

Abstract
Clinical brain imaging and postmortem studies provide evidence of structural and functional abnormalities of key limbic and cortical structures in depressed patients, suggesting that spine synapse connectivity is altered in depression. Characterization of the cellular determinants underlying these changes in patients are limited, but studies in rodent models demonstrate alterations of dendrite complexity and spine density and function that could contribute to the morphological and functional alterations observed in humans. Rodent studies demonstrate region specific effects in chronic stress models of depression, including reductions in dendrite complexity and spine density in the hippocampus and prefrontal cortex (PFC) but increases in the basolateral amygdala and nucleus accumbens. Alterations of spine synapse connectivity in these regions are thought to contribute to the behavioral symptoms of depression, including disruption of cognition, mood, emotion, motivation, and reward. Studies of the mechanisms underlying these effects demonstrate a role for altered brain derived neurotrophic factor (BDNF) signaling that regulates synaptic protein synthesis. In contrast, there is evidence that chronic antidepressant treatment can block or reverse the spine synapse alterations caused by stress. Notably, the new fast acting antidepressant ketamine, which produces rapid therapeutic actions in treatment resistant MDD patients, rapidly increases spine synapse number in the PFC of rodents and reverses the effects of chronic stress. The rapid synaptic and behavioral actions of ketamine occur via increased BDNF regulation of synaptic protein synthesis. Together these studies provide evidence for a neurotophic and synaptogenic hypothesis of depression and treatment response and indicate that spine synapse connectivity in key cortical and limbic brain regions is critical for control of mood and emotion.
Keywords: stress, neurotrophic factor, ketamine, antidepressant, glutamate
Introduction
Major depressive disorder (MDD) is a devastating illness that affects approximately 17 percent of the population in the United States causing enormous personal and economic consequences [53]. Moreover, the currently available monaminergic agents have significant limitations, including slow onset of action and low response rate [109]. Despite extensive efforts there have been no new therapeutic medications with novel mechanisms, in part due to the heterogeneity and complexity of depression. Therefore, rationale drug design is not possible without a more complete understanding of the underlying pathophysiology of depression.
Nevertheless, there has been significant progress from clinical and preclinical studies that have provided evidence that depression is associated with loss of neurotrophic factor support that leads to atrophy of neurons and reduced connectivity [23, 24]. Clinical brain imaging and postmortem studies demonstrate structural and functional alterations of several limbic and cortical regions in MDD, including the prefrontal cortex (PFC), hippocampus, cingulate cortex, amygdala, and basal ganglia [75, 99]. These studies demonstrate decreased function and atrophy of certain brain regions, including the PFC and hippocampus, but increased function and altered morphology of other regions, including the subcallosal cingulate and amygdala. Altered connectivity of these regions could contribute to the symptoms of depression, in part via reduced function of the PFC (e.g., decreased reaction time and cognitive function), and increased function of the amygdala (loss of control of emotion and mood, and increased fear, anxiety and hypothalamic-pituitary-adrenal axis reactivity). Altered function and connectivity of PFC and the ventral striatum could also underlie reduced motivation and reward in MDD.
The most consistent structural and functional alterations have been observed in the PFC and hippocampus, where reduced volume is inversely correlated with length of illness, time of treatment, and severity of depression [22, 67]. There is limited evidence from postmortem studies demonstrating decreased neuronal cell body size and atrophy of dendritic processes, although there is one report of decreased synapse number in a small cohort of depressed subjects [47]. Additional postmortem studies are needed to confirm and further characterize the synaptic alterations in MDD as well as related illnesses (e.g., psychotic depression) that could also involve disruption of synaptic connections. However, detailed studies of preclinical models of depression have provided extensive evidence demonstrating that chronic stress causes alterations of the density and function of spine synapses in key limbic and cortical brain regions implicated in depression. Here we provide a review of this literature, as well the mechanisms underlying the regulation of spine synapses by stress. In addition, we discuss the opposing effects of antidepressant treatments, notably novel rapid acting agents that increase spine synapses in the PFC and the functional consequences of these changes.
Spine Structure and Function
The small protrusions of the dendritic surface referred to as “spines” are the principal site of most cortical excitatory synapses. The existence of spines greatly increases the surface area available for synaptic transmission, allowing for a high density of synaptic connections onto individual dendrites and neurons [84]. In addition to providing a physical substrate for synapse formation, the spine functions as a subcellular compartment for postsynaptic responses. Spine heads contain postsynaptic components and are connected to the dendrite via a neck-like structure. The overall shape and volume of spines are important determinants of the extent to which transduced signals are spread within the dendrite, emphasizing a critical relationship between spine morphology and neuronal function [38, 122].
Spines are commonly categorized into morphological subtypes according to size and relative proportions of the spine head and neck. The 3 most commonly categorized types are stubby, thin and mushroom spines [38, 90]. Stubby spines lack a clear distinction between head and neck and are considered an immature type. Thin spines, considered immature with a narrow neck and a relatively small head, are prevalent during development and have a high turnover rate [43]. Large head diameter “mushroom” spines are the most mature and stable, and spine head volume is positively correlated with synapse strength and age. Mushroom spines generally receive synaptic input from large diameter presynaptic terminals, while small thin spines are motile, unstable and form comparatively weak synapses [39, 48, 91, 100]. In addition to these three subtypes there are filopodia, which are long thin spines that lack a distinct head [40, 112]. Filopodia are spontaneously generated spines or spine precursors that are short-lived and thought to provide a substrate for activity-dependent growth and strengthening of spines [37, 65].
Spine synapses are the primary sites for dynamic structural plasticity of excitatory transmission [38, 42]. Spines undergo activity-dependent enlargement and stabilization and persist according to their use, while inactive spines are eliminated [40, 74]. Because different spine types are thought to contribute differently to dendritic excitability, it is important to understand how a particular experimental influence can alter relative proportions of spine types and by extension, influence synaptic function. As illustrated in live imaging studies, spine structure and function are not fixed but are continually changing in a way that is responsive to the current state of the animal and the neuronal environment. Live imaging studies have been particularly interesting in illustrating morphological transitions of spines that occur on a rapid timescale. These studies highlight the reactive and dynamic nature of spine morphology and imply corresponding functional plasticity [40, 87].
Stress Models of Depression
A complete understanding of the underlying disease processes in depression is lacking making it difficult to recapitulate the complete pathophysiology of depression in animal models. Currently used models of depression attempt to produce quantifiable correlates of human symptoms in experimental animals. Examples of measures that can be assessed in rodent behavioral models include motor responses to stress, reward-related responding and social interaction, with the rationale that they reflect levels of helplessness or despair, anhedonia, and social withdrawal, respectively, all relevant to human depression. These measures are most often quantified following chronic stress paradigms. Exposure to stress is a key environmental risk factor associated with the occurrence of depression in humans [51, 52] and stress exposure may interact with genetic risk factors to increase susceptibility to depression [10, 50]. For these reasons, stress exposure, particularly chronic stress, has been used in animal models to reproduce some core components of major depressive disorder. Chronic restraint stress (CRS), chronic unpredictable stress (CUS) and social defeat stress (SDS) are chronic stress paradigms that have been used in adult rodents to produce depressive-like features that are sensitive to antidepressant treatment. Notably, the neurobiological alterations associated with these behavioral models include alterations of the size or volume of cortical and limbic brain structures implicated in depression as well as the number of spine synapses.
Chronic stress exposure alters the structure of cortical and limbic neurons
Many of the brain areas implicated in depression such as hippocampus, prefrontal cortex (PFC), amygdala and nucleus accumbens (NAC) are highly plastic regions that undergo morphological changes as a result of stress exposure. The hippocampus shows a high degree of functional and structural plasticity in response to many types of stimuli including stress [76]. Along with the PFC and amygdala, the hippocampus is an important part of the neurocircuitry that regulates the hypothalamic-pituitary-adrenal (HPA) axis response to stress [110]. HPA axis dysregulation is a common feature of depression and disruption of hippocampal neuronal morphology plays a role in the dysregulation of HPA axis-controlled glucocorticoid release that results from stress exposure [15, 105].
Hippocampus
A prominent morphological effect of chronic stress in the hippocampus is the atrophy of CA3 pyramidal cells, characterized by decreased length and branching of apical dendrites [68, 117]. Stress-induced dendritic atrophy has also been shown in dentate gyrus granule cells and CA1 pyramidal cells of the hippocampus [6, 106]. Stress-induced hippocampal dendritic atrophy is mimicked by and depends on glucocorticoids [71, 119]. Chronic stress also induces deficits in hippocampal-dependent behaviors such as spatial memory in male rats [66] and alters electrophysiologic measures in association with CA3 dendritic atrophy [88, 104]. Behavioral deficits as well as dendritic atrophy are reversible upon termination of stress exposure, emphasizing the plasticity and possibly adaptive nature of these responses [16, 106]. Chronic corticosterone exposure induces basal dendritic atrophy in CA1 pyramidal cells, although this effect does not normalize with a washout period [35].
Prefrontal cortex
The prefrontal cortex exhibits a high degree of experience-dependent plasticity including vulnerability to stress exposure [77]. The medial PFC (mPFC) is functionally important for the integration of cognitive and emotional information and plays a role in attention and response selection [2]. The efferent and afferent connections of mPFC with limbic, striatal and basal forebrain structures emphasize the potential significance of mPFC to the functioning of a broad cortical-limbic network [2]. Stress-induced atrophy of dendrites has been shown for pyramidal cells in layers II/III and V in mPFC. The duration and intensity of the stress exposures producing these effects range from long-term intensive stress (e.g., 6 hr. restraint/day for 21 days) [18, 95, 97] to short-term mild restraint stress (e.g., 20 min restraint/day for 7 days) [9, 46, 62] and chronic mild/variable stress [60, 93]. A general feature of this stress effect is that the apical dendrites are more sensitive to stress than the basal dendrites for pyramidal neurons in layers II/II and also layer V. A similar selective sensitivity of apical dendrites is caused by corticosterone treatment [11, 118]. Interestingly, chronic corticosterone treatment causes a shift in the organization of dendrites towards more proximal regions [11, 118]. Stress-induced dendritic changes in PFC are reversible by the removal of stress. A 21-day stress-free recovery period has been shown to reverse the apical dendritic atrophy in layer II/II cells (prelimbic and cingulate), and was also shown to promote proximal apical dendritic extension in layer V cells in the infralimibic PFC [33, 94].
Other brain regions
The effects of stress on dendritic morphology are not similar in all brain regions examined and instead appear to be region and circuit specific. In contrast to hippocampus and PFC, repeated restraint stress causes dendritic hypertrophy in some areas. In one study, the same CRS that induced apical dendritic atrophy in the mPFC, induced apical dendritic hypertrophy in the lateral orbitofrontal cortex (OFC) [61]. The dendrite change in mPFC, but not OFC was accompanied by a corresponding decrease in function. CRS also increases dendritic arborization in the principal neurons of the basoalteral amygdala and this hypertrophy persists following stress termination [114, 115]. These findings have suggested that increased synaptic efficacy in the amygdala could facilitate the development of increased anxiety-like behavior resulting from stress exposure [113]. Furthermore, CRS induces hypertrophy in the BNST, which is a downstream target of the BLA that participates in stress responses [27, 114].
Another study showed divergent effects of CUS on neuronal structure in associative vs. sensorimotor corticostriatal circuits and that these effects were related to behavioral consequences of the CUS. In this study, CUS resulted in apical dendritic atrophy of layer II/III pyramidal cells in mPFC along with evidence for dendritic atrophy in the dorsal medial striatum that receives input from mPFC [20]. The CUS caused hypertrophy of a sensorimotor striatal circuit that was accompanied by a corresponding shift in behavior away from goal-directed (mPFC-dorsomedial striatum-dependent) toward habitual strategies (sensorimotor cortex-dorsolateral striatum-dependent) [20]. Circuit specificity of stress effects has also been demonstrated within the mPFC; a subpopulation of pyramidal cells in the infralimbic region that project to the BLA, does not show the expected dendritic retraction in response to CRS exposure [101].
Chronic stress alters the number and function of spine synapses
Since spines are integral to neuronal function, it is obviously important to assess the effects of chronic stress on these synaptic structures. As discussed below, the density, morphology, and function of spine synapses are altered in chronic stress models of depression.
Hippocampus
The reported effects of chronic stress on dendritic spines in the hippocampus vary. Stress exposure has been shown to result in spine loss in hippocampus CA3 cells [69, 89, 92, 98, 106, 107], although increased or no change in spine density has also been reported [72, 108]. Stress-induced spine loss has been shown in CA1 (and CA3) along with impaired synaptic transmission and depressive behaviors [49, 92]. Decreased spine densities in CA1 neurons has been associated with depression-like behaviors in a light-induced depression model, in the absence of spine changes in CA3 or dendritic changes in CA3 or CA1 [4]. Chronic corticosterone administration, which results in a depressive phenotype, reduces spine density in CA1 and both the spine and behavioral deficits recover with chronic fluoxetine administration [35, 116]. These changes occurred mainly in thin and stubby spines [116]. Given the differential incidence of depression in women it is notable that spine morphologic changes in response to CRS are opposite in male and female rats [103]. Further studies are needed to determine if sex steroid state, notably fluctuations during the menstrual cycle, postpartum, or postmenopausal lead to increased vulnerability to stress and loss of synapses.
Developmental models of stress exposure have been examined to replicate some of the changes in brain circuitry and subsequently in behavior that may be related to risk for pathology in adults. Studies of early-life stress suggest sexually dimorphic outcomes of developmental stress exposure and temporal dependence. CA1 and CA3 spine densities are sensitive to prenatal restraint stress [73]. In addition, CA1 spine density is negatively correlated with spatial memory in a study of chronic stress and estradiol treatment in female rats, suggesting that experimental influences on spines in CA1 were more important to spatial ability than CA3 dendritic arbor regulation [17].
Medial prefrontal cortex: Layer II/III cells
In the PFC, studies of spine density suggest a loss of synaptic efficacy in response to chronic stress. Layer II/III pyramidal neurons in the prelimbic and anterior cingulate regions show reduced spine density following 21 day CRS [95, 96]. CRS-induced reduced spine density was shown to be partially reversed by a 21 day stress-free recovery period [7].
Similar to the effects of chronic stress on dendrites that are preferential for apical vs. basal, stress-induced reductions in spine density are also more pronounced in apical dendrites and are most prominent distally. Limited spine reductions however, have been shown on basal dendrites following chronic stress [78, 96]. Analysis of spine morphology has shown decreased spine volume and surface area along with an overall shift in the spine population from large to small spines in layer II/III cells after CRS [96]. A CRS-induced loss of thin and stubby spines has also been shown [7]. CUS also results in a preferential loss of large spines in layer II/III cells [6, 78, 93]. Vulnerability of mushroom spines to CUS was demonstrated to occur selectively in a subpopulation of prelimbic cells that project to the bed nuclei of the stria terminalis [93]. The association of this effect with decreased Fos expression and enhanced HPA activation suggested that deficits in this afferent pathway from the prelimbic PFC could be important in regulations of the HPA axis after stress.
A shift in spine characteristics from large to small spines, suggests that stress may impair the ability of spines to mature to a stable state. Functionally, a loss of mature stable spines could translate into a loss in synaptic efficacy. A stress-induced reduction in the population of large-diameter mushroom spines has also been demonstrated in a series of studies in layer V pyramidal cells and this is associated with reduced levels of synaptic proteins and functional deficits as described below [60].
Medial prefrontal cortex: Layer V cells
Liu and Aghajanian have directly investigated the functional correlates of stress-induced dendritic atrophy and spine loss in layer V pyramidal cells in the PFC. Using a combination of whole-cell recording and high-resolution spine imaging they have shown that stress-induced apical dendritic atrophy accompanied by spine loss correlates with decreased physiologic responses to apically targeted excitatory inputs in the same cells [62]. Further studies have shown that stress-induced spine loss and reduced synaptic currents occur in association with reductions in levels of synaptic proteins and increased depression-like behavior in a rodent model [60]. These studies provide compelling support for a role of synaptic changes in depression-related function by demonstrating that rapid-acting antidepressants increased spine diameter/number and reverse stress-induced deficits in synaptic number, function and behavior [60, 63, 111]. The relationship of larger diameter spine populations to enhanced synaptic and behavioral function in these studies emphasizes the relationship of spine morphology to synaptic efficacy and the plastic nature of the changes induced by stress and antidepressants and their functional importance.
Layer V pyramidal cells in the infralimbic region also show apical dendritic retraction and spine loss following CRS, along with impaired D1 receptor facilitated synaptic plasticity [33]. This study found that spine density per layer remained constant despite dendritic remodeling in response to stress exposure, prompting the authors to conclude that spine loss accompanied dendritic retraction after stress and that spine growth co-occurred along with proximal dendritic expansion following a recovery period.
Early life stress models also have shown alterations in dendritic spines in mPFC. Prenatal restraint stress has been shown to exert sexually dimorphic changes in spine densities in mPFC [81–83]. Postnatal stress (maternal separation) has been shown to decrease basal dendritic spine densities in PFC pyramidal neurons in layers II/III and V [8, 34]. Maternal separation stress was also shown to result in basal dendritic atrophy and reduced basal and apical dendritic spine densities in layer II/III [12].
Other brain regions: BLA and NAc
Stress-induced increases in spine density are reliably demonstrated in amygdala. Chronic stress exposure increases spine density in spiny pyramidal neurons of the BLA and given the role of the BLA in anxiety and fear responses, this may contribute along with stress-induced dendritic hypertrophy to affective dysregulation in response to chronic stress [113, 114]. Experiments showed that spine formation in the BLA could be dissociated from dendritic remodeling [79]. An increase in spine density in BLA pyramidal neurons was also seen after chronic corticosterone exposure [35].
The nucleus NAc is another brain region that shows increases in spine densities following chronic stress exposure. NAc medium spiny neurons receive afferents from dopamine neurons of the mesolimbic dopamine pathway that contributes to the regulation of motivation and reward, as well as social interaction. Many of the studies of NAc use social defeat stress (SDS), a model of depression that produces social avoidance and anhedonia in mice. SDS was shown to result in increased spine density in NAc medium spiny neurons, due to increased numbers of stubby spines with smaller postsynaptic densities [13]. The frequency of mini excitatory postsynaptic currents (EPSCs) was also increased in mice susceptible to chronic SDS suggesting a greater number of functional glutamate synapses. These changes were correlated with social avoidance behavior [13]. These authors also showed that the social avoidance phenotype and the formation of new stubby spines depended on the inhibitor of kappaB kinase (IkK)-nuclear factor kappaB (NFkB) pathway and that constitutively active IkK was sufficient to promote immature spine synapse formation in mice vulnerable to SDS [14].
Mechanisms underlying dendrite and spine deficits caused by chronic stress
In addition to providing fundamental information regarding the regulation spine synapses, characterization of the signaling pathways underlying the loss of spines in response to chronic stress could lead to novel targets for drug development. As discussed above, there is evidence that the effects of stress are mediated in part by elevated levels of adrenal glucocorticoids as many of these effects can be replicated by corticosterone administration. Here we will focus on neurotrophic factor signaling mechanisms involved in regulation of spines in the hippocampus and mPFC. In addition to these mechanisms, there is also evidence for transcriptional pathways that contribute to neuronal atrophy in the PFC [47]. The mechanisms underlying dendrite and spine alterations in other regions (i.e., BLA and OFC) have not been examined in detail and require further study.
Role of brain derived neurotrophic factor (BDNF)
Acute and chronic stress decrease the expression of BDNF in the hippocampus and mPFC raising the possibility that this neurotrophic factor contributes to the dendrite and spine deficits observed in these brain regions [24]. Direct evidence for this hypothesis is provided by studies in BDNF mutant mice. Studies have examined several different lines of mice, and one of the first available was a constitutive deletion mutant line. However, homozygous deletion mutants are lethal, demonstrating the importance of this neurotrophic factor, so heterozygous mutants, BDNF+/−, have been examined. The BDNF+/− mice have reduced length and branching of apical dendrites of CA3 pyramidal neurons in the hippocampus, similar to what is observed with stress [70]. Moreover, exposure to stress causes no further loss of dendrites, resulting in occlusion of the stress response, suggesting that the stress response occurs via a reduction in BDNF [70]. Spine density and morphology were not examined in this study.
Another BDNF mutant line that is relevant to clinical studies is a knock-in of a small nucleotide polymorphism (SNP), referred to as BDNF Val66Met that is carried by approximately 25 percent of humans [57]. This is a functional SNP that reduces the processing of pro-BDNF to mature BDNF decreases BDNF transport to terminals, and thereby blocks activity dependent release of BDNF. The BDNF Met allele has been associated with reduced hippocampal volume and executive function in humans [28, 29] and increased risk for depression in patients exposed to early life stress or trauma [32, 45, 54]. BDNF Met knock-in mice display a reduction in the number and length of branch points of CA3 pyramidal neurons in the hippocampus (spine density was not examined) [57]. In addition, these mice show reduced dendrite length in the layers II/III [120, 121] and V [64] pyramidal neurons in the mPFC, as well as decreased number and function of spines [64]. These dendritic and spine changes area also associated with increased anxiety and depression-like behaviors in rodent models [64, 120].
Role of the mTORC1 signaling pathway
There are several downstream signaling cascades that mediate the actions of BDNF, but one that has been examined with regard to stress and spine density is the mechanistic target of rapamycin complex 1 (mTORC1). The mTORC1 pathway regulates activity dependent protein synthesis and is required for protein synthesis dependent synaptic plasticity [41, 56]. We have found that CUS decreases levels of the signaling components of this pathway in the PFC, including decreased levels of the phosphorylated and activated levels of mTOR and ribosomal p70S6 kinase (S6K) [86]. Moreover, reduced mTORC1 signaling is associated with decreased expression of synaptic proteins and decreased number and function of spine synapses [59, 86].
The phosphorylation and function of mTORC1 signaling is regulated by a number of upstream signaling pathways, notably strong inhibition by the tuberous sclerosis complex (TSC1 and TSC2). The TSC1/2 complex is stabilized by another protein, REDD1 (regulated in DNA damage and repair 1) that is induced by cellular stress and glucocorticoid. We found that chronic stress increases REDD1 expression and that viral expression of REDD1 was sufficient to decrease mTORC1 signaling, decrease spine density in layer V cells in the PFC, and to produce depression like behaviors in rodent models [86]. Moreover, mice with a deletion of REDD1 are resilient to chronic stress, including the deficits in spine density and behavior caused by stress exposure. Finally, the relevance of REDD1 to atrophy of PFC in humans was examined in postmortem tissue from depressed subjects. The results demonstrate that levels of REDD1 are significantly increased in depressed subjects relative to psychiatric controls matched for age, sex, and race [86]. These studies are consistent with the hypothesis that stressful life events lead to increased expression of REDD1 that inhibits mTORC1 signaling and causes loss of spines in the PFC.
Antidepressant effects on spines and synaptic markers
Alterations of dendritic structure, spine number/morphology and synaptic proteins caused by stress and association of these changes with deficits in synaptic function and depression related behavior has focused significant research attention on synaptic mechanisms and synaptogenesis as critical targets of antidepressant treatment. These studies demonstrate differential effects of typical antidepressants that increase synaptic monoamines and new rapid acting agents that influence glutamatergic neurotransmission.
Monoaminergic antidepressants
The monoaminergic antidepressants increase the amount of serotonin and/or norepinephrine in the synapse by blocking the reuptake or breakdown of these neurotransmitters. These agents are modestly effective, producing a therapeutic response in approximately one third of patients after the first agent tested and up to two thirds after multiple drug trials [109]. Notably, there is a therapeutic time lag of several weeks with these agents, which can extend to many months or even years until an effective agent is identified. In preclinical studies, these agents do not appear to have a major impact on spine number when administered to naïve animals but there are reports that monoaminergic drugs are capable of blocking the effects of chronic stress.
Effects of antidepressant agents on dendritic spines have been studied using a CUS model to induce behavioral features of depression [6]. In the CUS model the behavioral deficits were reversible by chronic administration of antidepressant drugs with different primary mechanisms of action (imipramine, fluoxetine, CP 156,526, a type 1 corticotropin releasing hormone receptor antagonist, and SSR 1494515, a type 1b arginine vasopressin receptor antagonist). CUS also produced atrophy of granule cell dendrites and CA3 apical dendritic atrophy in hippocampus along with proximal spine loss. All of the antidepressants tested were effective in reversing these changes. In the PFC, the apical dendritic atrophy in layer II/II pyramidal cells was reversed by the antidepressants tested except for fluoxetine. In the PFC cells, CUS induced spine loss in proximal and distal dendrites was also reversed by the antidepressant treatments. Also in this study, the antidepressants promoted hippocampal neurogenesis, but the neurogenic action was not critical for antidepressant efficacy. Instead, the dendritic and synaptic remodeling were the primary changes associated with behavioral deficits caused by CUS and the improvements resulting from antidepressant treatments. Although fluoxetine alone had no effect, another study found that chronic fluoxetine administration increases spine density in retrosplenial granular cortex [1]. This study also reported that chronic fluoxetine treatment that produced antidepressant behavioral responses in rodent models of depression and anxiety, resulted in up-regulation of N-methyl-D-aspartate (NMDA) receptor subunit NR2A, and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor subunits GluR1 and 2 in forebrain [1]. GluR2 and NR2A were increased in synaptic membranes and postsynaptic densities suggesting synaptic localization of these changes. AMPA receptors are important to synapse maturation and the number and activity of AMPA receptors at the synapse is an important determinant of synaptic strength [19, 102]. The association of the altered subunit levels with increased mushroom spines regionally in the Ampeuro study [1], suggests stabilization of synaptic connections resulted from chronic fluoxetine.
The effect of chronic fluoxetine on synaptic proteins in the CA1 region of hippocampus has also been examined. Chronic fluoxetine was shown to increase expression of phospho-synapsin, PSD-95 and synaptic GluR1 in a rat model of reduced synapse density [85]. PSD-95 is a scaffolding protein that controls activity-dependent AMPA receptor incorporation in spines during experience-dependent synaptic strengthening and is considered to reflect synapse number and/or stabilization [25, 26]. The PSD-95 and associated GluR1 increases could suggest a synaptic maturation effect of chronic fluoxetine. This study also used a mutant mouse model to implicate involvement of BDNF and signaling at its receptor TrkB in the effect of fluoxetine on PSD-95.
The influence of a norepinephrine reuptake inhibitor, desipramine has also been examined in a learned helplessness model of depression. In this model, rats are exposed to inescapable foot shock and then subsequently are tested for their ability to escape a stressful event. Animals exposed to inescapable stress develop “helpless” behavior and show a dramatic decrease in escape behavior in response to a subsequent foot shock test. Exposure to this paradigm causes loss of synapses, analyzed in this study by electron microscopy, in CA1 and CA3 pyramidal and dentate gyrus granule neurons in the hippocampus [36]. Sub-chronic administration of desipramine (6 days) reversed the synaptic deficits as well as behavioral helplessness in this model.
Novel, rapid acting antidepressants: ketamine and scopolamine
Ketamine
Recent clinical studies have demonstrated that it is possible to achieve a rapid and efficacious antidepressant response in depressed patients. Most notable is the NMDA receptor antagonist ketamine, which produces a rapid antidepressant response even in treatment resistant depressed patients [5, 55, 123]. Ketamine is a nonselective NMDA receptor antagonist that when given at high doses produces sedation and is used as a dissociative anesthetic. At low doses it produces euphoria but also psychotomimetic effects, and has been used to study the neurobiology of schizophrenia [55]. In clinical studies to examine the antidepressant actions of ketamine, a single dose produces a brief dissociative and/or psychotomimetic effect that quickly dissipates within 60 min after administration. This is followed by a dramatic and significant improvement in depression ratings approximately 2 to 4 hrs. later that is sustained for approximately 7 to 10 days [55]. The discover that ketamine produces a rapid and efficacious antidepressant response by blocking a target, NMDA receptors, represents the most significant advance in drug development for depression in over 50 years.
We have examined the mechanisms, including spine synapse alterations that underlie the rapid actions of ketamine. These studies demonstrate that a single, low dose of ketamine increases the number of spine synapses in layer V neurons of the mPFC, and that the increase in spine number is accompanied by increased function measured by increased serotonin-induced ECPCs [59]. The spine changes are also accompanied by antidepressant behavioral responses in rodent models [59]. These synaptic and behavioral actions are observed with low doses of ketamine (3 to 10 mg/kg) that are non-sedating [59]. These studies also examine the mechanisms underlying the actions of ketamine, and demonstrate an up-regulation of the mTORC1 signaling pathway and increased levels of synaptic proteins in the PFC, including GluR1 and PSD95. A requirement for mTORC1 signaling has also been examined using the selective mTORC1 inhibitor rapamycin. Infusion of rapamycin into the lateral ventricle blocks the increase in spine synapses and the antidepressant behavioral actions of ketamine in rodents [59]. The involvement of the mPFC was further examined by direct infusions of rapamycin locally into this region, which completely blocked the behavioral actions of ketamine [59].
In addition to the studies, the influence of ketamine in a CUS model of depression has also been examined [60]. This is a critical test of the ability of ketamine to produce rapid antidepressant responses in rodents, as the actions of typical monoaminergic antidepressant require chronic administration. Three weeks exposure to CUS produced the expected decrease in sucrose preference, a measure of anhedonia, as well as loss of spine synapses in layer V pyramidal neurons in the mPFC. These behavioral and synaptic deficits were rapidly and completely reversed by a single dose of ketamine [60]. Moreover, the ability of ketamine to reverse the CUS induced deficits was blocked by infusions of rapamycin into the lateral ventricles. Together these studies demonstrate that ketamine rapidly reverses the spine synapse and behavioral deficits caused by CUS and that these effects are dependent on activation of the mTORC1 signaling pathway.
Interestingly, further studies have demonstrated a role for BDNF in the synaptic and behavioral actions of ketamine. Initial studies demonstrated that the antidepressant behavioral actions of ketamine were blocked in conditional BDNF deletion mutant mice [3], and confirmed by studies demonstrating that infusion of a function blocking anti-BDNF antibody into the mPFC blocks the behavioral actions of ketamine [58]. In addition, we have reported that the induction of spine synapses as well as antidepressant behavioral responses are blocked in BDNF Met allele knock in mice [64]. Blockade in the BDNF Met mice, which blocks activity dependent release of BDNF, as well as by the function-blocking antibody indicates that BDNF release and activity at extracellular receptors is required.
Scopolamine
Recent clinical studies have demonstrated that the muscarinic receptor antagonist scopolamine also produces rapid antidepressant responses in depressed patients [21, 30, 31]. A single low dose of scopolamine produces significant antidepressant effects at the first time of assessment 3 days later, and there are anecdotal reports of improvement as early as 24 hours after dosing. Because of the rapid actions similar to ketamine, we have also examined the influence of scopolamine on spine synapses and mTORC1 signaling in the mPFC. The results demonstrate that a single low dose of scopolamine increases the number and function of spine synapse in layer V pyramidal neurons and increases mTORC1 signaling in the PFC [111]. We also found that infusion of rapamycin blocked the behavioral effects of scopolamine, demonstrating a role for mTORC1 signaling.
The cellular mechanisms underlying the actions of ketamine and scopolamine are not obvious, but there is evidence that both NMDA and muscarinic receptor antagonists cause a burst of glutamate transmission. Earlier studies have shown that ketamine increases extracellular glutamate in the mPFC [80] and more recent preliminary studies demonstrate that a scopolamine causes a similar increase in levels of extracellular glutamate in this region [111]. This burst of glutamate is then thought to produce an activity dependent increase in spine synapse formation [23]. The cellular mechanisms underlying this paradoxical increase in glutamate by both NMDA and muscarinic receptor is thought to result from disinhibition of GABAergic interneurons that control the firing of glutamate neurons. This is supported by studies demonstrating that ketamine blocks the firing of GABA neurons in vivo [44]. Studies are currently being conducted to selectively delete NMDA and muscarinic receptor subtypes from GABAergic neurons to directly test this hypothesis.
Summary and Future Directions
Chronic stress and depression are associated with alterations of neuronal processes, including spine synapses, in a region dependent manner, resulting in disrupted connectivity of brain circuitry that contributes to depressive symptoms [75, 99]. Conversely, there is evidence that typical monoaminergic agents are capable of reversing the synaptic and behavioral deficits caused by stress, but that this requires chronic administration. Importantly, novel antidepressants such as the NMDA receptor antagonist ketamine cause a fast induction of spine synapses in the PFC and rapidly reverse the synaptic and behavioral deficits caused by chronic stress. Studies are needed to further characterize the molecular and cellular mechanisms that underlie the altered number and function of spine synapses in stress related illnesses. This will contribute to the development of novel fast acting antidepressant agents with fewer side effects, as well as treatment regimens that can sustain and stabilize new spine synapses required for proper connectivity and function of mood and emotion.
Figure 1.

Chronic stress causes atrophy of layer V pyramidal neurons in the medial PFC. Shown is the influence of repeated restraint stress (30 min per day for 7 d) on layer V pyramidal neurons in the medial PFC. The upper panels demonstrate that effects of stress on the length and branching of apical dendrites, and the lower panels show the effects of stress on the density of spines on apical dendrites of labeled layer V neurons. Neurobiotin labeled neurons were visualized by two-photon laser scanning miscrocopy (see Liu and Aghajanian, 2008).
Figure 2.
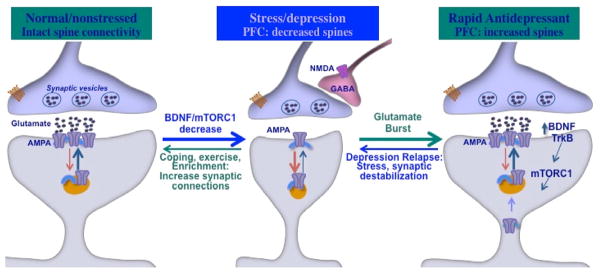
Schematic of the effects of stress/depression on spine synapses and reversal by rapid acting antidepressants. Under normal/nonstressed conditions spine synapse connections are intact and provide control over mood, emotion, and cognition. Chronic stress or depression leads to decreased levels of BDNF and downstream mTORC1 signaling, which contributes to decreased number and function of spine synapses in layer V pyramidal neurons of the medial PFC. Rapid acting antidepressant such as ketamine rapidly reverse the spine synapse deficits caused by stress via a burst of glutamate, which is thought to result from disinhibition of GABAergic interneurons that control glutamate transmission. This increase in glutamate-AMPA leads to activity dependent release of BDNF-TrkB and stimulation of mTORC1 signaling, resulting in increased synthesis of synaptic proteins required for new spine formation. The antidepressant response to a single dose of ketamine persists for approximately 7 day in humans and rodents before relapse. The new spines induced by ketamine also remain for a similar length of time, and the loss of spines could be related to relapse and reversal of antidepressant responses.
Footnotes
Disclosure
Dr. R.S. Duman reports having received honorarium fees from Lilly, Pfizer, Bristol Myers Squibb, Johnson & Johnson, Forest, and Lundbeck, consulting fees fromTaisho, and research support from Lilly, Lundbeck, Johnson & Johnson, and Forest. Dr. C.H. Duman reported no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ampuero E, Rubio FJ, Falcon R, Sandoval M, Diaz-Veliz G, Gonzalez RE, Earle N, Dagnino-Subiabre A, Aboitiz F, Orrego F, Wyneken U. Chronic fluoxetine treatment induces structural plasticity and selective changes in glutamate receptor subunits in the rat cerebral cortex. Neuroscience. 2010;169:98–108. doi: 10.1016/j.neuroscience.2010.04.035. [DOI] [PubMed] [Google Scholar]
- 2.Arnsten AF, Rubia K. Neurobiological circuits regulating attention, cognitive control, motivation, and emotion: disruptions in neurodevelopmental psychiatric disorders. J Am Acad Child Adolesc Psychiatry. 2012;51:356–367. doi: 10.1016/j.jaac.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Autry A, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM. NMDA receptor blockade at rest triggers rapid behavioural anatidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedrosian TA, Fonken LK, Walton JC, Haim A, Nelson RJ. Dim light at night provokes depression-like behaviors and reduces CA1 dendritic spine density in female hamsters. Psychoneuroendocrinology. 2011;36:1062–1069. doi: 10.1016/j.psyneuen.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 6.Bessa J, Ferreira D, Melo I, Marques F, Cerqueira J, Palha J, Almeida O, Sousa N. Hippocampal neurogenesis induced by antidepressant drugs: an epiphenomenon in their mood-improving actions. Mol Psych. 2009;14:739. doi: 10.1038/mp.2008.119. [DOI] [PubMed] [Google Scholar]
- 7.Bloss EB, Janssen WG, Ohm DT, Yuk FJ, Wadsworth S, Saardi KM, McEwen BS, Morrison JH. Evidence for reduced experience-dependent dendritic spine plasticity in the aging prefrontal cortex. J Neurosci. 2011;31:7831–7839. doi: 10.1523/JNEUROSCI.0839-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bock J, Gruss M, Becker S, Braun K. Experience-induced changes of dendritic spine densities in the prefrontal and sensory cortex: correlation with developmental time windows. Cereb Cortex. 2005;15:802–808. doi: 10.1093/cercor/bhh181. [DOI] [PubMed] [Google Scholar]
- 9.Brown SM, Henning S, Wellman CL. Mild, short-term stress alters dendritic morphology in rat medial prefrontal cortex. Cereb Cortex. 2005;15:1714–1722. doi: 10.1093/cercor/bhi048. [DOI] [PubMed] [Google Scholar]
- 10.Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 11.Cerqueira J, Mailliet F, Almeida O, Jay T, Sousa N. The prefrontal cortex as a key target of the maladaptive response tostress. J Neurosci. 2007;27:2781–2787. doi: 10.1523/JNEUROSCI.4372-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chocyk A, Bobula B, Dudys D, Przyborowska A, Majcher-Maslanka I, Hess G, Wedzony K. Early-life stress affects the structural and functional plasticity of the medial prefrontal cortex in adolescent rats. Eur J Neurosci. 2013;38:2089–2107. doi: 10.1111/ejn.12208. [DOI] [PubMed] [Google Scholar]
- 13.Christoffel D, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF, Krishnan V, Reyes CM, Han MH, Ables JL, Eisch AJ, Dietz DM, Ferguson D, Neve RL, Greengard P, Kim Y, Morrison JH, Russo SJ. IκB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci. 2011a;31:314–321. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christoffel DJ, Golden SA, Heshmati M, Graham A, Birnbaum S, Neve RL, Hodes GE, Russo SJ. Effects of inhibitor of kappaB kinase activity in the nucleus accumbens on emotional behavior. Neuropsychopharmacology. 2012;37:2615–2623. doi: 10.1038/npp.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conrad C. Chronic stress-induced hippocampal vulnerability: the glucocorticoid vulnerability hypothesis. Rev Neurosci. 2008;19:395–411. doi: 10.1515/revneuro.2008.19.6.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conrad C, Magarinos AM, LeDoux JE, McEwen BS. Repeated restraint stress facilitates fear conditioning independently of causing hioopcampal CA3 dendritic atrophy. Behav Neurosci. 1999;113:902–913. doi: 10.1037//0735-7044.113.5.902. [DOI] [PubMed] [Google Scholar]
- 17.Conrad CD, McLaughlin KJ, Huynh TN, El-Ashmawy M, Sparks M. Chronic stress and a cyclic regimen of estradiol administration separately facilitate spatial memory: relationship with hippocampal CA1 spine density and dendritic complexity. Behav Neurosci. 2012;126:142–156. doi: 10.1037/a0025770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. 2004;60:236–248. doi: 10.1002/neu.20025. [DOI] [PubMed] [Google Scholar]
- 19.Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- 20.Dias-Ferreira E, Sousa JC, Melo I, Morgado P, Mesquita AR, Cerqueira JJ, Costa RM, Sousa N. Chronic stress causes frontostriatal reorganization and affects decision-making. Science. 2009;325:621–625. doi: 10.1126/science.1171203. [DOI] [PubMed] [Google Scholar]
- 21.Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67:432–438. doi: 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213:93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duman R, Aghajanian G. Synaptic Dysfunction in Depression. Novel Therapeutic Targets Science. 2012;338:68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feldman S, Conforti N, Saphier D. The preoptic area and bed nucleus of the stria terminalis are involved in the effects of the amygdala on adrenocortical secretion. Neuroscience. 1990;37:775–779. doi: 10.1016/0306-4522(90)90107-f. [DOI] [PubMed] [Google Scholar]
- 28.Frodl T, Schule C, Schmitt G, Born C, Baghai T, Zill P, Bottlender R, Rupprecht R, Bondy B, Reiser M, Moller H-J, Meisenzahl EM. Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depressio. Arch Gen Psych. 2007;64:410–416. doi: 10.1001/archpsyc.64.4.410. [DOI] [PubMed] [Google Scholar]
- 29.Frodl T, Skokauskas N, Frey EM, Morris D, Gill M, Carballedo A. BDNF Val66Met genotype interacts with childhood adversity and influences the formation of hippocampal subfields. Hum Brain Mapp. 2014;35:5776–5783. doi: 10.1002/hbm.22584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63:1121–1129. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furey ML, Zarate CA., Jr Pulsed intravenous administration of scopolamine produces rapid antidepressant effects and modest side effects. J Clin Psychiatry. 2013;74:850–851. doi: 10.4088/JCP.13ac08584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR, Gordon E, Kemp AH, Williams LM. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Molecular Psychiatry. 2009;14:681–695. doi: 10.1038/mp.2008.143. [DOI] [PubMed] [Google Scholar]
- 33.Goldwater D, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, MJH Structural and functional alterations to rat medial prefrontal cortexfollowing chronic restraint stress and recovery. Neurosci. 2009;164:798–808. doi: 10.1016/j.neuroscience.2009.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gos T, Bock J, Poeggel G, Braun K. Stress-induced synaptic changes in the rat anterior cingulate cortex are dependent on endocrine developmental time windows. Synapse. 2008;62:229–232. doi: 10.1002/syn.20477. [DOI] [PubMed] [Google Scholar]
- 35.Gourley SL, Swanson AM, Koleske AJ. Corticosteroid-induced neural remodeling predicts behavioral vulnerability and resilience. J Neurosci. 2013;33:3107–3112. doi: 10.1523/JNEUROSCI.2138-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hajszan T, Dow A, Warner-Schmidt JL, Szigeti-Buck K, Sallam NL, Parducz A, Leranth C, Duman RS. Remodeling of hippocampus spine synapses in the rat learned helplessness model of depressant and antidepressant therapy. Biol Psych. 2008;65 doi: 10.1016/j.biopsych.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harris K. Structure, development, and plasticity of dendritic spines. Curr Opin Neurobiol. 1999;9:343–348. doi: 10.1016/s0959-4388(99)80050-6. [DOI] [PubMed] [Google Scholar]
- 38.Harris KM, Kater SB. Dendritic Spines - Cellular Specializations Imparting Both Stability and Flexibility to Synaptic Function. Annual Review of Neuroscience. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- 39.Harris KM, Stevens JK. Dendritic Spines of Ca-1 Pyramidal Cells in the Rat Hippocampus - Serial Electron-Microscopy with Reference to Their Biophysical Characteristics. Journal of Neuroscience. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2001;2:880–888. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- 41.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 43.Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 44.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hosang GM, Shiles C, Tansey KE, McGuffin P, Uher R. Interaction between stress and the BDNF Val66Met polymorphism in depression: a systematic review and meta-analysis. Bmc Medicine. 2014;12:7. doi: 10.1186/1741-7015-12-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Izquierdo A, Wellman CL, Holmes A. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci. 2006;26:5733–5738. doi: 10.1523/JNEUROSCI.0474-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, Lepack A, Majik MS, Jeong LS, Banasr M, Son H, Duman RS. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. 2012;18:1413–1417. doi: 10.1038/nm.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- 49.Kassem MS, Lagopoulos J, Stait-Gardner T, Price WS, Chohan TW, Arnold JC, Hatton SN, Bennett MR. Stress-Induced Grey Matter Loss Determined by MRI Is Primarily Due to Loss of Dendrites and Their Synapses. Molecular Neurobiology. 2013;47:645–661. doi: 10.1007/s12035-012-8365-7. [DOI] [PubMed] [Google Scholar]
- 50.Kaufman J, Yang BZ, Douglas-Palumberi H, Grasso D, Lipschitz D, Houshyar S, Krystal JH, Gelernter J. Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol Psychiatry. 2006;59:673–680. doi: 10.1016/j.biopsych.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 51.Keller M, Neale M, Kendler K. Association of different adverse life events with distinct patterns ofdepressive symptoms. Am J Psych. 2007;164:401. doi: 10.1176/appi.ajp.2007.06091564. [DOI] [PubMed] [Google Scholar]
- 52.Kessler RC. The effects of stressful life events on depression. Annu Rev Psychol. 1997;48:191–214. doi: 10.1146/annurev.psych.48.1.191. [DOI] [PubMed] [Google Scholar]
- 53.Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim JM, Stewart R, Kim SW, Yang SJ, Shin IS, Kim YH, Yoon JS. Interactions between life stressors and susceptibility genes (5-HTTLPR and BDNF) on depression in Korean elders. Biological Psychiatry. 2007;62:423–428. doi: 10.1016/j.biopsych.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 55.Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73:1133–1141. doi: 10.1016/j.biopsych.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leal G, Comprido D, Duarte CB. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology. 2014;76(Pt C):639–656. doi: 10.1016/j.neuropharm.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 57.Lee Y, Duman RS, Marek GJ. The mGlu2/3 receptor agonist LY354740 suppresses immobilization stress-induced increase in rat prefrontal cortical BDNF mRNA expression. Neurosci Lett. 2006;398:328–332. doi: 10.1016/j.neulet.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 58.Lepack A, Fuchikami M, Dwyer J, Banasr M, Aghajanian G, Duman R. BDNF release is required for the behavioral actions of ketamine. Neuropsychopharmacol. 2014 doi: 10.1093/ijnp/pyu033. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, Li XY, Aghajanian G, Duman RS. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, Morrison JH, McEwen BS. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26:7870–7874. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu RJ, Aghajanian GK. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci U S A. 2008;105:359–364. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu RJ, Fuchikami M, Dwyer JM, Lepack AE, Duman RS, Aghajanian GK. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38:2268–2277. doi: 10.1038/npp.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu RJ, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. 2012;71:996–1005. doi: 10.1016/j.biopsych.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lohmann C, Finski A, Bonhoeffer T. Local calcium transients regulate the spontaneous motility of dendritic filopodia. Nat Neurosci. 2005;8:305–312. doi: 10.1038/nn1406. [DOI] [PubMed] [Google Scholar]
- 66.Luine VN, Spencer RL, McEwen BS. Effects of chronic corticosterone ingestion on spatial memory performance and hippocampal serotonergic function. Brain Res. 1993;616:65–70. doi: 10.1016/0006-8993(93)90193-q. [DOI] [PubMed] [Google Scholar]
- 67.MacQueen G, Frodl T. The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol Psychiatry. 2011;16:252–264. doi: 10.1038/mp.2010.80. [DOI] [PubMed] [Google Scholar]
- 68.Magarinos A, McEwen B. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neurosci. 1995a;69:83–88. doi: 10.1016/0306-4522(95)00256-i. [DOI] [PubMed] [Google Scholar]
- 69.Magarinos AM, Verdugo JM, MBS Chronic stress alters synaptic terminal structure in hippocampus. PNAS. 1997;94:14002–14008. doi: 10.1073/pnas.94.25.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Magarinos AM, Li CJ, Gal Toth J, Bath KG, Jing D, Lee FS, McEwen BS. Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus. 2011;21:253–264. doi: 10.1002/hipo.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Magarinos AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995b;69:89–98. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- 72.Magarinos AM, McEwen BS, Flugge G, Fuchs E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci. 1996;16:3534–3540. doi: 10.1523/JNEUROSCI.16-10-03534.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martinez-Tellez RI, Hernandez-Torres E, Gamboa C, Flores G. Prenatal stress alters spine density and dendritic length of nucleus accumbens and hippocampus neurons in rat offspring. Synapse. 2009;63:794–804. doi: 10.1002/syn.20664. [DOI] [PubMed] [Google Scholar]
- 74.Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mayberg HS. Targeted electrode-based modulation of neural circuits for depression. J Clin Invest. 2009;119:717–725. doi: 10.1172/JCI38454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McEwen BS. Invited review: Estrogens effects on the brain: multiple sites and molecular mechanisms. J Appl Physiol (1985) 2001;91:2785–2801. doi: 10.1152/jappl.2001.91.6.2785. [DOI] [PubMed] [Google Scholar]
- 77.McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79:16–29. doi: 10.1016/j.neuron.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Michelsen KA, van den Hove DL, Schmitz C, Segers O, Prickaerts J, Steinbusch HW. Prenatal stress and subsequent exposure to chronic mild stress influence dendritic spine density and morphology in the rat medial prefrontal cortex. BMC Neurosci. 2007;8:107. doi: 10.1186/1471-2202-8-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mitra R, Jadhav S, McEwen BS, Vyas A, Chattarji S. Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc Natl Acad Sci U S A. 2005;102:9371–9376. doi: 10.1073/pnas.0504011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Muhammad A, Kolb B. Maternal separation altered behavior and neuronal spine density without influencing amphetamine sensitization. Behav Br Res. 2011;223:7–16. doi: 10.1016/j.bbr.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 82.Murmu MS, Salomon S, Biala Y, Weinstock M, Braun K, Bock J. Changes of spine density and dendritic complexity in the prefrontal cortex in offspring of mothers exposed to stress during pregnancy. Eur J Neurosci. 2006;24:1477–1487. doi: 10.1111/j.1460-9568.2006.05024.x. [DOI] [PubMed] [Google Scholar]
- 83.Mychasiuk R, Gibb R, Kolb B. Prenatal stress alters dendritic morphology and synaptic connectivity in the prefrontal cortex and hippocampus of developing offspring. Synapse. 2012;66:308–314. doi: 10.1002/syn.21512. [DOI] [PubMed] [Google Scholar]
- 84.Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- 85.O’Leary OF, Wu X, Castren E. Chronic fluoxetine treatment increases expression of synaptic proteins in the hippocampus of the ovariectomized rat: role of BDNF signalling. Psychoneuroendocrinology. 2009;34:367–381. doi: 10.1016/j.psyneuen.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 86.Ota K, Liu R, Voleti B, Maldonado-Aviles J, Duric V, Iwata M, Dutheil S, Duman C, Boikess S, Lewis D, Stockmeier C, DiLeone R, Rex C, Aghajanian G, Duman R. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat Med. 2014;20:531–535. doi: 10.1038/nm.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parnass Z, Tashiro A, Yuste R. Analysis of spine morphological plasticity in developing hippocampal pyramidal neurons. Hippocampus. 2000;10:561–568. doi: 10.1002/1098-1063(2000)10:5<561::AID-HIPO6>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 88.Pavlides C, Nivón L, McEwen B. Effects of chronic stress on hippocampal long-term potentiation. Hippocampus. 1992;12:245–257. doi: 10.1002/hipo.1116. [DOI] [PubMed] [Google Scholar]
- 89.Pawlak R, Rao BS, Melchor JP, Chattarji S, McEwen B, Strickland S. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc Natl Acad Sci U S A. 2005;102:18201–18206. doi: 10.1073/pnas.0509232102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peters A, Kaiserman-Abramof IR. The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat. 1970;127:321–355. doi: 10.1002/aja.1001270402. [DOI] [PubMed] [Google Scholar]
- 91.Petrak LJ, Harris KM, Kirov SA. Synaptogenesis on mature hippocampal dendrites occurs via filopodia and immature spines during blocked synaptic transmission. J Comp Neurol. 2005;484:183–190. doi: 10.1002/cne.20468. [DOI] [PubMed] [Google Scholar]
- 92.Qiao H, An SC, Ren W, Ma XM. Progressive alterations of hippocampal CA3-CA1 synapses in an animal model of depression. Behav Brain Res. 2014;275C:191–200. doi: 10.1016/j.bbr.2014.08.040. [DOI] [PubMed] [Google Scholar]
- 93.Radley J, Anderson R, Hamilton B, Alcock J, Romig-Martin S. Chronic stress-induced alterations of dendritic spine subtypes predict functional decrements in an hypothalamo-pituitary-adrenal-inhibitoryprefrontal circuit. J Neurosci. 2013;33:14379–14391. doi: 10.1523/JNEUROSCI.0287-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Radley JJ, Rocher AB, Janssen WG, Hof PR, McEwen BS, Morrison JH. Reversibility of apical dendritic retraction in the rat medial prefrontal cortex following repeated stress. Exp Neurol. 2005;196:199–203. doi: 10.1016/j.expneurol.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 95.Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, McEwen BS, Morrison JH. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 2006;16:313–320. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- 96.Radley JJ, Rocher AB, Rodriguez A, Ehlenberger DB, Dammann M, McEwen BS, Morrison JH, Wearne SL, Hof PR. Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. J Comp Neurol. 2008;507:1141–1150. doi: 10.1002/cne.21588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR, McEwen BS, Morrison JH. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2004;125:1–6. doi: 10.1016/j.neuroscience.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 98.Sandi C, Davies HA, Cordero MI, Rodriguez JJ, Popov VI, SMG Rapid reversal of stress induced loss of synapses in CA3 of rat hippocampus following water maze training. Eur J Neurosci. 2003;17:2447–2456. doi: 10.1046/j.1460-9568.2003.02675.x. [DOI] [PubMed] [Google Scholar]
- 99.Savitz J, Drevets WC. Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci Biobehav Rev. 2009;33:699–771. doi: 10.1016/j.neubiorev.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shansky RM, Morrison JH. Stress-induced dendritic remodeling in the medial prefrontal cortex: effects of circuit, hormones and rest. Brain Res. 2009;1293:108–113. doi: 10.1016/j.brainres.2009.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 103.Shors TJ, Falduto J, Leuner B. The opposite effects of stress on dendritic spines in male vs. female rats are NMDA receptor-dependent. Eur J Neurosci. 2004;19:145–150. doi: 10.1046/j.1460-9568.2003.03065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shors TJ, Seib TB, Levine S, Thompson RF. Inescapable versus escapable shock modulates long-term potentiation in the rat hippocampus. Science. 1989;244:224–226. doi: 10.1126/science.2704997. [DOI] [PubMed] [Google Scholar]
- 105.Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature. 2011;476:458–461. doi: 10.1038/nature10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sousa N, Lukoyanov NV, Madeira MD, Almeida OF, Paula-Barbosa MM. Reorganization of the morphology of hippocampal neurites and synapsed after stress-induced damage correlates with behavioral improvements. Neurosci. 2000;97:253–266. doi: 10.1016/s0306-4522(00)00050-6. [DOI] [PubMed] [Google Scholar]
- 107.Stewart MG, Davies HA, Sandi C, Kraev IV, Rogachevsky VV, Peddie CJ, Rodriguez JJ, Cordero MI, Donohue HS, GPL Stress suppresses and learning induces plasticity in CA3 of rat hippocampus: a three-dimensional ultrastructural study of thorny excrescences and their postsynaptic densities. Neurosci. 2005;131:43–54. doi: 10.1016/j.neuroscience.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 108.Sunanda MS, Rao TR, Raju Effect of chronic restraint stress on dendritic spines and excrescences of hippocampal CA3 pyramidal neurons--a quantitative study. Brain Res. 1995;694:312–317. doi: 10.1016/0006-8993(95)00822-8. [DOI] [PubMed] [Google Scholar]
- 109.Trivedi M, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, FM SDS Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psych. 2006;163:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 110.Ulrich-Lai Y, Herman J. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Voleti B, Navarria A, Liu R, Banasr M, Li N, Terwilliger R, Eid T, Sanacora G, Aghajanian G, Duman R. Scopolamine rapidly increases mTORC1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psych. 2013;74:742–749. doi: 10.1016/j.biopsych.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.von Bohlen U, Halbach O. Structure and function of dendritic spines within the hippocampus. Ann Anat. 2009;191:518–531. doi: 10.1016/j.aanat.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 113.Vyas A, Jadhav S, Chattarji S. Prolonged behavioral stress enhances synaptic connectivity in the basolateral amygdala. Neuroscience. 2006;143:387–393. doi: 10.1016/j.neuroscience.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 114.Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vyas A, Pillai AG, Chattarji S. Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience. 2004;128:667–673. doi: 10.1016/j.neuroscience.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 116.Wang GH, Cheng YF, Gong MF, Liang BF, Zhang MZ, Chen YP, Zhang C, Yuan X, Xu JP. Systematic correlation between spine plasticity and the anxiety/depression-like phenotype induced by corticosterone in mice. Neuroreport. 2013;24:682–687. doi: 10.1097/WNR.0b013e32836384db. [DOI] [PubMed] [Google Scholar]
- 117.Watanabe Y, Gould E, Mcewen BS. Stress Induces Atrophy of Apical Dendrites of Hippocampal Ca3 Pyramidal Neurons. Brain Research. 1992a;588:341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- 118.Wellman CL. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol. 2001;49:245–253. doi: 10.1002/neu.1079. [DOI] [PubMed] [Google Scholar]
- 119.Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531:225–231. doi: 10.1016/0006-8993(90)90778-a. [DOI] [PubMed] [Google Scholar]
- 120.Yu H, Wang DD, Wang Y, Liu T, Lee FS, Chen ZY. Variant brain-derived neurotrophic factor Val66Met polymorphism alters vulnerability to stress and response to antidepressants. J Neurosci. 2012;32:4092–4101. doi: 10.1523/JNEUROSCI.5048-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yu H, Wang Y, Pattwell S, Jing D, Liu T, Zhang Y, Bath KG, Lee FS, Chen ZY. Variant BDNF Val66Met polymorphism affects extinction of conditioned aversive memory. J Neurosci. 2009;29:4056–4064. doi: 10.1523/JNEUROSCI.5539-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yuste R, Majewska A, Holthoff K. From form to function: calcium compartmentalization in dendritic spines. Nat Neurosci. 2000;3:653–659. doi: 10.1038/76609. [DOI] [PubMed] [Google Scholar]
- 123.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]