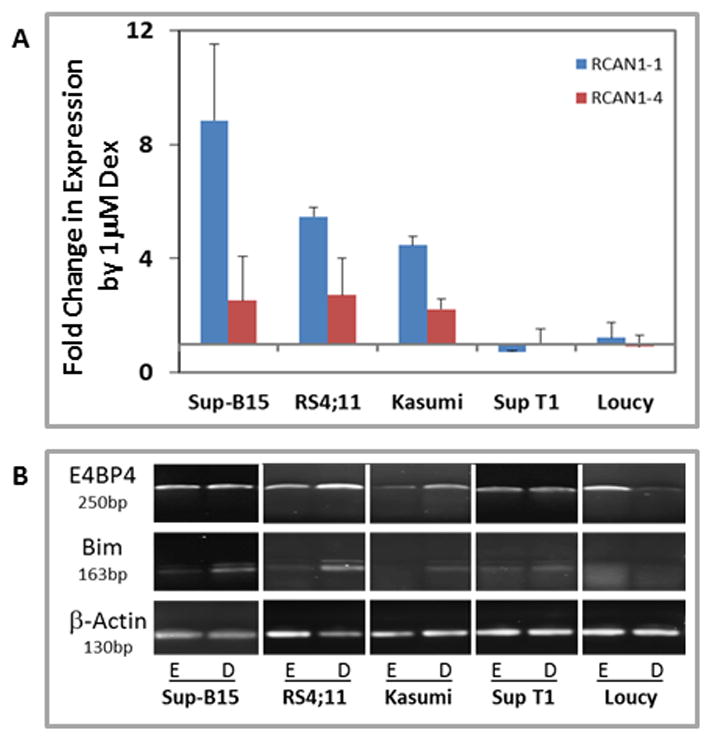
Sup-B15, RS4;11, Kasumi-1, Sup T1 and Loucy cells were treated with ethanol or 1μM Dex and real-time qPCR (Panel A) or endpoint PCR (Panel B) was performed using primers (Table 1) specific for RCAN1-1 and RCAN1-4 (Panel A), and E4BP4 and BIM (Panel B). For Panel A, fold change in expression of each transcript by 1μM Dex was calculated by the Pfaffl method using β-Actin as a reference. Data represent averages ± SD from three independent experiments. Panel B shows representative qualitative endpoint PCR data from one of two independent experiments using β-Actin as a reference.
Dex-mediated upregulation of BIM and E4BP4 in sensitive cells
We [9] and others [6, 21] have previously demonstrated that Dex-evoked apoptosis of leukemic cells is facilitated by upregulation of BIM, with E4BP4 acting as an upstream regulator in the CCRF-CEM human T-lymphoblastic leukemia model [9]. To extend our observations to other T-, B- and myeloid leukemia cell lines, we evaluated the ability of Dex to regulate BIM and E4BP4 in our panel of human leukemia cell lines, using an endpoint PCR assay. We found that E4BP4 expression was clearly upregulated by Dex in the sensitive RS4;11 and Kasumi-1 cells, but slightly induced in the sensitive B-leukemic cell line Sup-B15. There was a clear Dex-induced upregulation of BIM in all three GC-sensitive cell lines (Figure 2B). The GC-refractory Sup T1 and Loucy cells showed no Dex-mediated effect on either BIM or E4BP4 expression. Our data suggest a correlation between GC-evoked apoptosis and upregulation of RCAN1-1, BIM and E4BP4, except in Sup-B15 cells where E4BP4 upregulation was minimal.
Crosstalk between E4BP4 and RCAN1-1
Our laboratory is evaluating the hypothesis that E4BP4 is an upstream transcriptional regulator that modulates expression of key pro- and anti-apoptotic genes. Since RCAN1-1 is regulated coordinately with E4BP4, we sought to determine whether there was any crosstalk between RCAN1-1 and E4BP4. Interestingly, the RCAN1-1 promoter has been reported to have putative E4BP4 response elements (EBPRE) [20]. We searched the RCAN1-1 promoter for sequences resembling consensus EBPRE (Figure 3B) and found five putative EBPREs, including two that have been previously reported (Figure 3A). It is likely that GC-mediated RCAN1-1 expression is driven by E4BP4 via these sequences, along with GRE-driven responses. In addition to the half GRE reported by Sun et al. [20], we identified a full GRE sequence in the RCAN1-1 promoter, suggesting that GCs can directly regulate RCAN1-1 expression. In a preliminary experiment to test whether E4BP4 might regulate RCAN1-1 expression, we utilized the mouse E4BP4 overexpressing CEM C1-15mE#3 cell line [9], derived from the parental GC-resistant CEM C1-15 line, which is sensitized to GC-evoked apoptosis. [9]. Here we demonstrate that CEM C1-15mE#3 have a restored ability for Dex-mediated RCAN1-1 upregulation (Figure 3C). RCAN1-1 transcript levels were induced 13.2-fold in CEM C7-14 cells, 1.9-fold in CEM C1-15 cells, and 11.7-fold in CEM C1-15mE#3 cells, suggesting that E4BP4 overexpression restores GC-dependent RCAN1-1 upregulation. Indeed, RCAN1 expression is regulated in a circadian fashion, in the same phase as E4BP4, according to a study that did not distinguish between RCAN1-1 and RCAN1-4 [22]. In cardiomyocytes, the oscillatory expression of RCAN1-4, but not RCAN1-1, serves as a cardioprotector and a vital mediator of the circadian rhythmicity of cardiovascular susceptibility to ischemia/reperfusion injury [23, 24].
Figure 3. E4BP4 may be a regulator of RCAN1-1 expression.
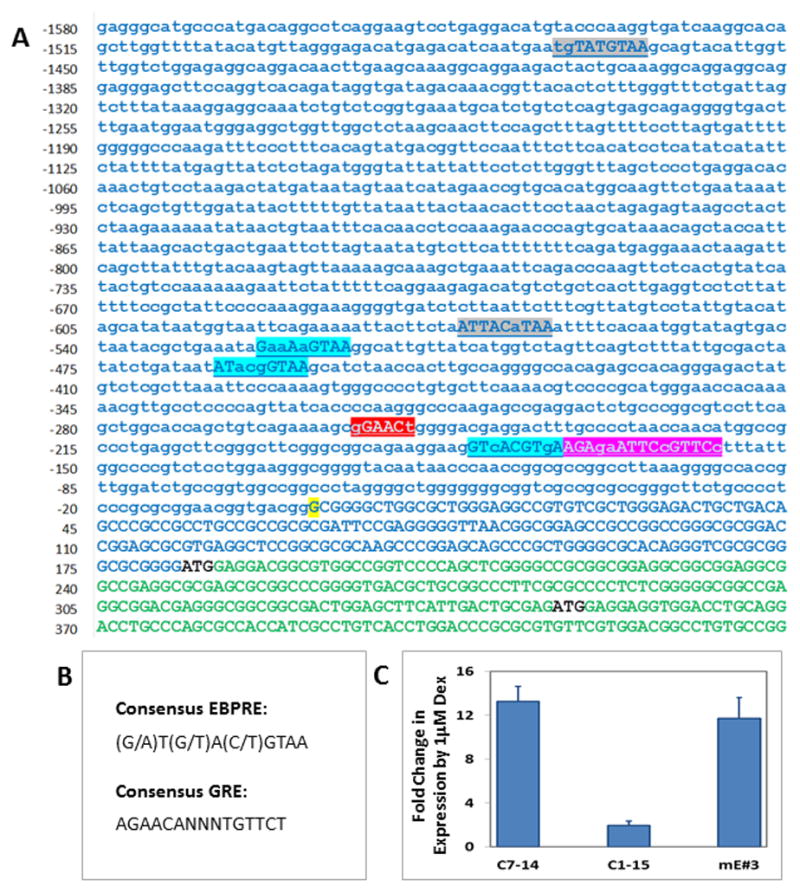
Panel A: Nucleotide sequence of the human RCAN1-1 gene promoter and first exon. Sequence is derived from GenBank Accession #NG_007071. Transcription start site (+1) is highlighted in yellow, and is according to Sun et al.[20]. The first exon is in uppercase green, with the two start codons representing the RCAN1-1L and RCAN1-1S isoforms are in black. The putative GRE (Red and Pink) and EBPRE sequences (grey and blue) are highlighted. The red and grey sequences are those reported by Sun et al.[20]. Panel B: Consensus GRE and EBPRE sequences. Panel C: CEM C7-14, CEM C1-15 and CEM C1-15mE#3cells were treated with ethanol or 1μM Dex and real-time qPCR was performed using primers (Table 1) specific for RCAN1-1. Fold change in expression was calculated by the Pfaffl method using β-Actin as a reference. Data represent averages ± SD from three independent experiments.
Proposed model for genetic coordination of GC-evoked apoptosis
A schematic outlining the cross-talk among the three GC-induced genes, BIM, E4BP4 and RCAN1-1, and their effect on lymphoid cell apoptosis, is presented in Figure 4. GCs are known to modulate their actions via binding to GR in the cytoplasm. The activated GR translocates to the nucleus where it binds as a dimer to conventional GRE sequences on target genes [2] or through other mechanisms. Although the BIM promoter lacks a traditional GRE, GCs regulate its expression via a novel intronic GR binding region [25], and through a region in the 3′UTR, by relieving miRNA mediated post-transcriptional repression [26]. GR-mediated upregulation of E4BP4, and subsequent induction of BIM, has been reported in correlation with apoptosis in lymphoid and osteoblastic cells [9, 27]. GR has been shown to regulate E4BP4 transcription via a GR binding sequence (GBS) located ~5kb upstream of the transcription start site [28, 29]. We have demonstrated that E4BP4 regulates BIM expression [9], however, the mechanism of this regulation is currently being investigated. GREs and EBPREs have been reported in the RCAN1-1 promoter [20], suggesting that RCAN1-1 is both a primary and secondary target of GC-dependent regulation. A fundamental step in GC-evoked apoptosis is the release of cytochrome c from the mitochondria. There are primarily two types of channels that facilitate this process [30]. The mitochondrial apoptosis induced channel (MAC) is formed in the outer mitochondrial membrane by oligomerization of pro-apoptotic members of the Bcl-2 family (Bax and Bak), in the absence of active inhibitory members (Bcl-2, Bcl-xL) [11]. Bim catalyzes the formation of MAC by facilitating Bax oligomerization [11]. Although the precise composition of the mitochondrial permeability transition pore (mPTP) is not clear, there is evidence that adenine nucleotide translocator (ANT), located on the inner mitochondrial membrane, and VDAC (voltage dependent anion channel), located on the outer mitochondrial membrane, form the mPTP [31]. RCAN1-1 is a homolog of the Drosophila protein nebula, which binds to ANT [32]. RCAN1-1L also has been proposed to bind to ANT and its sustained overexpression has been shown to open mPTP, stimulate mitochondrial degradation, and reduce cell survival [17]. RCAN1 is known to bind to and inhibit its PP3C phosphatase activity [16]. Since calcineurin is known to protect T- cells from GC-evoked apoptosis [33], RCAN1-1-mediated inhibition of calcineurin activity promotes apoptosis. Overexpression of RCAN1 has also been shown to activate caspase-3 activity to induce apoptosis [20]. In some models, such as cardiomyocytes, calcineurin facilitates Ca2+ dependent apoptosis, hence activation of RCAN1-4 has been shown to protect cells from apoptosis [23, 24].
Figure 4. Genetic control of GC-evoked apoptosis.
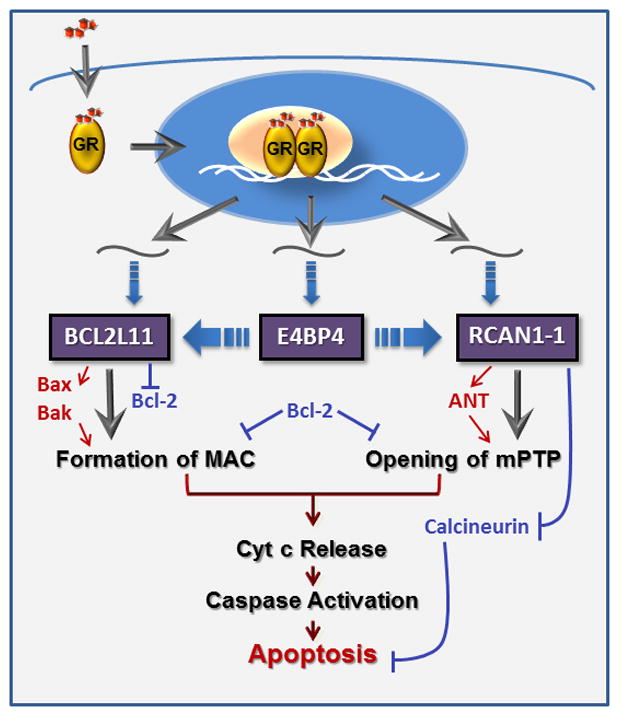
GCs bind to the GR, which dimerizes and translocates to the nucleus (blue oval), where it alters expression of target genes in conjunction with a coregulatory complex (beige oval). Three genes we focus on here are Bcl2L11 (BIM), E4BP4 and RCAN1-1, all of which are direct targets of GR. E4BP4 may modulate BIM and RCAN1-1 transcription. Bim plays a crucial role in formation of MAC, via activation of Bax/Bak and inhibition of Bcl-2. RCAN1-1 binds to and activates ANT to facilitate opening of the mPTP. Bcl-2 inhibits both MAC and mPTP formation. Opening of both channels leads to release of cytochrome c, driving the classic apoptosis cascade.
Acknowledgments
Supported in part by NIH MBRS-SCORE (5SC3GM 081099) grant awarded to RDM, the CSUN Office of Graduate Studies, Research and International Programs, and the CSUN College of Science & Mathematics. GJS performed some of this work as part of his MS thesis project at CSUN.
Contributor Information
G. Jonatan Saenz, Email: Gustavo.J.Saenz@kp.org.
Rebeka Hovanessian, Email: rebeka888@gmail.com.
Andrew D. Gisis, Email: andrew.d.gisis@my.csun.edu.
Rheem D. Medh, Email: rheem.medh@csun.edu.
References
- 1.Pui CH, Costlow ME, Dahl GV, et al. Response of recurrent acute lymphoblastic leukemia to glucocorticoids: serial studies of receptor content, in vivo cytokinetic changes and clinical responses. Leukemia Res. 1983;7:747–753. doi: 10.1016/0145-2126(83)90068-1. [DOI] [PubMed] [Google Scholar]
- 2.Schaaf MJM, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol. 2002;83:37–48. doi: 10.1016/s0960-0760(02)00263-7. [DOI] [PubMed] [Google Scholar]
- 3.Haarman EG, Kaspers GJL, Veerman AJP. Glucocorticoid resistance in childhood leukaemia: mechanisms and modulation. Br J Hematol. 2003;120:919–929. doi: 10.1046/j.1365-2141.2003.04189.x. [DOI] [PubMed] [Google Scholar]
- 4.Beach JA, Nary LJ, Hovanessian R, Medh RD. Correlation of glucocorticoid-mediated E4BP4 upregulation with altered expression of pro- and anti-apoptotic genes in CEM human lymphoblastic leukemia cells. Biochem Biophys Res Commun. 2014;451:382–388. doi: 10.1016/j.bbrc.2014.07.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Medh RD, Webb MS, Miller AL, et al. Gene expression profile of human lymphoid CEM cells sensitive and resistant to glucocorticoid-evoked apoptosis. Genomics. 2003;81:543–555. doi: 10.1016/s0888-7543(03)00045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Malone MH, He H, et al. Microarray analysis uncovers the induction of the proapoptotic BH3-only protein BIM in multiple models of glucocorticoid-induced apoptosis. J Biol Chem. 2003;278:23861–23867. doi: 10.1074/jbc.M301843200. [DOI] [PubMed] [Google Scholar]
- 7.Miller AL, Komak S, Webb MS, et al. Gene expression profiling of leukemic cells and primary thymocytes predicts a signature for apoptotic sensitivity to glucocorticoids. Cancer Cell International. 2007;7 doi: 10.1186/1475-2867-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirakawa Y, Nary LJ, Medh RD. Glucocorticoid evoked upregulation of RCAN1-1 in human leukemic CEM cells susceptible to apoptosis. J Mol Signaling. 2009;4:6. doi: 10.1186/1750-2187-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beach JA, Nary LJ, Hirakawa Y, et al. E4BP4 facilitates glucocorticoid-evoked apoptosis of human leukemic CEM cells via upregulation of BIM. J Mol Signaling. 2011;6:13. doi: 10.1186/1750-2187-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vieira HLA, Boya P, Cohen I, et al. Cell permeable BH3-peptides overcome the cytoprotective effect of Bcl-2 and Bcl-X(L) Oncogene. 2002;21:1963–1977. doi: 10.1038/sj.onc.1205270. [DOI] [PubMed] [Google Scholar]
- 11.Dejean LM, Ryu SY, Martinez-Caballero S, et al. MAC and Bcl-2 family proteins conspire in a deadly plot. Biochim Biophys Acta. 2010;1797:1231–8. doi: 10.1016/j.bbabio.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohno T, Onishi Y, Ishida N. The negative transcription factor E4BP4 is associated with circadian clock protein PERIOD2. Biochem Biophys Res Commun. 2007;354:1010–1015. doi: 10.1016/j.bbrc.2007.01.084. [DOI] [PubMed] [Google Scholar]
- 13.Kamizono S, Duncan GS, Seidel MG, et al. Nfil3/E4bp4 is required for the development and maturation of NK cells in vivo. J Exp Med. 2009;206:2977–2986. doi: 10.1084/jem.20092176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Junghans D, Chauvet S, Buhler E, et al. The CES-2-related transcription factor E4BP4 is an intrinsic regulator of motoneuron growth and survival. Development. 2004;131:4425–4434. doi: 10.1242/dev.01313. [DOI] [PubMed] [Google Scholar]
- 15.Fuentes JJ, Pritchard MA, Estivill X. Genomic organization, alternative splicing, and expression patterns of the DSCR1 (Down syndrome candidate region 1) gene. Genomics. 1997;44:358–361. doi: 10.1006/geno.1997.4866. [DOI] [PubMed] [Google Scholar]
- 16.Fuentes JJ, Genesca L, Kingsbury TJ, et al. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum Mol Gen. 2000;9:1681–1690. doi: 10.1093/hmg/9.11.1681. [DOI] [PubMed] [Google Scholar]
- 17.Ermak G, Davies KJA. Chronic high levels of the RCAN1-1 protein may promote neurodegeneration and Alzheimer disease. Free Rad Biol Med. 2013;62:47–51. doi: 10.1016/j.freeradbiomed.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ermak G, Davies KJA. DSCR1(Adapt78)--a Janus gene providing stress protection but causing Alzheimer’s disease? IUBMB life. 2003;55:29–31. doi: 10.1080/1521654031000066820. [DOI] [PubMed] [Google Scholar]
- 19.Ermak G, Hench KJ, Chang KT, et al. Regulator of calcineurin (RCAN1-1L) is deficient in Huntington disease and protective against mutant huntingtin toxicity in vitro. J Biol Chem. 2009;284:11845–11853. doi: 10.1074/jbc.M900639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun X, Wu Y, Chen B, et al. Regulator of calcineurin 1 (RCAN1) facilitates neuronal apoptosis through caspase-3 activation. J Biol Chem. 2011;286:9049–62. doi: 10.1074/jbc.M110.177519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Insel PA. The pro-apoptotic protein BIM is a convergence point for cAMP/protein kinase A- and glucocorticoid-promoted apoptosis of lymphoid cells. J Biol Chem. 2004;279:20858–20865. doi: 10.1074/jbc.M310643200. [DOI] [PubMed] [Google Scholar]
- 22.Bray MS, Shaw CA, Moore MWS, et al. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Phys. 2008;294:H1036–1047. doi: 10.1152/ajpheart.01291.2007. [DOI] [PubMed] [Google Scholar]
- 23.Rotter D, Grinsfelder DB, Parra V, et al. Calcineurin and its regulator, RCAN1, confer time-of-day changes in susceptibility of the heart to ischemia/reperfusion. J Mol Cell Cardiol. 2014;74:103–111. doi: 10.1016/j.yjmcc.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan L, Yang H, Li Y, et al. Regulator of calcineurin 1-1L protects cardiomyocytes against hypoxia-induced apoptosis via mitophagy. J Cardiovascular Pharmacol. 2014;64:310–317. doi: 10.1097/FJC.0000000000000121. [DOI] [PubMed] [Google Scholar]
- 25.Jing D, Bhadri VA, Beck D, et al. Opposing regulation of BIM and BCL2 controls glucocorticoid-induced apoptosis of pediatric acute lymphoblastic leukemia cells. Blood. 2015;125:273–283. doi: 10.1182/blood-2014-05-576470. [DOI] [PubMed] [Google Scholar]
- 26.Molitoris JK, McColl KS, Distelhorst CW. Glucocorticoid-mediated repression of the oncogenic microRNA cluster miR-17~92 contributes to the induction of BIM and initiation of apoptosis. Mol Endocrinol. 2011;25:409–420. doi: 10.1210/me.2010-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen F, Zhang L, OuYang Y, et al. Glucocorticoid induced osteoblast apoptosis by increasing E4BP4 expression via up-regulation of BIM. Calcified Tissue International. 2014;94:640–647. doi: 10.1007/s00223-014-9847-6. [DOI] [PubMed] [Google Scholar]
- 28.Carey KT, Tan KH, Ng J, et al. Nfil3 is a glucocorticoid-regulated gene required for glucocorticoid-induced apoptosis in male murine T cells. Endocrinol. 2013;154:1540–1552. doi: 10.1210/en.2012-1820. [DOI] [PubMed] [Google Scholar]
- 29.So AYL, Bernal TU, Pillsbury ML, et al. Glucocorticoid regulation of the circadian clock modulates glucose homeostasis. Proc Natl Acad Sci USA. 2009;106:17582–17587. doi: 10.1073/pnas.0909733106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryu SY, Peixoto PM, Teijido O, et al. Role of mitochondrial ion channels in cell death. BioFactors. 2010;36:255–263. doi: 10.1002/biof.101. [DOI] [PubMed] [Google Scholar]
- 31.Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. 2002;84:153–166. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- 32.Chang KT, Min KT. Drosophila melanogaster homolog of Down syndrome critical region 1 is critical for mitochondrial function. Nature Neurosci. 2005;8:1577–85. doi: 10.1038/nn1564. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Y, Tozawa Y, Iseki R, et al. Calcineurin activation protects T cells from glucocorticoid-induced apoptosis. J Immunol. 1995;154:6346–6354. [PubMed] [Google Scholar]