

Background: MCM10 regulates initiation of eukaryotic genome replication.
Results: HIV-1 Vpr enhances proteasomal degradation of MCM10.
Conclusion: HIV-1 Vpr-mediated degradation of MCM10 is essential for induction of G2/M cell cycle arrest.
Significance: Our study reveals a novel protein target for HIV-1 Vpr whose degradation is involved in the induction of G2/M cell cycle arrest by the virus.
Keywords: cell cycle, E3 ubiquitin ligase, human immunodeficiency virus (HIV), proteasome, protein degradation, ubiquitin, HIV-1 Vpr, MCM10, VprBP
Abstract
Human immunodeficiency virus type 1 Vpr is an accessory protein that induces G2/M cell cycle arrest. It is well documented that interaction of Vpr with the Cul4-DDB1[VprBP] E3 ubiquitin ligase is essential for the induction of G2/M arrest. In this study, we show that HIV-1 Vpr indirectly binds MCM10, a eukaryotic DNA replication factor, in a Vpr-binding protein (VprBP) (VprBP)-dependent manner. Binding of Vpr to MCM10 enhanced ubiquitination and proteasomal degradation of MCM10. G2/M-defective mutants of Vpr were not able to deplete MCM10, and we show that Vpr-induced depletion of MCM10 is related to the ability of Vpr to induce G2/M arrest. Our study demonstrates that MCM10 is the natural substrate of the Cul4-DDB1[VprBP] E3 ubiquitin ligase whose degradation is regulated by VprBP, but Vpr enhances the proteasomal degradation of MCM10 by interacting with VprBP.
Introduction
Viruses exploit cellular machinery to overcome restrictions and adverse conditions posed by host cells. Ubiquitin-proteasome machinery is usurped by a growing list of viruses to degrade certain cellular proteins. Some viruses encode proteins that function as E3 ubiquitin ligase to target new substrate proteins (1, 2). For example, herpes simplex virus encodes a viral E3 ubiquitin ligase, called infected cell protein 0 (ICP0), that interacts with components of the ubiquitin-proteasome pathway to degrade several proteins such as catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) and the centromeric proteins CENP-A, -B, and -C (3–5). A number of viruses encode proteins that redirect the host E3 ubiquitin ligases toward new protein targets. For example, protein V of paramyxovirus simian virus 5 (SV5) interacts with the CUL4A E3 ligase and directs the complex toward proteasome degradation of STAT1 (1, 2, 6).
Similarly to other viruses, retroviruses have evolved accessory proteins that take advantage of the E3 ubiquitin ligases to render the cellular environment permissive for the virus. For instance, retroviral protein Vpx, which is encoded by HIV-2 and a number of closely related simian immunodeficiency viruses, redirects the E3 ubiquitin ligase for proteasomal degradation of SAMHD1 (7–9). Retroviral Vif protein is also well documented to hijack the cellular CUL5 E3 ubiquitin ligase to direct APOBEC3G and APOBEC3F proteins toward proteasomal degradation (10, 11). Degradation of SAMHD1 and APOBEC proteins is beneficial for retroviruses (7, 9, 10).
HIV-1 Vpr is a small accessory protein that arrests the cell cycle at G2/M phase. The activity of Vpr in arresting the cell cycle has been attributed to its ability to interact with the Cul4-DDB1[VprBP] E3 ubiquitin ligase (12, 13). Vpr interaction with the E3 ubiquitin ligase has been shown to promote degradation of several proteins, including UNG2, Dicer, and telomerase. Instead of introducing new protein targets to the E3 ubiquitin ligase, Vpr seems to enhance proteasomal degradation of the natural substrates of the E3 ubiquitin ligase (14–17). Nonetheless, proteasomal degradation of these proteins does not seem to be related to Vpr-induced G2/M arrest.
Recently, interaction of Vpr with SLX4 has been shown to be essential for the induction of G2/M arrest. It was shown that Vpr directly interacts with SLX4 and induces recruitment of Vpr-binding protein (VprBP)2 and kinase-active PLK1, enhancing the cleavage of DNA by SLX4-associated MUS81-EME1 endonucleases (18). The initial events through which Vpr mediates G2/M arrest are followed by activation of ataxia-telangiectasia-mutated kinase (ATM) and ATM and Rad3-related kinase (ATR) that detect DNA damage and trigger downstream signaling cascades. It is believed that the end result of activation of ATM and ATR is induction of G2/M cell cycle arrest (13, 19, 20).
A recent study reported that VprBP interacts with minichromosome maintenance complex component 10 (MCM10) to recruit it to the E3 ubiquitin ligase for proteasomal degradation (21). MCM10 is one of the highly conserved MCM proteins that govern the initiation of eukaryotic genome replication (22, 23). MCM10 binds the origin of replication and plays an essential role in initiation and elongation of DNA replication by stabilizing DNA polymerase-α to maintain its association with chromatin (23, 24). Mutations in MCM10 result in stalled replication forks, followed by cell cycle arrest (25, 26).
The Vpr-mediated proteasomal degradation of natural substrates of VprBP and the involvement of MCM10 in cell cycle regulation prompted us to examine whether Vpr induces the enhanced degradation of MCM10 and whether this degradation is related to Vpr-mediated G2/M arrest. We observed an enhanced ubiquitination and proteolysis of MCM10 that were Vpr- and VprBP-dependent. Furthermore, depletion of MCM10 was found to be related to Vpr-mediated G2/M arrest.
Experimental Procedures
Cell Lines
HeLa and HEK293T cells were obtained from ATCC and maintained in DMEM supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FBS. MT4 T cells were obtained from the National Institutes of Health AIDS Reagent Program and maintained in RPMI 1640 media supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FBS.
Doxycycline inducible HeLa cell lines (HeLa-iFlag-Vpr, HeLa-iFlag-Q65R, and HeLa-iFlag-R80A) and a control cell line (HeLa-iMock) were engineered using a blasticidin-resistant construct expressing the reverse tetracycline transactivator (rtTA) from the CMV promoter (pLenti CMV rtTA3 Blast; Addgene). Briefly, N-terminally FLAG-tagged wild-type Vpr and the Q65R and R80A mutants were cloned into pLenti CMV rtTA3 Blast using XbaI and ApaI restriction sites. Lentiviral vectors were produced by co-transfecting 6 × 106 HEK293T cells with 30 μg of pLenti CMV rtTA3 Blast, 15 μg of psPAX2, and 6 μg of pCMV-VSV-G. Lentiviral particles were collected after 48 h and used to transduce HeLa cells. After 2 days, the transduced cells were selected by adding 2 μg/ml blasticidin for 1 week. Expression of Vpr was induced by adding 1 μg/ml doxycycline.
Antibodies and Reagents
MCM10 (PIPA126586) antibody was purchased from Thermo Fisher Scientific. HA (ab59076), actin (ab1801), histone H3 (ab70550), DDB1 (ab124672), and p24 (ab9071) antibodies were purchased from Abcam. VprBP (A301-888A) antibody was from Bethyl Laboratories. FLAG (F3165) antibody, monoclonal anti-HA agarose (A2095), and anti-FLAG M2 affinity gel were from Sigma-Aldrich. Vpr (NP_057852) antibody was from Proteintech. MCM2 (4007), MCM7 (4018), and rabbit IgG isotype control (2729) were from Cell Signaling. Mouse and rabbit HRP-conjugated antibodies were from Abcam. Protein A-Sepharose beads were from Amersham Biosciences. HA and FLAG peptides were from AnaSpec and Sigma-Aldrich, respectively. Benzonase nuclease was from Novagen. Caffeine, ethidium bromide, DMSO, doxycycline, cycloheximide, and blasticidin were from Sigma-Aldrich. MG132 was from Millipore. Protease inhibitor cocktail was from Roche Applied Science. VprBP siRNA and non-targeting siRNA were from Dharmacon. MCM10 siRNA was from Santa Cruz Biotechnology. SYBR Select Master Mix was from Life Technologies.
Constructs
HIV Gag-iGFP was obtained from the National Institutes of Health AIDS Research and Reference Reagent Program (NIAID). To generate HIV Gag-iGFP ΔVpr, the start codon of the vpr gene was inactivated and an additional stop codon was inserted in the 3rd codon of vpr using site-directed mutagenesis. To generate N-terminally FLAG-tagged Vpr, the vpr gene of HIV Gag-iGFP was cloned into FLAG-tagged pcDNA3.1. The Q65R and R80A mutations were induced into the construct using site-directed mutagenesis. pWPI vector was obtained from Addgene, and FLAG-tagged wild-type Vpr and Q65R and R80A mutants from FLAG-tagged pcDNA3.1 constructs were cloned into the pmeI site of pWPI. psPax2, pCMV-VSV-G, and HA-ubiquitin were obtained from Addgene. To generate C-terminally HA-tagged full-length MCM10 and ΔCTD MCM10, the cDNAs of full-length and ΔCTD MCM10 were amplified from HeLa cell total mRNA and cloned into HA-tagged pcDNA3.1.
Lentiviral Vectors and Virus Production
VSV-G-pseudotyped lentiviral particles were produced in HEK293T cells by co-transfecting 40 μg of pWPI-FLAG-Vpr/Q65R, R80A, or empty vector with 15 μg of psPax2 and 6 μg of pCMV-VSV-G using the standard calcium phosphate protocol in 15-cm Petri dishes. Lentiviral vector particles were collected 48 and 72 h post-transfection by ultracentrifugation at 35,000 rpm for 2 h. HIV-1 virus particles were produced by transfecting 30 μg HIV of Gag-iGFP (WT/ΔVpr) into HEK293T using the standard calcium phosphate protocol. Viruses were collected 48 and 72 h post-transfection by ultracentrifugation at 35,000 rpm for 2 h.
Transfection
siRNA depletion of VprBP or MCM10 in inducible HeLa and HEK293T cells, respectively, was performed using Lipofectamine RNAiMAX reagent according to the manufacturer's instructions. Co-transfection of plasmids into HEK293T cells was performed using Lipofectamine 2000 according to the manufacturer's instructions.
Cell Fractionation
Inducible HeLa cells were harvested and resuspended in 2 ml of 0.5% Triton lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 0.5% Triton) that contained protease inhibitor. Cells were lysed for 10 min with mild agitation at 4 °C and then centrifuged at 6000 rpm for 10 min at 4 °C to pellet chromatin and other large insoluble debris. Supernatant was transferred to a new Eppendorf tube (this constituted the soluble fraction). Pellet was resuspended in 2 ml of Benzonase buffer (50 mm Tris, 1.5 mm hydrated MgCl2, 0.1 mg/ml BSA, pH 8) that contained protease inhibitor. The resuspended pellet was centrifuged, and supernatant was discarded. One μl of Benzonase (25 units/μl) was added to 1 ml of Benzonase buffer (containing protease inhibitor). One ml of Benzonase-containing buffer was added to the pellet and resuspended by pipetting and then incubated on ice for 60 min. The Benzonase-treated pellet was centrifuged at 13,000 rpm for 10 min at 4 °C. The supernatant represented the fraction containing chromatin-bound proteins. Supernatants were transferred to new tubes. Integrity of the fractions was verified by the presence of actin mainly in the soluble protein fraction and histone H3 mainly in the chromatin-bound protein fraction.
Cycloheximide Chase Assay
To measure the half-life of MCM10, inducible HeLa cell lines were induced by adding 1 μg/ml doxycycline. After 10 h, cells were treated with 100 μg/ml cycloheximide to block protein synthesis. Cells were harvested at time 0, 2, 4, 6, 8, and 10 h post-treatment and analyzed using Western blot.
Western Blotting and Immunoprecipitation
For Western blotting, cells were lysed directly in Laemmli buffer (106 cells/ml) and boiled for 5 min. For immunoprecipitation, cells were lysed in 150 mm NaCl, 0.5% Triton, 50 mm Tris, 1.5 mm (pH 7.5) MgCl2, 25 units/ml Benzonase, and protease inhibitor cocktail and incubated for 1 h at 4 °C. Anti-HA or -FLAG immunoprecipitations were performed using monoclonal anti-HA or -FLAG agarose beads. For VprBP or MCM10 immunoprecipitations, 50 μl of protein A-Sepharose beads were conjugated overnight to the respective antibodies (2 μg per immunoprecipitation) in 1 ml of PBS supplemented with 5% FBS at 4 °C. All immunoprecipitations were performed in the presence of 150 mm NaCl and 0.5% Triton X-100 for 4 h at 4 °C. After thorough washes with 0.5% Triton lysis buffer, the anti-HA- and -FLAG-immunoprecipitated proteins were eluted by adding 100 μg/ml HA or FLAG peptides, respectively. Anti-VprBP- or MCM10-immunoprecipitated proteins were released by treating the beads with 0.1 m glycine, pH 2.0, for 5 min on ice. Immunoprecipitated proteins and cell lysates (30 μg) were resuspended in Laemmli buffer, heat-denatured for 5 min, and separated on 12% SDS-PAGE gels. After migration, proteins were transferred to nitrocellulose membranes (Bio-Rad). Specific proteins in cell lysates or immunocomplexes were probed by overnight incubation of the membranes with primary antibodies. After thorough washes of the membranes, primary antibodies were probed with secondary mouse or rabbit HRP-conjugated antibodies. Unbound secondary antibodies were washed, and the membranes were developed using ECL substrate and photographed on Amersham Biosciences ECL films. Bands corresponding to the probed proteins in the immunoprecipitated fractions were scanned, and densitometric quantitation of the bands was performed using ImageJ software (National Institutes of Health).
Quantitative PCR
To measure changes in the mRNA level of MCM10, total RNA was extracted using RNeasy mini kit (Qiagen, Germantown, MD) and reverse-transcribed into cDNA using SuperScript II reverse transcriptase (Invitrogen). Quantitative PCR was performed using SYBR Select master mix (Life Technologies). Changes in the expression levels were normalized to GAPDH and calculated using the 2−ΔΔCt method.
Flow Cytometry and Sorting
To analyze the cell cycle profile of HEK293T and inducible HeLa cells, 106 cells were resuspended in 1 ml of Krishan modified buffer (0.1% sodium citrate, 0.3% NP-40, 0.05 mg/ml propidium iodide, 0.02 mg/ml RNase A) and incubated on ice for 30 min. The DNA contents of the cells were then measured using BD FACSCalibur. ModFit LT 3.2 was used to analyze the cell cycle profile.
To sort infected MT4 cells, they were infected with pNL4.3 IRES_GFP_Nef-(WT/ΔVpr). After 48 h, the GFP-positive cells were sorted in PBS using an Influx cell sorter (BD Biosciences) and analyzed using Western blot.
Statistical Analyses
Student's t test was used to examine statistical significance in the experiments using GraphPad Prism 6.0. Effects with p < 0.05 were considered significant.
Results
HIV-1 Vpr Down-regulates MCM10
To test whether expression of Vpr has a significant effect on the level of MCM10 protein, we induced expression of FLAG-tagged Vpr in the doxycycline-inducible HeLa-iFlag-Vpr cells by adding doxycycline. As shown in Fig. 1A, Vpr was detectable at 5 h post-induction (h.p.i.) and 20% depletion of MCM10 was observed at 10 h.p.i. After 20 h, 50% of MCM10 had been depleted and the depletion still continued. At 40 h.p.i., 70% of MCM10 had been depleted and only 30% was remaining. At this time point, the experiment was stopped due to the cytotoxicity of doxycycline induction and expression of Vpr.
FIGURE 1.

HIV-1 Vpr down-regulates MCM10. A, HeLa-iFlag-Vpr cells were treated with doxycycline to induce expression of FLAG-Vpr. Cells were harvested at different time points, ranging from 0 to 40 h.p.i. as indicated in the figure. Equal numbers of cells were directly lysed in Laemmli buffer and analyzed using Western blot. Protein bands were quantified and normalized to actin. The experiment was repeated 3 times, and one representative Western blot is shown. B, a fraction of cells in A was spared for cell cycle analysis. The DNA content of the cells was labeled using propidium iodide and analyzed using flow cytometry. The data represent 3 independent experiments. C, HeLa-iFlag-Vpr cells were treated with doxycycline to express FLAG-Vpr, and 30 h later, cells were harvested. A fraction of the cells was used for Western blot analysis, and total RNA was extracted from 106 cells and reverse-transcribed into cDNA. Quantitative real-time PCR was performed on the cDNA and normalized to GAPDH. The experiment was repeated 3 times, and one representative Western blot is shown. ns, not significant. D, HeLa-iFlag-Vpr cells were induced to express FLAG-Vpr. 2.5 mm caffeine was added to the cells at the same time of induction. After 20 h, cells were harvested and analyzed for depletion of MCM10. E, cell cycle analysis was performed using propidium iodide. The experiment was repeated 3 times, and one representative experiment is shown. Statistical significance was calculated using the student t test. Asterisk indicates statistical significance.
Along with MCM10, we assessed whether Vpr could impact the expression levels of other MCM proteins. As shown in Fig. 1A, expression of Vpr did not affect the expression levels of MCM2 and MCM7.
In addition to the protein levels of MCM10, the cell cycle profile of the induced cells was analyzed to ensure that the observed depletion is not a result of G2/M arrest. A slight G2/M arrest was observed at 10 h.p.i. that shifted the G2+M:G1 ratio from 0.33 to 0.55 (Fig. 1B). A significant change in the G2+M:G1 ratio (5.4) was observed at 20 h.p.i. and continued to rise up to 17.5 at 40 h.p.i. Induction of G2/M arrest did not precede the Vpr-induced depletion of MCM10, suggesting that depletion of MCM10 did not result from G2/M arrest.
We examined whether Vpr could affect the mRNA levels of MCM10 at 30 h.p.i. before the induction causes a strong cytotoxicity. As shown in Fig. 1C, expression of Vpr induced depletion of 60% of MCM10 (40% remaining), but it did not affect MCM10 at transcriptional level.
To further explore whether depletion of MCM10 is upstream or downstream of G2/M arrest, caffeine, an ATR inhibitor, was added to the induced cells to inhibit the Vpr-induced G2/M arrest (Fig. 1, D and E). Caffeine treatment efficiently inhibited Vpr-induced G2/M arrest, but depletion of MCM10 was still observed at similar levels in the presence or absence of caffeine. This further confirmed that Vpr-induced depletion of MCM10 was upstream of G2/M arrest. Furthermore, it showed that Vpr does not affect MCM10 abundance indirectly through affecting cell cycle profile as Vpr induced depletion of MCM10 in the absence of G2/M arrest.
Vpr-induced Depletion of MCM10 Is Proteasome-dependent and Related to G2/M Arrest Activity of Vpr
We took advantage of the Q65R and R80A mutants of Vpr that are defective for induction of G2/M arrest. Q65R is not able to bind VprBP, the binding of which is essential for induction of G2/M arrest. The reason for the lack of G2/M arrest activity of R80A, however, is not well described. We hypothesized that the G2/M-defective mutants should not be able to induce degradation of G2/M target.
To avoid two different drug treatments in the same experiment, doxycycline induction and proteasomal inhibition by MG132, normal HeLa cells were used to avoid doxycycline induction. HeLa cells were transduced with lentiviral vectors that expressed the WT Vpr and Q65R and R80A mutants. Cells were treated with MG132 10 h before harvesting. Cells were harvested 48 h after transduction and subjected to Western blot analysis (Fig. 2A). The wild-type Vpr was able to deplete the majority of endogenous MCM10, whereas only 40% of MCM10 was remaining. The Q65R and R80A mutants, however, did not significantly affect the levels of MCM10. MG132 treatment significantly restored the level of MCM10 such that the wild-type Vpr only depleted 20% of the endogenous MCM10. This suggested that Vpr-induced depletion of MCM10 was proteasome-dependent.
FIGURE 2.

G2/M-defective mutants of Vpr are not able to induce proteasomal degradation of MCM10. A, HeLa cells were transduced with lentiviral vectors for expression of FLAG-tagged WT Vpr or the G2/M-defective mutants of Vpr (Q65R and R80A) at a multiplicity of infection of 1.0. One sample was transduced with empty vector (Emp). After 40 h, cells were treated with 10 μm MG132 or DMSO, and after 8 h (total of 48 h post-transduction), cells were harvested and lysed in Laemmli buffer. Cell lysates were analyzed using Western blot. Asterisk indicates statistical significance. B, HEK293T cells were co-transfected with plasmids for the expression of HA-tagged ubiquitin (HA-UB) and FLAG-tagged WT Vpr or FLAG-tagged Q65R mutant (Q65R). After 48 h, cells were lysed and immunoprecipitated using anti-HA antibody conjugated to agarose beads. C, HeLa-iMock, HeLa-iFlag-Q65R, HeLa-iFlag-R80A, and HeLa-iFlag-Vpr cells were treated with doxycycline for 10 h and then treated with cycloheximide (CHX). Cells were then harvested and lysed at 2-h intervals as indicated. The lysates were analyzed using Western blot. The experiment was repeated 3 times, and one representative Western blot is shown.
To further examine whether the depletion of MCM10 by Vpr is through a proteasomal pathway, we tested the ubiquitination of MCM10 by Vpr. HEK293T cells were co-transfected with HA-tagged ubiquitin and wild-type Vpr or Q65R mutant. After 48 h, cells were lysed and subjected to immunoprecipitation against HA to pull down ubiquitinated proteins (Fig. 2B). Vpr did not affect the total ubiquitination of cellular proteins in the lysate but enhanced ubiquitination of the pulled down MCM10 2.4-fold. This further confirmed that Vpr depletes MCM10 through a proteasomal pathway.
Additionally, we performed a cycloheximide chase assay to examine the effect of Vpr on the half-life of MCM10. HeLa-iFlag-Vpr and the respective mutants, HeLa-iFlag-Q65R and HeLa-iFlag-R80A, were induced to express Vpr. Ten hours after induction, cells were treated with cycloheximide to block protein synthesis and harvested at 2-h intervals. As shown in Fig. 2C, the half-life of MCM10 in HeLa cells was determined to be ∼6 h, and this was not significantly affected by the Q65R and R80A mutant forms of Vpr. However, the wild-type Vpr reduced the half-life of MCM10 to 4 h. Of note, the half-life of Vpr was longer than that of MCM10 and was not significantly affected during the 10-h period of the experiment.
Vpr-induced Depletion of MCM10 Is VprBP-dependent
We showed that Vpr induces depletion of MCM10 in a proteasome-dependent manner. From here, we revisited the interaction between VprBP and MCM10 and how Vpr affects this interaction. HeLa-iFlag-Vpr and HeLa-iFlag-Q65R cells were doxycycline-induced to express Vpr. Induction lasted for only 10 h to prevent a significant depletion of MCM10 that could affect the binding experiments. After the induction, cells were lysed and immunoprecipitated with anti-FLAG antibodies to pull down complexes of FLAG-tagged Vpr/Q65R (Fig. 3A). The wild-type Vpr, but not the Q65R mutant, immunoprecipitated both MCM10 and VprBP, suggesting that the interaction between Vpr and MCM10 is VprBP-dependent. To further examine the role of VprBP as the mediator of the interaction, VprBP was depleted using siRNA and cells were induced to express Vpr/Q65R. As shown in Fig. 3B, depletion of VprBP significantly prevented the basal depletion of MCM10, compare lanes 1 and 3, as well as the Vpr-induced depletion of MCM10, compare lanes 2 and 4. In addition, when VprBP was depleted, Vpr lost its ability to bind MCM10 (lane 4). It was also shown that the half-life of MCM10 is VprBP-dependent because depletion of VprBP increased the half-life of MCM10 (Fig. 3C). Interestingly, Vpr shortened the half-life of MCM10 only in the presence of VprBP.
FIGURE 3.

Vpr relies on the Cul4-DDB1[VprBP] E3 ubiquitin ligase to deplete MCM10. A, HeLa-iMock, HeLa-iFlag-Vpr, and HeLa-iFlag-Q65R were treated with doxycycline only for 10 h to have enough MCM10 for immunoprecipitation (IP) before it was degraded. The induced cells were harvested and lysed. The lysates were immunoprecipitated with anti-FLAG antibody conjugated to agarose beads, and the pulled down complexes were eluted with FLAG peptides. The eluates were analyzed using Western blot. B, HeLa-iMock and HeLa-iFlag-Vpr were transfected with siRNA against VprBP or non-targeting siRNA (NT). After 48 h, expression of Vpr in transfected cells was induced by doxycycline treatment for 10 h, and then cells were lysed and immunoprecipitated with anti-FLAG antibody conjugated to agarose beads. C, HeLa-iMock and HeLa-iFlag-Vpr were transfected with siRNA against VprBP or non-targeting siRNA (NT). After 48 h, expression of Vpr in transfected cells was induced by doxycycline treatment. Ten hours post-induction, the induced cells were treated with cycloheximide and harvested at 2-h intervals as indicated. Cells were lysed and analyzed using Western blot. The experiment was repeated 3 times, and one representative Western blot is shown. CHX, cycloheximide. D, VprBP binds MCM10 independently of Vpr. HeLa cells were lysed and immunoprecipitated with anti-VprBP antibody- or IgG-conjugated Sepharose beads. The pulled down proteins were released by treating the Sepharose beads with 0.1 m glycine, pH 2.0, and the eluates were then analyzed using Western blot. E, MCM10 binds VprBP and DDB1 independently of Vpr. HeLa cells were lysed and immunoprecipitated with anti-MCM10- or IgG-conjugated Sepharose beads. All experiments were repeated at least 2 times.
To test whether interaction of VprBP with MCM10 is dependent on the presence of Vpr, the endogenous VprBP was pulled down with antibodies against VprBP and the eluates were examined. As shown in Fig. 3D, VprBP alone was able to bind MCM10. Furthermore, immunoprecipitation of the endogenous MCM10 indicated that MCM10 was able to bind VprBP and DDB1 as the main components of the Cul4-DDB1[VprBP] E3 ubiquitin ligase (Fig. 3E).
Vpr Binds and Depletes MCM10 on Chromatin
Both MCM10 and Vpr are DNA-binding proteins. We asked whether Vpr targets MCM10 for degradation on chromatin. To determine the location in which MCM10 is targeted by Vpr, HeLa-iFlag-Vpr cells were doxycycline-induced to express FLAG-Vpr. After 10 h, cells were fractionated into Triton-X-soluble proteins (here called soluble fraction) and an insoluble fraction that was treated with Benzonase DNase to release the chromatin-associated proteins (chromatin fraction). Both soluble and chromatin fractions were immunoprecipitated with anti-FLAG antibody in the presence and absence of ethidium bromide. As shown in Fig. 4, Vpr was present in both fractions but mainly localized to the chromatin fraction. MCM10 was only present in the chromatin fraction and consistently Vpr-depleted MCM10 in the chromatin fraction. Furthermore, Vpr was found to interact with MCM10 only in chromatin fraction. The presence of ethidium bromide in the immunoprecipitations did not affect the binding, suggesting that the binding of Vpr to MCM10 is not through DNA intermediates.
FIGURE 4.
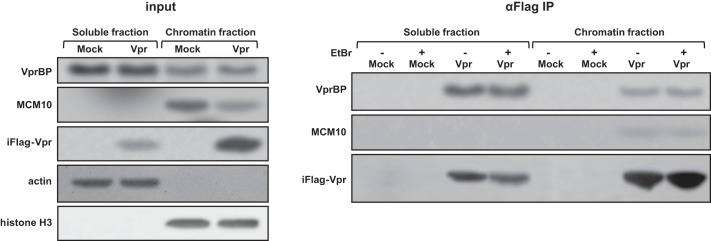
HIV-1 Vpr interacts with MCM10 on chromatin. HeLa-iMock and HeLa-iFlag-Vpr cells were treated with doxycycline for 20 h. Cells were then harvested and lysed in 0.5% Triton lysis buffer and centrifuged to obtain a clear soluble fraction. Pellet was washed and treated with Benzonase DNase to release the chromatin-associated proteins (chromatin fraction). Half of each fraction was treated with 25 μg/ml ethidium bromide, and all the treated and non-treated fractions were subjected to immunoprecipitation (IP) using anti-FLAG antibody conjugated to agarose beads.
Depletion of MCM10 Induces G2/M Arrest, and Supplementation of MCM10 Prevents Vpr-induced G2/M Arrest
To investigate the relationship between depletion of MCM10 and induction of G2/M arrest, MCM10 was depleted in HEK293T cells using siRNA and the cell cycle was analyzed. As shown in Fig. 5A, depletion of MCM10 induced a strong G2/M arrest by efficiently shifting the G2+M:G1 ratio from 0.28 in control to 2.17 in the MCM10-depleted cells.
FIGURE 5.

Depletion of MCM10 induces G2/M arrest and complementation of wild-type MCM10 inhibits Vpr-induced G2/M arrest. A, HEK293T cells were double-transfected with siRNA against MCM10 or non-targeting siRNA (NT) with 24-h intervals. Forty-eight hours after the second transfection, cells were analyzed for depletion of MCM10 and the cell cycle was analyzed by propidium iodide staining. B, complementation of MCM10 inhibits Vpr-induced G2/M arrest. HEK293T cells were co-transfected with increasing amounts of MCM10-HA expressor and a constant amount of FLAG-Vpr expressor. The total amount of transfected DNA was kept constant by adding empty vector. Cell cycle was analyzed after 48 h. C, complementation of ΔCTD MCM10 does not inhibit Vpr-induced G2/M arrest. HEK293T cells were co-transfected with increasing amounts of ΔCTD MCM10-HA expressor and a constant amount of FLAG-Vpr. After 48 h, the cell cycle was analyzed. D, ΔCTD MCM10 does not bind Vpr and VprBP. HEK293T cells were co-transfected with plasmids for expression of FLAG-Vpr and the full-length HA-tagged MCM10 (WT) or CTD truncated MCM10 (ΔCTD). After 48 h, cells were lysed and immunoprecipitated with anti-HA antibody conjugated to agarose beads. All experiments were repeated at least two times, and one representative experiment is shown.
We hypothesized that if Vpr induces G2/M arrest by depleting MCM10, complementation of MCM10 should prevent the G2/M arrest induced by Vpr. HEK293T cells were co-transfected with steady amounts of Vpr and increasing amounts of HA-tagged MCM10 expressors (Fig. 5B). Although expression of MCM10 alone did not affect the cell cycle profile of the transfected cells, complementation of MCM10 inhibited Vpr-induced G2/M arrest in a dose-response manner. In fact, restoring the cell cycle profile of the co-transfected cells to a normal profile started at very low concentrations of MCM10 (15 ng of MCM10 expressor). Increasing concentrations of MCM10 continued to restore the cell cycle at higher concentrations until the cell cycle was completely restored to normal condition, as compared with the control, at 500 ng of MCM10 expressor.
We challenged our hypothesis by asking whether restoration of the cell cycle profile by the exogenous MCM10 was an artifact resulting from co-transfection or an active function of MCM10. Previously, it has been shown that the C-terminal domain (CTD) of MCM10 binds VprBP (21). We took advantage of this truncated form of MCM10 and co-transfected HEK293T cells with ΔCTD MCM10 (Fig. 5C). Co-transfection of ΔCTD MCM10 did not significantly affect Vpr-induced G2/M arrest.
To characterize the domain of MCM10 involved in binding to Vpr and VprBP, HEK293T cells were transfected with the full-length and ΔCTD MCM10 in the presence or absence of Vpr (Fig. 5D). Pulldown of the full-length MCM10, but not ΔCTD MCM10, immunoprecipitated VprBP. Similarly, only the full-length MCM10 was able to bind Vpr, suggesting that the C-terminal domain of MCM10 is responsible for its binding to VprBP and therefore Vpr.
HIV-1 Down-regulates MCM10
After showing the effect of overexpressed Vpr on MCM10, we decided to examine the effect of viral encoded Vpr on the cellular levels of MCM10 in a T cell line. MT4 cells were infected with GFP reporter wild-type or ΔVpr viruses, and the infected cells were sorted using the GFP signal. Levels of MCM10 were assessed in the GFP-expressing cells (Fig. 6). The wild-type virus was able to deplete 35% of MCM10 (65% was remaining) in the infected cells, whereas the Vpr-defective virus, ΔVpr, did not down-regulate MCM10. This demonstrated that the de novo expression of Vpr was also able to efficiently deplete endogenous MCM10.
FIGURE 6.
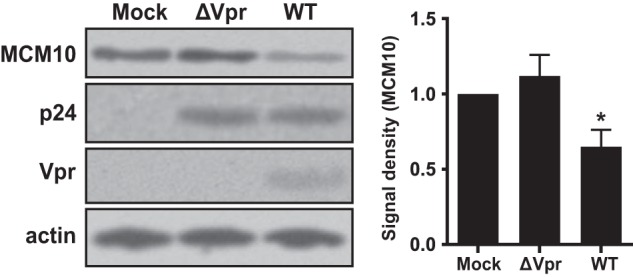
HIV-1 down-regulates MCM10. MT4 cells were infected with wild-type or ΔVpr GFP-marked HIV-1 at a multiplicity of infection of 0.1. Forty eight hours after infection, GFP-positive cells were sorted for immunoblot analysis. The data represent 5 independent experiments. Asterisk indicates statistical significance.
Discussion
In this study, we demonstrated that HIV-1 Vpr enhances proteasomal degradation of MCM10. Moreover, we showed that this effect is VprBP-dependent and that MCM10 is able to bind the components of the Cul4-DDB1[VprBP] E3 ubiquitin ligase. Our results also show that Vpr enhances ubiquitination of MCM10. Based on our results, we propose a model in which Vpr binds the Cul4-DDB1[VprBP] E3 ubiquitin ligase through VprBP and enhances ubiquitination of MCM10 (Fig. 7). We also showed that Vpr binds and depletes MCM10 on chromatin. To the best of our knowledge, this is the first study that reports targeting of a protein by an HIV-1 accessory protein on chromatin. It is worth determining the location in which Vpr binds and depletes its other targets.
FIGURE 7.
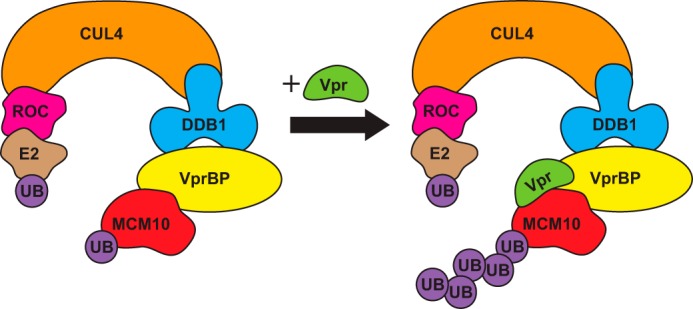
HIV-1 Vpr enhances proteasomal degradation MCM10. The Cul4-DDB1[VprBP] E3 ubiquitin ligase naturally targets MCM10 for proteasomal degradation. However, in the presence of Vpr, ubiquitination of MCM10 is significantly enhanced.
In addition to the binding of Vpr to MCM10 and mediating its depletion, we also propose the role of Vpr-mediated depletion of MCM10 in induction of G2/M arrest. Recently, it was shown that the premature activation of SLX4 complex by Vpr induces G2/M arrest (18). Our study reports a new mechanism for induction of G2/M arrest by Vpr that may not necessarily be in contrast to the recent report on the role of SLX4. These two mechanisms may co-exist in parallel or may be part of the same pathway/complex. Further studies are needed to explain or link the two mechanisms.
By looking at the body of evidence that so far the protein targets of Vpr have provided, one can envisage that Vpr removes the tight regulation of the Cul4-DDB1[VprBP] E3 ubiquitin ligase for ubiquitination of its natural substrates. This seems to be the case for UNG2, telomerase, Dicer (14–17), and here for MCM10. It seems logical to propose that Vpr enhances degradation of the natural targets of the Cul4-DDB1[VprBP] E3 ubiquitin ligase through VprBP, and there may be more unreported targets of VprBP whose degradations could be enhanced by Vpr.
Author Contributions
B. R., N. S. B., and E. A. designed and performed the experiments. E. A. and M. R. A. supervised the project and applied for funding. Figures were prepared by B. R. and N. S. B. BR wrote the manuscript. All authors read and approved the final revision of the manuscript.
Acknowledgments
HIV Gag-iGFP plasmids and MT4 cells were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program. We thank our colleagues in the University of Jundishapur and the University of Isfahan for helpful discussions.
The authors declare that they have no conflicts of interest with the contents of this article.
- VprBP
- Vpr-binding protein
- ATM
- ataxia-telangiectasia-mutated kinase
- ATR
- ATM and Rad3-related kinase
- rtTA
- reverse tetracycline transactivator
- VSV-G
- vesicular stomatitis virus G
- CTD
- C-terminal domain
- iGFP
- interdomain GFP
- DMSO
- dimethyl sulfoxide
- h.p.i.
- hours post-induction.
References
- 1. Banks L., Pim D., Thomas M. (2003) Viruses and the 26S proteasome: hacking into destruction. Trends Biochem. Sci. 28, 452–459 [DOI] [PubMed] [Google Scholar]
- 2. Isaacson M. K., Ploegh H. L. (2009) Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 5, 559–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith M. C., Boutell C., Davido D. J. (2011) HSV-1 ICP0: paving the way for viral replication. Future Virol. 6, 421–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lomonte P., Morency E. (2007) Centromeric protein CENP-B proteasomal degradation induced by the viral protein ICP0. FEBS Lett. 581, 658–662 [DOI] [PubMed] [Google Scholar]
- 5. Lomonte P., Sullivan K. F., Everett R. D. (2001) Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 276, 5829–5835 [DOI] [PubMed] [Google Scholar]
- 6. Precious B., Childs K., Fitzpatrick-Swallow V., Goodbourn S., Randall R. E. (2005) Simian virus 5 V protein acts as an adaptor, linking DDB1 to STAT2, to facilitate the ubiquitination of STAT1. J. Virol. 79, 13434–13441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hrecka K., Hao C., Gierszewska M., Swanson S. K., Kesik-Brodacka M., Srivastava S., Florens L., Washburn M. P., Skowronski J. (2011) Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Ségéral E., Yatim A., Emiliani S., Schwartz O., Benkirane M. (2011) SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lahouassa H., Daddacha W., Hofmann H., Ayinde D., Logue E. C., Dragin L., Bloch N., Maudet C., Bertrand M., Gramberg T., Pancino G., Priet S., Canard B., Laguette N., Benkirane M., Transy C., Landau N. R., Kim B., Margottin-Goguet F. (2012) SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 13, 223–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Romani B., Engelbrecht S., Glashoff R. H. (2009) Antiviral roles of APOBEC proteins against HIV-1 and suppression by Vif. Arch. Virol 154, 1579–1588 [DOI] [PubMed] [Google Scholar]
- 11. Yu X., Yu Y., Liu B., Luo K., Kong W., Mao P., Yu X. F. (2003) Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302, 1056–1060 [DOI] [PubMed] [Google Scholar]
- 12. Hrecka K., Gierszewska M., Srivastava S., Kozaczkiewicz L., Swanson S. K., Florens L., Washburn M. P., Skowronski J. (2007) Lentiviral Vpr usurps Cul4–DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. U.S.A. 104, 11778–11783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wen X., Duus K. M., Friedrich T. D., de Noronha C. M. C. (2007) The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J. Biol. Chem. 282, 27046–27057 [DOI] [PubMed] [Google Scholar]
- 14. Casey Klockow L., Sharifi H. J., Wen X., Flagg M., Furuya A. K. M., Nekorchuk M., de Noronha C. M. C. (2013) The HIV-1 protein Vpr targets the endoribonuclease Dicer for proteasomal degradation to boost macrophage infection. Virology 444, 191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wen X., Casey Klockow L., Nekorchuk M., Sharifi H. J., de-Noronha C. M. C. (2012) The HIV1 protein Vpr acts to enhance constitutive DCAF1-dependent UNG2 turnover. PLoS One 7, e30939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X., Singh S., Jung H. Y., Yang G., Jun S., Sastry K. J., Park J.-I. (2013) HIV-1 Vpr protein inhibits telomerase activity via the EDD-DDB1-VPRBP E3 ligase complex. J. Biol. Chem. 288, 15474–15480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jung H. Y., Wang X., Jun S., Park J.-I. (2013) Dyrk2-associated EDD-DDB1-VprBP E3 ligase inhibits telomerase by TERT degradation. J. Biol. Chem. 288, 7252–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Laguette N., Brégnard C., Hue P., Basbous J., Yatim A., Larroque M., Kirchhoff F., Constantinou A., Sobhian B., Benkirane M. (2014) Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 156, 134–145 [DOI] [PubMed] [Google Scholar]
- 19. Belzile J. P., Duisit G., Rougeau N., Mercier J., Finzi A., Cohen É. A. (2007) HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 3, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zimmerman E. S., Chen J., Andersen J. L., Ardon O., Dehart J. L., Blackett J., Choudhary S. K., Camerini D., Nghiem P., Planelles V. (2004) Human immunodeficiency virus type 1 Vpr-mediated G2 arrest requires Rad17 and Hus1 and induces nuclear BRCA1 and γ-H2AX focus formation. Mol. Cell. Biol. 24, 9286–9294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaur M., Khan M. M., Kar A., Sharma A., Saxena S. (2012) CRL4–DDB1–VPRBP ubiquitin ligase mediates the stress triggered proteolysis of Mcm10. Nucleic Acids Res. 40, 7332–7346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Homesley L., Lei M., Kawasaki Y., Sawyer S., Christensen T., Tye B. K. (2000) Mcm10 and the MCM2–7 complex interact to initiate DNA synthesis and to release replication factors from origins. Genes Dev. 14, 913–926 [PMC free article] [PubMed] [Google Scholar]
- 23. Ricke R. M., Bielinsky A. K. (2006) A conserved Hsp10-like domain in Mcm10 is required to stabilize the catalytic subunit of DNA polymerase-α in budding yeast. J. Biol. Chem. 281, 18414–18425 [DOI] [PubMed] [Google Scholar]
- 24. Ricke R. M., Bielinsky A. K. (2004) Mcm10 regulates the stability and chromatin association of DNA polymerase-α. Mol. Cell 16, 173–185 [DOI] [PubMed] [Google Scholar]
- 25. Merchant A. M., Kawasaki Y., Chen Y., Lei M., Tye B. K. (1997) A lesion in the DNA replication initiation factor Mcm10 induces pausing of elongation forks through chromosomal replication origins in Saccharomyces cerevisiae. Mol. Cell. Biol. 17, 3261–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wohlschlegel J. A., Dhar S. K., Prokhorova T. A., Dutta A., Walter J. C. (2002) Xenopus Mcm10 binds to origins of DNA replication after Mcm2–7 and stimulates origin binding of Cdc45. Mol. Cell 9, 233–240 [DOI] [PubMed] [Google Scholar]