

Abstract
Purpose
Blocking the interaction between the programmed cell death (PD)-1 protein and one of its ligands, PD-L1, has been reported to have impressive antitumor responses. Therapeutics targeting this pathway are currently in clinical trials. Pembrolizumab and nivolumab are the first of this anti-PD-1 pathway family of checkpoint inhibitors to gain accelerated approval from the US Food and Drug Administration (FDA) for the treatment of ipilimumab-refractory melanoma. Nivolumab has been associated with improved overall survival compared with dacarbazine in patients with previously untreated wild-type serine/threonine-protein kinase B-raf proto-oncogene BRAF melanoma. Although the most mature data are in the treatment of melanoma, the FDA has granted approval of nivolumab for squamous cell lung cancer and the breakthrough therapy designation to immune-checkpoint inhibitors for use in other cancers: nivolumab, an anti-PD-1 monoclonal antibody, for Hodgkin lymphoma, and MPDL-3280A, an anti-PD-L1 monoclonal antibody, for bladder cancer and non–small cell lung cancer. Here we review the literature on PD-1 and PD-L1 blockade and focus on the reported clinical studies that have included patients with melanoma.
Methods
PubMed was searched to identify relevant clinical studies of PD-1/PD-L1–targeted therapies in melanoma. A review of data from the current trials on clinicaltrial.gov was incorporated, as well as data presented in abstracts at the 2014 annual meeting of the American Society of Clinical Oncology, given the limited number of published clinical trials on this topic.
Findings
The anti-PD-1 and anti-PD-L1 agents have been reported to have impressive antitumor effects in several malignancies, including melanoma. The greatest clinical activity in unselected patients has been seen in melanoma. Tumor expression of PD-L1 is a suggestive, but inadequate, biomarker predictive of response to immune-checkpoint blockade. However, tumors expressing little or no PD-L1 are less likely to respond to PD-1 pathway blockade. Combination checkpoint blockade with PD-1 plus cytotoxic T-lymphocyte antigen (CTLA)-4 blockade appears to improve response rates in patients who are less likely to respond to single-checkpoint blockade. Toxicity with PD-1 blocking agents is less than the toxicity with previous immunotherapies (eg, interleukin 2, CTLA-4 blockade). Certain adverse events can be severe and potentially life threatening, but most can be prevented or reversed with close monitoring and appropriate management.
Implications
This family of immune-checkpoint inhibitors benefits not only patients with metastatic melanoma but also those with historically less responsive tumor types. Although a subset of patients responds to single-agent blockade, the initial trial of checkpoint-inhibitor combinations has reported a potential to improve response rates. Combination therapies appear to be a means of increasing response rates, albeit with increased immune-related adverse events. As these treatments become available to patients, education regarding the recognition and management of immune-related effects of immune-checkpoint blockade will be essential for maximizing clinical benefit.
Keywords: melanoma, MPDL3280A, nivolumab, PD-1, PD-L1, pembrolizumab, pidilizumab, programmed cell death 1
INTRODUCTION
For decades, research has attempted to stimulate an antitumor immune response to fight cancer. However, there are inhibitory pathways that regulate the function of T lymphocytes and cause these attempts to be generally unsuccessful. These “immune checkpoints” not only normally function to control excessive immune activation but also appear to be a means by which tumors evade the immune system. Blockade of these immune checkpoints, such as with programmed cell death (PD)-1 protein and one of its ligands, PD-L1 (also known at B7-H1 and CD274), has demonstrated clinical activity in several types of solid tumors.1-5 Much of the preclinical and clinical benefit has been described in melanoma.
Metastatic melanoma is an aggressive disease with a 16% 5-year survival rate and responds poorly to most standard chemotherapies.6 Over 80% of cases of melanoma are localized. Yet patients with regional lymph node involvement have a high rate of recurrence, and the number of deaths from this disease per 100,000 persons has remained stable between 1992 and 2011. Interferon and interleukin (IL)-2 have both been approved by the US Food and Drug Administration for the treatment of melanoma.7,8 Both mediate their benefit by stimulating an antitumor immune response. However, toxicity and low response rates have limited their use significantly. The first immune-checkpoint inhibitor approved by the US Food and Drug Administration (FDA) was ipilimumab, a fully human immunoglobulin (Ig) G1 monoclonal antibody (mAb) that blocks cytotoxic T-lymphocyte antigen (CTLA)-4 for the treatment of metastatic melanoma in 2011.9 Although not yet compared in a randomized clinical trial, ipilimumab is generally considered more tolerable than high-dose IL-2. Both have promising durable response in melanoma.10 Of note, the response rate of ipilimumab may be less than that cited for IL-2.11,12 A recent follow up of 1861 melanoma patients treated with ipilimumab showed that about 20% survived 3 years but most impressively, at this time the survival curve flattens and most patients alive at 3 years are alive up to 10 years after therapy has been completed.10 Atypical patterns of tumor response to immunotherapies, including ipilimumab, make comparisons of response rates less informative; thus, milestone survival (for example, at 3 years) may be a more appropriate measure of response to immunotherapy.13
With the promising tolerability and efficacy seen with PD-1/PD-L1 checkpoint inhibitors in Phase I/II trials, multiple Phase III clinical trials have opened. A Phase III trial that compared nivolumab and dacarbazine reported better overall survival in the nivolumab arm and was stopped early to allow chemotherapy-treated patients to cross over to PD-1 blockade.14 Currently, nivolumab is being compared to ipilimumab in metastatic melanoma in a Phase III trial. PD-1 pathway blockade has become a major focus in anticancer drug development beyond melanoma. In addition to benefiting patients with renal cell carcinoma, it has reported benefit in patients with tumors previously not considered sensitive to immunotherapies, including non–small cell lung cancer. This finding has stimulated the investigation of numerous combinations in ongoing Phase I and II trials. While immunotherapy combinations often have been limited by their toxicity, ipilimumab + nivolumab was the first reported checkpoint-inhibitor combination and has been associated with response in melanoma.5 Given the tolerability of many of the PD-1 pathway inhibitors, these combination regimens will likely play a major role in the future of immune-checkpoint blockade in oncology.5 The current goal is to find the treatment with the best balance of high efficacy and low toxicity.
MATERIALS AND METHODS
PubMed was searched for articles published before July 1, 2014, using the search terms PD-1 AND melanoma and PD-L1 AND melanoma. This key-word search yielded 171 and 119 references, respectively, totaling 207 nonduplicate references. In a PubMed search of clinical trials containing the terms PD-1 and PD-L1, there were 11 publications. These publications were reviewed, as were additional publications referenced. The clinicaltrial.gov database was searched for interventional trials of PD-1 or PD-L1 inhibitors that were “actively recruiting,” “active, not recruiting,” and “not yet recruiting.” This search found 46 and 42 studies of PD-1 and PD-L1 inhibitors, respectively.
RESULTS
Role of PD-1 Pathway in Tolerance and Chronic Infection
PD-1 was originally isolated from a T-cell hybridoma undergoing T-cell receptor activation–induced cell death, hence its name, programmed death 1.15 Despite its name, PD-1 does not appear to directly engage a cell-death pathway like CD95, but indirectly effects cell death through diminished cell growth factors and survival signals. The interaction between PD-1 and its ligands PD-L1 and PD-L2 reduces T-lymphocyte function (Figure 1).16 PD-1 signaling inhibits T-cell activation, leading to reduced proliferation, cytokine production, and T-cell cytolysis.17-19 The in vitro effects of PD-1 mAb blockade are generally just 2-fold increases in cytokine production, so the potent in vivo activity likely also depends on effects on T-cell motility and the duration of interaction with antigen-presenting cells and target cells.20,21 PD-1 expression is induced by the activation of T lymphocytes and declines after a successful immune response eliminates antigen.22,23 If the immune response is unsuccessful, prolonged antigen stimulation leads to elevated PD-1 expression and is associated with an “exhausted” T-cell phenotype, originally described in a lymphocyticchoriomeningitis virus model.22,23 The regulation of T-lymphocyte activation lies in requiring 2 separate signals, which involves specific recognition by the T-cell receptor of an antigen presented on major histocompatibility complex (MHC) and concurrent coactivation with CD28 by its ligands B7-1 or B7-2 on antigen-presenting cells. The B7/CD28 family includes members involved in T-cell tolerance as well as activation.24 CTLA-4, PD-1, PD-L1, and PD-L2 are members of this ligand/receptor superfamily. CTLA-4 is a potent co-inhibitor, as evident by the fatal phenotype of the CTLA-4–knockout mouse model.25 It appears to be involved in early T-lymphocyte tolerance. CTLA-4–knockout mice have lymphocyte hyperproliferation and typically die from massive lymphocyte tissue infiltration and organ failure 2 to 3 weeks after birth. On the other hand, the PD-1 pathway plays more subtle roles in maintaining peripheral T-lymphocyte tolerance and regulating inflammation.26 PD-1 signaling also inhibits the CD3:CD28-mediated upregulation of glucose metabolism within the T lymphocyte.27 Therefore, it is reasonable to expect that PD-1 pathway–blocking agents would be better tolerated in patients than would CTLA-4 blockade.
Figure 1.
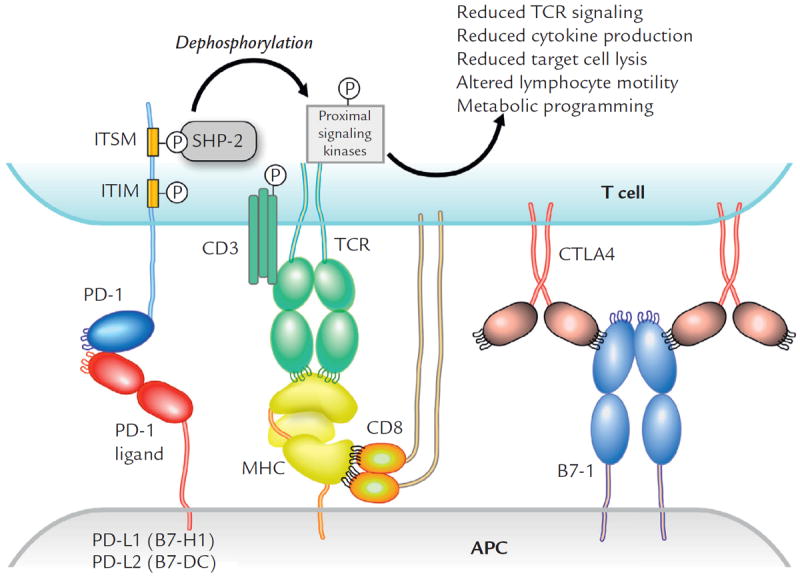
The interaction of PD-1 and PD-L1 reduces T-lymphocyte function. APC = antigen presenting cell; CTLA = cytotoxic T-lymphocyte antigen; ITIM = immunoreceptor tyrosine-based inhibitory motif; ITSM = immunoreceptor tyrosine-based switch motif; MHC = major histocompatibility complex; P = phosphoryation site; PD = programmed cell death protein; SHP = Src homology 2 domain–containing phosphatase; TCR = T cell receptor.
The role of the PD-1/PD-L1 pathway in peripheral tolerance is evident in the autoimmune phenotype in knockout mice on autoimmune backgrounds. Unlike knockout mouse models of Ctla-4, knockout of PD-1 and PD-L1 is not fatal in mice, and an autoimmune phenotype in these mice appears to require some additional initiator. In a nonobese diabetic mouse model, homozygous disruption of the PD-L1 gene, Pdcd1, accelerated the development of immune-mediated diabetes.28 Okazaki et al. found that a dilated cardiomyopathy can develop in Balb/c PD-1 knockout mice.29 On the autoimmune-prone background with the lupus-prone lpr mutation, Pdcd1 knockout mice develop nephritis and a lupus-like arthritis.30 PD-1 blockade improves lupus-like nephritis in New Zealand black × New Zealand white F1 (hybrid) mice.31 When the PD-L1 knockout mouse is crossed with the mouse strain 129S4/SvJae, which is resistant to experimental immune encephalitis, immunized mice develop an early-onset, rapidly progressive, severe experimental immune encephalitis.32 Thus, checkpoint blockade not only may block co-inhibitory signaling on effector T cells but also may shift the threshold at which antigen-specific T lymphocytes activate. This pathway appears also to play a significant role in human autoimmunity. In some patients with rheumatoid arthritis, splice variants of the PD-1 receptor that delete the transmembrane domain exon produce a soluble PD-1, and it is elevated in serum.33-35
The PD-1 pathway regulates the inflammatory response in infection. In patients with HIV, Day et al36 described that both an increased percentage of PD-1+ cells and the level of PD-1 expression on HIV-specific CD8 T cells were associated with increased disease severity, as measured by viral load and decreased CD4 count. This finding is clearly evident in multiple mouse models of infection. In a mouse model of liver infection, PD-1 knockout mice are able to clear the adenovirus quicker than are mice with intact PD-1.37 However, the knockout mice also develop worse hepatotoxicity than do infected wild-type mice. In herpes simplex virus keratitis, the expression of PD-L1 is upregulated on CD11b+ macrophages.38 PD-L1 blockade results in more severe keratitis and increased herpes simplex virus 1–specific T-cell proliferation. Whether the proinflammatory effect of PD-L1 blockade is a result of blocking the ligand on macrophages within the site of infection or on the antigen-presenting cells in the draining lymph node is difficult to distinguish. Not only do viruses and bacteria exploit this pathway but also parasitic infections appear to as well. Schistosoma mansoni induces T-cell anergy via upregulation of PD-L1 on macrophages.39,40 Homozygous PD-L1 knockout mice are viable and fertile but exhibit resistance to the parasite Leishmania mexicana.41
PD-1/PD-L1 Blockade as a Therapeutic Strategy in Cancer
The negative regulation of lymphocytes by PD-1 is mediated by the interaction with its ligands and B7-like proteins, PD-L1 and PD-L2.17,18,42 Many tumors have increased expression of PD-L1, including squamous cell carcinoma, colon adenocarcinoma, and breast adenocarcinoma. Transgenic expression of PD-L1 on tumors increase tumorigenesis and invasiveness in vivo; its overexpression also makes tumor cells less susceptible to specific CD8 T cell–mediated lysis in vitro.43 In mouse melanoma models44, tumor growth is transiently abrogated in PD-1 knockout mice or with treatment with antibodies blocking the interaction between PD-L1 and its receptor PD-1. Initial studies have reported not only better tolerability than with prior immunotherapies but also more impressive clinical efficacy than would be expected from preclinical mouse models.
Early Studies in Solid Malignancy: Anti-PD-1 Blockade
Nivolumab
The first in-human trial of an anti-PD-1 therapeutic agent in melanoma was the pilot Phase I, dose-escalation study of the fully human IgG4-blocking mAb against PD-1, nivolumab (formerly, BMS-936558, MDX-1106, and ONO-4538); this study included 10 patients with melanoma.1 It reported 1 complete response in a patient with colon adenocarcinoma and 2 partial responses in patients with melanoma and renal cell carcinoma. These first 3 responders developed durable responses.4 A patient with colon adenocarcinoma continued to have a complete response 3 years after treatment. A patient with kidney cancer had a partial response on therapy that converted to a complete response after therapy was completed. A patient with melanoma with a partial response was stable for 16 months, and after progression was successfully retreated with nivolumab. Although responses were few in number, the impressive durations of these responses suggest that, like ipilimumab and high-dose IL-2, this approach to immune-checkpoint blockade might be able to produce responses that continue after the completion of therapy and may result in tumor remissions.
In a larger-scale, Phase I, dose-escalation trial of nivolumab, 94 patients with melanoma were treated every 2 weeks for up to 96 weeks.45 The maximal tolerated dose was not found at doses up to 10 mg/kg. Five percent of patients stopped treatment due to adverse events. Due to the immune-related toxicity seen with the first checkpoint inhibitor, ipilimumab, the immune-related adverse events of nivolumab were specifically reported. These events included pneumonitis, vitiligo, colitis, hepatitis, hypophysitis, and thyroiditis. These immune-related adverse events were not correlated with dose. Diarrhea and transaminitis were reversible with treatment interruption and corticosteroids in severe cases; endocrinopathies were managed with supplementation. Pneumonitis was seen in 3% of patients (9/296), 3 episodes of which were associated with death (none in patients with melanoma). This finding prompted stricter vigilance of potential immune-related adverse events and prompted the administration of corticosteroids, which was associated with no additional deaths secondary to pneumonitis in later studies. That trial reported rates of response to nivolumab in melanoma and renal cell carcinoma of 28% and 27%, respectively. In patients with non–small cell lung cancer, 17% responded.
The antitumor effect of nivolumab in the melanoma cohort was also published after additional follow-up.4 Thirty-one percent of the 107 patients (n = 33) had an objective response, and the estimated median duration of response was 2 years. Forty-five percent of responders (n = 15) did so by the first assessment at 8 weeks. Seventeen patients had responses but stopped therapy for reasons other than progression (6 due to adverse events, 2 due to complete response, and 4 for other reasons; 5 completed the total 96 weeks of treatment). Twelve of these patients (71%) had continued responses for at least 16 weeks after terminating therapy.
The first published Phase III trial of PD-1 blocking agents was a comparison of nivolumab to dacarbazine in previously untreated patients with melanoma without the serine/threonine-protein kinase B-raf proto-oncogene BRAF mutation.14 Four hundred eighteen patients were randomized to receive either treatment. The prevalences of grade 3/4 drug-related adverse events were 11.7% in the nivolumab arm and 17.6% in the dacarbazine arm. The objective response rate was 40% with nivolumab compared with 13.9% in the dacarbazine group. The milestone survival rates at 1 year were 72.9% with nivolumab and 42.1% in the dacarbazine group (hazard ratio for death, 0.42; 99.79% CI, 0.25 to 0.73; P<0.001). Although the objective response rate with nivolumab was greater in the PD-L1+ subgroup (52.7%) than in the PD-L1−subgroup (33.1%), a survival benefit compared with dacarbazine was seen in both subgroups.
Pembrolizumab
Pembrolizumab (formerly, MK-3475) is a very high-affinity humanized anti-PD-1 IgG4 isotype antibody (Table I). A Phase I trial that evaluated 3 different dose regimens in 135 patients with metastatic melanoma included both ipilimumab-naive patients and those with progression on prior treatment with ipilimumab.3 Low-grade symptoms were frequent, and grade 3/4 drug-related adverse events occurred in 13%. The greatest prevalence of adverse events was seen with the highest dose (10 mg/kg q2w) compared with the 10- and 2-mg/kg q3w dose regimens. Although the study was not powered to compare the efficacy between arms, the highest response rates were seen in this highest-dose arm (52%–56%) compared with the q3w arms (25%–37%). One of the 17 patients with stable disease on treatment achieved a partial response after 48 weeks of treatment. However, of the 52 patients who responded, the majority did so within the first 12 weeks of treatment. The median duration progression-free survival was >7 months. Median overall survival was not reached at the time of reporting. Both ipilimumab-naive patients and those who had received ipilimumab previously responded to pembrolizumab (confirmed response rate, 37% and 38%, respectively). In the follow-up clinical report presented at the 2014 annual meeting of the American Society of Clinical Oncology, the overall response rate was 41%, complete response was 9%, and there was less of a difference between the dose levels.51
Table I.
Binding affinity of B7/CD28 family members to their ligands and targeted blocking antibodies.*
| B7/CD28 Family Member: Antibody (Kd) | Study |
|---|---|
| B7-1 | |
| B7-1:CD28 (4300 nM) | van der Merwe et al46 (Scatchard plots analysis) |
| B7-1:PD-L1 (1540–1990 nM) | Butte et al47 (Scatchard plots analysis) |
| B7-1:CTLA-4 | |
| 400 nM | van der Merwe et al46 (Scatchard plots analysis); |
| 310 nM | Butte et al47 (equilibrium binding†) |
| PD-1 | |
| PD-1:PD-L1 | |
| 270–526 nM | Youngnak et al 48 (Scatchard plots analysis) |
| 590–770 nM | Butte et al47 (Scatchard plots analysis) |
| 770 nM | Butte et al47 (equilibrium binding†) |
| PD-1:PD-L2 | |
| 89–106 nM | Youngnak et al 48 (Scatchard plots analysis) |
| 590 nM | Butte et al47 (equilibrium binding†) |
| PD-1:nivolumab (2.6 nM) | Brahmer et al1 (Scatchard plots analysis) |
| PD-1:pembrolizumab (0.028 nM)‡ | Hamid et al3 |
| PD-1:pidilizumab (20 nM) | Atkins et al49 |
| PD-L1:MPDL-3280A (0.4 nM) | Herbst et al50 |
PD = programmed cell death protein.
Significant differences were found in the binding affinity of different checkpoint inhibitors to their targets.
Biacore (GE Healthcare Life Sciences, Pittsburgh, Pennsylvania).
Although Kd 0.028 nM, 50% effective binding concentration of pembrolizumab was 0.1 to 0.3 nM.
In a pooled analysis including later arms of this Phase I study, the overall response rate in 411 patients with melanoma was 34%. Patient who were previously untreated or ipilimumab-naive had greater response rates than did the ipilimumab-treated patients (40%, 44%, and 28%, respectively). However, the only significant difference in response on multivariate subgroup analysis was that patients with less baseline tumor burden were more likely to respond to pembrolizumab.52
Pidilizumab
Pidilizumab (formerly, CT-011) is a humanized anti-PD-1 IgG1 isotype antibody and was one of the first anti-PD-1 agents used in patients with cancer. In a Phase I dose-ranging (0.2–6 mg/kg) trial in patients with hematologic malignancies, single-dose administration of CT-011 was well tolerated.53 In a Phase II, multicenter study of pidilizumab in metastatic melanoma, patients were randomized to receive either 1.5 or 6 mg/kg q2w for up to 54 weeks and were stratified by ipilimumab-experience status.49 The treatment was well tolerated, with 4% experiencing at least 1 adverse event and four grade 3/4 adverse events (appendicitis, arthritis, hepatitis, and pneumonitis), with no treatment-related deaths reported. Whether patients had previous experience with ipilimumab did not affect response rates. The overall response rate was 6%. Although this response rate did not meet the expectations about the primary end point of this study, the with regard to the secondary end point—overall survival at 1 year—the study drug exceeded expectations, at 64.5%. Although pidilizumab appears to be associated with lower response rates in melanoma than does nivolumab or pembrolizumab, the 1-year overall survival rate is similar to that reported in studies of nivolumab (62%).
Differences among the Anti-PD-1 Antibodies
Although all of the therapeutic agents in the anti-PD-1 antibody family target the binding of PD-L1 to PD-1, there may be differences in their clinical benefit. Two major differences between the 3 anti-PD-1 antibodies are the affinity and the isotype of the antibody (Tables I1,3,46 and II, respectively). Of the PD-1–directed antibodies, pembrolizumab has the highest affinity for PD-1 (Kd, 20 pM), which may result in a difference in clinical benefit. Each is reported to block the interaction of PD-1 with ligand, but greater affinity should allow a mAb to still be efficacious at the lower concentrations seen long after the end of administration of the mAb or at sites with low penetrance of mAb. Notably, pidilizumab has a lower affinity than do the other anti-PD-1 antibodies. However, longer follow-up of patients treated with pidilizumab may show that slower onset of tumor regression occurs with less avid binding of PD-1–blocking antibodies. An unanswered question in the field is whether the duration and extent of a response correlate with durable response and overall survival. This question is particularly relevant because the antibodies with greater affinity are also those associated with increased toxicity. Which metric is best for measuring clinical outcome in clinical trials of these agents is also unclear. Because immunotherapies can produce prolonged stable disease and inflammatory immune responses, which may be categorized as progression in the Response Evaluation Criteria In Solid Tumors, the clinical benefit may be significantly underestimated if response rate is evaluated as a primary end point.13
Table II.
Isotype of immune-checkpoint inhibitors.
| Checkpoint Inhibitor | Killer Isotype | Nonkiller Isotype |
|---|---|---|
| Anti-CTLA-4 | Ipilimumab (IgG1) | Tremelimumab (IgG2) |
| Anti-PD-1 | Pidilizumab (IgG1) | Nivolumab (IgG4), pembrolizumab (IgG4) |
| Anti-PD-L1 | – | BMS-936559 (IgG4), MPDL-3280A (mutated IgG1 that eliminates ADCC and CDC) |
ADCC = antibody-dependent cell-mediated cytotoxicity; CDC = complement dependent cytotoxicity; CTLA = cytotoxic T-lymphocyte antigen; Ig = immunoglobulin; PD = programmed cell death protein.
Unlike nivolumab or pembrolizumab, pidilizumab is an IgG1 isotype antibody. The Fc (fragment, crystallizable) domain of IgG1 contains recognition sites for Fc receptors that mediate antibody-dependent, cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). In correlative studies, treatment with pidilizumab was not associated with a change in PD-1+ CD4 or CD8 lymphocytes, suggesting that ADCC may not be the cause of its lower response rates.54,55 Nivolumab and pembrolizumab both contain IgG4 Fc (mutated in the hinge to maintain dimeric structure) rather than the IgG1 Fc. The IgG4 Fc has significantly less potential for ADCC than does the IgG1 Fc of ipilimumab or pidilizumab.56 Similarly, MPDL-3280A (see subsequent discussion) has a mutated IgG1 that eliminates ADCC and CDC. An unaddressed question is whether the mutations in the Fc of these IgG1 and IgG4 antibodies elicit any anti-antibody responses that limit their later efficacy.
Early Studies of PD-L1 Blockade
BMS-936559
Blocking PD-L1, the ligand of PD-1, has also been reported to have clinical benefit in patients. The first published report of in-human anti-PD-L1 antibody was the fully human IgG4-blocking mAb against PD-L1, BMS-936559, which inhibits the binding of both PD-1 and CD80/B7-1.2 In a Phase I, dose-escalation trial, the maximal tolerated dose was not found at doses up to 10 mg/kg. Because PD-1 is expressed on circulating T lymphocytes, some have proposed that anti-PD-L1 blockade would produce fewer adverse events than would anti-PD-1 blockade. Although there were no cases of pneumonitis with BMS-936559 use, immune-mediated events occurred in 39% of patients; these events included rash and hypothyroidism, as well as individual cases of sarcoidosis, endophthalmitis, diabetes mellitus, and myasthenia gravis. Six percent of patients discontinued therapy due to treatment-related adverse events. Of the subset of patients with melanoma, who were heavily pretreated, 16% had objective responses.
MPDL-3280A
MPDL-3280A is an engineered human IgG1 anti-PD-L1 mAb in which the IgG1 Fc domain is mutated to completely abrogate ADCC and CDC. In an in vitro assay, the engineered IgG1 of MPDL-3280A was associated with less ADCC than was the modified IgG4 isotype of nivolumab and pembrolizumab.57 MPDL-3280A was well tolerated at doses ranging up to 20 mg/kg. In a Phase Ia extension arm, a group with advanced cutaneous melanoma had a response rate of 29%, which was greater than the overall response rate of 21% seen in a group with other tumor types.57 With longer follow-up, the overall response rate across tumor types was similar to that in the earlier report and in the melanoma cohort (21% and 30%, respectively).50 Patients with melanoma accounted for one fourth of the 175-patient cohort.
Although PD-L1 expression on tumor cells has the strongest association with response to nivolumab, response to MPDL-3280A was associated with high PD-L1 expression, particularly on the tumor-infiltrating immune cells (by immunohistochemistry [IHC] analysis with rabbit anti-human mAb, clone SP142, Ventana platform).50 With this mAb, PD-L1 expression on immune cells, myeloid cells (macrophage and dendritic cells), and T lymphocytes was much more common than on tumor cells. The activated CD8 cell and type 1 T helper cell gene-expression profile also was associated with response to MPDL-3280A, which fits with the hypothesis that PD-1 pathway blockade benefits patients with inflamed tumors most.
Investigation of Potential Biomarkers
PD-L1 is expressed by many cancer cells42 and has been associated with worse prognosis in lung adenocarcinoma and renal cell carcinoma.58,59 In renal cell carcinoma, both immune-cell expression and tumor-cell expression of PD-L1 on IHC with the anti-PD-L1 clone 5H1 were markers of poor prognosis.59 Patients with renal cancer who have ≥10% tumor expression have a 3-fold increased risk for cancer-associated death.59 The extent of PD-L1 expression on tumor cells has been associated with vertical growth of primary melanoma tumors and poor prognosis in some studies, but its role as a prognostic biomarker in melanoma remains controversial.60-62
One issue that makes tumor tissue expression of PD-L1 difficult to use as a biomarker is the accessibility of tissue and the variability between tumor samples within an individual patient. Tumor heterogeneity is a significant issue in many tumors, making smaller biopsy samples less reliable for tissue-based biomarkers, such as PD-L1 tumor expression.63 Furthermore, PD-L1 is a dynamic marker that can be upregulated by local inflammation and some oncogenic mutations.64-66 Tumors may evade the immune system by upregulation not only of PD-L1 on the tumor cell but also of PD-L1 in its microenvironment. Infections can induce PD-L1 expression on macrophages. Similarly, the expression of PD-L1 within a tumor and its microenvironment also may change with treatment.
Much effort has been spent on determining potential biomarkers predictive of response to PD-1 pathway therapy. PD-L1 tumor expression has produced much enthusiasm since the preclinical data and early clinical correlative studies reported that it was associated with clinical benefit of PD-1 blockade. Preclinical models support that tumors can evade tumor-specific effector T-cell cytotoxicity by expressing PD-L1.45 In the pilot Phase I study of nivolumab, 9 patients’ tumor specimens were available for exploratory analysis of PD-L1 (B7-H1) expression on tumors on IHC analysis with the murine anti-human B7-H1, clone 5H1 (previously described67). This analysis found that PD-L1 expression within tumor specimens was associated with response to treatment.1 The expression of PD-L1 had 1 of 3 patterns: cytoplasmic, membranous, or none. Three of four patients (75%) with membranous pattern on manual staining with the 5H1 antibody responded to treatment; none of the tumors of the 5 nonresponders expressed membranous PD-L1. These findings were further explored in a larger cohort with a lower true-positive rate of response. In this cohort of nivolumab-treated patients, none of the patients with PD-L1− tumor cells responded; 9 of 25 patients (36%) with PD-L1+ tumor cells had an objective response.45 The assay used for subsequent nivolumab trials was transitioned to an automated assay with a different anti-PD-L1 antibody (clone 28-8). With this assay, response rates of up to 67% were seen with nivolumab in the PD-L1+ subset. Taube et al64 explored the expression of PD-1, PD-L1, and PD-L2 on tumor cells and the immune infiltrate. Although the expression of PD-L1 on tumor cells and immune cells was associated with PD-1 expression on lymphocytes, the expression of PD-L1 on tumor cells had the strongest association with response to nivolumab. However, there may be little value in using PD-L1 as a predictive marker in melanoma; in a Phase III, randomized trial, a survival benefit was seen across all prespecified subgroups regardless of PD-L1 status.14 The objective rate of response to nivolumab was greater in the PD-L1+ subgroup than in the PD-L1− subgroup (52.7% and 33.1%, respectively). Clearly, the PD-L1−subgroup also has significant benefit. PD-L1 can also be expressed on infiltrating lymphocytes, monocytes, and macrophages. Therefore, the role of PD-L1 expression may vary, depending on tumor type and whether alternative combination therapies are employed.5,68 (Table II)
Lack of Definitive Criteria for PD-L1 Positivity across Types of Tumor or PD-1 Blockade
Although the initial exploratory studies in this field suggested that PD-L1 IHC was both a specific and a sensitive predictive biomarker, later studies revealed it to be less reliable. Since the publication of the initial studies that used IHC analysis of tumor tissue with the anti-PD-L1 antibody (5H1), clinical responses have been reported in both PD-L1+ and PD-L1− tumors in 10 studies of 3 different checkpoint inhibitors (Table III). With the automated anti-PD-L1 IHC assay (28-8), PD-L1+ tumors derived a greater benefit than did PD-L1− tumors.69,70 However, PD-L1− melanomas had a 17% to 19% rates of response to nivolumab.69,70 Similarly, 4 trials of pembrolizumab reported 11% to 13% response rates in PD-L1− tumors (see Table III). However, the greater rates of response to treatment with nivolumab and pembrolizumab were correlated with PD-L1 expression on the surface of the tumor cells on 2 different IHC assays (28-8 and 22C3).
Table III.
Clinical response rates in PD-L1+* and PD-L1– tumors after treatment with checkpoint inhibitors. Data are given as %.
| Antibody/Tumor Type/Treatment | Unselected† | PD-L1+ | PD-L1– |
|---|---|---|---|
| Anti-PD-1 antibody‡ | |||
| Melanoma specific (28-8 clone) | |||
| Nivolumab§ | |||
| Grosso et al69 (n = 34) | 29 | 44 | 17 |
| Weber et al70 (n = 44) | 32 | 67 | 19 |
| Pembrolizumab∥ | |||
| Daud et al71 (n = 113) | 40 | 49 | 13 |
| Ribas et al52 (n = 411) | 40 | 49 | 13 |
| All/multiple tumors | |||
| Nivolumab§ | |||
| Topalian et al4 (solid tumor [5H1 clone]; n = 42) | 21 | 36 | 0 |
| Pembrolizumab∥ | |||
| Gandhi et al72 (NSCLC; n = 129) ¶ | 19 | 37 | 11 |
| Seiwert et al73 (head and neck; n = 55)# | 18 | 46 | 11 |
| Anti-PD-L1 antibody** | |||
| Melanoma specific | |||
| MPDL-3280A | |||
| Hamid et al57 (n = 30) | 29 | 27 | 20 |
| All/multiple tumors | |||
| MPDL-3280A | |||
| Herbst et al50 (solid tumor; n = 94) | 23 | 46 | 15 |
| Powles et al68 (bladder; n = 65) | 26 | 43 | 11 |
| Soria et al74 (NSCLC; n = 53) | 21 | 36 | 13 |
NSCLC = non–small cell lung cancer; PD = programmed cell death protein.
Adapted with permission from Callahan.80
Positive intratumoral PD-L1 expression may refer to either a pattern of expression on tumor cells or the immune infiltrate, depending on the assay.41,43,54,62,63,65,75-79
In some studies, “unselected” included patients with tumors not assayable for PD-L1.
For membranous pattern on tumor cells on PD-L1 assay.
Cutoff: ≥ 5%.
Cutoff: ≥ 1% or stroma; 22C3 clone.
The cutoff of PD-L1 positivity, as determined by the Youden index, was not disclosed.75
Patients with ≤ 1% expression of PD-L1 were excluded, and the cutoff for PD-L1 positivity, as determined by the Youden index, was not disclosed.76
Immune infiltrate on PD-L1 assay (proprietary).
Four trials of the anti-PD-L1 antibody MPDL-3280A also reported that greater response rates also correlated with PD-L1+ tumors (see Table III). However, in this assay, developed in parallel with the clinical trials of MPDL-3280A, PD-L1 expression on the tumor immune infiltrate, not on the tumor cells, correlated better with response.81 Similarly, although the extent of PD-L1 positivity on immune cells was associated with greater rates of response to MPDL3280A in non–small cell carcinoma,68 11% to 20% of PD-L1− tumors responded to MPDL-3280A.
Is PD-L1 Expression in Tumors a Predictive Biomarker?
The objective of a predictive biomarker is to define the subset of patients who will and will not derive benefit from a given therapy. Across different treatments and assays for PD-L1 expression on tumor cells or immune infiltrate, response rates are greater in PD-L1+ tumors than in PD-L1− tumors. Given the clinical benefit seen in patients with PD-L1− tumors, the lack of tumor PD-L1 expression does not appear to be a biomarker appropriate for excluding patients from receiving therapy. PD-L1 status may be more appropriately used for distinguishing which patients may respond to single-checkpoint blockade and which patients should be directed to clinical trials of combinations of immune-checkpoint inhibitors. This hypothesis is particularly well illustrated by the additional benefit seen in patents with PD-L1− melanoma who receive the combination of nivolumab + ipilimumab.5 Clearly, PD-L1 is far from a definitive predictor of response to PD-1 pathway–blocking agents. Establishing a suitable predictive biomarker for single-agent immune-checkpoint inhibition will allow for the deferral of alternative treatments and potentially avoid their toxicity in those who develop a remission. The identification of which patients most likely to respond to a single immune-checkpoint inhibitor may avoid the additional toxicity seen with combination checkpoint blockade.
One goal of a predictive biomarker is to determine which patients will benefit from a treatment (true positives). However, an effective predictable biomarker test also minimizes the number of patients who have a false-negative result and who can respond to therapy. Unfortunately, decreasing the number of false-negative results of a test will increase the false-positive results of a test. The threshold, or cutoff, of a positive test result defines the positive and negative predictive values of an assay. The threshold of PD-L1 positivity, ≥1%, in patients treated with pembrolizumab was selected to reduce the number of patients with a false-negative result. On the other hand, in studies of nivolumab, the threshold of positive PD-L1 generally reported is ≥5% (with both 5H1 and 28-8). Weber et al70 illustrated the effect of changing the threshold on the predictive characteristics of a biomarker in patients with melanoma treated with dose-ranging nivolumab. In that study, ≥5% and ≥1% cutoffs for PD-L1 positivity were compared. When the threshold of PD-L1+ was lowered to ≥1%, 1 of the 6 PD-L1− patients (at 5% threshold) was included in the PD-L1+ cohort. There were still 5 patients who responded to nivolumab in whom <1% PD-L1 was expressed on the tumor. However, a lesser cutoff was associated with many more patients with a false-positive result (ie, specificity was decreased). Thus, lowering the threshold to include 1 more true positive was associated with also adding 10 false positives. Lowering the cutoff to ≥1% reduced the difference in overall response rates between PD-L1+ and PD-L1−. The role that PD-L1 expression in tumors will play as a biomarker predictive of PD-1 blockade is unclear. The majority of melanoma in this study express PD-L1, and a lack of expression does not exclude the potential to respond to treatment.70 However, for other tumors in which the expression of PD-L1 is lesser and rates of response to PD-1 blockade are lesser, PD-L1 tumor expression may be a clinically significant biomarker.
Although patients with melanoma appear more sensitive to PD-1 blockade than do patients with other tumors, still 30% to 70% of PD-L1+ tumors do not respond to monotherapy with checkpoint blockade. Therefore, the true-positive rate (sensitivity) of the assay is also lacking. Whether there is a subset of PD-L1+ patients who will benefit from combination therapy is unclear. In a trial of combination nivolumab + ipilimumab, concurrent therapy was associated with increased Ki67+ and inducible T-cell co-stimulator ICOS+ lymphocytes.82 The depletion of T-regulatory cells with ipilimumab may affect the extent of proliferating and activated lymphocytes. Whether these factors are predictive of response is yet to be determined. A better understanding of other active co-inhibitory molecules in PD-L1+ tumors may also help to distinguish which PD-L1+ patients may benefit from combination therapy and may help to direct the rational design of combination trials. The ongoing melanoma biomarker study comparing ipilimumab or nivolumab monotherapy to combination nivolumab + ipilimumab (clinicaltrials.gov identifier: NCT01621490) will help to establish whether PD-L1 expression on tumors is a biomarker predictive of response to nivolumab monotherapy in melanoma.
Combination Therapies and New Agents in Melanoma
There are currently many trials pairing PD-1 pathway blockade with novel and FDA-approved agents to improve the response rates relative to monotherapy (Table IV). CTLA-4 and PD-1 play distinct roles in regulating adaptive immunity. The combination of CTLA-4 and PD-1 blockade has reported synergistic antitumor activity in the preclinical B16 mouse melanoma model.83 Korman et al84 described these results in seminars beginning in 2007. These results prompted the first Phase I combination of nivolumab + ipilimumab in advanced melanoma. Fifty-three patients were treated in the combination arm with 4 doses of a combination of nivolumab + ipilimumab followed by 4 doses of nivolumab.5 Thirty-three patients who had received prior ipilimumab treatment were treated with nivolumab q2w for up to 48 doses. The cohort that received nivolumab 3 mg/kg + ipilimumab 3 mg/kg exceeded the maximum tolerated toxicity (persistent asymptomatic grade 3/4 elevated lipase in 3 of 6 patients). In the combination cohorts, 6 of 28 patients (21%) experienced dose-limiting toxicity; 53% of patients experienced grade 3/4 adverse events, most commonly elevated lipase and transaminitis. Clinical activity was seen in all treatment groups. Forty percent of the combination arm had a confirmed objective response (not including 4 objective responses by immune-related response criteria).13 No treatment-related deaths were reported, and clinical benefit (including conventional, nonconventional, immune-related response, or stable disease over 6 months) was seen in 65% of the combination cohort. Patients who received the maximal doses (nivolumab 1 mg/kg + ipilimumab 3 mg/kg) with acceptable adverse events had a greater response rate (53%), with deeper responses at the first scheduled assessment than in other patients. As seen in the ipilimumab-refractory arm of the trial reported by Weber et al,70 patients previously treated with ipilimumab also may respond to nivolumab (sequentially treated, 20% had objective responses).5,70
Table IV.
Anti-PD-1 and PD-L1 blockade agents currently* in clinical trials.
| Target/Treatment | Fc Domain |
|---|---|
| PD-1 (blocks interaction between PD-L1 and PD-L2) | |
| Nivolumab, BMS-936558, MDX-1106, ONO-45381 | Human IgG4, stabilizing mutation S228P |
| Pembrolizumab3 | Humanized IgG4, S228P |
| Pidilizumab, CT-01149 | Humanized IgG1 |
| AMP-224 (PD-1 targeting therapy) | PD-L2-Fc fusion protein (blocking) |
| AMP-514, MEDI-0680 | IgG, details unpublished |
| PD-L1 (inhibits binding to PD-1 and CD80) | |
| BMS-9365592 | Human IgG4, S228P |
| MEDI-4736 | Engineered human IgG1 |
| MPDL-3280A57 | Engineered human IgG1 |
| MSB-0010718C | IgG1, details unpublished |
Fc = fragment, crystallizable; Ig = immunoglobulin; PD = programmed cell death protein.
Registered on clinicaltrials.gov as of July 7, 2014.
Toxicity has proven to be an issue in immunotherapy, making Phase I trials of combinations essential for evaluating the tolerability of these regimens. Ipilimumab in combination with vemurafenib in metastatic melanoma can produce significant hepatotoxicity.85 Because PD-1 pathway inhibitors are better tolerated than is ipilimumab, combinations based on the PD-1 blockade may be better tolerated. Early tolerability data from the Phase Ib trial of a combination of vemurafenib + MPDL-3280A suggested transaminitis as a common adverse event. However, it was found to be reversible after vemurafenib was withheld and patients tolerated restarting vemurafenib treatment.57
Despite the clinical efficacy of immune-checkpoint blockade thus far, there is much room for improvement. The combination of nivolumab + ipilimumab has shown impressive duration and rates of response in melanoma, prompting combination with multiple other immune modulators, both novel and FDA approved (anti–lymphocyte-activation gene 3 [anti-LAG-3], 4-1BB, or killer Ig-like receptors [KIR], and IL-21, pegylated interferon, or lenalidomide). Chronic activation of lymphocytes can result in T-cell exhaustion, in which lymphocytes express multiple co-inhibitory receptors and essentially become nonfunctional (Figure 2).87 We now recognize cancer as a disease that can take years to evolve and develop, with constant immune interactions. In many cases, T cells attack the tumor but are unsuccessful, and the antitumor T cells develop this “exhausted” phenotype. Weber et al70 found that patients who had greater levels of tumor-specific lymphocytes before therapy were actually less likely to benefit from nivolumab. They proposed that these might be exhausted tumor-specific lymphocytes, which would require multiple-checkpoint inhibitors to regain function.
Figure 2.
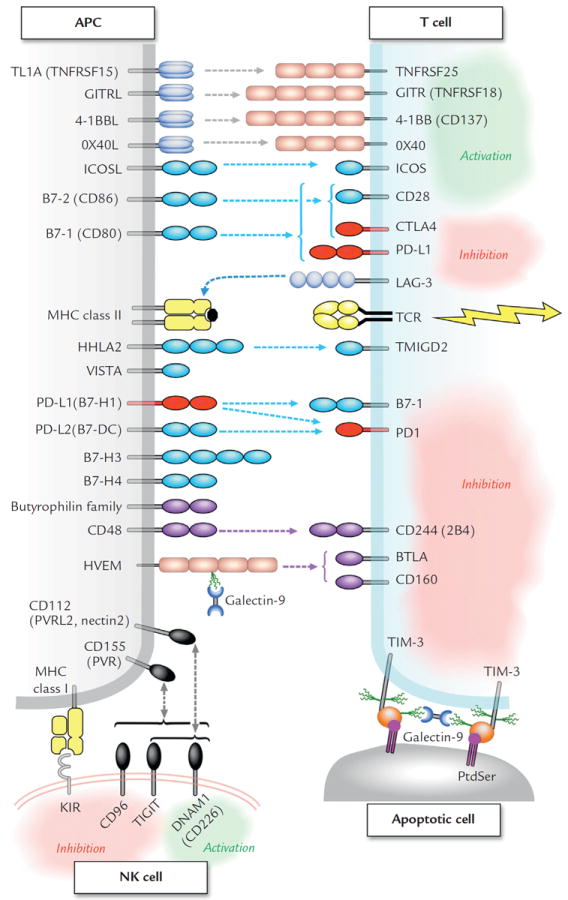
Tumor-infiltrating lymphocytes and natural killer cells can express multiple co-stimulatory and co-inhibitory receptors, which may be potential therapeutic targets. The butyrophilin gene family consists of ~30 B7-like proteins, associated with some autoimmune diseases, and some appear to negatively regulate lymphocyte activation.86 To avoid confusion, B7-H5 has been omitted because it has been assigned to both proteins VISTA and HHLA-2. APC = antigen presenting cell; BTLA = B- and T-lymphocyte attenuator; DNAM = DNAX Accessory Molecule-1; GITR = glucocorticoid-induced tumor necrosis factor receptor; HHLA = HERV–H LTR-associating protein 2; HVEM = herpesvirus entry mediator; ICOS = inducible co-stimulator; KIR = killer Ig-like receptors; L = ligand; LAG-3 = Lymphocyte-activation gene 3; MHC = major histocompatibility complex; NK = natural killer; PD = programmed cell death protein; PtdSer = phosphotidylserine; PVR = poliovirus receptor; TCR = T cell receptor; TIGIT = T cell immunoreceptor with Ig and ITIM domains; TIM = T-cell immunoglobulin and mucin domain; TL1A = TNF-like cytokine; a TNF-like ligand for DR3 and TR6/DcR3; TMIGD = Transmembrane and Immunoglobulin Domain-containing Protein 1; TNFRSF = TNF receptor superfamily; VISTA = V-domain Ig suppressor of T cell activation.87
Without predictive biomarkers, understanding the mechanism by which a treatment works is essential for deciding which agents would be best combined to benefit patients. There are 5 anti-PD-1 and 4 anti-PD-L1 agents in clinical trials registered on clinicaltrials. gov (Table V). There are 2 Phase III clinical trials: (1) nivolumab versus chemotherapy in previously ipilimumab-treated or -untreated patients (clinical-trials.gov identifiers: NCT01721746 and NCT01721772), and (2) a 3-arm study comparing nivolumab, ipilimumab, and nivolumab + ipilimumab in untreated melanoma (clinicaltrials.gov identifier: NCT01844505). A Phase III clinical trial randomizing patients with untreated BRAF (wild-type) melanoma to receive nivolumab or dacarbazine was stopped early because an independent data-monitoring committee found improved overall survival in the nivolumab arm.88 There are an additional 31 registered clinical trials, which include those in patients with melanoma. There are 11 trials of combinations enrolling patients with melanoma, as well as many trials in patients with other solid and hematologic malignancies. Because of the differences in cellular expression of PD-1 and PD-L1, the tolerability of combination PD-1 + PD-L1 blockade is also being investigated (clinicaltrials.gov identifier: NCT02118337).
Table V.
Current* clinical trials of anti-PD-1 and anti-PD-L1 blockades, including patients with metastatic melanoma.
| Blockade/Tumor Type/Treatment | clinicaltrials.gov Identifier |
|---|---|
| Anti-PD-1 | |
| Melanoma specific | |
| Nivolumab | |
| Nivolumab monotherapy | NCT01621490 |
| Nivolumab monotherapy vs nivolumab + ipilimumab vs ipilimumab monotherapy | NCT01844505† |
| Nivolumab vs chemotherapy | NCT01721746,† NCT010721772† |
| Nivolumab + peptide‡ | NCT01176474, NCT01176461 |
| Pembrolizumab | |
| Pembrolizumab vs chemotherapy | NCT01704287 |
| Pembrolizumab vs ipilimumab | NCT01866319 |
| Pembrolizumab + pegIFN | NCT02112032 |
| All/multiple tumors | |
| Nivolumab | |
| Nivolumab monotherapy | NCT00729664, NCT00836888 |
| Nivolumab + anti-KIR | NCT01714739 |
| Nivolumab + anti-LAG-3 | NCT01968109 |
| Nivolumab + IL-21 | NCT01629758 |
| Pembrolizumab | |
| Pembrolizumab monotherapy | NCT01295827, NCT01848834 |
| Pembrolizumab + 4-1BB agonist | NCT02179918 |
| AMP-224 monotherapy | NCT01352884 |
| AMP-514 | |
| AMP-514 monotherapy (MEDI-0680) | NCT02013804 |
| AMP-514 + MEDI-4736 | NCT02118337 |
| CT-0116 | NCT01386502 (w/d) |
| Anti-PD-L1 | |
| Melanoma specific | |
| BMS-936559 | NCT01455103 (w/d) |
| MEDI-4736 + dabrafenib/trametinib | NCT02027961 |
| MPDL-3280A + vemurafenib | NCT01656642 |
| All/multiple tumors | |
| BMS-936559 | NCT00729664 |
| MEDI-4736 | |
| MEDI-4736 monotherapy | NCT01938612 |
| MEDI-4736 + AMP-514 | NCT02118337 |
| MEDI-4736 + tremelimumab | NCT01975831 |
| MPDL-3280A | |
| MPDL-3280A monotherapy | NCT01375842 |
| MPDL-3280A + bevacizumab | NCT01633970 |
| MSB-0010718C | NCT01772004, NCT01943461 |
IL = interleukin; KIR = killer immunoglobulin-like receptor; LAG-3 = lymphocyte-activation gene 3; PD = programmed cell death protein; pegIFN = pegylated interferon; w/d = withdrawn.
Registered on clinicaltrials.gov as of July 7, 2014.
Phase III.
The peptide vaccine was not included as a therapeutics, but to monitoring T-lymphocyte immune response in this dose-ranging nivolumab trial.
Even within a family of immune-checkpoint inhibitors, there can be distinct differences. For example, the checkpoint inhibitor ipilimumab and the IgG2 mAb tremelimumab both block the interaction between CTLA-4 on lymphocytes and its B7 receptor on antigen-presenting cells. Although tremelimumab had promising findings in Phase I and II studies, Phase III reported no significant difference in response rates or overall survival over standard-of-care chemotherapy. The mechanism of ipilimumab may not depend on blocking binding to the B7 receptor but instead may depend on killing the cells that express the highest level of CTLA-4: intratumoral T-regulatory cells.75,76 When the effect of anti-mouse CTLA-4 is compared between antibodies with cytotoxic Fc and a non-ADCC/CDC Fc in mouse models, only the antibody that kills is effective in antitumor therapy. T-regulatory cells express CTLA-4 constitutively, rather than transiently as on most T cells, and levels are highest within the tumor.77 Ipilimumab is an IgG1 isotype antibody (killer) while tremelimumab is an IgG2 isotype (nonkiller). The mechanism of action of PD-1 antibodies is believed to be primarily through conventional T cells and involve reactivation, not T-cell depletion. Consequently, in terms of mechanism of action, CTLA-4 and PD-1 are well positioned to synergize.
Future Clinical Considerations
Over the upcoming years, with longer follow-up periods, the completion of ongoing trials, and the development of new agents, the landscape of immunotherapy likely will become richer, but more complicated.78,79 Although there are many questions unanswered, ongoing clinical trials with immune checkpoint–blocking therapeutics are expected to add insight in this field.89 We are entering a field with many of the difficulties inherent to allogeneic stem cell transplantation in balancing the benefit of graft-versus-leukemia effect and graft-versus-host disease. As we try to increase the antitumor effect of immune-modulatory therapeutics to produce remissions, we risk increasing immune-related adverse events. Early trials have reported that increased response rates in patients appear to correlate with increased adverse events. Toxicity also may vary depending on the sequence of therapy. Clearly, an effort is needed for educating oncologists on the symptoms and management of the immune-related adverse events that can occur with immunotherapy.
A major strength in studying the clinical benefit of PD-1/PD-L1 blockade and its combinations is the rapidity of response relative to other immunotherapies, including ipilimumab, IL-2, and allogeneic stem cell transplantation. Tumor-specific T-lymphocyte activity often requires months to develop with these other agents. The majority of responders to PD-1 pathway blockade appear to do so in the first 8 to 12 weeks of treatment, although some respond slowly.
Meanwhile, the investigation for novel biomarkers predictive of rapid response is currently ongoing (reviewed by Mahoney and Atkins90). PD-L1 expression clearly correlates with greater rates of response to monotherapy with PD-1 pathway blockade. Although it is far from a perfect predictive biomarker, PD-L1 expression may play a role in directing the treatment choice or sequence for patients. In the cohort of melanoma patients treated with pembrolizumab and having evaluable tumor (n = 71), 77% of patients were identified as PD-L1+, which was associated with improved overall response rates by Response Evaluation Criteria In Solid Tumors (51% vs 6%; P = 0.0012 [Fisher exact test]) and progression-free survival (median, 12 vs 3 months; HR, 0.31; 95% CI, 0.16–0.61; P = 0.0004 [log rank test]).51 Perhaps the threshold that defines a positive result should be raised to increase the positive predictive value of the test. By raising the threshold of a positive result, we may better distinguish patients who will respond to monotherapy. This biomarker also may be useful for determining patients who are less likely to respond to monotherapy and who may achieve greater clinical benefit from combination trials. Identifying these patients also may reduce the number of patients exposed to the risk for greater toxicity seen with combination immune-checkpoint blockade. As we learn more about the safety profiles of immunotherapy combinations, it is hoped that the role of PD-L1 will become clearer.79
DISCUSSION
Immune-checkpoint blockade has been reported to have antitumor effects with multiple agents in a wide variety of Phase I, II, and III clinical trials. With accelerated approval of pembrolizumab and nivolumab, it is essential to realize that vigilance is required in the Phase IV setting. Despite nivolumab not being FDA approved for the treatment of melanoma before ipilimumab, with the positive results of the Phase III trial that compared nivolumab to chemotherapy in previously untreated patients with BRAF− melanoma, some suggest that it should be offered before ipilimumab given the greater response rates and lower toxicity with nivolumab compared with ipilimumab. Multiple Phase III trials in patients with melanoma are currently underway and will likely shed light on the optimal sequencing versus combining of PD-1– and CTLA-4–directed therapy.
Over the next few years, results from ongoing clinical trials and expected trials of combinations also will shed light on how we can increase response rates, minimize toxicity, and develop more durable responses in patients. Oncologists need to learn the optimal means of managing toxicity to maximize the benefit of targeted inhibitors, such as vemurafenib, erlotinib, and sunitinib. Similarly, treating patients with checkpoint blockade will have its own learning curve. This blossoming field of anticancer therapy has shown clinical benefit across tumor types. Although the checkpoint inhibitors may be significantly less toxic than many prior immunotherapies, we still are learning more about the side effects of these agents. For example, it will be important to monitor how these agents affect patients in the setting of both acute and chronic infection (eg, HIV, hepatitis C virus) moving forward. These agents can produce life-threatening immune-related adverse events about which physicians and patients must be educated, particularly because early recognition and treatment of these immune-related toxicities could be reversible. With education and management of immune-related effects of checkpoint blockade, we hope to achieve the greatest clinical benefit possible from these treatments: remission in patients with melanoma as well as other cancers.
CONCLUSIONS
This family of immune-checkpoint inhibitors not only benefits patients with metastatic melanoma, but also historically less responsive tumor types. Although a subset of patients responds to single-agent blockade, the initial trial of checkpoint-inhibitor combinations has shown potential to improve response rates. Combination therapies appear to be a means of increasing response rates, albeit with increased immune-related adverse events. As these treatments become available to patients, education regarding the recognition and management of the immune-related effects of immune-checkpoint blockade will be essential for maximizing clinical benefit.
Acknowledgments
This research was financially supported by a grant from the National Institutes of Health–National Cancer Institute (P50CA101942; BIDMC); the Claudia Adams Barr Program for Innovative Cancer Research; an American Association for Cancer Research Basic Cancer Research Fellowship (14-40-01-MAHO); the American Society of Clinical Oncology Young Investigator Award from the Kidney Cancer Association (to K.M.); and grants U54CA163125, P01AI054456, U54CA16312, and R01AI089955 (to G.J.F.; DFCI).
Footnotes
CONFLICTS OF INTEREST
Dr. Freeman holds patents, and receives patent royalties from, on the PD-1 pathway from Bristol-Myers Squibb, Roche, Merck, EMD-Serono, Boehringer-Ingelheim, Amplimmune, and Novartis. Dr. McDermott has served on the advisory boards of Genentech, Bristol-Myers Squibb, Merck, and Prometheus. The authors have indicated that they have no other conflicts of interest with regard to the content of this article.
References
- 1.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-pd-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.http://seer.cancer.gov/statfacts/html/melan.html. (as of 3/15/2015).
- 7.Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet. 1999;353:14–17. [PubMed] [Google Scholar]
- 8.Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–913. [PubMed] [Google Scholar]
- 9.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schadendorf D, Hodi FS, Robert C, et al. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol. 2015 doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joseph RW, Eckel-Passow JE, Sharma R, et al. Characterizing the clinical benefit of ipilimumab in patients who progressed on high-dose IL-2. J Immunother. 2012;35:711–715. doi: 10.1097/CJI.0b013e3182742c27. [DOI] [PubMed] [Google Scholar]
- 12.Payne R, Glenn L, Hoen H, et al. Durable responses and reversible toxicity of high-dose interleukin-2 treatment of melanoma and renal cancer in a community hospital biotherapy program. J Immunother Cancer. 2014;2:13. doi: 10.1186/2051-1426-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 14.Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 15.Ishida Y, Agata Y, Shibahara K, Honjo T, et al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freeman GJ. Structures of PD-1 with its ligands: sideways and dancing cheek to cheek. Proc Natl Acad Sci U S A. 2008;105:10275–10276. doi: 10.1073/pnas.0805459105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 19.Rodig N, Ryan T, Allen JA, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003;33:3117–3126. doi: 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]
- 20.Zinselmeyer BH, Heydari S, Sacristan C, et al. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med. 2013;210:757–774. doi: 10.1084/jem.20121416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Honda T, Egen JG, Lammermann T, et al. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity. 2014;40:235–247. doi: 10.1016/j.immuni.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vibhakar R, Juan G, Traganos F, et al. Activation-induced expression of human programmed death-1 gene in T-lymphocytes. Exp Cell Res. 1997;232:25–28. doi: 10.1006/excr.1997.3493. [DOI] [PubMed] [Google Scholar]
- 23.Zajac AJ, Blattman JN, MuraliKrishna K, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 25.Khattri R, Auger JA, Griffin MD, et al. Lymphoproliferative disorder in CTLA-4 knockout mice is characterized by CD28-regulated activation of Th2 responses. J Immunol. 1999;162:5784–5791. [PubMed] [Google Scholar]
- 26.Keir ME, Butte MJ, Freeman GJ, Sharpe AH, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Yoshida T, Nakaki F, et al. Establishment of NOD-Pdcd1-/-mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A. 2005;102:11823–11828. doi: 10.1073/pnas.0505497102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okazaki T, Tanaka Y, Nishio R, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. 2003;9:1477–1483. doi: 10.1038/nm955. [DOI] [PubMed] [Google Scholar]
- 30.Nishimura H, Nose M, Hiai H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 31.Kasagi S, Kawano S, Okazaki T, et al. Anti-programmed cell death 1 antibody reduces CD4+PD-1+ T cells and relieves the lupus-like nephritis of NZB/W F1 mice. J Immunol. 2010;184:2337–2347. doi: 10.4049/jimmunol.0901652. [DOI] [PubMed] [Google Scholar]
- 32.Chang TT, Jabs C, Sobel RA, et al. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J Exp Med. 1999;190:733–740. doi: 10.1084/jem.190.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nielsen C, Ohm-Laursen L, Barington T, et al. Alternative splice variants of the human PD-1 gene. Cell Immunol. 2005;235:109–116. doi: 10.1016/j.cellimm.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 34.Ueda H, Howson JM, Esposito L, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 35.Wan B, Nie H, Liu A, et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J Immunol. 2006;177:8844–8850. doi: 10.4049/jimmunol.177.12.8844. [DOI] [PubMed] [Google Scholar]
- 36.Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 37.Iwai Y, Terawaki S, Ikegawa M, et al. PD-1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med. 2003;198:39–50. doi: 10.1084/jem.20022235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jun H, Seo SK, Jeong HY, et al. B7-H1 (CD274) inhibits the development of herpetic stromal keratitis (HSK) FEBS Lett. 2005;579:6259–6264. doi: 10.1016/j.febslet.2005.09.098. [DOI] [PubMed] [Google Scholar]
- 39.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ, et al. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 40.Smith P, Walsh CM, Mangan NE, et al. Schistosoma mansoni worms induce anergy of T cells via selective up-regulation of programmed death ligand 1 on macrophages. J Immunol. 2004;173:1240–1248. doi: 10.4049/jimmunol.173.2.1240. [DOI] [PubMed] [Google Scholar]
- 41.Liang SC, Greenwald RJ, Latchman YE, et al. PD-L1 and PD-L2 have distinct roles in regulating host immunity to cutaneous leishmaniasis. Eur J Immunol. 2006;36:58–64. doi: 10.1002/eji.200535458. [DOI] [PubMed] [Google Scholar]
- 42.Brown JA, Dorfman DM, Ma FR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 43.Iwai Y, Ishida M, Tanaka Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iwai Y1, Terawaki S, Honjo T, et al. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133–144. doi: 10.1093/intimm/dxh194. [DOI] [PubMed] [Google Scholar]
- 45.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Merwe PA, Bodian DL, Daenke S, et al. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med. 1997;185:393–403. doi: 10.1084/jem.185.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butte MJ, Keir ME, Phamduy TB, et al. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun. 2003;307:672–677. doi: 10.1016/s0006-291x(03)01257-9. [DOI] [PubMed] [Google Scholar]
- 49.Atkins MB, Kudchadkar RR, Sznol M, Kudchadkar RR, Sznol M, et al. Phase 2, multicenter, safety and efficacy study of pidilizumab in patients with metastatic melanoma. J Clin Oncol. 2014;32(suppl; abstr 9001) [Google Scholar]
- 50.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kefford R, Ribas A, Hamid O, et al. Clinical efficacy and correlation with tumor PD-L1 expression in patients (pts) with melanoma (MEL) treated with the anti-PD-1 monoclonal antibody MK-3475. J Clin Oncol. 2014;32(suppl; abstr 3005̂) [Google Scholar]
- 52.Ribas A, Hodi FS, Kefford R, et al. Efficacy and safety of the anti-PD-1 monoclonal antibody MK-3475 in 411 patients (pts) with melanoma (MEL) J Clin Oncol. 2014;32(suppl; abstr LBA9000̂) [Google Scholar]
- 53.Berger R, Rotem-Yehudar R, Slama G, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14:3044–3051. doi: 10.1158/1078-0432.CCR-07-4079. [DOI] [PubMed] [Google Scholar]
- 54.Armand P, Nagler A, Weller EA, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31:4199–4206. doi: 10.1200/JCO.2012.48.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Westin JR, Chu F, Zhang M, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014;15:69–77. doi: 10.1016/S1470-2045(13)70551-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-pd-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2:846–856. doi: 10.1158/2326-6066.CIR-14-0040. [DOI] [PubMed] [Google Scholar]
- 57.Hamid O, Sosman JA, Lawrence DP, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM) J Clin Oncol. 2013;31(suppl; abstr 9010) [Google Scholar]
- 58.Mu CY, Huang JA, Chen Y, et al. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol. 2011;28:682–688. doi: 10.1007/s12032-010-9515-2. [DOI] [PubMed] [Google Scholar]
- 59.Thompson RH, Gillett MD, Cheville JC, et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci U S A. 2004;101:17174–17179. doi: 10.1073/pnas.0406351101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gadiot J, Hooijkaas AI, Kaiser AD, et al. Overall survival and PD-L1 expression in metastasized malignant melanoma. Cancer. 2011;117:2192–2201. doi: 10.1002/cncr.25747. [DOI] [PubMed] [Google Scholar]
- 61.Hino R, Kabashima K, Kato Y, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 2010;116:1757–1766. doi: 10.1002/cncr.24899. [DOI] [PubMed] [Google Scholar]
- 62.Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taube JM, Klein AP, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–3277. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu C, Fillmore CM, Koyama S, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25:590–604. doi: 10.1016/j.ccr.2014.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thompson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006;66:3381–3385. doi: 10.1158/0008-5472.CAN-05-4303. [DOI] [PubMed] [Google Scholar]
- 68.Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 69.Grosso J, Horak CE, Inzunza D, et al. Association of tumor PD-L1 expression and immune biomarkers with clinical activity in patients (pts) with advanced solid tumors treated with nivolumab (anti-PD-1; BMS-936558; ONO-4538) J Clin Oncol. 2013;31(suppl; abstr 3016) [Google Scholar]
- 70.Weber JS, Kudchadkar RR, Yu B, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013;31:4311–4318. doi: 10.1200/JCO.2013.51.4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Daud AI, Hamid O, Ribas A, et al. Antitumor activity of the anti-PD-1 monoclonal antibody MK-3475 in melanoma(MEL): Correlation of tumor PD-L1 expression with outcome. AACR Annual Meeting; 2014. p. CT104. [Google Scholar]
- 72.Gandhi L, Balmanoukian A, Hui R, et al. MK-3475 (anti-PD-1 monoclonal antibody) for non-small cell lung cancer (NSCLC): Antitumor activity and association with tumor PD-L1 expression. AACR Annual Meeting; 2014. p. CT105. [Google Scholar]
- 73.Seiwert T, Burtness B, Weiss J, et al. A phase Ib study of MK-3475 in patients with human papillomavirus (HPV)-associated and non-HPV–associated head and neck (H/N) cancer. J Clin Oncol. 2014;32(suppl; abstr 6011) [Google Scholar]
- 74.Soria JC, Cruz C, Bahleda R, et al. Clinical activity, safety and biomarkers of PD-L1 blockade in non-small cell lung cancer (NSCLC): Additional analyses from a clinical study of the engineered antibody MPDL3280A (anti-PDL1) ECCO. 2013 Abstract no: 3408. [Google Scholar]
- 75.Simpson TR, Li F, Montalvo-Ortiz W, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Selby MJ, Engelhardt JJ, Quigley M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 77.Alegre ML, Noel PJ, Eisfelder BJ, et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol. 1996;157:4762–4770. [PubMed] [Google Scholar]
- 78.Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res. 2013;19:4917–4924. doi: 10.1158/1078-0432.CCR-12-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Curr Opin Immunol. 2015;33C:23–35. doi: 10.1016/j.coi.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 80.Callahan MK. Understanding the Biology Behind Responses to Immunotherapy. J Clin Oncol. 2014 oral presentation. [Google Scholar]
- 81.Powderly JD, Koeppen H, Hodi FS, et al. Biomarkers and associations with the clinical activity of PD-L1 blockade in a MPDL3280A study. J Clin Oncol. 2013;31(suppl; abstr 3001) [Google Scholar]
- 82.Callahan MK, et al. Peripheral and tumor immune correlates in patients with advanced melanoma treated with combination nivolumab (anti-PD-1, BMS-936558, ONO-4538) and ipilimumab. J Clin Oncol. 2013;31(suppl; abstr 3003) [Google Scholar]
- 83.Curran MA, Montalvo W, Yagita H, Allison JP, et al. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Korman A, Chen BJ, Changyu W, et al. Activity of anti-pd-1 in murine tumor models: role of “host” PD-L1 and synergistic effect of anti-PD-1 and anti-CTLA-4. J Immunol. 2007;178:48.37. [Google Scholar]
- 85.Ribas A, Hodi FS, Callahan M, et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–1366. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 86.Afrache H, Gouret P, Ainouche S, et al. The butyrophilin (BTN) gene family: from milk fat to the regulation of the immune response. Immunogenetics. 2012;64:781–794. doi: 10.1007/s00251-012-0619-z. [DOI] [PubMed] [Google Scholar]
- 87.Freeman GJ, Sharpe AH. A new therapeutic strategy for malaria: targeting T cell exhaustion. Nat Immunol. 2012;13:113–115. doi: 10.1038/ni.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bristol-Myers Squibb. Phase 3 First-Line Melanoma Study of Nivolumab, an Investigational PD-1 Checkpoint Inhibitor, Demonstrates Superior Overall Survival Compared with Dacarbazine; Study Stopped Early. [February 24, 2015];2014 http://news.bms.com/press-release/phase-3-first-line-melanoma-study-nivolumab-investigational-pd-1-checkpoint-inhibitor-
- 89.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015 doi: 10.1200/JCO.2014.59.4358. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mahoney KM, Atkins MB. Prognostic and predictive markers for the new immunotherapies. Oncology (Williston Park) 2014;28(11 Suppl 3) [PubMed] [Google Scholar]