

Background: Multimodularity is essential in most plant cell wall recycling enzymes.
Results: The CBM22-1–CBM22-2 tandem possesses extraordinary plasticity and two sites with different affinities. CBM22-2 exhibits novel specificity within CBM22s.
Conclusion: Depolymerization of highly substituted arabinoxylans by Xyn10C and a delivery strategy mediated by Xyn10C-XBD are proposed.
Significance: Elucidating the mechanisms regulating specificity through multimodularity is crucial for producing efficient biocatalysts for biomass deconstruction.
Keywords: carbohydrate-binding protein, enzyme structure, glycoside hydrolase, plant cell wall, x-ray crystallography, CBM22 tandem, GH10, xylanase
Abstract
Elucidating the molecular mechanisms regulating multimodularity is a challenging task. Paenibacillus barcinonensis Xyn10C is a 120-kDa modular enzyme that presents the CBM22/GH10/CBM9 architecture found in a subset of large xylanases. We report here the three-dimensional structure of the Xyn10C N-terminal region, containing the xylan-binding CBM22-1–CBM22-2 tandem (Xyn10C-XBD), which represents the first solved crystal structure of two contiguous CBM22 modules. Xyn10C-XBD is folded into two separate CBM22 modules linked by a flexible segment that endows the tandem with extraordinary plasticity. Each isolated domain has been expressed and crystallized, and their binding abilities have been investigated. Both domains contain the R(W/Y)YYE motif required for xylan binding. However, crystallographic analysis of CBM22-2 complexes shows Trp-308 as an additional binding determinant. The long loop containing Trp-308 creates a platform that possibly contributes to the recognition of precise decorations at subsite S2. CBM22-2 may thus define a subset of xylan-binding CBM22 modules directed to particular regions of the polysaccharide. Affinity electrophoresis reveals that Xyn10C-XBD binds arabinoxylans more tightly, which is more apparent when CBM22-2 is tested against highly substituted xylan. The crystal structure of the catalytic domain, also reported, shows the capacity of the active site to accommodate xylan substitutions at almost all subsites. The structural differences found at both Xyn10C-XBD domains are consistent with the isothermal titration calorimetry experiments showing two sites with different affinities in the tandem. On the basis of the distinct characteristics of CBM22, a delivery strategy of Xyn10C mediated by Xyn10C-XBD is proposed.
Introduction
Plant cell walls are highly complicated constructions containing an intricate network of polymers that are recalcitrant to degradation. Moreover, they are very dynamic and change their structures and composition depending on cell development and environmental conditions. To cope with this extraordinary complexity, nature has created a large variety of enzymes that act synergistically to deconstruct biomass (1). An interesting molecular mechanism developed by many plant cell wall hydrolases is their modular composition, with additional domains that increase accessibility of its catalytic partner to the substrate. Most of these supplementary domains are carbohydrate-binding modules (CBMs).3 Their presence in a broad range of hydrolases clearly highlights their functional importance, although their precise mode of action is still unknown. In fact, the occurrence of several copies of homologous CBMs within some enzymes suggests an additional subtle mechanism of regulation in enzymatic specificity. Structural studies from isolated CBMs have revealed that substrate recognition and binding by these domains are extremely specific. To understand the mechanism by which modularity enhances the interaction of enzymes with their substrate and to determine the functional relationship between carbohydrate binding and catalysis, more structural information on multiplicity is required at the atomic level.
CBMs are generally considered as noncatalytic modules that potentiate the activity of carbohydrate-active enzymes by bringing them in close proximity to their substrates. Most CBMs are part of modular carbohydrate hydrolases, although several examples of CBMs linked to cellulosomal scaffolding proteins or not linked to a hydrolase at all, i.e. the so-called “orphan CBMs,” have been described (2). Based on amino acid sequence similarity, they are classified into different families (CAZy); currently 71 are known, and the number is still increasing (3). CBMs show substantial differences in their ligand specificity and binding properties (4, 5). They recognize and bind a diversity of plant carbohydrates, including crystalline and noncrystalline cellulose, soluble and insoluble xylan, chitin, β-glucans, mannan, galactan, and starch; moreover, some members of a few CBM families can recognize mammalian glycans (6). CBMs target enzymes to their specific substrates, enhancing carbohydrate degradation as a result of the increased local concentration of each enzyme around its substrate (4, 7). The frequent occurrence of CBMs with specificity for cellulose in enzymes, such as xylanases, without activity on this polysaccharide, can be explained by the close proximity of these two carbohydrates, cellulose and xylan, in cell wall structure in biomass (7). In a similar way, CBMs of scaffolding proteins play an important role in the deconstruction of the plant cell wall by keeping the clustered catalytic components of cellulosomes in close proximity to the cell wall, yielding an enhanced and synergistic degradation of polysaccharides (8, 9). A previous proposal that CBMs may act by a disruptive mechanism that separates glycan chains from the cell wall (10) was supported by the finding of some CBM33s, not attached to enzymes, which disrupted chitin structure and promoted hydrolysis by chitinases (11). However, it has been recently shown that CBM33s constitute a new type of carbohydrate depolymerases that act by an oxidative mechanism and are reclassified in CAZy as auxiliary activity family 10 (12). Furthermore, the analysis of some CBM35s showed that they display specificity for Δ4,5-anhydrogalacturonic acid, a signature molecule of plant cell wall degradation by pectate lyases, suggesting that CBMs of this family could direct the enzymes toward regions of the plant cell wall that are being actively degraded or remodeled (13). In addition to targeting the catalytic modules to their substrates, CBMs probably have a more complex role in polysaccharides depolymerization, with additional functions to enable the deconstruction of the cell wall and the catalytic degradation of carbohydrates.
CBMs of several families show specificity for xylan (2, 14–16). Among them, family 22 CBMs were previously considered as thermostabilizing domains of enzymes from thermophiles (17). The finding that they were not restricted to thermophilic enzymes prompted the analysis of their binding properties, showing that they promote binding to xylan and xylo-oligosaccharides (18, 19). They were regarded as xylan binding domains that were reclassified as the new CBM22 (20, 21). Xylanase Xyn10C belongs to the secretome of Paenibacillus barcinonensis, a powerful xylanolytic microorganism (22) that produces a complex set of enzymes, including xylanases of families GH10, GH11, and GH30 that have been cloned and characterized (16, 23, 24). Xyn10C was previously characterized showing a distinctive modular structure containing an N-terminal tandem of two CBM22s and a duplicated CMB9 at its C terminus (25). In our study, we have solved the three-dimensional structure of the CBM22-1–CBM22-2 tandem, and its binding abilities have been investigated by soaking experiments, affinity gel electrophoresis, and ITC. In addition, the structure and binding features of the individual CBM22-1, CBM22-2, and the catalytic GH10 modules are reported. The results found contribute to deciphering the function of CBM22s and their contribution to xylan degradation. Further studies will be required to fully ascertain the role of CBM22 duplicity in catalytic depolymerization of xylan by their partner enzymes and in deconstruction of biomass.
Experimental Procedures
Cloning, Expression, and Purification
The respective forward and reverse primers used for all constructs are given in Table 1. The cDNA of Xyn10C was amplified from P. barcinonensis BP-23 genomic DNA (UniProt accession number O69230). The resulting plasmid pET28Xyn10C produced the full-length enzyme linked to an N-terminal His6 tag (Xyn10C). To maintain the His tag motif in the N terminus of the expressed protein, the first 28 residues of the sequence were not included in the DNA amplification because they correspond to the signal peptide that is cleaved in Escherichia coli hosts during protein translation. The plasmid vector pET28 was used to clone the GH10 catalytic domain fused to an N-terminal His6 tag (Xyn10C-CD). Both constructs (Xyn10C and Xyn10C-CD) were expressed and purified using a similar protocol. BLR (DE3) E. coli strain was transformed with the corresponding plasmid and grown in LB medium at 37 °C for 4 h until the A600 reached 0.8. Protein expression was induced by adding 0.3 mm isopropyl β-d-thiogalactopyranoside followed by incubation for 15 h at 16 °C. Cells were disrupted by using a French press. The recombinant His6 tag proteins were purified from cell extracts by immobilized metal affinity chromatography using HisTrap HP columns of 5 ml (GE Healthcare) and eluted in 20 mm sodium phosphate buffer, pH 7.4, 500 ml of NaCl with a 0–500 mm imidazole gradient. For crystallization, it was necessary to cleave the His6 tag of Xyn10C-CD with thrombin (Sigma).
TABLE 1.
Oligonucleotides employed
Forward (F) and reverse (R) sequences include the restriction sites in boldface letters.
| Protein | Orientation | Sequence |
|---|---|---|
| Xyn10C | F | 5′-GGAGAACATATGGCAAGCGCAGCG-3′ |
| R | 5′-TGGCGGATCCGAGCTTGAACA-3′ | |
| Xyn10C-CD | F | 5′-GATTCATATGGAAAAAAATATTCCG G-3′ |
| R | 5′-TGCGGATCCCTAATTTCGATATACG-3′ | |
| Xyn10C-XBD | F | 5′-CGCGGATCCATGGCAAGCGCAGCGAAGGCGG-3′ |
| R | 5′-GGCCTCGAGTTATTCAATAGCAATCGCTTCGG-3′ | |
| CBM22–1 | F | 5′-CGCGGATCCATGGCAAGCGCAGCGAAGGCGG-3′ |
| R | 5′-GGCCTCGAGTTACGCAGCTTTAACGAGACGAATC-3′ | |
| CBM22–2 | F | 5′-CGC GGATCCGGTGAGCCGGGAGAAGCAGGAC-3′ |
| R | 5′- GGCCTCGAGTTATTCAATAGCAATCGCTTCGG-3′ |
The regions encoding for the N-terminal domain, Xyn10C-XBD, and the CBM22-1 and CBM22-2 modules were also amplified and cloned into the pGEX-4T-2 vector (GE Healthcare). The GST-fused protein expression was performed under similar conditions to those described above, except the BL21 (DE3) E. coli strain was employed. All constructs were purified using a similar protocol. Cell pellets were resuspended in buffer A (20 mm Tris/HCl, pH 8, 100 mm NaCl) and disrupted through sonication. The clarified cell lysates were mixed with 10 ml of glutathione-Sepharose 4B resin (GE Healthcare) equilibrated in buffer A. After washing with buffer A, the target protein was cleaved from resin-bound GST by incubating with thrombin overnight. Eluted protein was diluted to 50 mm NaCl and loaded to a 5-ml anionic exchange column (HiTrap Q HP; GE Healthcare) equilibrated in buffer B (50 mm NaCl, 20 mm Tris/HCl, pH 8). After washing with buffer B, the protein was eluted using a gradient between 50 and 500 mm NaCl. In the case of Xyn10C-XBD, the protein was further purified by size exclusion chromatography using a 16/60 Superdex 200 column (GE Healthcare) equilibrated in buffer A. The protein purity was assessed by SDS-PAGE in all cases. Pure proteins were concentrated and frozen at −80 °C until use.
Binding to Insoluble Polysaccharides
Binding activity to insoluble polysaccharides was assessed as described by Hogg et al. (26) with some modifications. Briefly, 250 μg of purified Xyn10C, Xyn10C-XBD, or Xyn10C-CD was mixed with 25 mg of Avicel or insoluble oat spelt xylan in a final volume of 500 μl of 50 mm Tris, pH 7, in 1.5-ml microcentrifuge tubes. The samples were incubated for 1 h at 4 °C with gentle orbital mixing. Samples were then centrifuged at 18,000 × g for 20 min, and supernatants, containing unbound protein, were carefully removed. Pellets were washed three times with 400 μl of the same buffer, before being resuspended in 400 μl of 10% SDS and boiled for 10 min to release bound protein. Samples were then analyzed by SDS-PAGE on 8 or 12% (w/v) polyacrylamide gels.
Affinity Gel Electrophoresis and Zymogram Analysis
Affinity gel electrophoresis was performed by following the method of Correia et al. (27). Continuous native polyacrylamide gels containing 10% acrylamide in 25 mm Tris, 250 mm glycine buffer, pH 8.3, were used. Soluble xylan (4 mg/ml) was included in gels before polymerization. Gels, with and without xylan, were polymerized at the same time and run in the same gel tank. About 6 μg of target protein was loaded in each well at room temperature, and gels were run at 10 mA/gel for 2 h. Bovine serum albumin (BSA) was used as a negative noninteracting control.
To analyze the electrophoretic homogeneity of purified proteins, SDS-PAGE was performed in 15% (w/v) (for CBM22-1 and CBM22-2) and 12% (w/v) (for Xyn10C-XBD) polyacrylamide gels essentially as described by Laemmli (28). For detection of xylanase activity, zymogram analysis was performed in SDS-polyacrylamide gels containing 0.2% (w/v) birchwood xylan. Samples were heated for 10 min at 45 °C in sample buffer before being applied to gels. After electrophoresis, gels were soaked in 2.5% (w/v) Triton X-100 for 30 min, washed in 50 mm acetate buffer, pH 5.0, for 30 min, and incubated at 45 °C for 2 h in the same buffer. Gels were then stained with 0.1% (w/v) Congo red for 15 min and washed with 1 m NaCl until xylanase bands became visible, as described previously (29). Gels were then immersed in 10% (v/v) acetic acid and photographed.
Isothermal Titration Calorimetry
ITC measurements were performed at 25 °C using a MicroCal VP-ITC (GE Healthcare). The samples were dialyzed against 50 mm sodium phosphate, pH 7, and ligand solutions were prepared using the same buffer. All samples were thoroughly degassed before use. Titrations were carried out by injecting consecutive aliquots of 10–20 mm oligosaccharide or 4 mg/ml xylan oat spelt polysaccharide into the sample cell loaded with protein at 50–65 μm. For mixed oligosaccharide and xylan oat spelt, two consecutive titrations were carried out with the same protein solution, and the CONCAT32 program (GE Healthcare) was used to concatenate the raw data of both series. Heat developed on ligand dilution was determined separately and subtracted when required. The binding constants (K) and the enthalpies (ΔH) of binding were calculated by analyzing the binding isotherms with the MicroCal ITC Origin software, assuming one carbohydrate-binding site per domain, based on the structural data and xylan binding capacity of the isolated domains. The entropic contribution to ligand binding was calculated using Gibbs equation (ΔG = −RTlnK = ΔH − TΔS). The molar concentration of binding sites in the polysaccharide was set to the value that after iteration gave a number of sites per CBM22 domain equal to 1. The oligosaccharides were purchased from Megazyme. The mixed oligosaccharides used were 1,3:1,4-β-glucotetraose A (Glc-3Glc-4Glc-4Glc), B (Glc-4Glc-4Glc-3Glc), and C (Glc-4Glc-3Glc-4Glc).
Analytical Ultracentrifugation
Sedimentation velocity experiments were run at 45,000 rpm using cells with double sector Epon-charcoal centerpieces. Differential sedimentation coefficients were calculated by least squares boundary modeling of the experimental data with the program SEDFIT. Sedimentation equilibrium experiments were carried out at different rotor speeds, as described previously (30), and the weight-average molecular weights were calculated using the heteroanalysis program (Biotechnology-Bioservices Center, University of Connecticut). Measurements were performed at 20 °C in an Optima XL-A analytical ultracentrifuge (Beckman Coulter) using Xyn10C-XBD (5.3–24.3 μm) or CBM22-1 (17.3 μm) in ITC buffer (with and without 3 mm xylotetraose). Partial specific volumes were calculated from the amino acid sequences with the SEDNTERP program.
Crystallization and Data Collection
Crystals of Xyn10C-XBD were grown from 47 mg/ml protein, 25% (w/v) PEG 3350, 0.2 m NaCl, 2% (v/v) 1,6-hexanediol, 0.1 m BisTris, pH 5.5, using the streak-seeding technique. Very thin plates grew by vapor diffusion at room temperature, as described before (31). For data collection, crystals were transferred to cryoprotectant solutions consisting of mother liquor plus 20% (v/v) glycerol before being cooled in liquid nitrogen.
Screening of crystallization conditions for the isolated CBM22s and Xyn10C-CD domains was performed with thePACT and the JCSG Suites from Qiagen and INDEX and SALTRXfrom Hampton Research. Crystals from CBM22-1 appeared in solutions containing either PEG 1500 or PEG 3350 with pH 5.5–7.0. Further optimization was performed at room temperature and the vapor diffusion method by mixing 29 mg/ml protein sample (20 mm Tris, pH 8.0, and 125 mm NaCl) and an equal volume of different reservoir solutions in a sitting-drop procedure. Large crystals grew from 20% (w/v) PEG 3350 and 0.2 m NaNO3, with a 1.5:1 ratio of protein/precipitant, reaching maximum size in 10 days. Treatment with different cryoprotectants as glycerol, PEG 400, or xylose seemed to greatly affect the crystal integrity, with only a very fast soaking into 15% (v/v) ethylene glycol being tolerated and thus allowing collection of high quality data (Table 2). Soaking with ligand solutions damaged crystals, and co-crystallization experiments were unsuccessful, therefore precluding analysis of complexes.
TABLE 2.
Crystallographic data
Values in parentheses are for the high resolution shell.
| Crystal data | Xyn10C-XBD | CBM22-1 | CBM22-2 | CBM22-2/Xyl4 | CBM22-2/Xyl3 | CBM22-2/Glc-4Glc-4Glc-3Glc | Xyn10C-CD |
|---|---|---|---|---|---|---|---|
| Space group | P 21 | H3 | P32 | P32 | P32 | P32 | P32 |
| Unit cell | |||||||
| a (Å) | 84.17 | 85.35 | 92.54 | 92.77 | 92.46 | 92.48 | 126.69 |
| b (Å) | 110.36 | 85.35 | 92.54 | 92.77 | 92.46 | 92.48 | 126.69 |
| c (Å) | 118.52 | 108.46 | 48.40 | 48.57 | 48.43 | 48.38 | 57.70 |
| γ (°) | 90.63 | ||||||
| Data collection | |||||||
| Beamline | ID29(ESRF) | ID23–2(ESRF) | ID23–2(ESRF) | ID23–2(ESRF) | BL13-XALOC (ALBA) | BL13-XALOC (ALBA) | ID23–1 (ESRF) |
| Wavelength (Å) | 0.979235 | 0.8729 | 0.8729 | 0.8729 | 0.9795 | 0.9795 | 0.9801 |
| Resolution (Å) | 84.16-2.43 (2.48-2.43) | 30.54-1.70 (1.79-1.70) | 33.45-1.75 (1.84-1.75) | 33.55-1.67 (1.76-1.67) | 48.42-1.95 (2.05-1.95) | 48.38-1.80 (1.90-1.80) | 36.57-1.76 (1.86-1.76) |
| Data processing | |||||||
| Total reflections | 510618 (29165) | 184811 (26283) | 272776 (39214) | 307899 (41596) | 315365 (44621) | 393762 (54642) | 567570 (81227) |
| Unique reflections | 81116 (4468) | 32396 (4730) | 46784 (6810) | 54496 (7920) | 33856 (4948) | 42863 (6228) | 102672 (14987) |
| Multiplicity | 6.3 (6.5) | 5.7 (5.6) | 5.8 (5.8) | 5.6 (5.3) | 9.3 (9.0) | 9.2 (8.8) | 5.5 (5.4) |
| Completeness (%) | 99.4 (99.3) | 100 (100) | 100 (100) | 100 (100) | 99.9 (99.7) | 100 (100) | 100.0 (100.0) |
| I/σ (I) | 4.8 (1.3) | 2.4 (1.7) | 5.8 (1.9) | 6.1 (1.7) | 6.2 (2.5) | 5.8 (1.7) | 5.1 (1.4) |
| Mean I/σ (I) | 8.6 (2.2) | 9.7 (4.2) | 13.4 (4.4) | 13.1 (3.4) | 17.5 (7.6) | 14.8 (4.6) | 9.1 (3.2) |
| Rmergea (%) | 13.0 (65.2) | 13.5 (41.6) | 9.4 (40.3) | 9.1 (45.1) | 9.0 (28.7) | 9.2 (43.2) | 10.6 (49.4) |
| Rpimb (%) | 8.5 (34.9) | 6.2 (19.3) | 4.3 (18.4) | 4.2 (21.8) | 3.1 (10.1) | 3.2 (15.3) | 5.0 (23.5) |
| Molecules per a.u. | 6 | 2 | 3 | 3 | 3 | 3 | 3 |
| Matthews coefficient (Å3 Da−1) | 2.54 | 2.24 | 2.23 | 2.25 | 2.23 | 2.23 | 2.26 |
| Solvent content (%) | 51.5 | 45.2 | 44.9 | 45.0 | 44.8 | 44.8 | 45.5 |
| Overall B-factor Wilson plot (Å2) | 37.24 | 18.79 | 12.64 | 11.79 | 16.39 | 18.16 | 15.55 |
| Refinement | |||||||
| Rwork/Rfreec (%) | 22.52/26.05 | 21.09/24.71 | 15.14/18.20 | 15.78/19.37 | 17.92/21.60 | 20.41/22.57 | 21.79/25.15 |
| No. of atoms/average B (Å2) | |||||||
| Protein | 12529/49.18 | 2398/25.29 | 3789/17.74 | 3789/16.40 | 3789/21.39 | 3789/24.20 | 8355/16.55 |
| Carbohydrate | 0 | 0 | 0 | 101/26.22 | 66/33.23 | 23/41.73 | 0 |
| Other | 22/41.13 | 2/19.9 | 3/16.69 | 3/18.41 | 3/20.21 | 3/23.53 | 21/23.63 |
| Water molecules | 29/30.30 | 59/24.16 | 231/21.69 | 94/15.76 | 84/19.39 | 113/23.45 | 523/21.60 |
| Ramachandran | |||||||
| Favored (%) | 98.00 | 98.05 | 97.69 | 97.48 | 97.48 | 97.48 | 95.43 |
| Outliers (%) | 0.00 | 0.32 | 0.00 | 0.00 | 0.00 | 0.00 | 0.38 |
| r.m.s.d. | |||||||
| Bonds (Å) | 0.0108 | 0.0106 | 0.0123 | 0.0190 | 0.0153 | 0.0146 | 0.007 |
| Angles (°) | 1.374 | 1.405 | 1.451 | 1.622 | 1.425 | 1.593 | 1.147 |
| Protein Data Bank codes | 4XUP | 4XUO | 4XUN | 4XUR | 4XUQ | 4XUT | 4W8L |
a Rmerge = ΣhklΣi|Ii(hkl) − (I(hkl))|/Σhkl Σi Ii(hkl), where Ii(hkl) is the ith measurement of reflection hkl and (I(hkl)) is the weighted mean of all measurements.
b Rpim = Σhkl(1/(N − 1))1/2 Σi|Ii(hkl) − (I(hkl))|/Σhkl Σi Ii (hkl), where N is the redundancy for the hkl reflection.
c Rwork/Rfree = Σhkl|Fo − Fc|/Σ hkl|Fo|, where Fc is the calculated and Fo is the observed structure factor amplitude of reflection hkl for the working/free (5%) set, respectively.
Rod-shaped crystals from the CBM22-2 construct were obtained by mixing 0.5 μl of 19 mg/ml protein solution (20 mm Tris, pH 8.0, and 150 mm NaCl) with 1 μl of a solution containing 1.85 m sodium malonate, pH 6.0, and equilibrating by vapor diffusion at room temperature. Complexes were obtained by the soaking technique using xylotriose, xylotetraose, or 1,3:1,4 β-glucotetraose B. The crystals were soaked for 5–30 min in solutions made of mother liquor plus 10 mm of the corresponding ligand. For data collection, all crystals were transferred to cryoprotectant solutions containing 3 m sodium malonate before being cooled in liquid nitrogen.
Rod-shaped crystals from Xyn10C-CD were grown by mixing equal amounts of 10 mg/ml protein solution (in 20 mm Tris, pH 7.5, 0.5 m NaCl) with 23% (w/v) PEG 3350, 0.2 m KSCN, 0.1 m NaAc, pH 5.0, and equilibrating at root temperature. For data collection, 20% glycerol was added to mother liquor before cooling.
X-ray data from all crystals were collected using different Synchrotron sources at Alba (Spain) and ESRF (France) on the beamlines given in Table 2. Diffraction images were processed with iMOSFLM (32) or XDS (33) and merged using the CCP4 package (34).
Structure Solution and Refinement
The structure of the Xyn10C-XBD tandem was solved by molecular replacement using the PHASER program (35). The search models were the CBM22-1 and CBM22-2 domains from the Rhuminoclostridium thermocellum xylanase 10B (RthXyn10B; PDB codes 2W5F and 1DYO, (20, 36)), each sharing 21/25 and 23/23% sequence identity, respectively, with respect to the CBM22-1 and CBM22-2 domains. Nine units of the search models were placed in the asymmetric unit by PHASER. Then an additional unit was manually included on the basis of the observed association pattern, which increased the likelihood score and showed appropriate packing. The solution means six molecules of the Xyn10C-XBD tandem within the asymmetric unit (a.u.), with two of them being incomplete. This initial model was subjected to restrained refinement with REFMAC (37), within the CCP4 suite, with flat bulk-solvent correction and local noncrystallographic symmetry. Then, the calculated electron density maps allowed unambiguous assignment of the CBM22-1 and CBM22-2 sequences. Further refinement with REFMAC, combined with manual building using the program COOT (38), led to a model showing an almost continuous density for the whole polypeptide chain in four molecules (residues 1–334), a small fragment of the linker region showing poor or chopped density as follows: A (residues 164–166), B (residues 162–171), D (residues 161–170), and F (residues 165–170), whereas only the chain comprising residues 3–162 could be modeled in molecules D and F. Many attempts to complete this model by manual and automatic rebuilding were unsuccessful. At the later stages, water molecules were included, which, combined with more rounds of restrained refinement, led to a final R-factor of 22.7 (Rfree = 26.0) for all data set up to 2.43 Å resolution. Refinement parameters are reported in Table 2.
The structure of CBM22-1 and CBM22-2 crystals was solved by molecular replacement using MOLREP (39) and the coordinates of the 1–159 and 171–334 portion of Xyn10C-XBD, respectively, as the search models. Crystallographic refinement was performed using REFMAC (37) combined with model building with COOT (38) and the addition of water molecules, which led to the final R-factors given in Table 2. The structures of the CBM22-2 complexes were solved by difference Fourier synthesis using the refined coordinates of the CBM22-2 domain. The ligands were manually built in the electron density maps and were refined similarly, to reach the R-factors listed in Table 2.
The structure of XynC-CD was solved by molecular replacement using MOLREP (39) and the intracellular xylanase from Geobacillus stearothermophilus (PDB code 2Q8X) as search model. The model was refined with REFMAC (37) and completed with COOT (38). The crystals belong to the P32 space group with three independent molecules within the a.u. that are essentially identical, showing root mean square deviation (r.m.s.d.) in the range 0.17–0.24 Å when superimposing their 352 α-carbon atoms Cαs.
Stereochemistry of the models was checked with PROCHECK (40) and MOLPROBITY (41). The figures were generated with PyMOL (42). r.m.s.d. analyses were made using the program SUPERPOSE within the CCP4 package (34). The interfaces were analyzed by PISA (43).
Results
N-terminal Xyn10C Region Binds Xylan
Specificity of Xyn10C binding to insoluble polysaccharides was investigated. Avicel and insoluble oat spelt xylan were incubated withthe complete Xyn10C and its isolated Xyn10C-XBD and Xyn10C-CD domains, and their bound and unbound fractions were separated by centrifugation and analyzed by SDS-PAGE (Fig. 1a). Binding to Avicel is observed only in the complete enzyme, whereas the isolated Xyn10C-XBD and Xyn10C-CD domains only bind insoluble xylan, apparently less efficiently than the full-length protein. Consequently, only the C-terminal CBM9--1–CBM9-2 region has cellulose-binding ability.
FIGURE 1.

Binding specificity of Xyn10C. a, SDS-PAGE analysis of binding to insoluble polysaccharides. Proteins (P) were mixed with Avicel or with the insoluble fraction of oat spelt xylan for 1 h; unbound (U) and bound (B) fractions were separated by centrifugation; pellets were washed three times (W). b, nondenaturing polyacrylamide gels containing no ligand (control) or soluble polysaccharides. Lane 1, BSA; lane 2, Xyn10C; lane 3, Xyn10C-CD; lane 4, Xyn10C-XBD.
Binding of the three samples to soluble fractions of xylan (wheat arabinoxylan, oat spelt xylan, and beechwood xylan), barley β-glucan, and hydroxyethylcellulose was also evaluated using affinity gel electrophoresis (Fig. 1b). Migration of full-length Xyn10C and Xyn10C-XBD in gels containing soluble xylans and barley β-glucan was markedly retarded with respect to migration in gels without the substrates, whereas no binding to hydroxyethylcellulose was observed by any of them. Arabinoxylans from oat spelt and wheat produced the largest effect, whereas beechwood xylan and barley β-glucan show a comparable and smaller effect. Therefore, Xyn10C and its Xyn10C-XBD region present higher affinity for arabinoxylans than for glucuronic-substituted xylan or mixed β-glucans. In contrast, migration of Xyn10C-CD domain was not retarded by any substrate.
Xyn10C-XBD Crystal Structure Shows a Multimeric Arrangement
The crystallization of a construct containing the xylan-binding N-terminal portion (residues 1–334) of xylanase 10C from P. barcinonensis (Xyn10C-XBD) has been previously reported (31). We present here its three-dimensional structure determined by molecular replacement at 2.4 Å resolution. Details are given under “Experimental Procedures” and in Table 2. The structure of Xyn10C-XBD reveals the presence of two separate domains, corresponding to CBM22-1 (residues 1–159) and CBM22-2 (residues 171–334) repeats, both adopting a β-sandwich fold, as explained below.
The final model contains six Xyn10C-XBD molecules in the a.u., although two of them could not be fully traced. The six molecules are arranged in two trimers, ABC and DEF, and the CBM22-2 portion of subunits C and E was not visible in the electron density maps (Fig. 2a). However, structural superimposition of the two observed trimers allows the depiction of a putative “full” trimer that would be represented by molecules A/DBF (Fig. 2b). Crystal packing shows cavities where the CBM22-2 moiety of molecules C and E could be allocated, but the electron density maps only showed residual noise despite the many attempts to perform manual and automatic building. Taking into account that mass spectrometry excludes proteolysis of the polypeptide chain, we assume that the missing parts are disordered in the crystals.
FIGURE 2.

Crystal structure of the Xyn10C-XBD tandem. a, asymmetric unit contains six independent molecules forming two trimers, ABC (left) and DEF (right); CBM22-1 domains are highlighted with a transparent surface. Molecules C and E could not be full-traced in the electron density maps. b, superposition of the two trimers showing that both are arranged by a common association pattern of their CBM22-1 moieties, whereas the second CBM22-2 domain is asymmetrically located. c, different conformations of the tandem observed in the crystals, with the linker being partially disordered.
As shown in Fig. 2a, both trimers lack noncrystallographic 3-fold symmetry. i.e. each protomer adopts a different molecular conformation (Fig. 2c) within the trimers. However, it should be noted that each trimer is built up from a regular association of three CBM22-1 domains (Fig. 2a) that bury 700 Å2 (p value 0.59). In contrast, the observed asymmetrical orientation of the corresponding CBM22-2 moieties is probably determined by the highly intricate crystal packing interactions. The association pattern observed in Xyn10C-XBD is commented on below.
Two Differing CBM22 Modules Are Attached with a Great Flexibility
The main consequence of these results is the high flexibility presented by the N-terminal domain of Xyn10C. Thus, the structural comparison of the four full-traced molecules, illustrated in Fig. 2d, reveals a broad molecular conformational landscape in which CBM22-2 may adopt different dispositions with respect to CBM22-1. This flexibility is allowed by the long proline-glycine-rich 160PENPGEPGEAG170 segment linking both modules, which is partially disordered in the crystal (a segment varying from 4 to 10 residues, depending on the different molecules within the a.u., is unobserved) and must provide high mobility to the polypeptide chain.
Each CBM22 domain is folded into a classical lectin-like β-jelly roll composed, mainly, of two antiparallel β-sheets, each with six (β9, β6, β11, β1, β4, and β3) and five β-strands (β8, β7, β10, β5, and β2) (Fig. 3, a and b). One of these β-sheets shapes an extended cleft encompassing the three aromatic residues from the substrate-binding motif, Trp-55/Tyr-217, Tyr-104/Tyr-268, and Tyr-136/Tyr-303 (in CBM22-1/CBM22-2 numbering), the two first contouring a sandwiched hydrophobic platform (Fig. 3, b and c). The cleft also presents two conserved residues, an arginine (Arg-27/Arg-189) and a glutamate (Glu-138/Glu-305) that have been proved to be absolutely required for xylan recognition in CBM22-2 from RthXyn10b (21). Structural comparison of CBM22-1 and CBM22-2 shown in Fig. 3b reveals an r.m.s.d. of 1.9 Å, based on superposition of 146 Cα atoms. Despite their rather modest sequence similarity (39%) and identity (23%), the topology og the two domains is essentially conserved. The main differences are located in some loops on the concave face of the β-sandwich, which are longer in the CBM22-2 domain; these are Arg-261–Leu-266, at the end of strand β7, and the loops Pro-291–Ser-298 and Trp-308–Glu-316, before and after strand β10 (Fig. 3d). It is known that differences in these loops may alter radically the substrate affinity on analogous CBMs, and in the case of Xyn10C-XBD, they generate a significantly deeper groove in CBM22-2. Particularly remarkable is the long loop Trp-308–Glu-316 inserted at the end of strand β10 that contains Trp-308, an additional aromatic residue next to Tyr-217 that is solvent-exposed and extends the hydrophobic platform, shaping new subsites at the binding cleft (Fig. 3c) as described below. Finally, and in common with most β-sandwich CBMs, there is a conserved calcium ion having a structural role, located on the surface at the opposite face of the groove. This calcium ion is coordinated by the carboxylates of Glu-17/Glu-179 and Asp-149/Asp-325, both conserved within the CBM22 family (Fig. 3e). The coordination sphere is completed with the main chain carbonyls from Ser-15, Gln-41, and Gln-44, in domain CBM22-1, and by the carbonyls from Asp-177 and Arg-206, and the side chain of Ser-204, in the case of domain CBM22-2. Both calcium ions are also coordinated to a water molecule configuring an octahedral arrangement.
FIGURE 3.

Topology of the CBM22-1 and CBM22-2 domains. a, secondary structure of the Xyn10C-XBD tandem in rainbow sequence code. The two calcium ions are shown as white spheres. b, superimposition of CBM22-1 (blue) onto CBM22-2 (red) showing the five residues previously reported to be required for xylan binding. Topological differences between both domains are concentrated at some loops surrounding the binding cleft. c, molecular surface of CBM22-1 (top) and CBM22-2 (bottom), highlighting the hydrophobic platform defined by the aromatic residues located at the binding cleft. d, structure-based sequence alignment of CBM22-1 and CBM22-2 with the CBM22-1 (PDB code 2W5F) and CBM22-2 (PDB code 1DYO) modules of RthXyn10B (20, 36). Residues involved in ligand binding are highlighted in yellow, and residues coordinating the calcium ion are marked in green. e, zoom of the calcium ion-binding site of CBM22-1 (blue) and CB22–2 (red) showing the coordination pattern at each domain.
Because of the difficulties in handling the very thin and fragile plates, and the limited resolution shown by many of them, each domain was expressed independently with the aim to undertake soaking experiments. However, only the CBM22-2 crystals provided suitable complexes to investigate its binding specificity, as explained below.
Isolated CBM22-1 Domain Keeps in the Crystal the Same Association Trend of the Tandem
A construct encoding the N-terminal CBM22-1 domain was produced, and its structure has been determined at 1.7 Å resolution (Table 2). The crystals belong to the H3 space group, with two molecules in the a.u., A and B, which are essentially equivalent, with an r.m.s.d. of 0.172 Å after superposition of 135 Cα atoms. The electron density maps showed the full polypeptide chain, including Ala-4–Ala-159. Interestingly, analysis of the structure of the CBM22-1 crystals shows an identical association pattern to that described in the Xyn10C-XBD crystals, i.e. crystal packing of each of the two independent molecules A/B generates a similar trimer (Fig. 4a) that is equivalent to that presented by the tandem construct (Fig. 2a). Thus, the CBM22-1 domains from the ABC tandem trimer superimposes onto the AAA trimers with an r.m.s.d. of 0.673 Å for 410 Cα atoms. Interestingly, the number of polar interactions between subunits is moderate (eight H-bonds and three salt-bridges), but the number of van der Waals contacts (56 contacts shorter than 4 Å), and the evident shape complementarity among subunits is remarkable. Furthermore, an inspection of the CBM22-1 trimer interface, shown in Fig. 4b, reveals that the aromatic residues from the substrate-binding motif are located close to the interface, and the N-terminal segment from the adjacent subunits is filling the binding cleft. Consequently, diffusion of the ligands into the binding site probably disrupts the interface, which might be deleterious for crystal packing. This might be the reason why soaking experiments with ligands dislocates most crystals or led to highly twinned data, precluding the structural characterization of the complex.
FIGURE 4.
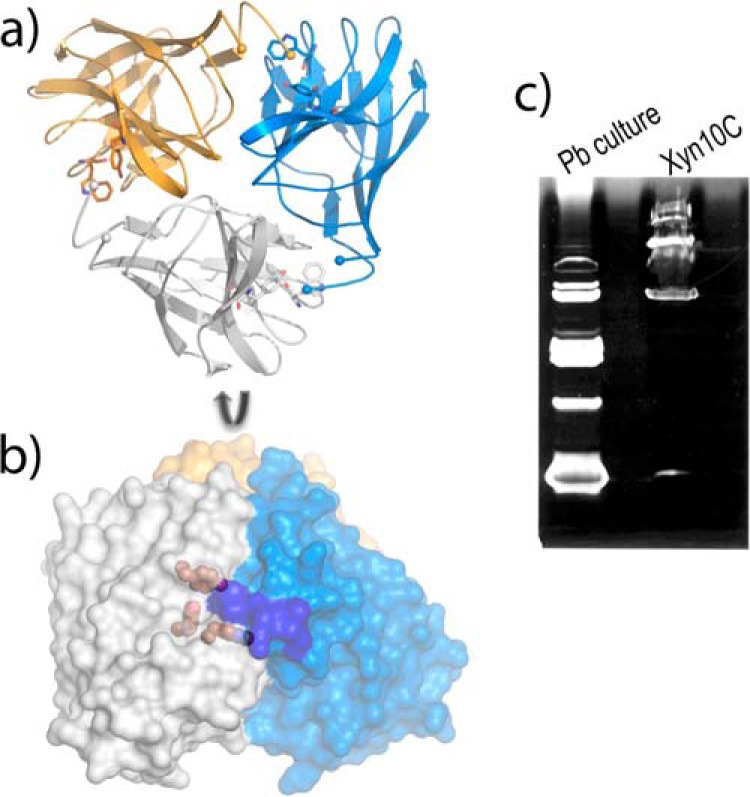
Crystal structure of the isolated CBM22-1 domain. a, packing of each independent molecule within the a.u. shows a trimeric association, where the subunits are colored in blue, gold, and white. b, perpendicular view of this trimer represented as molecular surface, showing the aromatic residues from the binding motif as gold spheres, and the region occupied by the N-terminal segment of the adjacent subunit highlighted in blue. c, zymogram analysis of Xyn10C; 1st lane, xylanases identified from a P. barcinonensis culture; 2nd lane, recombinant sample of Xyn10C showing the presence of different active forms.
The physiological significance of a putative trimeric association of the CBM22-1 domain is unclear. The association buries only 21% of the total molecular surface (4209/20,129 Å2), and apparently, the trimer formation might constrain the binding ability of CBM22-1 due to the proximity of the binding cleft to the interface. In fact, only terminal xylooligosaccharides could be allocated within each binding cleft in the trimer. However, the absolute conservation of this assembly through Xyn10C-XBD and its isolated CBM22-1 domain crystals is remarkable. The outstanding feature is that the association trait is conserved in the CBM22-1 crystals in the absence of the CBM22-2 moiety, which was doing many crystal contacts in the tandem. This observation might suggest that the association trend observed in both crystals might be a trait of the CBM22-1 domain. Although ultracentrifugal analysis has shown that both the Xyn10C-XBD tandem and the CBM22-1 domain are primarily monomeric in solution (data not shown), active oligomeric forms have been detected by zymogram analysis of full-length Xyn10C (Fig. 4c). Whether these observations assign a role to CBM22-1 in oligomerization of Xyn10C requires further investigation.
CBM22-2 Complexes Illustrate Its Ligand-binding Mode
A construct encoding the truncated CBM22-2 domain, which encompasses residues 172–332, was produced, and its structure has been determined at 1.75 Å resolution. Crystals belong to the P32 space group with three molecules within the a.u. that do not associate and are essentially equivalent (r.m.s.d. 0.281/0.318 Å for 143/145 Cα atoms when superimposing molecules B or C, respectively, to A). The only significant differences are observed at loop 308–313, within the insertion located at the end of β10, which is involved in substrate binding, as explained below.
To investigate the molecular basis for ligand specificity, soaking experiments of the crystals were performed with xylotriose, xylotetraose, and 1,3:1,4 β-glucotetraose B. As shown in Fig. 3c, CBM22-2 contains a deep cleft, 20 Å length, 9 Å deep, running across the concave surface of the β-sheet that conforms the substrate-binding site. In the middle of this cleft, three protruding aromatic residues define a sandwich hydrophobic platform to allocate the substrate (Fig. 5). The side chains of Tyr-217 and Trp-308, at one face of the platform, are making an angle of 140°, thereby shaping a somehow twisted configuration for ligand binding. The electron density of the xylotetraose-soaked crystals show four ordered xylose units within this binding cleft (Fig. 5a), which have been ascribed to subsites S1 to S4 going from the reducing to the nonreducing end, and covers the whole surface along the crevice. The direction of the sugar chain was assigned by evaluating the temperature factors of the two alternative O5/C5 positions and the hydrogen-bonding pattern of O5. The xylooligosaccharide binds in the predicted 3-fold helical conformation previously determined by x-ray fiber diffraction analysis of xylan (44) and characterized by the internal hydrogen bonds from O3n to O5 n + 1, which is especially evident in subsites S1 to S3. This conformation is stabilized by Tyr-217 and Trp-308, both positioning the central xylose units, whereasTyr-303, at the other face of the platform, is stacking to the glycosidic bond linking both xyloses at subsites S2 and S3.
FIGURE 5.

Structure of the CBM22-2 complexes. a, xylotetraose; b, xylotriose; c, the mixed glucan Glc-4Glc-4Glc-3Glc, bound at the CBM22-2 binding cleft. The ligands are represented as yellow sticks and the electron density 2Fo − Fc maps are contoured at 1σ. The residues involved in ligand binding are labeled. The schematic representation (right) shows the aromatic residues defining the hydrophobic platform in red.
Apart from the hydrophobic stacking interactions, there are relatively few polar contacts involved in ligand binding, as shown in Fig. 5a. The xylose unit located at subsite S1 only makes a single direct polar interaction through its O3 hydroxyl with the Arg-189 peptide carbonyl. Also, the sugar O2 atom makes some packing polar interactions in molecules A and C, but not in molecule B, in which the xylose ring occupying this subsite is not clearly seen in the electron density map. Consequently, it is apparent that the xylose observed in molecules A and C may be partially fixed by packing interactions and not by a tight interaction within this subsite. On the contrary, subsite S2 is mainly defined by stacking to Tyr-217 but is further stabilized by the O5 hydrogen bond to Tyr-268-OH and Arg-189-NH2. Similarly, the xylose unit at subsite S3 is positioned by stacking to Trp-308, but its binding is further enhanced by tight interactions of the O2 hydroxyl to Tyr-303 and Arg-189 side chains, and a bifurcated hydrogen bond of Glu-305 carboxylate to both the O2 and O3 hydroxyls. Finally, the xylose at subsite S4is only interacting through its O2 hydroxyl with the peptide CO of the Tyr-268 main chain. Again, some packing interactions are also observed in molecule A, which creates a different environment possibly producing the diverse conformations observed at this subsite and commented below.
Soaking with xylotriose led to the sugar occupying subsites S2 to S4 (Fig. 5b). The interaction pattern is conserved with that described for xylotetraose. However, only molecule A shows the oligosaccharide at full occupancy in all three subsites, as molecules B and C present poor density at subsite S4, which may be attributed to the different packing environment described above. These results led to the conclusion that positions S2 and S3 are tightly held at the binding cleft, whereas xyloses at subsites S1 and S4 are more loosely bound. Also, an inspection to the complex reveals that only O2 and O3 hydroxyls are engaged in polar interactions with the protein at subsite S3, whereas all other xylose units situate these hydroxyls completely exposed to the solvent. Consequently, and with the sole exception of subsite S3, the CBM22-2 binding cleft appears able to allocate the full repertoire of side chains evident in xylans.
Moreover, soaking experiments with the tetrasaccharide Glc-4Glc-4Glc-3Glc yielded rather disordered crystals in which only the central portion, including two β-1,4-linked glucose units, could be modeled in molecule A (Fig. 5c). The two glucose rings are located at subsites S2 and S3 as described previously, but their positions are shifted along the hydrophobic platform with respect to that observed in the xylooligosaccharides. First, the glucose occupying subsite S2 moves away from Arg-189 to allocate its C6-OH hydroxyl group while losing the polar interactions made by the cyclic O5 from xylose. In this way, Tyr-217 is stacking against the C3-C4-C5 portion of the glucose ring. Second, the glucose at subsite S3 is now stacking against Tyr-268, at the other face of the platform, but maintains hydrogen bonds through its O2 and O3 hydroxyls to Arg-189, Tyr-303, and Glu-305, although with a different interaction pattern (Fig. 5c). Nevertheless, and because of the poor density maps, we can no exclude the presence of other conformations bound at the cleft.
Superposition of the different ligand complexes allows depiction of the molecular mechanism of substrate recognition by CBM22-2. As shown on Fig. 6a, one face of the cleft is formed by a series of polar residues that form a network of hydrogen bonds, which maintains a tight arrangement of the residues essential for binding. Thus, the Arg-189 side chain is fixed by hydrogen links to Tyr-217 and Glu-256 through its NH1 and NH2, whereas the position of Glu305 is fixed by interaction with Ser-254. Moreover, the stacking pattern interaction through Ser-254, Tyr-303, and Arg-189 contributes to fixing this conformation. On the contrary, the other face of the cleft is defined by a plastic hydrophobic platform formed by Tyr-217 and Trp-308. In fact, mobility of Trp-308 seems to be facilitated by flexibility of the loop 308–313, whose conformation has been observed to be variable in the crystals. Therefore, the inserted loop, including Trp-308, is an essential determinant bringing specificity to CBM22-2. Furthermore, the plasticity of the hydrophobic platform base, and the looser binding observed at subsites S1 and S4, might be the structural basis explaining why CBM22-2 is able to accommodate mixed glucans.
FIGURE 6.
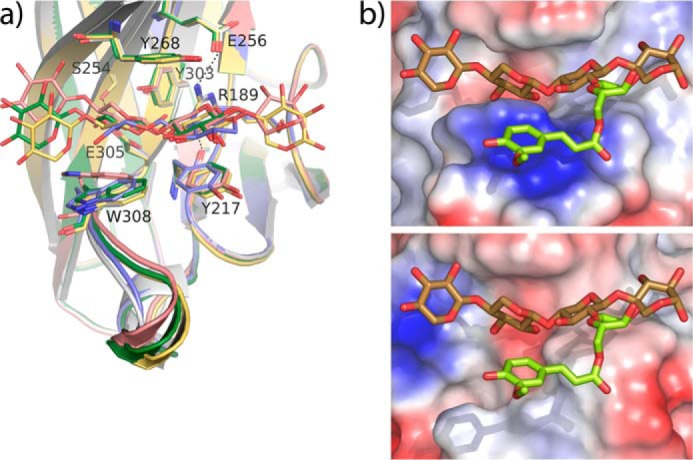
CBM22-2-binding site architecture. a, superposition of the CBM22-2 complexes onto the free CBM22-2 (white). Two positions of xyl4 (pink and yellow), xyl3 (green), and Glc-4Glc-4Glc-3Glc (blue) are shown. A face of the binding cleft is fixed through polar interactions of the involved residues, although the opposite face is defined by a plastic hydrophobic platform able to fit the substrate. b, top, CBM22-2 binding cleft showing the experimental position of xyl4 and a modeled arabinose-ferulate moiety (lime) decorating O2 at subsite S2. Bottom, CBM22-1 binding cleft showing a putative position of the same ligand, inferred from structural superimposition of both domains.
CBM22-2 Domain Presents Novel Xylan Binding Determinants within the CBM22 Family
The 23% sequence identity shared by CBM22-1 and CBM22-2 is similar to the values obtained when comparing each domain to other CBM22 members, with sequence identities ranging from 15 to 30%. Yet topology is rather conserved, and both domains show root mean square deviations of only 1.8–1.9 Å (over 145–149 Cα atoms) when superimposed on the two other structurally known, CBM22-1/CBM22-2 from RthXyn10B (PDB codes 1DYO and 2W5F). This prominent topology conservation is not related to specificity as RthCBM22-1, like many CBM22 members, does not contain the five residues identified as essential for ligand binding in RthCBM22-2, thus explaining the inability of some CBM22 modules to bind xylan (21). In this work, we have identified Trp-308, as an additional determinant directly involved in CBM22-2 binding to ligands. A Blast search shows that this Trp is found only in a low number of the closest CBM22-2 homologues (less than one-fourth), mostly present in modular GH10-containing enzymes from Paenibacillus or Clostridium species that also contain the (W/Y)YY motif. The finding that this Trp is missing in CBM22-1 and RthCBM22-2 sequences, both known to bind xylan, suggests that subtle differences in sequence may allow a fine-tuning of substrate targeting among xylan-binding CBM22 domains.
Fig. 6b shows the profile of the CBM22-2 binding cleft, as compared with its partner CBM22-1. As shown in the figure, the flexible loop comprising residues 308–313 protrudes from the CBM22-2 binding crevice in a way that, apart from fitting the xylose unit at subsite S3, it also contours a platform that could help in creating a site to allocate the xylan substitutions of the xylose bound at subsite S2. As in CBM22-1, this loop is much shorter in most CBM22s that, consequently, leave room to tolerate large xylan substitutions at subsite S2. The special feature of the CBM22-2-type domains is that they might be able to recognize, or even select, a particular substitution of xylan. This ability is illustrated by a putative arabinose-ferulate decoration modeled at O2, represented in Fig. 6b, which seems to be accommodated nicely onto the binding platform. Thus, the loop insertion and the presence of an aromatic residue at the position equivalent to Trp-308 might be a common trait in some CBM22 members accounting for preferential binding to a particular region of the heterogeneous multivalent polymer, which has yet to be discovered.
Binding Affinity of Xyn10C-XBD and Its Isolated Modules
The binding ability of Xyn10C-XBD for short-chain oligosaccharides was evaluated by ITC. The binding isotherms for each ligand were fitted by nonlinear regression assuming two independent sites (one per domain) in the tandem (Table 3 and Fig. 7a). The stoichiometry of binding was experimentally confirmed for K values on the order of 104 m−1 or higher. Both domains bind xylotriose with similar affinity, but one of them recognizes oligosaccharides with four or more xylose units with higher affinity and more favorable enthalpy. Moreover, the binding of xylopentaose or xylohexose was clearly favored over that of xylotetraose. The presence of four subsites at the binding cleft of CBM22-2 can account for the stronger interaction of xylotetraose, further enhanced by the extra contribution of Trp-308 to the complex stabilization when compared with CBM22-1. However, the higher affinity of Xyn10C-XBD against xylopentaose or xylohexaose with respect to xylotetraose might be attributed to a higher conservation of the preferred 3-fold helical conformation in the longer oligosaccharides, resulting in higher binding efficiencies, as further subsites were not evident from the CBM22-2 complexes. Interestingly, titration experiments also showed that the CBM22 tandem of Xyn10C-XBD binds xylooligosaccharides with higher affinity than mixed glucans. This reduced affinity for glucans is consistent with the disorder observed in the crystals by soaking experiments with the tetrasaccharide Glc-4Glc-4Glc-3Glc, and it probably reflects a statistical occupancy of different suboptimal binding modes. Binding against a polymeric substrate, the oat spelt arabinoxylan was also assessed. The titration curve was best fitted by assuming two types of sites whose affinities for the polysaccharide exceeded those of xylopentaose or xylohexose, but the ratios between the binding constants of both sites were comparable. To further characterize the individual Xyn10C-XBD-binding sites, binding of each independent CBM22 domain was tested against xylotetraose by ITC. Affinities were similar and around 2–5 times lower than for the tandem highest affinity site, excluding remarkable cooperative effects between both binding sites within the tandem (Table 3).
TABLE 3.
Thermodynamic parameters of Xyn10C-XBD to different ligands at 25 °C
| Ligand | K1 ×10−3 | −ΔH1 | −TΔS1 | K2 × 10−3 | −ΔH2 | −TΔS2 | |
|---|---|---|---|---|---|---|---|
| [iscap]m−1 | kcal/mol | kcal/mol | [iscap]m−1 | kcal/mol | kcal/mol | ||
| Xyn10C-XBD | Xylotriosea | 1.26 ± 0.01 | 7.82 ± 0.04 | 3.60 ± 0.04 | |||
| Xylotetraose | 7.8 ± 0.4 | 15.2 ± 0.9 | 9.9 ± 0.5 | 2.1 ± 0.2 | 7.5 ± 0.9 | 2.98 ± 0.8 | |
| Xylopentaose | 31 ± 9 | 16 ± 2 | 10 ± 2 | 2 ± 1 | 8 ± 1 | 3.5 ± 0.7 | |
| Xylohexaose | 36 ± 6 | 11.5 ± 0.8 | 5.31 ± 0.7 | 5.0 ± 0.4 | 8.6 ± 0.8 | 3.6 ± 0.8 | |
| Xylan oat spelt (0.381 mmb | 64 ± 7 | 5.3 ± 0.6 | −1.2 ± 0.5 | 12 ± 1 | 24.8 ± 0.5 | 19.3 ± 0.4 | |
| G3G4G4Ga | ∼0.2c | ∼5.9 | 2.6 | ||||
| G4G4G3Ga | ∼0.2 | ∼5.0 | 1.9 | ||||
| G3G3G4Ga | ∼0.2 | ∼6.5 | 3.4. | ||||
| CBM22-1 | Xylotetraose | 3.6 ± 0.2 | 17.42 ± 0.04 | 12.58 ± 0.01 | |||
| CBM22-2 | Xylotetraose | 1.6 ± 0.2 | 10.07 ± 0.08 | 5.71 ± 0.01 |
a Data were fit to the onset of sites model with a 1:1 stoichiometry per CM22 domain.
b Millimolar concentration of the two types of sites in the polysaccharide that gives a value of binding sites per CM22 domain equal to 1.
c Reported values are estimations of the thermodynamic parameters due to the incomplete (∼50–60%) saturation of sites reached in ITC with the mixed oligosaccharides.
FIGURE 7.

Binding specificity of Xyn10C-XBD and its isolated domains. a, representative binding of different carbohydrates to Xyn10C-XBD by ITC. The ligands in the syringe (20 mm oligosaccharides, 4 mg/ml polysaccharide) were titrated into Xyn10C-XBD loaded in the cell (46–60 μm). Measurements were performed at 25 °C in 50 mm sodium hydrogen/dihydrogen phosphate, pH 7. The upper panels show the raw data upon injection of 1, 5 × 5, 8 × 10, and 8 × 20 μl for xylotetraose; 1, 5 × 2, 5 × 3, 5 × 5, 4 × 10, and 9 × 20 μl for xylopentaose; and 1, 15 × 5, 4 × 10, and 21 × 20 μl for oat spelt arabinoxylan. The bottom panels show the integrated areas (symbols) obtained from respective raw data and their theoretical fit (continuous lines) using the two-sets of sites binding-model of the Origin ITC software with the best fitting thermodynamic parameters summarized in Table 3. b, nondenaturing polyacrylamide gels containing no ligand (control) or soluble xylans. Lane 1, BSA; lane 2, Xyn10C-XBD; lane 3, CBM22-1; lane 4, CBM22-2.
Binding ability to xylans and barley of each isolated CBM22 domain was qualitatively evaluated using affinity gel electrophoresis (Fig. 7b). Migration of both CBM22-1 and CBM22-2 was markedly retarded with respect to migration in gels without the substrates, especially when arabinoxylans are tested; in this case, larger retardation to the highly substituted wheat arabinoxylan was apparent in CBM22-2. Moreover, the binding observed in gels for each domain was similar and equivalent to that observed for the tandem when tested against beechwood glucuronoxylan. On the contrary, Xyn10C-XBD was more retarded in barley β-glucan than each isolated CBM22.
Finally, a fact worth mentioning is that CBM22-1 migrates in absence/presence of substrates similarly to Xyn10C-XBD and is distinctly compared with that observed for its partner CBM22-2. It is tentatively suggested that the reason for this observation might be attributed to the common association state suggested by the crystal analysis presented here.
Catalytic Domain May Bind Highly Decorated Substrates
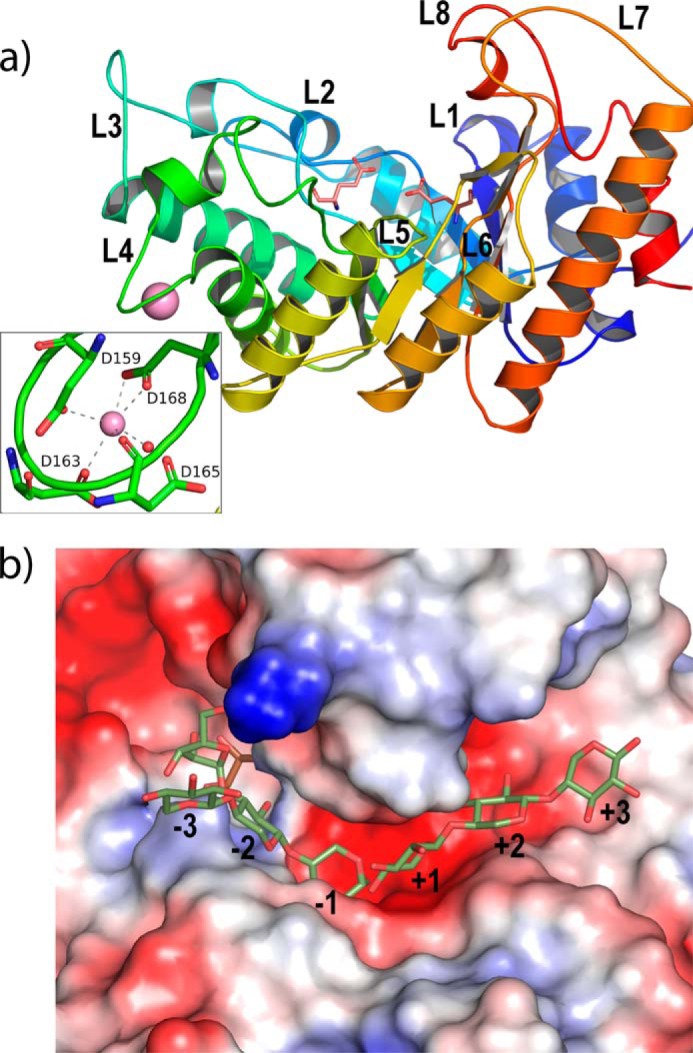
To further elucidate the catalytic functionality of Xyn10C, a construct containing its GH10 domain (residues 340–691) has been solved at 1.76 Å resolution. The structure of the catalytic domain corresponds to the expected topology of a single (β/α)8 barrel, which is common in the GH10 family. The barrel is closely packed on the face corresponding to the short loops linking the different β/α repeats (Fig. 8a), whereas the long segments at the C terminus of the β-strands (L1 to L8) present additional elements of secondary structure and form the active site. The catalytic residues are located at the end of β4 (the general acid/base Glu-475) and β7 (the nucleophile Glu-593) strands, respectively. A special feature of Xyn10C-CD is the 10-residue insertion found in loop L4. Most GH10 members present a short α-helix just prior to α4, but an aspartate-rich loop (498DVDGDGDDSD507) is inserted between these two helices in Xyn10C-CD, constituting a nonconserved calcium-binding site (Fig. 8a). A structural calcium ion conferring stability to Cellvibrio japonicus Xyn10A has been reported (45), but that metal ion is bound by residues from loop L7 (PDB code 1W2P).
FIGURE 8.
Structure of Xyn10C-CD. a, catalytic domain (represented in rainbow sequence code) folds into a (β/α)8 barrel common to GH10 family, with the catalytic Glu-136 and Glu-254 being at the end of β4 and β7. The long segments at the end of the β-strands (L1 to L8) are shaping an open active site cleft. An insertion at loop L4 is defining a calcium ion (violet sphere)-binding site, zoomed in the inset. b, mapping of the active site by superimposition of Xyn10C-CD onto CmXyn10b (46) complexed with arabinose-α-1,3 xylotriose/xylotriose at the glycone (minus)/aglycone (plus) subsites. A MPD molecule (pink) from the crystallization buffer is occupying subsite −2 in Xyn10C-CD crystals.
As observed in GH10 xylanases, the shape of the α/β-barrel is elliptical, with the long L7 and L8 loops shaping an extended open cleft consistent with its endo-mode of attack and able to accommodate polymeric substrates. The subsite mapping of the binding cleft is illustrated by superimposition of the XynC-CD coordinates to those of the xylanase Xyn10b from Cellvibrio mixtus (46) (37% sequence identity), in complex with arabinose-substituted xylo-oligosaccharides (PDB code 1UR2). In this complex, arabinose-α-1,3-xylotriose and xylotriose were observed at the glycone (minus) and aglycone (plus) subsites, respectively, following the nomenclature used for sugar-binding subsites, which defines that the cleavage occurs between subsite −1 and +1 (Fig. 8b). The inspection to the Xyn10C-CD binding cleft reveals that the enzyme may allocate at least six xylose units at the crevice. Previous studies on C. mixtus xylanase Xyn10b (46) and other GH10 xylanases (47) bound to decorated xylo-oligosaccharides have shown that xylan substitutions can be accommodated only in selected subsites in which the O2 and O3 of xylose point into the solvent, i.e. −3, +1, and +3, whereas in the case of the −1 and +2 subsites, the hydroxyls are directed into the protein surface precluding the binding of side-chain xylan decorations. In subsite −2, only O3 is solvent-exposed, whereas most of the GH10s have a glutamate at loop L2 that makes a hydrogen bond to O2, which consequently avoids decoration at this position. However, Xyn10C-CD is one of the reported GH10s having the Glu/Gly substitution that provides limited access to O2 (48). In the case of Xyn10C-CD, a short L1 loop leaves additional room that seems able to harbor large substituents at both O2/O3 of subsite −2. A similar broad cavity is observed at subsite +2, which is occupied by several ordered water molecules in the crystal. Consequently and similarly to what it is observed in the N-terminal domain, the catalytic module of Xyn10C seems able to bind highly decorated xylans by accommodation of substitutions in almost all its subsites.
Discussion
Structural studies on representative members of many CBMs and their complexes have provided insight into the underlying mechanism of CBM-ligand recognition and interaction. At present, one of the greatest challenges is to depict the concerted molecular mechanism that displays multimodular biomass-degrading enzymes that present multiple copies of ancillary noncatalytic domains. Homogeneous multimodularity has been related to multivalency and avidity effects, whereas the heterogeneous pattern has been proposed to provide distinct ligand-binding specificities. However, previous studies have revealed that tandem of homologous CBMs may present a more complex functional mechanism (49), thereby suggesting that the apparent functional distinction between the heterogeneous and homogeneous modular arrangement might not be as simple.
The xylanase Xyn10C from P. barcinonensis is a 120-kDa modular enzyme that presents the CBM22/GH10/CBM9 architecture found in a subset of large GH10 xylanases, mainly produced by saprophytic microorganisms (Fig. 9). Apart from the group of enzymes found in thermophiles, with a regular domain's composition, a more variable modular architecture is found in some members from Clostridium species where, in some cases, two CBM22s are flanking the catalytic GH10 domains as occurs in CthXyn10B. Interestingly, some of these CBM22s do not present the three-aromatic motif associated with xylan binding, therefore pointing to yet unknown functions. In a few cases, the CBM22 domain is present in bacterial enzymes involved in xylan degradation bearing catalytic domains from families other than GH10, as GH11 or GH43. Furthermore, a number of plant xylanases encompass one to four CBM22 repeats preceding their catalytic GH10. This diversity reflects the existence of common recognition mechanisms that must be combined with particular determinants specific to each function.
FIGURE 9.

Modular structure of representative CBM22 containing enzymes. P. barcinonensis xylanase C (PbXyn10C); Thermoanaerobacterium thermosulfurigenes xylanase A (TthXynA, Q60046); Thermotoga maritima xylanase A (TmXynA, Q60037); Clostridium thermocellum xylanase X (CthXynX, P38535); C. thermocellum xylanase 10B (CthXyn10B, P51584); Clostridium cellulolyticum xylanase (CcXyn, B8I5C0); Oryza sativa xylanase (OsXyn, Q84R74); Ruminococcus albus xylanase 11C (RaXyn11C, BAM46898.1); Amphibacillus xylanus arabinoxylan arabinofuranohydrolase (AxAxH). S, S-layer-like domain; DOCK, dockering domain; CE1, carboxyl esterase 1; FNIII, fibronectin type III. L1, loop linking CBM22-1 to CBM22-2; L2, loop linking CBM22-2 to GH10. +/−, the xylan binding motif (W/Y)YY is present/absent in CBM22 domain.
We report here the three-dimensional structure of the Xyn10C N-terminal region, containing the CBM22-1–CBM22-2 tandem, which was shown to bind xylan, and hence we called it Xyn10C-XBD. This represents the first crystal structure of two contiguous CBM22 domains solved to date. Xyn10C-XBD is folded into two separate CBM22 domains that are linked by a flexible 10-residue segment, which provides an extraordinary plasticity to the domain that consequently exhibits a broad conformational landscape. This feature must potentiate significantly the approach of each CBM moiety to their corresponding targets.
Each CBM22 domain of Xyn10C-XBD presents the expected β-jelly roll topology and both contain the R(W/Y)YYE motif that had previously been identified to be required for xylan binding. However, the soaking analysis of the isolated CBM22-2 crystals with ligands has allowed us to identify the Trp-308 as an additional determinant for binding. This residue is located in an insertion found in a low number of closest CBM22-2 homologues mostly integrated in modular GH10-containing enzymes from Paenibacillus or Clostridium species that also present the xylan-binding motif. Consequently, the CBM22-2 domain may define a subset of xylan-binding CBM22s directed to precise regions of the polysaccharide. There might be other structural determinants fine-tuning xylan binding functionality within the CBM22 family that have yet to be discovered.
The structural differences found in both Xyn10C-XBD-binding sites are consistent with the ITC experiments that show the presence of two types of sites with different affinity. Consequently, it is most likely that both domains account for a different functionality in vivo, explaining the need of domain multiplicity in Xyn10C. Moreover, the affinity gel results show that both the tandem and its isolated domains were noticeably retarded when tested with arabinoxylans. However, it did not show clear differences between the isolated CBM22s, apart from a slight preference of CBM22-2 for highly substituted arabinoxylan. Nevertheless, it has been previously reported that CBMs showing equivalent specificity against purified substrates exhibit significant differences in their recognition of plant cell walls (50). Therefore, the precise and distinct physiological role of CBM22-1 versus CBM22-2 may elude analyses performed with polysaccharides isolated from their natural environment.
The different structural determinants described in this work for each CBM22 moiety of Xyn10C suggest that the tandem might represent a sophisticated delivery strategy of Xyn10C in which CBM22-1 would represent a primary recognition site directing the enzyme to a particular region or a specific type of cell wall. Then the CBM22-2 domain would recognize specific regions of the cell wall that are going to be degraded by the GH10 domain, enhancing its catalytic efficiency. The great flexibility of the CBM22-1–CBM22-2 linker could enable the CBM22-2/GH10 domains to access a large extension of the cell wall providing a greater chance of target substrate capturing. In agreement with this, the shorter link attaching the CBM22 tandem to GH10 (332TEAIAIE338) is indicative of a more restricted conformational freedom at this segment and a more concerted action of the CBM22-2/GH10 domains, both of which are apparently able to target highly decorated glucuronoarabinoxylans. These polysaccharides are major components of the primary cell walls in cereals and, therefore, they probably are the most abundant substrate found in the natural habitat of Xyn10C.
As more structural studies on CBMs become available, it is more evident that the information obtained from the isolated modules must necessarily be placed in the context of the whole enzyme. In particular, it has been suggested that the size and chemical nature of linkers connecting different CBMs and/or their associated catalytic domains may reveal functional features on a putative integrated role between modules (2). In this sense, it is interesting to note that the Xyn10C homologues from Bacillus containing the same CBM22-CBM22-GH10-CBM9-CBM9 architecture present a conserved configuration in its N-terminal region with loops following the pattern L1 > L2 (Fig. 9), whereas, their homologues from thermophiles present L1 < L2. Therefore, the integrated role of CBM22-2/GH10 domains proposed for Xyn10C may be a trait of its homologues from mesophilic bacteria but not from thermophiles. However, the occurrence of the common additional C-terminal cellulose-directed CBM9 domain would confer to all of them the additional advantage of allowing the enzymes to remain in close contact with the cell wall materials, as CBM9s can diffuse over the surface of the cellulose microfibrils.
In conclusion, the analysis of Xyn10C and its CBM22-1–CBM22-2 tandem domains presented here provides novel features that may help to understand the intricate molecular mechanisms displayed by enzymes containing multiples copies of CBMs. Indeed, more work is necessary to have a clear picture of the sophisticated strategy used by nature to tailor specificity against the highly complex and heterogeneous polysaccharides forming the plant cell wall. This basic knowledge is crucial to take the full potential of biocatalysts to produce biofuel and more efficient bioprocessing industries.
Acknowledgments
We thank Randy Read and Paul Adams for their valuable help in solving the structure of the Xyn10C-XBD crystals. We thank the Spanish Synchrotron at Barcelona (ALBA, Spain) for assistance at the XALOC beamline. We also thank the staff of the European Synchrotron Radiation Facility at Grenoble (ESRF, France) for providing access and technical assistance at beamlines ID23-1, ID23-2, and ID29. We thank Dr. Douglas Laurents for kindly revising the manuscript.
This work was supported by Grants BIO2010-20508-C04-03, BIO2013-48779-C4-2-R, CTQ2013-48995-C2-2-R, and BFU2012-36825 from the Spanish Ministry of Science and Innovation. The authors declare that they have no conflicts of interest with the contents of this article.
- CBM
- carbohydrate-binding module
- a.u.
- asymmetric unit
- r.m.s.d.
- root mean square deviation
- RthXyn10B
- R. thermocellum xylanase 10B
- ITC
- isothermal titration calorimetry
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PDB
- Protein Data Bank.
References
- 1. Gilbert H. J. (2010) The biochemistry and structural biology of plant cell wall deconstruction. Plant Physiol. 153, 444–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abbott D. W., van Bueren A. L. (2014) Using structure to inform carbohydrate binding module function. Curr. Opin. Struct. Biol. 28, 32–40 [DOI] [PubMed] [Google Scholar]
- 3. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boraston A. B., Bolam D. N., Gilbert H. J., Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gilbert H. J., Knox J. P., Boraston A. B. (2013) Advances in understanding the molecular basis of plant cell wall polysaccharide recognition by carbohydrate-binding modules. Curr. Opin. Struct. Biol. 23, 669–677 [DOI] [PubMed] [Google Scholar]
- 6. Ficko-Blean E., Boraston A. B. (2012) Insights into the recognition of the human glycome by microbial carbohydrate-binding modules. Curr. Opin. Struct. Biol. 22, 570–577 [DOI] [PubMed] [Google Scholar]
- 7. Hervé C., Rogowski A., Blake A. W., Marcus S. E., Gilbert H. J., Knox J. P. (2010) Carbohydrate-binding modules promote the enzymatic deconstruction of intact plant cell walls by targeting and proximity effects. Proc. Natl. Acad. Sci. U.S.A. 107, 15293–15298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fontes C. M., Gilbert H. J. (2010) Cellulosomes: highly efficient nanomachines designed to deconstruct plant cell wall complex carbohydrates. Annu. Rev. Biochem. 79, 655–681 [DOI] [PubMed] [Google Scholar]
- 9. Vazana Y., Moraïs S., Barak Y., Lamed R., Bayer E. A. (2012) Designer cellulosomes for enhanced hydrolysis of cellulosic substrates. Methods Enzymol. 510, 429–452 [DOI] [PubMed] [Google Scholar]
- 10. Shoseyov O., Shani Z., Levy I. (2006) Carbohydrate binding modules: biochemical properties and novel applications. Microbiol. Mol. Biol. Rev. 70, 283–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vaaje-Kolstad G., Horn S. J., van Aalten D. M., Synstad B., Eijsink V. G. (2005) The non-catalytic chitin-binding protein CBP21 from Serratia marcescens is essential for chitin degradation. J. Biol. Chem. 280, 28492–28497 [DOI] [PubMed] [Google Scholar]
- 12. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., Eijsink V. G. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 [DOI] [PubMed] [Google Scholar]
- 13. Montanier C., van Bueren A. L., Dumon C., Flint J. E., Correia M. A., Prates J. A., Firbank S. J., Lewis R. J., Grondin G. G., Ghinet M. G., Gloster T. M., Herve C., Knox J. P., Talbot B. G., Turkenburg J. P., et al. (2009) Evidence that family 35 carbohydrate binding modules display conserved specificity but divergent function. Proc. Natl. Acad. Sci. U.S.A. 106, 3065–3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simpson P. J., Bolam D. N., Cooper A., Ciruela A., Hazlewood G. P., Gilbert H. J., Williamson M. P. (1999) A family IIb xylan-binding domain has a similar secondary structure to a homologous family IIa cellulose-binding domain but different ligand specificity. Structure 7, 853–864 [DOI] [PubMed] [Google Scholar]
- 15. Szabo L., Jamal S., Xie H., Charnock S. J., Bolam D. N., Gilbert H. J., Davies G. J. (2001) Structure of a family 15 carbohydrate-binding module in complex with xylopentaose. Evidence that xylan binds in an approximate 3-fold helical conformation. J. Biol. Chem. 276, 49061–49065 [DOI] [PubMed] [Google Scholar]
- 16. Sainz-Polo M. A., Valenzuela S. V., González B., Pastor F. I., Sanz-Aparicio J. (2014) Structural analysis of glucuronoxylan-specific Xyn30D and its attached CBM35 domain gives insights into the role of modularity in specificity. J. Biol. Chem. 289, 31088–31101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fontes C. M., Hazlewood G. P., Morag E., Hall J., Hirst B. H., Gilbert H. J. (1995) Evidence for a general role for non-catalytic thermostabilizing domains in xylanases from thermophilic bacteria. Biochem. J. 307, 151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meissner K., Wassenberg D., Liebl W. (2000) The thermostabilizing domain of the modular xylanase XynA of Thermotoga maritima represents a novel type of binding domain with affinity for soluble xylan and mixed-linkage β-1,3/β-1, 4-glucan. Mol. Microbiol. 36, 898–912 [DOI] [PubMed] [Google Scholar]
- 19. Sunna A., Gibbs M. D., Bergquist P. L. (2000) The thermostabilizing domain, XynA, of Caldibacillus cellulovorans xylanase is a xylan binding domain. Biochem. J. 346, 583–586 [PMC free article] [PubMed] [Google Scholar]
- 20. Charnock S. J., Bolam D. N., Turkenburg J. P., Gilbert H. J., Ferreira L. M., Davies G. J., Fontes C. M. (2000) The X6 “thermostabilizing” domains of xylanases are carbohydrate-binding modules: structure and biochemistry of the Clostridium thermocellum X6b domain. Biochemistry 39, 5013–5021 [DOI] [PubMed] [Google Scholar]
- 21. Xie H., Gilbert H. J., Charnock S. J., Davies G. J., Williamson M. P., Simpson P. J., Raghothama S., Fontes C. M., Dias F. M., Ferreira L. M., Bolam D. N. (2001) Clostridium thermocellum Xyn10B carbohydrate-binding module 22-2: the role of conserved amino acids in ligand binding. Biochemistry 40, 9167–9176 [DOI] [PubMed] [Google Scholar]
- 22. Sánchez M. M., Fritze D., Blanco A., Spröer C., Tindall B. J., Schumann P., Kroppenstedt R. M., Diaz P., Pastor F. I. (2005) Paenibacillus barcinonensis sp. nov., a xylanase-producing bacterium isolated from a rice field in the Ebro River delta. Int. J. Syst. Evol. Microbiol. 55, 935–939 [DOI] [PubMed] [Google Scholar]
- 23. Gallardo O., Pastor F. I., Polaina J., Diaz P., L̸ysek R., Vogel P., Isorna P., González B., Sanz-Aparicio J. (2010) Structural insights into the specificity of Xyn10B from Paenibacillus barcinonensis and its improved stability by forced protein evolution. J. Biol. Chem. 285, 2721–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Valenzuela S. V., Diaz P., Pastor F. I. (2014) Xyn11E from Paenibacillus barcinonensis BP-23: a LppX-chaperone-dependent xylanase with potential for upgrading paper pulps. Appl. Microbiol. Biotechnol. 98, 5949–5957 [DOI] [PubMed] [Google Scholar]
- 25. Blanco A., Díaz P., Zueco J., Parascandola P., Javier Pastor F. I. (1999) A multidomain xylanase from a Bacillus sp. with a region homologous to thermostabilizing domains of thermophilic enzymes. Microbiology 145, 2163–2170 [DOI] [PubMed] [Google Scholar]
- 26. Hogg D., Pell G., Dupree P., Goubet F., Martín-Orúe S. M., Armand S., Gilbert H. J. (2003) The modular architecture of Cellvibrio japonicus mannanases in glycoside hydrolase families 5 and 26 points to differences in their role in mannan degradation. Biochem. J. 371, 1027–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Correia M. A., Abbott D. W., Gloster T. M., Fernandes V. O., Prates J. A., Montanier C., Dumon C., Williamson M. P., Tunnicliffe R. B., Liu Z., Flint J. E., Davies G. J., Henrissat B., Coutinho P. M., Fontes C. M., Gilbert H. J. (2010) Signature active site architectures illuminate the molecular basis for ligand specificity in family 35 carbohydrate binding module. Biochemistry 49, 6193–6205 [DOI] [PubMed] [Google Scholar]
- 28. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 29. Valenzuela S. V., Díaz P., Javier Pastor F. I. (2010) Recombinant expression of an alkali stable GH10 xylanase from Paenibacillus barcinonensis. J. Agric. Food Chem. 58, 4814–4818 [DOI] [PubMed] [Google Scholar]
- 30. Monterroso B., Sáiz J. L., García P., García J. L., Menéndez M. (2008) Insights into the structure-function relationships of pneumococcal cell wall lysozymes, LytC and Cpl-1. J. Biol. Chem. 283, 28618–28628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sainz-Polo M. Á., González B., Pastor F. I., Sanz-Aparicio J. (2015) Crystallization and preliminary x-ray diffraction analysis of the N-terminal domain of Paenibacillus barcinonensis xylanase 10C containing the CBM22-1–CBM22-2 tandem. Acta Crystallogr. F Struct. Biol. Commun. 71, 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Najmudin S., Pinheiro B. A., Prates J. A., Gilbert H. J., Romão M. J., Fontes C. M. (2010) Putting an N-terminal end to the Clostridium thermocellum xylanase Xyn10B story: crystal structure of the CBM22-1-GH10 modules complexed with xylohexaose. J. Struct. Biol. 172, 353–362 [DOI] [PubMed] [Google Scholar]
- 37. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 38. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 39. Vagin A. A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 30, 1022–1025 [Google Scholar]
- 40. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 [Google Scholar]
- 41. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.3, DeLano Scientific, San Carlos, CA [Google Scholar]
- 43. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 44. Atkins E. D. T. (1992) Xylan and Xylanases: Progress in Biotechnology, Vol. 7, pp. 39–50, Elsevier Science Publishers, Amsterdam [Google Scholar]
- 45. Andrews S. R., Taylor E. J., Pell G., Vincent F., Ducros V. M., Davies G. J., Lakey J. H., Gilbert H. J. (2004) The use of forced protein evolution to investigate and improve stability of family 10 xylanases. The production of Ca2+-independent stable xylanases. J. Biol. Chem. 279, 54369–54379 [DOI] [PubMed] [Google Scholar]
- 46. Pell G., Taylor E. J., Gloster T. M., Turkenburg J. P., Fontes C. M., Ferreira L. M., Nagy T., Clark S. J., Davies G. J., Gilbert H. J. (2004) The mechanisms by which family 10 glycoside hydrolases bind decorated substrates. J. Biol. Chem. 279, 9597–9605 [DOI] [PubMed] [Google Scholar]
- 47. Vardakou M., Flint J., Christakopoulos P., Lewis R. J., Gilbert H. J., Murray J. W. (2005) A family 10 Thermoascus aurantiacus xylanase utilizes arabinose decorations of xylan as significant substrate specificity determinants. J. Mol. Biol. 352, 1060–1067 [DOI] [PubMed] [Google Scholar]
- 48. Pell G., Szabo L., Charnock S. J., Xie H., Gloster T. M., Davies G. J., Gilbert H. J. (2004) Structural and biochemical analysis of Cellvibrio japonicus xylanase 10C: how variation in substrate-binding cleft influences the catalytic profile of family GH-10 xylanases. J. Biol. Chem. 279, 11777–11788 [DOI] [PubMed] [Google Scholar]
- 49. Abbott D. W., Erin-López J. M., Boraston A. B. (2008) Insight into ligand diversity and novel biological roles for family 32 carbohydrate-binding modules. Mol. Biol. Evol. 25, 155–167 [DOI] [PubMed] [Google Scholar]
- 50. McCartney L., Blake A. W., Flint J., Bolam D. N., Boraston A. B., Gilbert H. J., Knox J. P. (2006) Differential recognition of plant cell walls by microbial xylan-specific carbohydrate-binding modules. Proc. Natl. Acad. Sci. U.S.A. 103, 4765–4770 [DOI] [PMC free article] [PubMed] [Google Scholar]