

Background: Anserine is an abundant dipeptide in vertebrate skeletal muscles.
Results: We identified UPF0586 protein C9orf41 homolog as a carnosine N-methyltransferase, responsible for anserine formation in rat muscle.
Conclusion: Besides being a carnosine N-methyltransferase, UPF0586 protein is likely to be a novel peptide or protein methyltransferase in eukaryotes.
Significance: This molecular identification will help to elucidate physiological functions of UPF0586 protein in eukaryotes.
Keywords: enzyme kinetics, enzyme purification, eukaryote, peptides, skeletal muscle metabolism, UPF0586 protein C9orf41 homolog, anserine, carnosine, carnosine N-methyltransferase
Abstract
Anserine (β-alanyl-N(Pi)-methyl-l-histidine), a methylated derivative of carnosine (β-alanyl-l-histidine), is an abundant constituent of vertebrate skeletal muscles. Although it has been suggested to serve as a proton buffer and radical scavenger, its physiological function remains mysterious. The formation of anserine is catalyzed by carnosine N-methyltransferase, recently identified in chicken as histamine N-methyltransferase-like (HNMT-like) protein. Although the HNMT-like gene is absent in mammalian genomes, the activity of carnosine N-methyltransferase was reported in most mammalian species. In the present investigation, we purified carnosine N-methyltransferase from rat muscles about 2600-fold. Three polypeptides of ∼45, 50, and 70 kDa coeluting with the enzyme activity were identified in the preparation. Mass spectrometry analysis of these polypeptides resulted in the identification of UPF0586 protein C9orf41 homolog as the only meaningful candidate. Rat UPF0586 and its yeast, chicken, and human orthologs were expressed in COS-7 cells and purified to homogeneity. Although all recombinant proteins catalyzed the formation of anserine, as confirmed by chromatographic and mass spectrometry analysis, rat UPF0586 was more active on carnosine than other orthologs. Confocal microscopy of HeLa cells expressing recombinant UPF5086 proteins revealed their presence in both cytosol and nucleus. Carnosine and Gly-His were the best substrates for all UPF0586 orthologs studied, although the enzymes also methylated other l-histidine-containing di- and tripeptides. Finally, cotransfection of COS-7 cells with rat or human UPF0586 and carnosine synthase transformed the cells into efficient anserine producers. We conclude that UPF0586 is mammalian carnosine N-methyltransferase and hypothesize that it may also serve as a peptide or protein methyltransferase in eukaryotes.
Introduction
Carnosine (β-alanyl-l-histidine) and its methylated derivative, anserine (β-alanyl-N-π-methyl-l-histidine), are dipeptides commonly found in excitable animal tissues, whereas they have never been detected in plants, fungi, or other eukaryotes (for a review, see Ref. 1). In contrast to carnosine, which is present at high concentrations (in the range of 0.6–30 mm) in both skeletal muscles and the central nervous system of almost all vertebrates, including human beings (2, 3), the storage of anserine is more selective. Anserine is abundant in skeletal muscles of most vertebrates (1), and only traces of this dipeptide have been occasionally detected in brain (4, 5). Anserine was reported as a major l-histidine-containing dipeptide in avian tissues (up to 43 mm in chicken pectoral muscle) (4), whereas much lower concentrations of this compound have been measured in muscles of mammals, such as rats (2–9 mm), cats (8 mm), and rabbits (17 mm) (1, 6). Intriguingly, no endogenous anserine has ever been detected in human muscles (1, 3).
A great effort has been made to understand the physiological role of both carnosine and anserine. Originally, these two dipeptides were postulated to serve as pH buffers neutralizing lactic acid produced in working muscle due to their abundance and pKa value that is close to the physiological pH (4). This hypothesis, however, provides no explanation for the synthesis of anserine, which shows buffer capacity similar to that of carnosine. Recently, histidine-containing dipeptides have been postulated to exert a more complex effect on cell and tissue metabolism via their antiglycemic (7), antiglycation (8), and antioxidant properties (9). Unfortunately, no definitive explanation for their physiological importance has been provided so far.
Knowledge of the enzymes catalyzing the formation of histidine-containing dipeptides is rather limited, and only recently have carnosine synthase and chicken carnosine N-methyltransferase been molecularly identified as ATPGD1 (ATP-grasp domain-containing protein 1) and histamine N-methyltransferase-like (HNMT-like)2 protein, respectively (10, 11). Unexpectedly, HNMT-like gene encoding anserine-producing methyltransferase in birds and reptiles was not detected in available mammalian genomes (11), although the activity of carnosine N-methyltransferase has been consistently reported in most mammalian species (1). This observation led us to hypothesize that (i) the HNMT-like gene has been lost in mammals, and (ii) mammalian carnosine N-methyltransferase must be a different protein from the “HNMT-like” one (11).
In the current investigation, we report the identification of mammalian carnosine N-methyltransferase as a protein of unknown function designated UPF0586 protein C9orf41 homolog. We have characterized this enzyme and proved that its overexpression in COS-7 cells results in their transformation into efficient anserine producers. We also provide data suggesting that UPF0596 protein may serve as a novel peptide and/or protein methyltransferase in eukaryotes.
Experimental Procedures
Materials
Reagents, of analytical grade whenever possible, were from Sigma-Aldrich (Poznań, Poland), Roche Diagnostics (Warsaw, Poland), or Merck (Darmstadt, Germany). S-[methyl-3H]Adenosyl-l-methionine and Ultima Gold LSC mixture were purchased from PerkinElmer Life Sciences. Di- and tripeptides were from Sigma-Aldrich or Caslo Laboratory (Lingby, Denmark) with the exception of GABA-l-His, which was a generous gift of Dr. Maria Veiga-da-Cunha (de Duve Institute, Brussels, Belgium) and was prepared as described previously (12). DEAE-Sepharose, Q-Sepharose, Superdex 200, HiScreen Blue FF, HisTrap HP (Ni2+ form), and PD-10 columns were obtained from GE Healthcare (Uppsala, Sweden). Dowex 50W-X4 (200-mesh) resin was from Sigma-Aldrich, and Vivaspin-500 and -20 centrifugal concentrators were from Sartorius Stedim (Kostrzyn, Poland). All other enzymes and DNA-modifying enzymes as well as the TurboFect transfection reagent were obtained from Thermo-Fermentas.
Assay of Carnosine N-Methyltransferase Activity
Carnosine N-methytransferase activity was determined by measuring the incorporation of [3H]methyl group from S-[methyl-3H]adenosyl-l-methionine ([3H]SAM) into carnosine. The standard incubation mixture (0.11 ml) contained 50 mm Hepes, pH 7.5, 10 mm KCl, 1 mm EGTA, 1 mm MgCl2, 1 mm DTT, 20 mm carnosine (or other acceptors), and 1 μm [1H + 3H]SAM (from about 400 × 103 to 800 × 103 cpm). When the effect of pH on recombinant enzyme activity was investigated, Hepes, pH 7.5, was replaced by MES, pH 6.0; Hepes, pH 7.0; or Tris, pH 8.0, 8.5, or 9.0. Blanks containing no carnosine were included in all assays. The reaction was started by the addition of enzyme preparation and carried out at 37 °C for 20 min unless otherwise described. Anserine production was linear for at least 30 min under all conditions studied. The incubation was stopped by the addition of 0.1 ml of the reaction medium to 0.2 ml of ice-cold 10% (w/v) HClO4. The samples were diluted with 0.12 ml of H2O and centrifuged at 13,000 × g for 10 min. After neutralization of the supernatant with 3 m K2CO3, the salts were removed by centrifugation (13,000 × g for 10 min); the clear supernatant was diluted 5 times with 20 mm Hepes, pH 7.5; and 2 ml were applied to Dowex 50W-X4 columns (1 ml, Na+ form), equilibrated with 20 mm Hepes, pH 7.5. The columns were washed with 5 × 2 ml of 20 mm Hepes, pH 7.5, to remove minor radioactive contaminants of the radioreagent. Anserine and methylated forms of l-histidine were eluted with 5 × 2 ml of 20 mm Hepes, pH 7.5, containing 0.5 m NaCl. To elute a non-consumed [1H + 3H]SAM, the columns were washed with 4 × 2 ml of 1 m NH4OH. In the experiments where Gly-Gly-His, Gly-His-Gly, Ala-His or His-Ala, and His-Gly were used as substrates, 20 mm Hepes, pH 7.5, in the equilibration and elution buffers for Dowex 50W-X4 columns was replaced by 20 mm Hepes, pH 7.0, or 20 mm MES, pH 5.5, respectively, to obtain retention of the methylated peptides on the cationite. In all cases, the samples to be counted were mixed with 6 volumes of scintillation fluid (Ultima Gold), and the incorporated radioactivity was analyzed with a Beckman LS6000 IC liquid scintillation counter.
Purification of Rat Carnosine N-Methyltransferase
Rat leg muscles (130 g) from four male Wistar rats, aged 4 months, were homogenized with 4 volumes (w/v) of buffer A (50 mm Hepes, pH 7.6, 10 mm KCl, 1 mm DTT, 1 mm EGTA, 1 mm MgCl2, 5 μg/ml leupeptin, and 5 μg/ml antipain) with a Waring blender 7011HS. The homogenate was centrifuged for 40 min at 20,000 × g at 4 °C. The resulting supernatant (400 ml) was filtered through three layers of gauze to remove fat particles and was applied to a DEAE-Sepharose column (300-ml bed volume) equilibrated with buffer A. The column was washed with 850 ml of buffer A and developed with a NaCl gradient (0–0.5 m in 715 ml) in buffer A, and fractions (6.5 ml) were collected. The most active fractions of the DEAE column (39 ml) were diluted to 220 ml with buffer B (50 mm Tris-HCl, pH 8.0, 10 mm KCl, 1 mm DTT, 1 mm EGTA, 1 mm MgCl2, 5 μg/ml leupeptin, and 5 μg/ml antipain) and applied to a Q-Sepharose column (50-ml bed volume) equilibrated with buffer B. The column was washed with 135 ml of buffer B containing 25 mm NaCl, and the retained protein was eluted with a NaCl gradient (25–500 mm in 350 ml in buffer B), and fractions (5 ml) were collected. The most active fractions (15 ml) were pooled, concentrated to 2.6 ml in Vivaspin-20 ultrafiltration devices, and loaded on a Superdex 200 16/60 column (120-ml bed volume) equilibrated with buffer A containing 100 mm NaCl. One-ml fractions were collected. To obtain more purified enzyme preparation for a tandem mass spectrometry, fractions 44–53 (10 ml) of the Superdex 200 purification step were pooled again, concentrated to 1 ml in Vivaspin-20 ultrafiltration devices, and loaded on a HiScreen blue Sepharose column (4.7-ml bed volume) equilibrated with buffer A. The column was washed with 25 ml of buffer A and developed with a NaCl gradient (0–1 m in 50 ml) in buffer A, and fractions (1 ml) were collected. All purification steps were performed at 4 °C, and the enzymatic preparation was stored at −70 °C between steps. Protein concentration was determined spectrophotometrically according to Bradford (13) using bovine γ-globulin as a standard. Protein content in the most active blue Sepharose fractions were determined following their 10-fold concentration in Vivaspin-500 ultrafiltration devices.
Identification of Rat Carnosine N-Methyltransferase by Tandem Mass Spectrometry
Because SDS-PAGE analysis of the peak activity fractions from the HiScreen blue Sepharose revealed a low protein content, resulting in very faint protein bands, fractions were 10-fold concentrated in a Vivaspin-500 ultrafiltration device and reanalyzed by SDS-PAGE. The bands co-eluting with carnosine N-methyltransferase activity in the blue Sepharose purification step were cut from a 10% gel and digested with trypsin. In-gel digestions of the peptides were performed as described previously (14). Peptides were analyzed by nano-UPLC-tandem mass spectrometry employing Acquity nano-UPLC coupled with a Synapt G2 HDMS Q-TOF mass spectrometer (Waters) fitted with a nanospray source and working in MS∧E mode under default parameters. Briefly, products of in-gel protein digestion were loaded onto a Waters Symmetry C18 trapping column (20 mm × 180 μm) coupled to the Waters BEH130 C18 UPLC column (250 mm × 75 μm). The peptides were eluted from these columns in a 1–85% gradient of acetonitrile in water (both containing 0.1% formic acid) at a flow rate of 0.3 μl/min. The peptides were directly eluted into the mass spectrometer. Data were acquired and analyzed using MassLynx version 4.1 software (Waters) and ProteinLynx Global Server version 2.4 software (Waters) with a false discovery rate of ≤4%, respectively. To identify rat carnosine N-methyltransferase, the complete rat (Rattus norvegicus) reference proteome was downloaded from UniProt, randomized, and used as a data bank of the MS/MS software.
Phylogenetic Analysis
Sequences homologous to rat UPF0586 protein C9orf41 homolog (GenBankTM accession number NP_001020145.1) were identified by Protein BLAST searches. A phylogenetic analysis was performed on the Phylogeny.fr platform (15). Amino acid sequences were aligned with MUSCLE (version 3.7) (16). After alignment, ambiguous regions were removed with Gblocks (version 0.91b) (17). Phylogenetic trees were generated using phylogenetic estimation using maximum likelihood (18) with the WAG model for amino acid substitution (19). The final tree was customized with the editing interface TreeDyn (20). A confidence level was assessed using the approximate likelihood ratio test (minimum of Shimodaira-Hasegawa-like procedure and χ2-based parametric) (21).
Overexpression and Purification of Recombinant UPF0586 Proteins
Yeast (Saccharomyces cerevisiae), chicken, and rat total RNA was prepared from 250 mg of cell pellet, 200 mg of pectoral muscle, and 200 mg of leg muscle, respectively, with the use of TriPure reagent according to the manufacturer's instructions. cDNA was synthesized using Moloney murine leukemia virus-reverse transcriptase (Thermo-Fermentas), with oligo(dT)18 primer and 2.5 μg of total RNA according to the manufacturer's instructions.
The open reading frames encoding yeast (GenBankTM accession number NM_001182930.1), chicken (XM_003643032.1), and rat UPF0586 protein (NM_001024974.1) were PCR-amplified using Pfu DNA polymerase in the presence of 1 m betaine, whereas the ORF coding for the human protein (BC034033.1) was amplified from cDNA clone HsCD00296470 (DNASU Plasmid Repository). UPF05086s were amplified using 5′ primers containing the initiator codon preceded by the Kozak consensus sequence (22) and an EcoRI site and 3′ primers in which the original stop codon was replaced by an amino acid coding codon flanked by an XbaI site (for primer sequences, see Table 1). The amplified DNA products of the expected size were digested with the appropriate restriction enzymes; cloned into the pEF6/Myc-His A expression vector (Invitrogen), which allows the production of proteins with a C-terminal His6 tag; and verified by DNA sequencing. For transfections, COS-7 cells (Cell Lines Service, Eppelheim, Germany) or HEK-293T cells (a kind gift of Dr. Maria Veiga-da-Cunha) were plated in 100-mm Petri dishes at a cell density of 1.7 × 106 or 2.1 × 106 cells/plate, respectively, in Dulbecco's minimal essential medium supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% (v/v) fetal bovine serum and grown in a humidified incubator under a 95% air and 5% CO2 atmosphere at 37 °C. After 24 h, each plate was transfected with 6 μg of either unmodified pEF6/Myc-His A vector or the same vector encoding HNMT-like protein using the TurboFect transfection reagent according to the protocol provided by the manufacturer. After 48 h, the culture medium was removed, and the cells were washed with 5 ml of phosphate-buffered saline and harvested in 1 ml of 50 mm Hepes, pH 7.5, containing 10 mm KCl, 1 mm MgCl2, 1 mm EGTA, 5 μg/ml leupeptin, and 5 μg/ml antipain. The cells were lysed by freezing in liquid nitrogen, and after thawing and vortexing, the extracts were centrifuged at 4 °C (20,000 × g for 30 min) to remove insoluble material.
TABLE 1.
Sequences of primers used in PCR and quantitative RT-PCR experiments
The nucleotides corresponding to the coding sequences are in capital letters, the Kozak consensus sequence is shown in boldface type, and the restriction sites added are underlined.
| Primer | Sequence | Restriction site | Plasmid | Protein expressed |
|---|---|---|---|---|
| Preparation of UPF0586 protein expression vectors | ||||
| 1 | gtggaattcgccaccATGCAGCGACGACGGCGC | EcoRI | pEF6/Myc-His A | C-terminal His6-tagged rat UPF0586 protein C9orf41 homolog |
| 2a | cagtctagaATATTGTGGCTTCCGGACCAC | XbaI | ||
| 3 | gtggaattcgccaccATGCAGCGACGGCGTCGC | EcoRI | pEF6/Myc-His A | C-terminal His6-tagged human UPF0586 protein C9orf41 |
| 4a | cagtctagaCCATTGTGGCTTACGGACCAC | XbaI | ||
| 5 | gtggaattcgccaccATGTCGGCCGCGTGCGCG | EcoRI | pEF6/Myc-His A | C-terminal His6-tagged chicken UPF0586 protein C9orf41 homolog |
| 6a | cagtctagaCCATTCAAAGCACTTCTCTTCTG | XbaI | ||
| 7 | gtggaattcgccaccATGGACGAGAATGAATTTGATAATC | EcoRI | pEF6/Myc-His A | C-terminal His6-tagged yeast UPF0586 protein C9orf41 homolog |
| 8a | cagtctagaATATGATTCATTGGTGGGGTTCG | XbaI | ||
| Subcellular localization of rat and human UPF0586 protein | ||||
| 9 | CACgaattccacgATGCAGCGACGGCGTCGC | EcoRI | pEGFP-N1 | C-terminal EGFP-tagged human UPF0586 protein C9orf41 |
| 10a | CATgtcgacGATTGTGGCTTACGGACCAC | SalI | ||
| 11 | CACgaattccacgATGCAGCGACGACGGCGC | EcoRI | pEGFP-N1 | C-terminal EGFP-tagged rat UPF0586 protein C9orf41 |
| 12a | CATgtcgacGATTGTGGCTTCCGGACCAC | SalI | ||
| Quantitative real-time PCR | ||||
| Rat UPF0586 | TAACGCGTTCCGCTACTACG | |||
| TCCGGATCTTGTCCAAGTGA | ||||
| Rat GAPDH | ACCACCAACTGCTTAGCCCC | |||
| CACAGCCTTGGCAGCACC | ||||
| Human UPF0586 | GGGAAAGCAGAAAGGGATGC | |||
| GCCAGTCTTCCTAGTCCAGC | ||||
| Human GAPDH | TTCCAGGAGCGAGATCCCT | |||
| CACCCATGACGAACATGGG | ||||
a Original stop codons were replaced by either ATA and CCA or CGA (shown in italic type and underlined) to allow C-terminal His tag or C-terminal EGFP translation, respectively.
In the experiments where COS-7 cells were co-transfected with two different plasmids, 1.7 × 106 seeded cells were transfected with 3 μg of each plasmid to reach a total of 6 μg of DNA. Plasmid encoding carnosine synthase (CARNS1) was a kind gift of Dr. Maria Veiga-da-Cunha and was prepared as described (10). Twenty-four hours after transfection, the culture medium was changed to a fresh one (10 ml) containing 0.1 mm β-alanine, and the cells were left for another 24 h before removing the medium, washing with PBS, and collecting from each plate in 0.5 ml of 50 mm Hepes, pH 7.6, 10 mm KCl, 1 mm DTT, 1 mm EGTA, 1 mm MgCl2, 5 μg/ml leupeptin, and 5 μg/ml antipain. The cells were lysed by freezing three times in liquid nitrogen, and insoluble material was removed by centrifugation (13,000 × g for 15 min). Part (0.2 ml) of soluble extract was deproteinized by the addition of 0.6 ml of acetonitrile, followed by centrifugation (13,000 × g for 15 min) and filtration (Whatman 0.2 μm PVDF filter), and used to determine carnosine and anserine content by HPLC-hydrophilic interaction chromatography (HILIC) (see “Product Analysis”). Another part (0.3 ml) was used to measure protein concentration and for expression analysis of the recombinant proteins by SDS-PAGE and Western blotting (11).
For the purification of recombinant UPF0586 proteins, the supernatant of COS-7 lysate (7–10 ml) was diluted 4-fold with buffer A (50 mm Hepes, pH 7.5, 300 mm NaCl, 10 mm KCl, 20 mm imidazole, 1 mm MgCl2, 5 μg/ml leupeptin and 5 μg/ml antipain) and applied on a HisTrap HP column (1 ml) equilibrated with the same buffer. The column was washed with 6 ml of buffer A, and the retained protein was eluted with a stepwise gradient of imidazole (7 ml of 30 mm, 7 ml of 60 mm, and 8 ml of 300 mm) in buffer A. The recombinant proteins were eluted with 300 mm imidazole in homogeneous form as confirmed by SDS-PAGE (Figs. 1 and 2). The enzyme preparations were desalted on PD-10 columns equilibrated with 50 mm Hepes, pH 7.5, 10 mm KCl, 1 mm DTT, 1 mm EGTA, and 1 mm MgCl2. Protein content in purified recombinant enzyme preparations was quantitated by densitometric analysis using Quantity One (Bio-Rad). The yield of recombinant proteins ranged between 0.07 mg (yeast) and 0.2–0.3 mg (vertebrates) of homogeneous protein per 20 mg of soluble COS-7 cell protein. The purified enzymes were supplemented with 1 mg/ml BSA and stored at −70 °C. When appropriate, the C-terminal His6-tagged recombinant proteins were detected by Western blot analysis as described previously (11).
FIGURE 1.

SDS-PAGE (A) and Western blot (B) analysis of fractions obtained from purification of recombinant rat UPF0586 protein. Rat UPF0586 protein was purified to homogeneity by affinity chromatography on nickel-Sepharose (HisTrap HP) as described under “Experimental Procedures.” For the SDS-PAGE analysis, 7.5 μl of sample from each fraction was loaded onto a 10% gel and electrophoresed, and the resulting gel was then stained with silver (11). For the Western blot analysis, 7.5 μl of each fraction was loaded onto a 10% gel, electrophoresed, and blotted to nitrocellulose membrane, which was then sequentially probed with a mouse primary antibody against His6 tag and a horseradish peroxidase-conjugated goat anti-mouse antibody. Secondary antibody was detected through autoradiography using chemiluminescence. M, prestained protein marker; L, cell-free lysate of COS-7 cells overexpressing the recombinant enzyme; AL, 4-fold diluted lysate applied on the column; FT, flow-through; W, wash. Fractions 30–300 were eluted with the indicated concentrations of imidazole.
FIGURE 2.
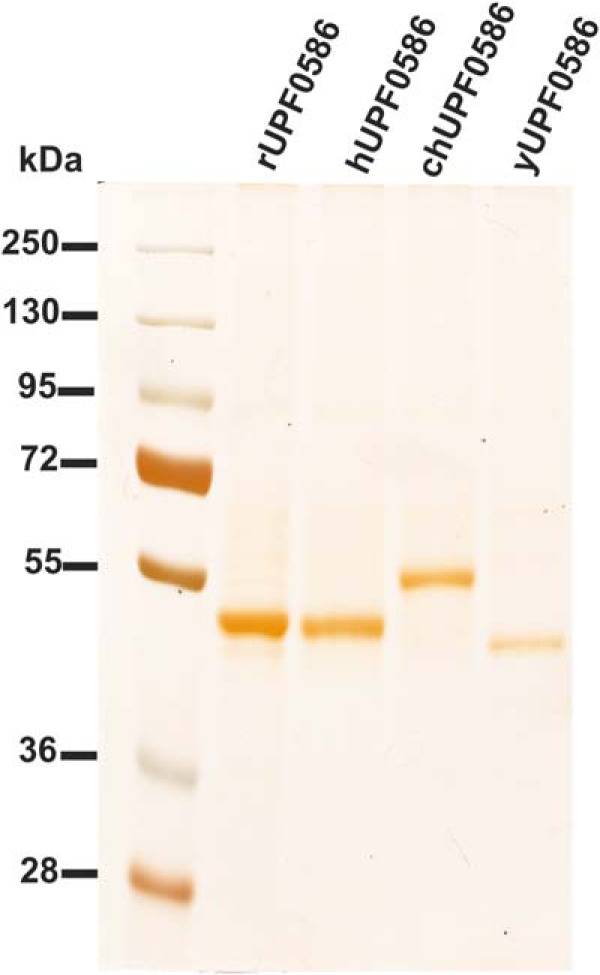
SDS-PAGE analysis of purified recombinant UPF0586 proteins. Rat (rUPF0586), human (hUPF0586), chicken (chUPF0586), and yeast (yUPF0586) proteins were purified to homogeneity by affinity chromatography on nickel-Sepharose (HisTrap HP), as described under “Experimental Procedures.” For the SDS-PAGE analysis, 10 μl of sample derived from each fraction was loaded onto a 10% gel and electrophoresed, and the resulting gel was then stained with silver (11).
Product Analysis
To obtain a sufficient amount of the methylated dipeptide formed in the reaction catalyzed by recombinant UPF0586 proteins for mass spectrometry analysis, the reaction mixture was scaled up. Briefly, 1 μg of yeast or 4 μg of vertebrate UPF0586 proteins were incubated for 12 h at 30 °C in 0.15 ml of a reaction mixture containing 25 mm Hepes, pH 7.5, 80–100 μg of BSA derived from the enzyme preparation, 10 mm KCl, 1 mm EGTA, 1 mm MgCl2, 1 mm DTT, 4 mm carnosine in the absence or presence of 0.5 mm SAM. The reaction was stopped by the addition of 0.45 ml of acetonitrile. Precipitated protein was removed by centrifugation (13,000 × g for 10 min), and the clear supernatants were analyzed by HPLC-HILIC according to a slightly modified method of Mora et al. (23). Briefly, carnosine and anserine were separated by the gradient mode on Ascentis Express HILIC (2.1 × 100 mm, 2.7 μm) using Acquity UPLC (Waters). Mobile phases consisted of solvent A, containing 1 mm ammonium acetate, pH 5.5, in water/acetonitrile (25:75), and solvent B, containing 4.9 mm ammonium acetate, pH 5.5, in water/acetonitrile (70:30). The separation was performed in a linear gradient from 0 to 100% of solvent B for 3.5 min at a flow rate of 0.3 ml/min, followed by the column wash in 100% of solvent B for 2.6 min and equilibration for 10 min under the initial conditions. The column eluate was monitored by a UV detector at λ = 214 nm, followed by a mass spectrometer. All mass spectral analysis were performed on a Synapt G2 HDMS Q-TOF mass spectrometer fitted with an electrospray source (Waters). The detector worked in positive MS/MS mode. The electrospray ionization-MS source was set at a temperature of 100 °C, capillary voltage of 3.5 kV, and cone voltage of 40 V. The flow rate of the nebulizer gas (nitrogen) was 700 liters/h. To confirm the structure of anserine precursor ion, collision-induced dissociation experiments were run by selecting the target ion (m/z 241). The trap collision energy was 20 eV. Quantification was achieved using external standards of carnosine and anserine.
Subcellular Localization of Recombinant UPF0586 Proteins in Transfected HeLa Cells
Expression plasmids for rat and human UPF0586s fused to a C-terminal EGFP tag were constructed by PCR amplification of the appropriate open reading frame, using Pfu DNA polymerase, the primers shown in Table 1, and either pEF6/rat UPF5086 or pEF6/human UPF5086 construct as a template. The amplified fragments were digested using EcoRI and SalI restriction enzymes, cloned into the pEGFP-N1 vector (Clontech), and verified by sequencing.
Subcellular localization of fused proteins were performed as described (24). Briefly, HeLa cells (European Collection of Cell Cultures) were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37 °C in a humidified atmosphere containing 5% CO2. For transfection with the FuGENE HD reagent (Roche, Mannheim, Germany), 1.5 × 105 cells were seeded on a 16-mm glass coverslip in a 35-mm plate, and the manufacturer's recommended protocol for transfection with plasmid DNA in the presence of serum was followed. Forty-eight hours after transfection, cells were washed with PBS and analyzed by confocal microscopy. For DNA staining, Hoechst 33342 had been added at the concentration of 1 μg/ml for 20 min just before cells were washed and analyzed.
Images of cells were acquired sequentially on a Zeiss LSM700 confocal laser scanning microscope equipped with a Plan-Apochromat ×63/1.40 oil objective. For detecting green fluorescence of EGFP-tagged UPF0586 proteins, a 488-nm excitation line was used, and for detecting blue fluorescence of Hoechst 33342, a 405-nm excitation line was employed.
Quantitative Real-time PCR Assays
Human kidneys from deceased donors were obtained from Eurotransplant. These kidneys were unsuitable for transplantation due to technical reasons. Human brain tissue was obtained at autopsy, and normal muscle tissue was obtained from the non-affected part of tumor resection samples. All tissues were coded and handled anonymously in accordance with the Dutch National Ethics Guidelines (Code for Proper Secondary Use of Human Tissue, Dutch Federation of Medical Scientific Societies).
Total RNA was isolated from fresh rat and frozen human kidney cortex, leg muscle, and brain with TRIzol according to the manufacturer's instructions. RNA was converted to cDNA by using avian myeloblastosis virus reverse transcriptase (Roche) with random hexamer priming. Gene-specific primers (Table 1) were designed to generate PCR products from UPF0586 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). A SYBR Green quantitative polymerase chain reaction (Bio-Rad) was performed to quantify the relative UPF0586 mRNA levels. The expression of enzyme was normalized to the expression of GAPDH using the 2ΔCt method (25).
Calculations
Vmax, Km, and kcat for the methyltransferase activity of studied enzymes were calculated with Prism version 4.0 (GraphPad Software) using a nonlinear regression.
Results
Purification and Identification of Rat Carnosine N-Methyltransferase
Carnosine N-methyltransferase activity was assayed by measuring the incorporation of [3H]methyl group of [3H]SAM into carnosine. The enzyme was purified ∼2600-fold from rat leg muscle by a procedure involving chromatography on DEAE-Sepharose, Q-Sepharose, Superdex 200, and HiScreen blue Sepharose (Table 2). The methyltransferase was eluted as a single peak in each of the purification steps (Fig. 3), indicating the presence of a single enzyme species. The gel filtration step on Superdex 200 disclosed that the molecular mass of native enzyme in the peak fraction (F49) was equal to about 95,000 (not shown). The overall yield of the purification was only about 1.5% (see Table 2) due to the fact that only fractions exhibiting at least 70% activity of the most active one were used for the next step procedure. The only exceptions to this rule were Superdex 200 fractions loaded on the blue Sepharose column containing almost all activity recovered from the gel filtration step (see Fig. 3).
TABLE 2.
Purification of carnosine N-methyltransferase from rat skeletal muscles
| Fraction | Volume | Total protein | Total activity | Specific activity | Purification | Yield |
|---|---|---|---|---|---|---|
| ml | mg | pmol min−1 | pmol min−1 mg−1 | -fold | % | |
| 20,000 × g supernatant | 400 | 12,466 | 1536 | 0.123 | 1 | 100 |
| DEAE-Sepharose | 39 | 207 | 1296 | 6.26 | 51 | 84.4 |
| Q Sepharose | 15 | 60 | 614 | 10.23 | 83 | 40.0 |
| Superdex 200 | 10 | 22 | 415 | 18.86 | 153 | 27.0 |
| HiScreen Bluea | 2 | 0.07 | 22.6 | 322.86 | 2625 | 1.5 |
a The data represent mean values for the two most purified fractions (fractions 73 and 74).
FIGURE 3.
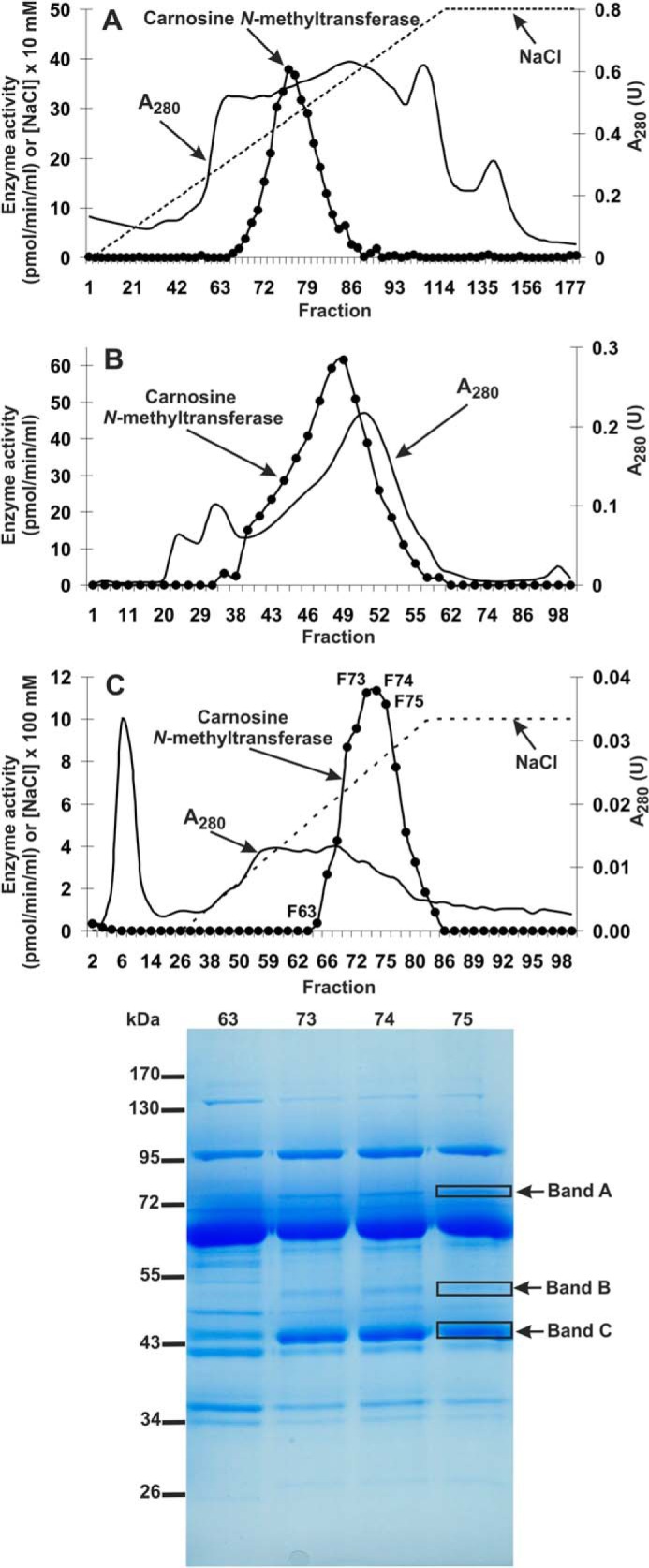
Purification of rat carnosine N-methyltransferase. The enzyme was purified by chromatography on DEAE-Sepharose (A), Q-Sepharose (not shown), Superdex 200 (B), and Cibacron Blue 3G Sepharose (C) as described under “Experimental Procedures.” Fractions were tested for carnosine N-methyltransferase activity. Protein concentration was determined in the most active fractions with the Bradford assay. The indicated fractions eluted from the Cibacron Blue 3G Sepharose column were concentrated 10-fold by ultrafiltration (Vivaspin 500) and analyzed by SDS-PAGE (0.8–1.2 μg of protein). The indicated bands were cut out of the gel, submitted to trypsin digestion, and analyzed by tandem mass spectrometry.
SDS-PAGE analysis of the peak activity fractions (F66–F80) derived from the HiScreen blue Sepharose revealed a low protein content, resulting in very faint protein bands, which prevented from the identification of protein bands coeluting with the enzyme activity (not shown). Thus, both peak activity fractions F73–F75 and fraction F63, displaying a residual enzyme activity, were concentrated about 10-fold and reanalyzed by SDS-PAGE. Carnosine N-methyltransferase activity was coeluted with three protein bands of about 45, 50, and 70 kDa present in fractions F73–F75 but not in the fraction F63 withdrawn from the last purification step (see Fig. 3). The bands were cut out from the gel and digested with trypsin, and the resulting peptides were analyzed by MS/MS and compared with the Uniprot reference proteome of rat. The analysis indicated that only bands B and C contained methyltransferase Smyd1 and a protein of unknown function designated UPF0586 protein C9orf41 homolog (Table 3), the methyltransferase identity of which could be identified only by its similarity to bacterial methyltransferases (not shown). To exclude the possibility of missing any potential methyltransferase in protein bands that did not coelute with the enzyme activity, we also performed MS/MS analysis of all bands visualized by SDS-PAGE. No methyltransferase was identified in the remaining protein bands (not shown).
TABLE 3.
Proteins identified in the gel bands submitted to trypsin digestion and MS/MS analysis
Identified proteins are listed for each band according to their score calculated by ProteinLynx Global Server software (PLGS). For each protein, its molecular weight (Mr) and the sequence coverage are also indicated. Occasional peptide hits corresponding to keratins have not been included.
| Gel band | Protein name | UniProt accession number | PLGS scorea | Mr | Coverage |
|---|---|---|---|---|---|
| % | |||||
| A | Glycine-tRNA ligase | G3V7G8 | 5061 | 81,740 | 50 |
| Serum albumin | P02770 | 2293 | 68,686 | 33 | |
| Anionic trypsin 1 | P00762 | 1070 | 25,942 | 8 | |
| Calpain 1 catalytic subunit | P97571 | 532 | 82,067 | 32 | |
| B | Smyd1 | D4A7U4, D4A3D2b | 2786 | 54,948 | 55 |
| Myosin light chain kinase 2 | G3V731 | 1507 | 65,907 | 29 | |
| F 5-hydroxytryptamine receptor 2B | F1LWG8 | 1497 | 95,672 | 29 | |
| Anionic trypsin 1 | P00762 | 1074 | 25,942 | 8 | |
| Aspartyl aminopeptidase | Q4V8H5 | 896 | 52,522 | 46 | |
| C | UPF0586 protein C9orf41 homolog | Q5BJZ6 | 7356 | 46,355 | 58.5 |
| Anionic trypsin 1 | P00762 | 575 | 25,942 | 8 |
a PLGS score is calculated by the Protein Lynx Global Server (version 2.4) software using a Monte Carlo algorithm to analyze all acquired mass spectral data and is a statistical measure of accuracy of assignation. A higher score implies a greater confidence of protein identity.
b Current primary accession number.
Although Smyd1 protein is a histone-lysine N-methyltransferase (26) and it was very unlikely that it would catalyze methylation of l-histidine residue in carnosine, we cloned and overexpressed recombinant rat Smyd1 (GenBankTM accession number NM_001106595.1) in COS-7 cells. As expected, Smyd1 did not catalyze methylation of carnosine (not shown). Thus, UPF0586 protein was the only meaningful candidate for rat carnosine N-methyltransferase (see Table 3). UPF0586 was a single protein identified in band C, and 11 matching peptides (underlined in Fig. 4) were found to cover about 59% of its sequence. Expression profiles of mRNA available in the EMBL Expression Atlas database (27) revealed a predominant expression of UPF0586 in leg muscles compared with other tissues of both rats and mice, as expected for carnosine N-methyltransferase. Taken together, these data strongly suggest that the rat UPF0586 protein homologous to human C9orf41 is carnosine N-methyltransferase.
FIGURE 4.

Amino acid sequence alignment of rat UPF0586 protein with its yeast, chicken, and human orthologues. Sequences were obtained with following GenBankTM accession numbers: yeast (S. cerevisiae, NP_014307.1), chicken (G. gallus, XP_003643080.1), rat (R. norvegicus, NP_001020145.1), and human (H. sapiens, NP_689633.1). All shown sequences have been confirmed by PCR amplification of the cDNA and sequencing. The percentage amino acid identities with rat UPF0586 protein are given at the top right. The highly conserved domain of unknown function (N-2227) is labeled above the alignment, whereas amino acids residues interacting with SAM are indicated by asterisks, as predicted by I-TASSER (29). The peptides identified by mass spectrometry in the protein purified from rat leg muscle are underlined in the rat sequence. The level of residue conservation is indicated as 100% (black background), 70% and more (dark gray background), and 50% and more (light gray background).
Analysis of UPF0586 Protein Sequences
Protein BLAST searches (28) with rat UPF0586 and phylogenetic analysis of resulting sequences indicated that orthologs of this protein were found in all eukaryotes, including vertebrates (∼90% identity with the rat sequence), insects (∼50% identity), and plants and fungi (both ∼30% identity) (Figs. 4 and 5). All taxa followed the expected lines of descent, indicating that the enzyme was already present in a common eukaryotic ancestor (see Fig. 5). Because a low similarity in amino acid sequence between eukaryotic UPF0586 orthologs and some bacterial proteins was also detected (∼20% identity, Chitinophaga pinensis, GenBankTM accession number YP_003120969.1), it is likely that the eukaryotic enzyme was acquired from an ancestral prokaryote.
FIGURE 5.

Phylogenetic tree of UPF0586 proteins. Protein sequences were aligned using Muscle (16), whereas the phylogentic tree was inferred with the use of phylogenetic estimation using maximum likelihood (18). Brunch support values assessed using the approximate likelihood ratio test are indicated (21). The protein sequences that were used for the analysis and corresponding GenBankTM accession numbers are as follows: A. carolinensis (XP_003216582.1); Arabidopsis thaliana (NP_180775.2); Bombus terrestris (XP_003400717.1); Bos taurus (XP_580351.6); Ceratitis capitata (XP_004530443.1); Columba livia (XP_005499843.1); Crassostrea gigas (EKC21396.1); Cucumis sativus (XP_004149525.1); Danio rerio (XP_005171825.1); Drosophila melanogaster (NP_726779.1); G. gallus (XP_003643080.1); H. sapiens (NP_689633.1); Kluyveromyces marxianus (BAP73908.1); Maylandia zebra (XP_004544272.1); Melopsittacus undulatus (XP_005155020.1); Oreochromis niloticus (XP_005473258.1); Oryza sativa (EEE64325.1); Oryzias latipes (XP_004072743.1); Pichia kudriavzevii (KGK39127.1); Python bivittatus (XP_007433028.1); R. norvegicus (NP_001020145.1); S. cerevisiae (NP_014307.1); Sus scrofa (XP_003122012.3); Taeniopygia guttata (XP_002190574.1); Xenopus tropicalis (NP_001119994.1); Zea mays (NP_001167849.1).
All identified proteins contain the N-2227 domain of unknown function at their C terminus, whereas an N-terminal sequence of 80–180 amino acids could plausibly form an additional domain (see Fig. 4). Although the amino acid sequence identity between orthologs from distinct species, such as rat and yeast, was rather low (about 30%), the alignment of their sequences showed the presence of motifs that were strictly or at least moderately conserved in both putative N-terminal and N-2227 domains (see Fig. 4). Protein structures of UPF0586 orthologs have not been reported so far. However, we performed their prediction employing the I-TASSER server (29), which indicated that UPF0586 proteins were methyltransferases, and at least seven conserved amino acid residues within the N-2227 domain were involved in the binding of S-adenosyl-l-methionine (see Fig. 4). No function was identified for the N-terminal part of UPF0586 proteins.
Characterization of Recombinant UPF0586 Proteins
To confirm the molecular identity of rat carnosine N-methyltransferase with that of rat UPF0586 protein and to compare the enzymatic activities among orthologs of UPF0586 protein from evolutionarily distinct species, rat, human, chicken, and yeast (S. cerevisiae) UPF0586 proteins were expressed in both COS-7 and HEK-293T cells as fusion proteins with the C-terminal polyhistidine tag (Fig. 6). The recombinant enzymes catalyzed synthesis of anserine, as determined by the radiochemical assay (see Fig. 6). Large amounts of recombinant enzymes for further studies were produced in COS-7 and purified to homogeneity (see Figs. 1 and 2).
FIGURE 6.

Time course of anserine production in lysates of control as well as UPF0586 protein-overexpressing COS-7 and HEK-293T cells. COS-7 and HEK-293T cells were transfected for 48 h with an empty vector (Control) or a vector encoding yeast, chicken, rat, or human UPF0586 protein (yUPF0586, chUPF0586, rUPF0586, or hUPF0586, respectively). Cell-free lysates (40 μg of protein) were incubated at 37 °C for the indicated times in the presence of 1 μm [1H + 3H]SAM (100 pmol, ∼400 × 103 cpm) and 20 mm carnosine. The formation of radiolabeled anserine was determined after its chromatographic separation from [3H]SAM. The presence of recombinant protein in tested lysates was verified by Western blot analysis, which was performed using 40 μg of total protein and an antibody against the His6 tag. The figure shows results of representative experiments.
The identity of the methylated product formed from carnosine by homogeneous recombinant rat UPF0586 protein was verified by HPLC-HILIC MS/MS. As shown in Fig. 7, chromatographic analysis of the generated product revealed its co-migration with a commercial standard of anserine. The addition of anserine to the reaction mixture resulted in a selective increase in the peak area of the product (from 11,627 to 57,122 units) without a noticeable disturbance in its peak symmetry, disclosing the product's identity as anserine (see Fig. 7). No anserine signal was detected in the control reaction with no SAM in the reaction mixture. Analysis of the product by electrospray mass spectrometry indicated the presence of a protonated molecular ion with m/z 241, as expected for anserine (not shown). As shown in Fig. 8, the tandem mass spectrometry analysis of this ion revealed a fragmentation pattern in agreement with the anserine structure, which was indeed identical with that of commercial anserine. Recorded fragmentation spectrum of anserine was also in a perfect agreement with that available for this dipeptide (MassBank Record PR100392). Similar results of HPLC-HILIC MS/MS analysis were obtained for all recombinant UPF0586 orthologs tested (not shown).
FIGURE 7.

HPLC-HILIC analysis of the product formed by rat UPF0586 protein. Shown are chromatograms of a standard mixture of carnosine and anserine (2.5 nmol) (A) and of deproteinized reaction mixtures obtained during incubation of homogeneous recombinant rat UPF0586 protein (4 μg) for 12 h with 4 mm carnosine in the absence (B) or the presence of 0.5 mm SAM (C) as well as following the supplementation of the former deproteinized reaction mixture with 1.88 nmol of anserine standard (D). The identities of all indicated compounds were confirmed by mass spectrometry. The sample processing and chromatographic conditions are described under “Experimental Procedures.”
FIGURE 8.

Tandem mass spectra of anserine and the product formed by rat UPF0586 protein. Homogeneous recombinant rat enzyme was incubated for 12 h with 4 mm carnosine in the absence or presence of 0.5 mm SAM. The produced methylated dipeptide was separated from substrates and analyzed by mass spectrometry. Both mass spectra, covering the mass range m/z 50–600, and tandem mass spectra (Q-TOF, precursor ion m/z 241) of commercial anserine (A) and the product generated by rat UPF0586 enzyme (B) were acquired.
Activities of the four homogeneous recombinant enzymes determined at various temperature and pH values with carnosine as substrate are presented in Fig. 9. They were maximal at pH between 7.0 and 7.5 (i.e. a typical value estimated for cytosolic enzymes) (30). Whereas the yeast enzyme showed an optimal activity at 30 °C, which was in fact consistent with the optimal temperature for yeast growth, mammalian proteins were most active at 50 °C, with ∼1.3- (rat) and 2-fold (human) higher activities than at the physiological temperature of 37 °C. The chicken enzyme exhibited an optimal activity at 40 °C (i.e. within the range of physiological temperature of the chicken body), and it was constant up to 50 °C (see Fig. 9). The ability of vertebrate enzymes to be catalytically active at relatively high temperatures suggests that structures of these proteins were subjected to significant changes when compared with yeast enzyme. More interestingly, all of these enzymes were still able to produce anserine. In the subsequent experiments, carnosine N-methyltransferase activity was assayed in the presence of Hepes buffer, pH 7.5, at 30 °C (yeast enzyme) or 40 °C (vertebrate enzymes).
FIGURE 9.

Effect of temperature and pH on UPF0586 protein activity. Temperature (A) and pH dependence (B) of enzymes were determined with the use of homogeneous recombinant yeast, chicken, rat, or human UPF0586 protein (yUPF0586, chUPF0586, rUPF0586, or hUPF0586, respectively). Enzyme preparations (0.10–0.35 μg of protein) were incubated for 20 min in the presence of 1 μm [1H + 3H]SAM (100 pmol, 400–600 × 103 cpm) and 20 mm carnosine, as described under “Experimental Procedures.” Values are the means ± range (error bars) of two separate experiments. When no error bar is shown, the error is smaller than the width of the line.
Substrate specificities of the four homogeneous recombinant enzymes are shown in Table 4. Several compounds structurally related to carnosine were tested as possible methyl group acceptors. A similar profile of specificity was found among all enzymes tested with carnosine (β-Ala-His) as the best substrate, followed by Gly-Gly-His, Gly-His, and homocarnosine (GABA-His). The highest activity of yeast enzyme on carnosine, when compared with the vertebrate enzymes, was due to its lowest Km value for SAM (Table 5). A substitution of non-standard amino acid by α-Ala resulted in a ∼5–10-fold decrease in the activity of all enzymes, whereas tripeptide Gly-Gly-His was a good methyl group acceptor. All peptides used as substrates by the enzymes contained C-terminal histidine, whereas methylation of free l-His was negligible. These findings suggest that C-terminal histidine of other peptides or proteins might be methylated, provided a correct amino acid sequence is present at their C terminus.
TABLE 4.
[3H]Methylation of both l-histidine and various l-histidine-containing peptides catalyzed by UPF0586 proteins of various species
The reaction of methyl group transfer from SAM into the indicated methyl group acceptors was determined with the use of homogeneous recombinant rat, human, chicken, or yeast UPF0586 proteins. Enzyme preparations (0.20–0.72 μg of protein) were incubated for 10 min at 30 °C (yeast) or 40 °C (vertebrates) in the presence of 1 μm [1H + 3H]SAM (100 pmol, ∼400 × 103 cpm) and a 10 mm concentration of the indicated methyl group acceptor. Values are the means ± S.E. of 3–9 separate experiments. ND, not determined; activity below the detection limit.
| Methyl group acceptor | UPF0586 protein |
|||
|---|---|---|---|---|
| Rat | Human | Chicken | Yeast | |
| pmol min−1 mg−1 | pmol min−1 mg−1 | pmol min−1 mg−1 | pmol min−1 mg−1 | |
| β-Ala-His (carnosine) | 1934 ± 58 | 1124 ± 33 | 1236 ± 36 | 2268 ± 73 |
| GABA-His (homocarnosine) | 1209 ± 44 | 567 ± 9 | 562 ± 47 | 1110 ± 90 |
| α-Ala-His | 404 ± 44 | 122 ± 7 | 134 ± 4 | 252 ± 110 |
| l-Histidine | 75 ± 17 | 172 ± 96 | 16 ± 10 | 99 ± 68 |
| Gly-His | 1802 ± 198 | 1047 ± 38 | 1080 ± 65 | 1696 ± 135 |
| Gly-Gly-His | 1993 ± 5 | 1044 ± 13 | 1102 ± 10 | 1449 ± 100 |
| Gly-His-Gly | 73 ± 21 | 9 ± 5 | 22 ± 10 | 155 ± 89 |
| His-Gly | 28 ± 4 | ND | 5 ± 5 | 144 ± 72 |
| His-α-Ala | 6 ± 6 | 11 ± 11 | 4 ± 4 | 33 ± 31 |
| β-Ala-N-π-methyl-His (anserine) | ND | 48 ± 27 | 9 ± 9 | 74 ± 58 |
TABLE 5.
Kinetic properties of rat, human, chicken, and yeast UPF0586 proteins
Kinetic properties were determined with the use of purified recombinant C-terminal His6-tagged UPF0586 proteins. Determinations for SAM were performed with enzyme preparations (0.20–0.62 μg protein) that were incubated for 10 min at either 30 °C (yeast) or 40 °C (vertebrates) in the reaction mixture containing 40 mm carnosine and variable concentrations of [1H + 3H] SAM (∼800 × 103 cpm), whereas the measurements for carnosine were done in the presence of non-saturating 1 μm concentration of [1H + 3H] SAM (100 pmol, ∼600 × 103 cpm). Values are the means of three separate experiments. The S.E. value is also given.
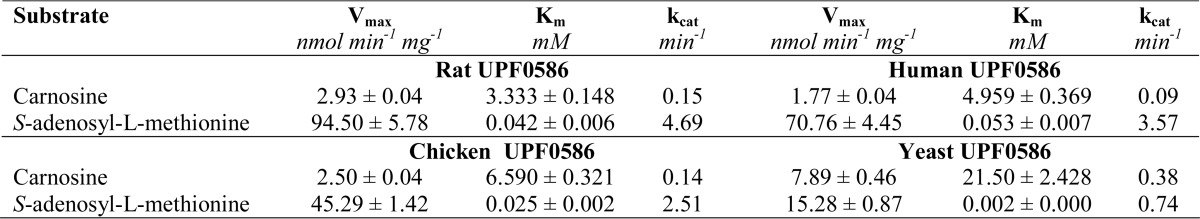
The kinetic properties of homogeneous recombinant UPF0586 proteins were studied in the presence of carnosine, which was the best substrate for all enzymes tested (see Table 4). Rat enzyme exhibited the highest affinity for carnosine, and all vertebrate enzymes displayed about 3–6-fold higher affinity for the dipeptide than yeast one (see Table 5), presenting Km values much lower than the physiological concentration of carnosine in vertebrate skeletal muscles (1). Similar differences between vertebrate and yeast enzymes were observed for Vmax values estimated at saturating concentrations of SAM, indicating that vertebrate enzymes are clearly better at methylating carnosine than the yeast enzyme. This was also true when the reaction rate was determined in the presence of intracellular SAM concentration (∼30 μm in both yeast and vertebrate cells). Yeast enzyme operated at V0 ≈ Vmax (i.e. 15 nmol × min−1 × mg−1 protein) due to its very high affinity for SAM (Km = 2 μm), whereas rat, human, and chicken enzymes exhibited 12–25-fold higher Km for SAM than did the yeast enzyme and catalyzed the reaction at V0 equal to about 38, 26, and 25 nmol × min−1 × mg−1 protein, respectively (data not shown).
Analysis of SAM reagent purity revealed the presence of 1% S-adenosyl-l-homocysteine (SAH). SAH contamination did not affect Km values obtained for the investigated recombinant enzymes, as judged by V0 values determined in the presence of rabbit SAH hydrolase and adenosine deaminase from Streptococcus thermophilus (data not shown).
Tissue Distribution of mRNA for Rat and Human UPF0586 Proteins
EMBL-EBI Expression Atlas searches with rat and human UPF0586 revealed that rat enzyme showed about 7-fold higher expression of its mRNA in skeletal muscle in comparison with all other tissues, whereas mRNA of human enzyme was present at a low level in all tissues with the exception of kidney and liver, where it doubled the basal level. A highly enriched expression of the mRNA for human enzyme was also present in kidney but not in liver, as shown by the Genevestigator database (31). To verify microarray results, we determined the level of UPF0586 mRNAs in both rat and human brain, leg muscle, and kidney cortex by quantitative real-time PCR using GAPDH transcript as a reference. As shown in Fig. 10, the expression of UPF0586 mRNA was different in human tissues from that in rat tissues. As for the human enzyme, the highest level of expression was observed in kidney. Brain and skeletal muscle showed about 25 and 9% of the level found in kidney, respectively. The rat enzyme was mainly expressed in skeletal muscle, followed by brain and kidney (about 38 and 3% of the muscle level, respectively). It is, however, worth noticing that the relative levels of UPF0586 mRNA observed in skeletal muscle may be underestimated due to a high expression of glycolytic GAPDH in muscle.
FIGURE 10.

Relative mRNA levels for rat and human UPF0586 protein as determined in brain, kidney, and muscle tissues by quantitative real time PCR. Cycle threshold (Ct) values were determined for rat and human UPF0586 and GAPDH. The expression of a target UPF0586 gene was normalized to the expression of the related GAPDH using the 2ΔCt method. Values are either the means ± S.E. (error bars) of three to four independent samples (rat tissues and human kidney) or the means of two independent samples ± range (error bars) (human brain and muscle).
Although our results are in a fairly good agreement with reported data showing that (i) activity of rat carnosine N-methyltransferase was associated with skeletal muscle (32) and (ii) no anserine was detected in human muscle tissue (3), the unexpectedly high expression of UPF0586 mRNA in human kidney suggests that in some species (e.g. human beings), the enzyme may be active in cells or tissues that have not been considered as a source of anserine so far.
Subcellular Localization of Recombinant UPF0586 Proteins
Analysis with WolF PSORT (33) and CELLO (34) predicted cytoplasmic and/or nuclear localization of vertebrate UPF0586 orthologs, whereas yeast enzyme was suggested to interact with the cytoskeleton or the plasma membrane of fungal cells. To verify subcellular localization of mammalian enzymes, confocal microscopy was used to analyze HeLa cells transfected with C-terminal EGFP-tagged rat or human UPF0586 proteins. The direct fluorescence microscopic observations indicated that these two mammalian UPF0586 proteins were present in both the cytoplasm and nucleus of HeLa cells, exhibiting a very similar diffuse pattern that excluded their localization in the nucleoli (Fig. 11). It was previously shown that both cytosolic MEK1 (mitogen-activated protein kinase 1) and nuclear ER (enhancer of rudimentary) protein fused to a C-terminal EGFP correctly localized in HeLa cells, confirming the validity of the employed reporting system (35). Both nuclear and cytoplasmic localization of human UPF0586 was also reported in normal human tissues (36), confirming our results.
FIGURE 11.
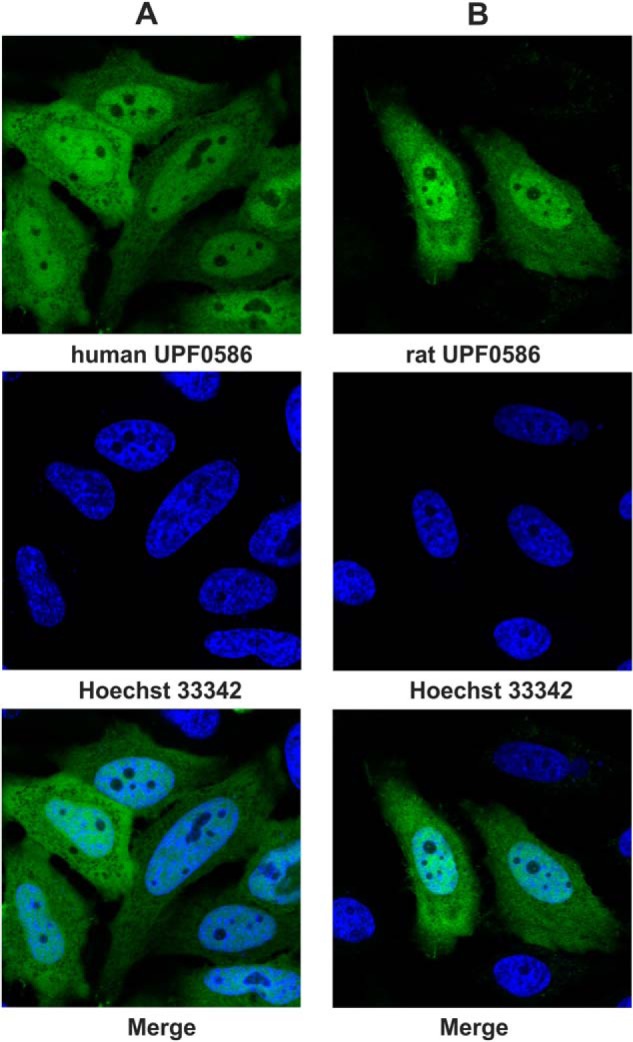
Subcellular localization of human and rat UPF0586 protein in HeLa cells visualized by confocal microscopy. HeLa cells were transiently transfected with plasmids encoding human UPF0586-EGFP (A) or rat UPF0586-EGFP (B). Localization of chimeric proteins was determined by the direct fluorescence of EGFP in living cells, while nuclei were visualized by staining with Hoechst 33342. Representative images obtained with a confocal laser-scanning microscope are shown.
Overexpression of Carnosine Synthase and UPF0586 Proteins in COS-7 Cells
To investigate anserine-producing activity of rat and human UPF0586 proteins in more physiological conditions, we transfected COS-7 cells with plasmids driving the expression of human carnosine synthase (12) with or without either rat or human UPF0586 protein. After 24 h of transfection, cell culture media were exchanged to fresh media that were supplemented with 0.1 mm β-alanine, and the cells were further incubated for 24 h. Next, acetonitrile extracts of cells were prepared, and the concentrations of both carnosine and anserine were determined in deproteinized samples by HPLC-HILIC.
As shown in Fig. 12, COS-7 cells transfected with human carnosine synthase synthesized carnosine, and the intracellular concentration of the dipeptide was about 11.8 ± 1.8 mm (n = 6) based on a water/protein ratio for COS-7 cells equal to 6.8 μl/mg, as determined by three independent measurements. The co-expression of carnosine synthase with rat or human UPF0586 protein triggered anserine production in the COS-7 cells that led to its accumulation up to about 2.2 ± 0.3 and 3.7 ± 0.4 mm (for rat and human UPF0586s, respectively, n = 6) accompanied by a decrease in the intracellular content of carnosine (by ∼30%), probably due to its conversion into anserine. Intriguingly, anserine concentration in COS-7 cells expressing human UPF0586 protein was 40% lower than in cells transfected with rat enzyme, which seems not to fully reflect a clearly higher difference in the expression of these two enzymes. This finding suggests that factors other than the level of UPF0586 expression may affect the production of anserine (e.g. a limited availability of SAM).
FIGURE 12.

Production of carnosine and anserine in COS-7 cells overexpressing carnosine synthase and carnosine N-methyltransferase. COS-7 cells were transfected for 48 h with an empty vector (Control) or vectors encoding various rat (r) or human (h) proteins. A, Western blot analysis of COS-7 cell extracts, showing the overexpression of recombinant human carnosine synthase (hCARNS1) and recombinant rat or human UPF0586 protein. The analysis was performed using 40 μg of total protein with an antibody against the His6 tag. B, effect of cotransfection of human carnosine synthase (hCARNS1) with either mouse or human UPF0586 protein on the accumulation of carnosine and anserine in the cells. The cell culture medium was supplemented with 0.1 mm β-alanine 24 h after transfection and incubated for another 24 h as described under “Experimental Procedures.” Values are the means ± S.E. (error bars) of 4–6 independent transfections.
Most importantly, both the concentrations of the dipeptides and anserine/carnosine ratio determined in the transfected COS-7 are in fairly good agreement with the corresponding data reported for rat leg muscle (6), indicating that UPF0586 proteins act as carnosine N-methyltransferase in mammalian cells, at least if present in sufficient amounts.
Discussion
Molecular Identity and Biochemical Properties of Rat Carnosine N-Methyltransferase
Recently, we have reported the identification of carnosine N-methyltransferase as chicken HNMT-like protein. To our surprise, the HNMT-like gene is absent in available mammalian genomes (11), although the activity of carnosine N-methyltransferase has been shown in most mammalian species (1). These observations led us to hypothesize that mammals possessed an anserine-forming enzyme completely different from the HNMT-like one (11). In the present study, we report the molecular identification of rat carnosine N-methyltransferase as UPF0586 protein C9orf41 homolog, disclosing the identity of the mammalian enzyme. This conclusion results from the following findings: (i) rat leg muscle is a rich source of mammalian carnosine N-methyltransferase (32) and UPF0586 protein is the only logical candidate for the enzyme that was identified in the most highly purified preparation from the rat tissue; (ii) the recombinant rat UPF0586 protein catalyzes the transfer of the methyl group from SAM onto carnosine, yielding anserine; (iii) the identity of the product made by the recombinant enzyme was confirmed by both hydrophilic interaction chromatography and tandem mass spectrometry; and (iv) the overexpression of carnosine synthase together with UPF0586 protein in COS-7 cells transformed them into efficient anserine producers.
The fact that COS-7 cells overexpressing carnosine synthase and rat UPF0586 protein efficaciously produce anserine is an argument supporting the identification of rat carnosine N-methyltransferase. However, the production of anserine by transfected COS-7 cells was observed under conditions of strong overexpression of UPF0586 protein, raising the question on the importance of those data. Taking into account that the half-lives of both carnosine and anserine are ∼3 weeks in the rat (37), there is no need for a rapid process, and much lower expression of the enzymes may be sufficient to reach and maintain physiological concentration of the dipeptides.
To the best of our knowledge, there are no published studies on the substrate specificity and kinetic properties of rat carnosine N-methyltransferase. In our hands, the recombinant rat UPF0586 acted best on carnosine and homocarnosine, whereas α-alanine-containing dipeptide was a poor substrate, confirming identification of the enzyme as carnosine N-methyltransferase. More interestingly, the enzyme also showed a high activity toward carnosine analogues containing glycine or glycine-glycine instead of β-alanine, suggesting that there might be other endogenous substrate(s) for it.
Considering that the intracellular concentrations of SAM and carnosine in rat leg muscle are about 30 μm and 9 mm, respectively (1, 38), the rat UPF0586 showed rather low affinity for SAM (Km = 42 μm) and a high affinity for carnosine (Km = 3.3 mm). These observations indicate that the enzyme works at a rate of about 40% of the Vmax in rat muscle, and the SAM concentration rather than carnosine availability affects its activity in vivo.
It is intriguing that both rat and human enzymes exhibit optimal activities at an unexpectedly high temperature (50 °C). This phenomenon may simply be a consequence of their protein structures without a clear physiological relevance, as shown for other enzymes (e.g. plant aspartate aminotransferases (39) or human l-isoaspartyl methyltransferase (40)). On the other hand, the high optimum temperature for the enzymes may be beneficial from a physiological standpoint. As the temperature of working muscle rises during even short term moderate exercise (41), a higher activity of carnosine N-methyltransferase could result in an increase in the rate of anserine formation, which in turn might raise a buffering capacity of working muscle.
Other Roles of UPF0586 Protein
Besides the identification of rat carnosine N-methyltransferase, the major finding reported in this work is demonstration that UPF0586 orthologs from evolutionarily distinct species (i.e. yeast, chicken, and human) are all anserine-producing methyltransferases. To the best of our knowledge, this is the first experimental evidence that N-2227 domain-containing enzymes do indeed catalyze the SAM-dependent transfer of methyl group, and the N-2227 domain is most likely involved in SAM binding. Furthermore, UPF0586 is a widespread protein in eukaryotes, and its orthologs are also found in organisms that are unable to produce carnosine due to the lack of carnosine synthase (plants, fungi including yeast, and several fishes). This indicates that the enzyme may have other function related to its ability to N-methylate histidine residues in peptides. This notion is further supported by our findings that (i) all tested UPF0586 orthologs are very sluggish enzymes, with a kcat of 1–5 min−1, resembling that estimated for protein methyltransferases rather than that measured for the enzymes catalyzing methylation of low molecular weight metabolites (38, 42), and (ii) anserine-producing UPF0586 protein is present in chicken as well as other avian and reptilian species that express, mainly in their muscles, HNMT-like protein, carnosine N-methyltransferase being about ∼22-fold more efficacious than UPF0586 protein (kcat/Km = 2224 and 100 min−1 mm−1, respectively) (11). Finally, a gene encoding a protein similar to UPF0586 is also present in genomes of some prokaryotic species (e.g. Haliangium ochraceum, GenBankTM accession number YP_003265816.1), where it is located together with a non-ribosomal peptide synthase in a single gene cluster, plausibly an operon. Thus, this prokaryotic methyltransferase may catalyze methylation of bacterial non-ribosomal peptides. We therefore hypothesize that UPF0586 protein may also serve as a peptide or protein histidine-methylating enzyme in eukaryotes. In fact, several eukaryotic proteins have been shown to contain methylhistidine residues, although yeast YIL110W enzyme responsible for the modification of Rpl3 protein has been the only identified protein histidine methyltransferase to date (43). Based on our results, a cytosolic and/or nuclear protein might be a plausible substrate that would be methylated at its C-terminal histidine. The presence of Gly or Gly-Gly upstream of histidine would facilitate its methylation, whereas α-Ala could be expected to inhibit the reaction. No such modification of protein C terminus has been reported so far, although there are several proteins in vertebrates that could be potential substrates (e.g. caspase-1 (GenBankTM accession number P43527) or inositol monophosphatase 3 (GenBankTM accession number D4AD37)). A plausible function of such post-translational modification might be to protect proteins from attack by carboxypeptidases. Interestingly, methylation of the N terminus has also been observed for a variety of proteins in both prokaryotes and eukaryotes (44), and protein N-terminal methyltransferases have been recently identified in yeast and humans (45). However, a clear physiological role of such protein modification remains unknown.
Evolution of Rat Carnosine N-Methyltransferase
Our present hypothesis that the function of UPF0596 in eukaryotes may be different from carnosine methylation and related to its ability to methylate peptides and/or proteins raises the question of how the enzyme gained carnosine N-methyltransferase activity in rats and other mammals. Because the enzyme is slow, to be an effective anserine producer, it would have to be expressed at a high level in the major carnosine-containing tissue (i.e. muscle). This condition is indeed fulfilled in rodents, as indicated by tissue expression profiles of the UPF0586 mRNA in rats and mice but not in chicken that in fact exhibits other form of carnosine N-methyltransferase (HNMT-like). Our findings therefore suggest that the acquisition of carnosine N-methyltransferase activity by UPF0586 protein may simply result from both its promiscuous activity (i.e. broad substrate specificity) and a large increase in the level of its expression in muscle tissue. In fact, this conclusion is in perfect agreement with both experimental data and theoretical analyses showing that the acquisition of a new enzyme activity frequently results from the utilization of its promiscuous activity and an up-regulation of its expression, which appears to be a key step in the divergence of a new function (46) (for a review, see Ref. 47).
Human UPF0586 Protein
Human beings are believed to be the only mammalian species that has no methylated carnosine in its tissues (1). This rare exception could be easily explained by a finding that the gene encoding mammalian carnosine N-methyltransferase is absent or is mutated in Homo sapiens. Alternatively, the absence of anserine in humans may be more apparent than real. In fact, our findings suggest that humans possess carnosine N-methyltransferase activity, and the enzyme may be considerably more active in kidney cortex than in skeletal muscle, leading to the accumulation of anserine in renal tissue. Because virtually all searches for anserine in humans were limited to the skeletal muscle (1, 3), the presence of anserine in kidney as well as in other tissues, which have not been considered as a source of anserine so far, might be easily overlooked. This hypothesis is supported by findings that storage of both carnosine and anserine are clearly detectable in the kidney cortex of human and mouse renal tissue by immunohistochemistry and by HPLC analysis (48),3 although the physiological role of these dipeptides in the kidney is unclear.
Recently, more than dozen patients have been reported with novel chromosome microdeletions at 9q21.13, presenting mental retardation, speech delay, and epilepsy (49, 50). The smallest region of deletion contained six genes, including that encoding human UPF0586 (also known as C9orf41). Although the authors suggested the RORB gene as a candidate for a neurological phenotype (49), the effect of UPF0586 gene loss remains undetermined. Intriguingly, almost half of the patients have suffered from urinary tract disorders (enuresis, pyelonephritis, and vesico-ureteral reflux) of unknown origin (49, 50), suggesting that some of the deleted genes may be important for renal physiology. We therefore hypothesize that these renal disorders may be related to the loss of UPF0586 protein, and it would be of interest to determine more systematically the renal functions in patients with deletion of the UPF0586 gene, which could be helpful to identify the physiological role of the enzyme.
Conclusions
In the current investigation, we have identified rat carnosine N-methyltransferase, which catalyzes the last step in the anserine biosynthesis pathway. The UPF0586 protein is a novel form of vertebrate carnosine N-methyltransferase that is unrelated to the reptilian and avian enzyme (HNMT-like) and seems to be typical of mammalian species. Because anserine-producing orthologs of UPF0586 are found in organisms that are unable to produce carnosine, we hypothesize that the enzyme might also catalyze methylation of histidine residues in some eukaryotic peptides or proteins.
Author Contributions
J. D. conceived and designed the study, wrote the paper, and partially performed and analyzed the experiments shown in Tables 1–5 and Figs. 1–9 and 12. M. P. provided technical assistance and partially performed and analyzed experiments shown in Tables 4 and 5 and Figs. 6, 9, and 12. O. P. provided technical assistance and partially performed and analyzed experiments shown in Tables 1 and 2 and Figs. 1 and 3. L. C. provided technical assistance and partially performed and analyzed experiments shown in Tables 1 and 2 and Figs. 2 and 3. P. K. designed, performed, and analyzed the experiments shown in Fig. 11. H. J. B. and E. H. designed, performed, and analyzed the experiments shown in Fig. 10. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Professor J. Bryla (University of Warsaw) for indispensable assistance during the preparation of the manuscript. We are also very grateful to Dr. Adam K. Jagielski (University of Warsaw) for the maintenance and service of nano-UPLC-MS/MS as well as to colleagues from the Department of Molecular Biology (University of Warsaw) for making the FPLC apparatus available for protein purification. Zeiss Poland is also acknowledged for providing LSM700.
This work was supported by Opus-6 Grant DEC-2013/11/B/NZ1/00078 from the Polish National Science Center. It was carried out with the use of the CePT infrastructure financed by the European Union European Regional Development Fund within the Operational Program “Innovative Economy” for 2007–2013. The authors declare that they have no conflicts of interest with the contents of this article.
C. Klessens, H. J. Baelde, K. Veraar, and E. de Heer, unpublished results.
- HNMT-like
- histamine N-methyltransferase-like
- HILIC
- hydrophilic interaction liquid chromatography
- SAH
- S-adenosyl-l-homocysteine
- SAM
- S-adenosyl-l-methionine.
References
- 1. Boldyrev A. A., Aldini G., Derave W. (2013) Physiology and pathophysiology of carnosine. Physiol. Rev. 93, 1803–1845 [DOI] [PubMed] [Google Scholar]
- 2. Crush K. G. (1970) Carnosine and related substances in animal tissues. Comp. Biochem. Physiol. 34, 3–30 [DOI] [PubMed] [Google Scholar]
- 3. Mannion A. F., Jakeman P. M., Dunnett M., Harris R. C., Willan P. L. (1992) Carnosine and anserine concentrations in the quadriceps femoris muscle of healthy humans. Eur. J. Appl. Physiol. Occup. Physiol. 64, 47–50 [DOI] [PubMed] [Google Scholar]
- 4. Boldyrev A. A. (2007) Carnosine and Oxidative Stress in Cells and Tissues, pp. 37–38, Nova Scientific Publisher, New York [Google Scholar]
- 5. Tomonaga S., Hayakawa T., Yamane H., Maemura H., Sato M., Takahata Y., Morimatsu F., Furuse M. (2007) Oral administration of chicken breast extract increases brain carnosine and anserine concentrations in rats. Nutr. Neurosci. 10, 181–186 [DOI] [PubMed] [Google Scholar]
- 6. Christman A. A. (1976) Factors affecting anserine and carnosine levels in skeletal muscles of various animals. Int. J. Biochem. 7, 519–527 [Google Scholar]
- 7. Kubomura D., Matahira Y., Nagai K., Niijima A. (2010) Effect of anserine ingestion on hyperglycemia and the autonomic nerves in rats and humans. Nutr. Neurosci. 13, 183–188 [DOI] [PubMed] [Google Scholar]
- 8. Szwergold B. S. (2005) Carnosine and anserine act as effective transglycating agents in decomposition of aldose-derived Schiff bases. Biochem. Biophys. Res. Commun. 336, 36–41 [DOI] [PubMed] [Google Scholar]
- 9. Bae O. N., Serfozo K., Baek S. H., Lee K. Y., Dorrance A., Rumbeiha W., Fitzgerald S. D., Farooq M. U., Naravelta B., Bhatt A., Majid A. (2013) Safety and efficacy evaluation of carnosine, an endogenous neuroprotective agent for ischemic stroke. Stroke 44, 205–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drozak J., Veiga-da-Cunha M., Vertommen D., Stroobant V., Van Schaftingen E. (2010) Molecular identification of carnosine synthase as ATP-grasp domain-containing protein 1 (ATPGD1). J. Biol. Chem. 285, 9346–9356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Drozak J., Chrobok L., Poleszak O., Jagielski A. K., Derlacz R. (2013) Molecular identification of carnosine N-methyltransferase as chicken histamine N-methyltransferase-like protein (hnmt-like). PLoS One 8, e64805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Veiga-da-Cunha M., Chevalier N., Stroobant V., Vertommen D., Van Schaftingen E. (2014) Metabolite proofreading in carnosine and homocarnosine synthesis: molecular identification of PM20D2 as β-alanyl-lysine dipeptidase. J. Biol. Chem. 289, 19726–19736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 14. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 15. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J. F., Guindon S., Lefort V., Lescot M., Claverie J. M., Gascuel O. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Castresana J. (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 [DOI] [PubMed] [Google Scholar]
- 18. Guindon S., Gascuel O. (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 [DOI] [PubMed] [Google Scholar]
- 19. Whelan S., Goldman N. (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699 [DOI] [PubMed] [Google Scholar]
- 20. Chevenet F., Brun C., Bañuls A. L., Jacq B., Christen R. (2006) TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics 7, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anisimova M., Gascuel O. (2006) Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552 [DOI] [PubMed] [Google Scholar]
- 22. Kozak M. (1987) An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15, 8125–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mora L., Sentandreu M. A., Toldrá F. (2007) Hydrophilic chromatographic determination of carnosine, anserine, balenine, creatine, and creatinine. J. Agric. Food. Chem. 55, 4664–4669 [DOI] [PubMed] [Google Scholar]
- 24. Banko M. I., Krzyzanowski M. K., Turcza P., Maniecka Z., Kulis M., Kozlowski P. (2013) Identification of amino acid residues of ERH required for its recruitment to nuclear speckles and replication foci in HeLa cells. PLoS One 8, e74885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 26. Li D., Niu Z., Yu W., Qian Y., Wang Q., Li Q., Yi Z., Luo J., Wu X., Wang Y., Schwartz R. J., Liu M. (2009) SMYD1, the myogenic activator, is a direct target of serum response factor and myogenin. Nucleic Acids Res. 37, 7059–7071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Petryszak R., Burdett T., Fiorelli B., Fonseca N. A., Gonzalez-Porta M., Hastings E., Huber W., Jupp S., Keays M., Kryvych N., McMurry J., Marioni J. C., Malone J., Megy K., Rustici G., Tang A. Y., Taubert J., Williams E., Mannion O., Parkinson H. E., Brazma A. (2014) Expression Atlas update: a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 42, D926–D932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Madshus I. H. (1988) Regulation of intracellular pH in eukaryotic cells. Biochem. J. 250, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hruz T., Laule O., Szabo G., Wessendorp F., Bleuler S., Oertle L., Widmayer P., Gruissem W., Zimmermann P. (2008) Genevestigator v3: a reference expression database for the meta-analysis of transcriptomes. Adv. Bioinformatics 2008, 420747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McManus I. R. (1962) Enzymatic synthesis of anserine in skeletal muscle by N-methylation of carnosine. J. Biol. Chem. 237, 1207–1211 [Google Scholar]
- 33. Horton P., Park K. J., Obayashi T., Fujita N., Harada H., Adams-Collier C. J., Nakai K. (2007) WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu C. S., Chen Y. C., Lu C. H., Hwang J. K. (2006) Prediction of protein subcellular localization. Proteins 64, 643–651 [DOI] [PubMed] [Google Scholar]
- 35. Smyk A., Szuminska M., Uniewicz K. A., Graves L. M., Kozlowski P. (2006) Human enhancer of rudimentary is a molecular partner of PDIP46/SKAR, a protein interacting with DNA polymerase delta and S6K1 and regulating cell growth. FEBS J. 273, 4728–4741 [DOI] [PubMed] [Google Scholar]
- 36. Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S., Wernerus H., Björling L., Ponten F. (2010) Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 28, 1248–1250 [DOI] [PubMed] [Google Scholar]
- 37. Harms W. S., Winnick T. (1954) Further studies of the biosynthesis of carnosine and anserine in vertebrates. Biochim. Biophys. Acta 15, 480–488 [DOI] [PubMed] [Google Scholar]
- 38. Clarke S., Banfield K. (2001) in Homocysteine in Health and Disease (Carmel R., Jacobsen D., eds), pp. 63–78, Cambridge University Press, Cambridge, UK [Google Scholar]
- 39. Martins M. L., Mourato M. P., de Varennes e Mendonça A. P. (2002) Characterization of aspartate aminotransferase isoenzymes from leaves of Lupinus albus L. cv. estoril. J. Biochem. Mol. Biol. 35, 220–227 [DOI] [PubMed] [Google Scholar]
- 40. Thapar N., Kim A. K., Clarke S. (2001) Distinct patterns of expression but similar biochemical properties of protein l-isoaspartyl methyltransferase in higher plants. Plant Physiol. 125, 1023–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wirth V. J., Van Lunen B. L., Mistry D., Saliba E., McCue F. C. (1998) Temperature changes in deep muscles of humans during upper and lower extremity exercise. J. Athl. Train. 33, 211–215 [PMC free article] [PubMed] [Google Scholar]
- 42. Osborne T. C., Obianyo O., Zhang X., Cheng X., Thompson P. R. (2007) Protein arginine methyltransferase 1: positively charged residues in substrate peptides distal to the site of methylation are important for substrate binding and catalysis. Biochemistry 46, 13370–13381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Webb K. J., Zurita-Lopez C. I., Al-Hadid Q., Laganowsky A., Young B. D., Lipson R. S., Souda P., Faull K. F., Whitelegge J. P., Clarke S. G. (2010) A novel 3-methylhistidine modification of yeast ribosomal protein Rpl3 is dependent upon the YIL110W methyltransferase. J. Biol. Chem. 285, 37598–37606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stock A., Clarke S., Clarke C., Stock J. (1987) N-terminal methylation of proteins: structure, function and specificity. FEBS Lett. 220, 8–14 [DOI] [PubMed] [Google Scholar]
- 45. Webb K. J., Lipson R. S., Al-Hadid Q., Whitelegge J. P., Clarke S. G. (2010) Identification of protein N-terminal methyltransferases in yeast and humans. Biochemistry 49, 5225–5235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miller B. G., Raines R. T. (2005) Reconstitution of a defunct glycolytic pathway via recruitment of ambiguous sugar kinases. Biochemistry 44, 10776–10783 [DOI] [PubMed] [Google Scholar]
- 47. Khersonsky O., Roodveldt C., Tawfik D. S. (2006) Enzyme promiscuity: evolutionary and mechanistic aspects. Curr. Opin. Chem. Biol. 10, 498–508 [DOI] [PubMed] [Google Scholar]
- 48. Peters V., Schmitt C. P., Zschocke J., Gross M. L., Brismar K., Forsberg E. (2012) Carnosine treatment largely prevents alterations of renal carnosine metabolism in diabetic mice. Amino Acids 42, 2411–2416 [DOI] [PubMed] [Google Scholar]
- 49. Boudry-Labis E., Demeer B., Le Caignec C., Isidor B., Mathieu-Dramard M., Plessis G., George A. M., Taylor J., Aftimos S., Wiemer-Kruel A., Kohlhase J., Annerén G., Firth H., Simonic I., Vermeesch J., Thuresson A. C., Copin H., Love D. R., Andrieux J. (2013) A novel microdeletion syndrome at 9q21.13 characterised by mental retardation, speech delay, epilepsy and characteristic facial features. Eur. J. Med. Genet. 56, 163–170 [DOI] [PubMed] [Google Scholar]
- 50. Baglietto M. G., Caridi G., Gimelli G., Mancardi M., Prato G., Ronchetto P., Cuoco C., Tassano E. (2014) RORB gene and 9q21.13 microdeletion: report on a patient with epilepsy and mild intellectual disability. Eur. J. Med. Genet. 57, 44–46 [DOI] [PubMed] [Google Scholar]