

Background: Amyloid-β (Aβ) oligomers are key in Alzheimer disease (AD) but are diverse and poorly characterized.
Results: Multiple Aβ forms were measured across the life span of AD model mice and human AD brain.
Conclusion: Aβ species interacting with prion protein were tightly linked to behavioral impairment.
Significance: An Aβ oligomer subset with defined biochemical properties is present in multiple AD-relevant samples.
Keywords: Alzheimer disease, amyloid-beta (AB), ligand-binding protein, prion, transgenic mice, learn, memory, oligomer
Abstract
Alzheimer disease (AD) is characterized by amyloid-β accumulation, with soluble oligomers (Aβo) being the most synaptotoxic. However, the multivalent and unstable nature of Aβo limits molecular characterization and hinders research reproducibility. Here, we characterized multiple Aβo forms throughout the life span of various AD mice and in post-mortem human brain. Aβo exists in several populations, where prion protein (PrPC)-interacting Aβo is a high molecular weight Aβ assembly present in multiple mice and humans with AD. Levels of PrPC-interacting Aβo match closely with mouse memory and are equal or superior to other Aβ measures in predicting behavioral impairment. However, Aβo metrics vary considerably between mouse strains. Deleting PrPC expression in mice with relatively low PrPC-interacting Aβo (Tg2576) results in partial rescue of cognitive performance as opposed to complete recovery in animals with a high percentage of PrPC-interacting Aβo (APP/PSEN1). These findings highlight the relative contributions and interplay of Aβo forms in AD.
Introduction
Alzheimer disease (AD),3 the most common cause of dementia in the elderly (1), is characterized by progressive synaptic dysfunction and neuronal loss resulting in memory impairment, disorientation, and ultimately death (2–4). Despite its prevalence and severity, the proximal causes of AD are still elusive (5). Substantial human genetic data along with in vivo and in vitro experimental studies highlight a key role of amyloid-β peptide (Aβ) in triggering AD pathogenesis (6–8). Aβ is released through proteolytic cleavage of transmembrane amyloid precursor protein (APP) by β- and γ-secretases and accumulates in the extracellular space (9). Elevated levels of Aβ42 favor its aggregation, resulting in a diverse set of assemblies ranging from dimers to soluble multimeric oligomers to large polymeric fibrils forming the core of senile plaques, a histopathological feature of AD. The levels of Aβ monomers and fibrils are elevated in AD brain, but it is the soluble Aβ oligomers (Aβo) that are the most synaptotoxic Aβ species, with the highest correlation to memory dysfunction in transgenic mice and in humans (10–15).
Although Aβ and its oligomeric forms are strongly implicated in triggering AD, the Aβo term encompasses a very broad array of molecular species. Different studies have described the correlation of an Aβ*56 species from SDS-PAGE across the behavioral progression of one transgenic mouse strain (16), from the presence of Aβ dimers in selected fractions from human AD brain extracts, from transfected cell-conditioned medium (17, 18), and the existence of high molecular weight Aβo species in transgenic mouse brain microdialysates (19). Lack of comprehensive assessment of a particular molecular species defined by functional tools across multiple models and human disease leads to poor experimental reproducibility and creates confusion and controversy in the field (20).
For example, experiments with a range of models have shown that Aβo toxicity is critically dependent on cellular prion protein (PrPC), a cell surface glycoprotein acting as a high affinity receptor with high selectivity for Aβo (21–28). Interaction between Aβo and PrPC is essential for development of an array of AD features, including activation of certain signaling cascades, synaptotoxicity, inhibition of long term potentiation, memory impairment, and decreased survival in mice (25, 27–43). However, it was also observed that Aβo can induce AD-like phenotypes independently of PrPC in J20 mice (44) and in some in vitro studies (23, 24, 45). Evidence suggests that certain Aβo species bind to and act via PrPC, although others do not (25, 36). An ability to distinguish Aβo species based on biological or structural features would advance the field. To date, the primary tools have been conformationally specific antibodies and denaturing gel analysis. However, studies using PrP-Aβo interaction as a biologically relevant selective filter for Aβo detection have demonstrated dramatic increase in PrPC-interacting Aβo in human AD patients, with virtual absence of such Aβo species in control subjects (27, 43). Together with a high functional importance of PrP-Aβo interaction for AD pathogenesis, it makes it particularly interesting to further characterize the properties of PrPC-interacting Aβo and their impact on AD progression.
Here, we utilized several oligomer-directed quantitative assays, including a high specificity binding assay (PrP-ELISA or PLISA) (27) allowing quantitation of PrPC-interacting Aβo and two Aβo-specific ELISAs allowing quantitation of all Aβo forms. We employed these assays across brain tissue from multiple mouse strains, mouse ages, and human disease. We show that among different Aβ measures, PrPC-interacting Aβo is at least as accurate as any other predictor of memory impairment in AD mouse models and human AD patients. We find that the fraction of PrPC-interacting Aβo in the total Aβo pool varies greatly between AD models and may determine the extent to which PrPC-dependent molecular mechanisms contribute toward the expression of AD-like symptoms. These observations provide further support for the hypothesis that PrPC-Aβo interaction mediates pathophysiology in AD; however, they also indicate that other forms of Aβo with lower affinity toward PrPC may contribute toward AD progression (although less effectively), especially when PrPC-interacting Aβo are scarce.
Experimental Procedures
Human Brain Tissue
Post-mortem human brain tissue (pre-frontal cortex) from AD patients and neurologically healthy individuals was collected as approved by the Institutional Review Board at Yale and stored at −80 °C immediately after autopsy. Samples of brain tissue from each patient were grossly and microscopically examined for neuropathological features of AD consistent with the clinical diagnosis of dementia, using previously described methods (46–48). The following areas were examined histologically in all cases: middle frontal gyrus, middle temporal gyrus, inferior parietal lobule, parahippocampal gyrus, cingulate gyrus and midbrain, including the substantia nigra and locus coeruleus. Immunohistology with anti-phospho-τ and anti-β-amyloid, as well as modified Bielschowsky silver stain, were applied for detection of neurofibrillary tangles and neuritic plaques. Many of the brains were collected and classified prior to 2012 (49, 50), so diagnosis was made using the National Institutes of Health, NIA, consensus method for the neuropathological diagnosis of AD at the time of autopsy (47, 51, 52). Each pathology-confirmed AD brain in this sample was Braak stage V or higher and had a CERAD neuritic plaque score classified as Frequent. Brains of all neurologically healthy control patients had no signs or minimal signs of AD-associated histopathology, with Braak stage 0-II and CERAD neuritic plaque of None or Sparse. Patient demographical data is presented in Table 1.
TABLE 1.
Human brain tissue used in the study
| Sample size | Age | Gender | Post-mortem interval | |
|---|---|---|---|---|
| years | % female | d | ||
| AD | 12 | 77 ± 12 | 58 | 13 ± 7 |
| Control | 10 | 71 ± 14 | 50 | 17 ± 6 |
Mice
Wild-type and APP/PSen mice on the C57B6/J background were as described (29). Tg2576 mice and 5× FAD mice were obtained from Taconic Biosciences and The Jackson Laboratory, respectively. The 3× Tg-AD mice (53) were a gift of Dr. F. LaFerla, via Dr. P. Lombroso at Yale University. Brain tissue from CRND8 mice was generously provided by Todd Golde. Both males and females were used in approximately equal numbers and none were excluded. All experiments were approved by the Institutional Animal Care and Use Committee of Yale University. For numbers of animals in each genotype and age-group used in the study please refer to Table 2.
TABLE 2.
Numbers of animals of each genotype used in the study
| Age | WT | APP/PSEN1 | 5× FAD | tg2576 | CRND8 | 3× Tg |
|---|---|---|---|---|---|---|
| months | ||||||
| 1 | ||||||
| 2 | 1 | 6 | 2 | |||
| 3 | 1 | 6 | 3 | |||
| 4 | 2 | |||||
| 5 | 4 | |||||
| 6 | 2 | 3 | 1 | |||
| 7 | ||||||
| 8 | ||||||
| 9 | ||||||
| 10 | 1 | 2 | 4 | |||
| 11 | ||||||
| 12 | 1 | 2 | ||||
| 13 | 3 | 3 | ||||
| 14 | ||||||
| 15 | ||||||
| 16 | 3 | |||||
| 17 | 3 | 6 | ||||
| 18 | 2 | 2 | ||||
| Total: 63 |
Behavioral Testing of Transgenic Mice
The experimenter performing the behavioral testing was blind to genotype throughout the course of the entire experiment. Starting 5 days prior to the start of the experiment, the mice were handled for 5 min to acclimate them to the experimenter. The mice were tested over the course of 3 days in a Morris water maze (MWM) (37, 54). The testing consisted of placing the mice in a tank of water that was about 1 m in diameter to find a submerged hidden platform. The platform's location remained constant throughout each 3-day experimental period. The order that the mice were tested in remained constant within each experiment. Each mouse was tested a total of eight times per day, which was divided into two blocks of four. One block was performed in the morning, and the second was performed in the afternoon. The mice were put into the tank at four distinct locations on the opposite side from the hidden platform. The order of the locations was randomized for each block. This was done to ensure that the mice had to rely on spatial cues to find the hidden platform rather than memorizing a specific route. Each mouse had 60 s per trial to find the platform. If a mouse could not find the platform within the allotted time, the mouse would be gently guided to the platform and allowed to rest on the platform for 15 s.
A probe trial was performed 24 h after the completion of the training in the MWM. The mice were placed in the same pool as before, but the platform was removed. The start location was directly diagonal from where the platform was originally located. Each mouse had 60 s to explore the pool. The MWM training and probe trials were all recorded on a JVC Everio G-series camcorder and tracked with Panlab's Smart software. To calculate the normalized spatial learning deficit (nSLD), the following calculation was performed: nSLD = (BTg − BWT)/(ATg/2 + AWT/2), where A and B are the times it takes for a transgenic (Tg) or genetic background-matched wild-type (WT) mouse to find the hidden platform at the beginning and the end of the training, respectively. To calculate the nSMD, the following formula was used: nSMD = (tWT − tTg)/tWT, where tWT and tTg is the time a genetic background-matched wild-type or transgenic animal (respectively) spent in the target quadrant of MWM.
Animal Treatment with LY-411575
γ-Secretase inhibitor LY-411575 was obtained from Sigma (SML0506). The compound was prepared in 10% (v/v) polysorbate in water at 1 mg/ml and administered to 12-month-old APP/PSEN1 mice (n = 4) through oral gavage at 10 mg/kg. As a control, another group of animals (n = 5) was administered a vehicle solution (10% polysorbate in water). Animals were treated at 12-h intervals for 7 days and sacrificed 3 h after the last dose.
Synthetic Aβ Preparation
Synthetic Aβ(1–42) peptide was obtained as lyophilized powder from Keck Large Scale Peptide Synthesis Facility (Yale University). Each batch of peptide was reconstituted in 1,1,1,3,3,3-hexafluoroisopropanol at 10 mg/ml, boiled for 1 h. at 70 °C, aliquoted in microcentrifuge tubes at 0.5 mg/tube, and allowed to dry overnight at room temperature. The remaining trace amounts of 1,1,1,3,3,3-hexafluoroisopropanol were removed by SpeedVac centrifugation for 1 h. To prepare oligomeric Aβ, each aliquot was thoroughly resuspended in 40 μl of dimethyl sulfoxide (DMSO), divided into 20-μl aliquots, and diluted in 1 ml of Ham's F-12 medium giving a final Aβ concentration of 55 μm. The tubes were incubated overnight at room temperature to allow oligomerization (21, 27, 37, 55). For monomeric Aβ preparations, the overnight incubation step was omitted, and the peptide was assayed immediately after F-12 dilution.
Recombinant PrP(23–111) Production
Purification of PrP(23–111) protein was described before (27, 56). DNA encoding amino acids 23–111 of human PrPC was cloned into pRSET-A vector with an N-terminal extension encoding a hexa-histidine tag and a thrombin cleavage site. Plasmid-transformed BL21 (DE3) Escherichia coli (Agilent) cells were cultured overnight in noninducing MDAG135 medium and then diluted 1:100 in ZYM-5052 auto-inducing medium and grown for 16 h at 37 °C. Bacteria were lysed in Buffer G (6 m guanidine HCl, 100 mm Na2HPO4, 10 mm Tris-HCl, pH 8) and then centrifuged at 100,000 × g for 1 h. The supernatant was applied to nickel-nitrilotriacetic acid resin. To refold bound protein, a 20–100% stepwise gradient of Buffer B (100 mm Na2HPO4, 10 mm Tris-HCl, 10 mm imidazole, pH 8) in Buffer G was applied. After washing the resin with 50 mm imidazole in Buffer B, bound protein was eluted with 100 mm Na2HPO4, 10 mm Tris-HCl, 500 mm imidazole, pH 5.8, and dialyzed against 10 mm Na2HPO4, pH 5.8, and then against water. Protein concentration was determined by A280 method (extinction coefficient for PrP 23–111 is 42,390). Final yields were 30–40 mg of protein per liter of culture.
Human and Mouse Brain Tissue Fractionation
Frozen whole mouse brains or human post-mortem pre-frontal cortex samples (27, 37) were stored at −80 °C until processed. Before extraction, each tissue fragment was weighed to maintain consistent w/v ratio of tissue to homogenization buffer. First, to extract soluble material, each piece of tissue was Dounce-homogenized on ice in 3 volumes of TBS, pH 7.4, with sequential sets of 20 strokes of loose and tight pestle. The homogenization buffer was supplemented with 1× Complete protease inhibitors and 1× PhosSTOP phosphatase inhibitors (Roche Applied Science) to prevent protein degradation. The TBS-insoluble material was pelleted by centrifugation at 100,000 × g for 1 h. The supernatant was then collected, aliquoted in microcentrifuge tubes, flash-frozen in liquid nitrogen, and stored at −80 °C until assayed. This supernatant was referred to as a TBS fraction. The pellets were resuspended in 4 volumes of TBSX (TBS, 1% Triton X-100, pH 7.4) supplemented with phosphatase and protease inhibitors and incubated on a nutator for 1 h at 4 °C to solubilize membrane-associated proteins. The TBSX-insoluble material was removed by centrifugation at 100,000 × g for 1 h. The supernatant was processed similarly to the TBS fraction and referred to as TBSX fraction. Finally, the TBSX pellets were resuspended in 3 volumes of 70% formic acid, incubated for 30 min at room temperature, and cleared by centrifugation at 100,000 × g for 1 h. The pellets were discarded, and the supernatant was diluted 5-fold with water, divided in 100-μl aliquots, frozen in liquid nitrogen, and lyophilized to remove formic acid. The lyophilized material was resuspended in 40 μl of 8 m urea, 20 mm Tris, pH 7.4, stored at −80 °C, and referred to as FA-extracted fraction.
Measuring the Levels of PrPC-interacting Aβ Oligomers and Total Aβ Oligomers
We modified a previously described (27) plate-based assay for measuring PrPC-interacting Aβo to achieve higher sensitivity, referred to as PLISA. Low volume high binding white microplates (Greiner 784074) were coated overnight with 20 μl/well of 250 nm human PrP AA23-111 in 30 mm Na2CO3, 80 mm NaHCO3, pH 9.6, at 4 °C. After washing two times with PBST (PBS, 0.05% Tween 20), the plates were blocked with 25 μl/well protein-free T20 PBS blocking buffer (Pierce) for 4 h at room temperature. After washing three times with PBST, 20 μl of TBS or TBSX fraction samples diluted 1:2–1:4 in PBSTB (PBS, 0.05% Tween 20, 0.5% bovine serum albumin) were applied to microplates in triplicate and incubated overnight at 4 °C. 2-Fold serial dilutions of synthetic Aβo in PBSTB (0–10 nm range) were included in each plate as a standard curve. Plates were then washed four times with PBST and incubated with D54D2 anti-Aβ antibody (epitope in the N terminus of Aβ peptide, Cell Signaling Technology, catalog no. 8243) diluted 1:2000 in PBSTB for 2 h. After washing four times in PBST to remove the unbound primary antibody, 20 μl/well of 1:8000 dilution of Eu-N1 goat anti-rabbit IgG (PerkinElmer Life Sciences) in DELFIA assay buffer (PerkinElmer Life Sciences) was applied for 1 h. Finally, after washing five times in PBST, 20 μl of DELFIA Enhancement Solution (PerkinElmer Life Sciences) was applied to each well, and time-resolved europium fluorescence was measured using a Victor 3V plate reader (PerkinElmer Life Sciences). To measure the levels of total Aβ oligomers, two ELISAs were developed. Overall processing of the plates and reagents used was the same as in PLISA, with a few modifications discussed below. In the first assay (82e1-8243), 82e1 mouse monoclonal antibody (IBL, 10323) recognizing the N terminus of the Aβ peptide was used for coating plates (1:500 dilution in 30 mm Na2CO3, 80 mm NaHCO3, pH 9.6, overnight at 4 °C), and D54D2 antibody was used for detection (1:2000 in PBSTB). In the second assay (8243-Nu4) plates were coated with D54D2 antibody (1:500 dilution) and conformation-specific anti-Aβ oligomer antibody Nu4 was used for detection (1:2000 in PBSTB).
Affinity Depletion of PrPC-interacting Aβ Species
1.5 ml of pooled TBS brain extracts from the brains of Alzheimer patients were incubated overnight at 4 °C with 100 μl of protein A-Sepharose CL-4B (GE Healthcare, 17-0780-01) preconjugated with 50 μg of recombinant human PrP-Fc or, alternatively, with human Fc (Jackson ImmunoResearch 009-000-008), which served as a negative control resin. The generated affinity-depleted and control lysates as well as Alzheimer and control lysates prior to the resin incubation were analyzed by PLISA and 82e1-8243 assays directly or following size-exclusion chromatography fractionation.
Aβ ELISA and Western Blot
For Western blot (WB) measurements, TBS, TBSX, and FA fractions were diluted 1:1 with 2× Laemmli Sample buffer, incubated for 5 min at 95 °C, and separated on 10–20% Criterion Precast Tris-Tricine gels (Bio-Rad). The heating step was omitted for the urea-containing FA fraction. Additionally, monomeric preparation of synthetic Aβ at 32, 64, and 128 nm was loaded on each gel alongside the samples to allow quantitation and inter-membrane comparison. Following the gel electrophoresis, proteins were transferred to nitrocellulose membranes using the iBlot semi-dry method (Life Technologies, Inc.). After transfer, the membranes for TBS and TBSX fractions were microwaved in PBS until boiling to unmask the epitopes and increase the immunoblot sensitivity (57). All membranes were blocked with Blocking Buffer for Fluorescent Western blotting (Rockland) for 30 min at room temperature and then incubated with 1:2000 dilution of D54D2 anti-Aβ antibody in PBST overnight at 4 °C. After washing three times with PBST, IRDye 800CW-conjugated donkey anti-rabbit secondary antibody (LiCor; 1:5000 in PBST) was added for 2 h at room temperature. Finally, the membranes were washed three times with PBST and imaged using Odyssey near-infrared scanner (LiCor). The signal intensity of Aβ-immunoreactive bands was quantitated using ImageJ software package. For ELISA measurements, a commercially available Aβ(1–42) detection kit was used (Life Technologies, Inc., KHB3442). The measurements were performed according to the manufacturer's recommendations. The TBS and TBSX fractions from both mice and humans were diluted in Sample Diluent Buffer 1:1, and the optimal dilution for FA fraction was empirically determined to be 1:800 for humans and 1:32,000 for mice. The absorbance at 450 nm was measured using a Victor 3V plate reader (PerkinElmer Life Sciences), and the absolute amount of Aβ was determined using the standard curve based on serial dilutions of monomeric preparation of synthetic Aβ.
Size-exclusion Chromatography
Oligomeric and monomeric preparations of synthetic Aβ at 100 nm, as well as TBS-extracted material from mouse brains and human post-mortem prefrontal cortex, were separated on TSKGel G3000SWxl gel filtration column (Tosoh) using AKTA purifier FPLC system (GE Healthcare). 200 μl of sample was injected at a flow rate of 0.75 ml/min. PBS, pH 7.4, was used as a mobile phase. Fractions of 0.75 ml were continuously collected throughout the whole run and analyzed the same day for Aβ content. The separation and fraction collections were performed at 4 °C. The molecular weight estimations were performed using the gel filtration standard kit (Bio-Rad) containing thyroglobulin, γ-globulin, ovalbumin, myoglobin, and vitamin B12 (molecular mass of 670, 158, 44, 17, and 1.3 kDa, respectively) as a reference.
Histology
Brains were obtained from 3- and 12-month-old APP/PSEN1 mice, 4- and 18-month-old Tg2576 mice, and 18-month-old 3× mice. The brains were embedded in gelatin. 40-μm sagittal sections were cut using A Leica VT 1000 S vibratome. Sections were stained for amyloid plaque deposition using thioflavin-T (ThT) and immunofluorescence. Sections used for ThT were washed in PBS for 5 min and then mounted on Superfrost Plus slides (Fisher) and allowed to dry completely. The slides were then incubated at room temperature for 15 min in a 0.1% ThT solution made in 70% ethanol. The slides were washed twice in 70% ethanol and then twice in distilled water, 1 min per each wash. The slides were then dried, covered in fluorescence mounting media (Vectashield) and a coverslip, and sealed.
Sections used for immunofluorescence were washed in PBS for 5 min and then washed in blocking buffer (10% normal donkey serum, 0.2% Triton X-100, in PBS) for 1 h. A primary antibody solution was made with a 1:400 concentration of β-amyloid antibody (catalog no. 2454, Cell Signaling Technology, made in rabbit) in PBS with 1% normal donkey serum and 0.2% Triton X-100. Sections were washed in this primary antibody solution overnight at 4 °C. The sections were then washed three times in PBS for 10 min per wash. A secondary antibody solution was made with a 1:600 concentration of an anti-rabbit antibody (made in donkey, Invitrogen, Alexa Fluor 594) in PBS with 1% normal donkey serum and 0.2% Triton X-100. Sections were washed in this secondary solution for 2 h at room temperature, and then three times in PBS for 10 min per wash. Sections were mounted on Superfrost Plus sides (Fisher), allowed to dry completely, covered in fluorescence mounting media (Vectashield) and a coverslip, and sealed.
Sections were imaged on an UltraVIEW VoX (PerkinElmer Life Sciences) spinning disc confocal microscope with a ×4 objective (CFI Achro Flat Field, NA 0.1, air). A solid state 488 nm laser (Coherent) was used to image the ThT-stained sections, and a solid state 561 nm laser (Cobolt) was used to image the sections with the β-amyloid antibody (catalog no. 2454). Images were taken of the frontal cortex.
Results
PrPC Binding Aβo Levels Are Increased in Old APP/PSEN1 Mice and in Human AD Patients
To verify that the presence of PrPC-interacting Aβ oligomers in biological samples is specifically confined to AD-relevant samples, but not to control samples, we assessed an optimized PLISA in aged (15–18 months) AD model APPswe/PSen1ΔE9 (APP/PSEN1) mice in comparison with wild-type animals. At this age, APP/PSEN1 mice have fully developed impairment of learning and memory as well as large amounts of fibrillar Aβ deposited in the form of plaques across the cerebral cortex and hippocampus (29, 58, 59). In whole-brain TBS extracts, PLISA measurement detected PrPC-interacting Aβ species only in APP/PSEN1 and not in WT brain homogenates (Fig. 1a; APP/PSEN1 mice, 33 ± 3.6 ng/g of brain, n = 7; WT, below 4 pg/ml detection limit, n = 5; p = 0.0001), thus confirming that the presence of PrPC-interacting Aβ is a distinctive feature of cognitively impaired AD model mice.
FIGURE 1.

Characterization of PrPC-interacting Aβo. Using PLISA, amounts of PrPC-interacting Aβ oligomers were quantified in brain lysates from wild-type (black dots) and APP/PSEN1 (red dots) transgenic mice (a) as well as from neurologically healthy (black dots) and AD-affected (red dots) human individuals (b). Human AD patient and APP/PSEN mouse samples showed considerably higher levels of Aβo compared with WT mice and human control samples, which had only marginally detectable levels of Aβo. Synthetic Aβ was oligomerized in F-12 medium and separated using size-exclusion chromatography (c). Aβo peak elutes in early fractions (*), whereas monomeric Aβ elutes at the very end of separation (monomeric Aβ peak corresponds to highly absorbing components of F-12 medium, large peak at Vc). SEC fractionation was performed on oligomeric (oAβ, straight lines) and monomeric preparations (mAβ, dashed lines) of synthetic Aβ (d), and fractions were assayed using PLISA (black lines) and conventional ELISAs (red lines). PLISA demonstrated strong preference toward high molecular weight Aβ assemblies showing strong peak in column void volume (Vo) and early fractions, particularly prominent in oligomeric preparation. ELISA was mostly specific toward monomeric forms of Aβ resulting in sharp peak close to fractions corresponding to total column volume (Vc). In analogy with synthetic Aβ preparations, SEC fractionation coupled to PLISA and ELISA was performed on TBS brain lysates of APP/PSEN1 (Tg, straight lines) and wild-type control (WT, dashed lines) mice (e) as well as TBS lysates from post-mortem brain tissue from human AD patients (AD, straight lines) and neurologically healthy control individuals (CTRL, dashed lines) (f). Similarly to synthetic material, PLISA was highly selective toward HMW Aβ assemblies only in AD-related but not in control samples, whereas ELISA was mostly detecting monomeric Aβ. PLISA activity forms a sharp peak in HMW fractions, whereas the activity of Aβ oligomer-specific ELISAs (8243-Nu4, 82e1-8243) distributes proportionally to the amount of eluting Aβo (g). Specificity of PLISA toward HMW Aβo was also true for TBS brain lysates from old APP/PSEN1 mice, whereas 82e1-8243 detected a broader range of Aβo species (h). Bio-Rad Gel Filtration protein standards were used to aid with the molecular weight determination of Aβ species: peak 1, thyroglobulin (bovine), 670 kDa; peak 2, γ-globulin (bovine), 158 kDa; peak 3, ovalbumin (chicken), 44 kDa; peak 4, myoglobin (horse), 17 kDa; peak 5, vitamin B12, 1.35 kDa. mAU, milliabsorbance units. Error bars represent S.E.
Next, we investigated whether the specific presence of PrPC-interacting Aβo in AD-relevant samples is generalizable to human brain. Our prior use of PLISA to assess human brain samples preceded assay optimization but demonstrated statistical separation of AD from control with some overlap of values near the detection limit of the assay (27). Here, we utilized an optimized PLISA on TBS extracts from human post-mortem prefrontal cortex from AD patients and age-matched controls free of AD pathology. All AD samples had nanogram amounts of Aβo per g of brain tissue, whereas the average amount of Aβo in control brain was more than 2 orders of magnitude lower (Fig. 1b; AD, 13.19 ± 3.19 ng/g of brain, n = 12; control, 0.10 ± 0.05 ng/g of brain, n = 9; p = 0.0017). Not surprisingly, due to interpersonal variation the human samples exhibited a greater variability of PLISA values than inbred APP/PSEN1 mice. However, even the lowest PLISA value for the AD sample (2.47 ng/g) was almost five times larger than the highest control value for the control human brain (0.50 ng/g), thus validating the level of PrPC-interacting Aβ in differentiating between samples from AD-affected and neurologically healthy individuals. Interestingly, the average absolute values of PLISA activity in human AD patients were similar (tens of nanograms/g of brain) to APP/PSEN1 mice at the stage when they exhibit an AD-like behavioral deficit, suggesting consistent potency of PrPc-interacting Aβo to induce AD-related changes in both APP/PSEN1 mice and humans.
PLISA Specifically Detects Soluble Oligomeric Aβ Assemblies of High Molecular Weight
The major distinction between the numerous previously described and presumed toxic Aβo assemblies is their size (20). Therefore, we sought to determine the hydrodynamic dimensions of PrPC-interacting Aβ by SEC. First, we fractionated 100 nm oligomeric and monomeric synthetic Aβ preparations and analyzed the fractions using PLISA and commercially available Aβ42 ELISA methods. The PLISA signal of F-12 oligomeric Aβ preparation peaked in the column's high molecular weight early eluting fractions (Fig. 1d). Thus, based on monitoring of UV absorbance at 280 nm, the fractions showing highest PLISA activity are consistent with the regions of chromatogram where Aβo elutes (Fig. 1c, 1st peak). The monomeric preparation of Aβ shows only a modest PLISA activity peak in high molecular weight fractions while demonstrating negligible signal in fractions corresponding to the Aβ monomer. Conversely, the Aβ42 ELISA signal shows a clear bias toward monomeric species of Aβ in both the monomeric and oligomeric preparations. It is possible that the Aβ42 ELISA is completely selective for monomer, and the small ELISA activity peak in high molecular weight fractions in oligomeric preparation could be explained by disassembly of oligomers, because the dynamic equilibrium is shifted toward the monomeric state after dilution (Fig. 1d).
To assess biological samples, we performed PLISA and Aβ42 ELISA on SEC fractions of TBS extracts from post-mortem brains of AD patients and aged APP/PSEN1 mice, with extracts from post-mortem brains of neurologically healthy humans and WT mice as controls, respectively. Lysates from both APP/PSEN1 mice and human AD patients had a single sharp PLISA activity peak eluting in early SEC fractions, consistent with PrPC selectivity toward large Aβ assemblies observed for synthetic Aβ preparations. PLISA activity in TBS extracts from the brains of WT mice and human controls was marginal or undetectable across all the fractions. Conversely, as with synthetic Aβ preparations, the major peak of Aβ42 ELISA activity eluted in late SEC fractions consistent with the size of monomeric Aβ. Although both WT mice and control human lysates demonstrated lower ELISA activity than their APP/PSEN1 and AD counterparts, the separation was not as dramatic as for high molecular weight PLISA activity (Fig. 1, e and f). Sequential TBS extractions from human and mouse tissues showed a progressive decrease in PLISA levels, indicating that PrPC binding activity comes from a distinct population of soluble Aβo assemblies rather than mechanical fragmentation of abundant insoluble Aβ fibrils (data not shown).
PrPC-interacting Aβo Represents a Distinct Pool of High Molecular Weight Aβ Assemblies
To determine whether or not PrPc is binding all types of Aβo indiscriminately or demonstrates selectivity for certain species, we compared the size distribution of PLISA activity to two oligomer-specific ELISAs developed here for this purpose. The first assay (82e1-8243 sandwich ELISA) relies on a pair of antibodies recognizing identical epitopes within the N terminus of Aβ and therefore detects multimeric Aβ assemblies exclusively. The second assay (8243-Nu4 sandwich ELISA) relies on the conformation-specific antibody Nu4, which recognizes oligomeric Aβ but not monomeric or fibrillar Aβ (60). We have tested the assay specificity by incubating the plates with monomeric Aβ, F-12 Aβo (ADDL) preparation, or a globulomer Aβo preparation. Both oligomer ELISAs are highly specific toward oligomeric Aβ; however, both 82e1-8243 and 8243-Nu4 demonstrated higher signals when incubated with globulomer Aβo, whereas PLISA demonstrated greater reactivity with the F-12 preparation of Aβo (Fig. 2, a–c). Globulomer preparation consists of highly homogeneous, smaller molecular weight oligomers (10–12-mers) (61), although the F-12 preparation is more polymorphic with a higher proportion of HMW assemblies (21). Therefore, it is likely that PrPC preferentially binds the HMW subset of Aβo, thus resulting in a higher signal when incubated with F-12 preparation. This observation is further supported by distribution of 82e1-8243 oligomer ELISA, 8243-Nu4 oligomer ELISA, and PLISA activity after SEC fractionation of Aβo. PLISA activity elutes as a sharp peak that is largely confined to HMW fractions, whereas 82e1-8243 and 8243-Nu4 activities are distributed proportionally to the amount of Aβo in the eluted fraction (based on absorbance at 280 nm; Fig. 1g). As a result, 82e1-8243 and 8243-Nu4 allow unbiased quantitation of all forms of Aβo in the sample, whereas PLISA specifically measures the fraction of PrPC-interacting HMW Aβo assemblies. A combination of these assays allows a more comprehensive picture of Aβo accumulation in biological samples.
FIGURE 2.
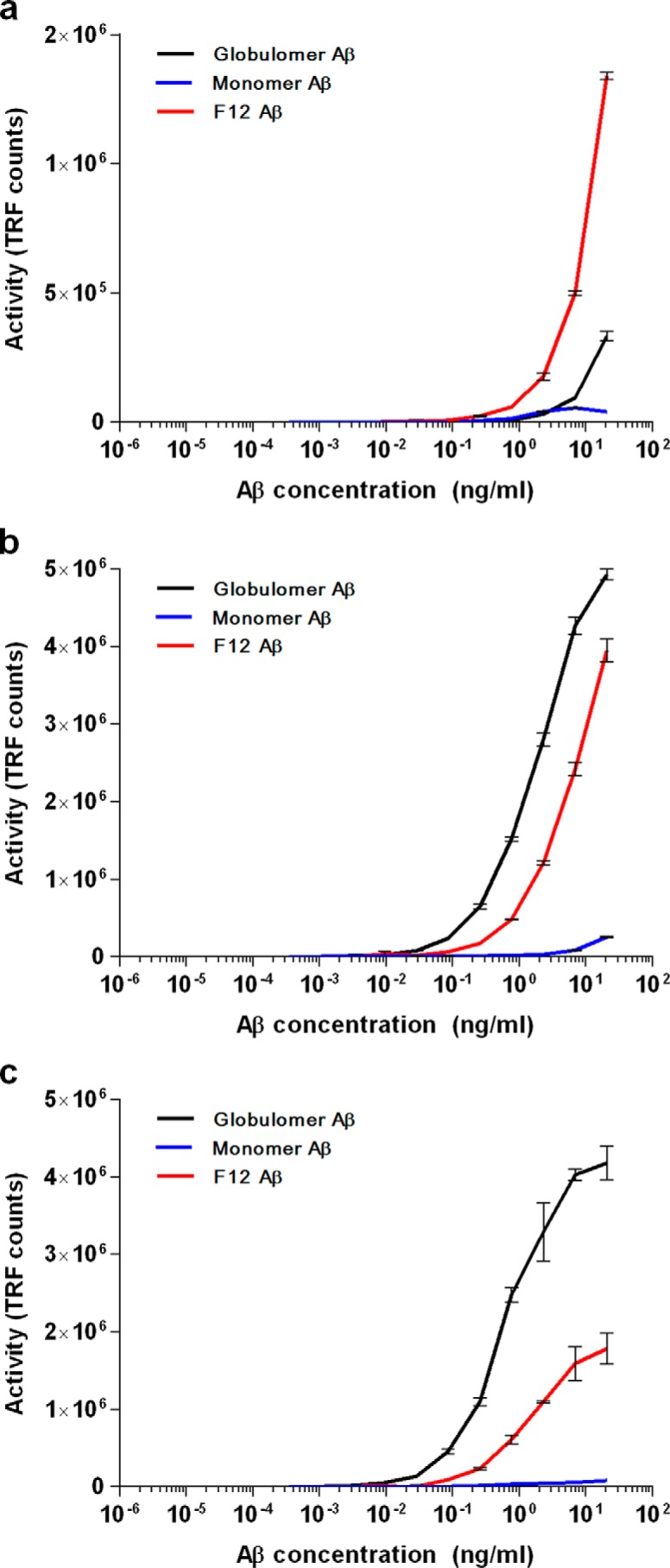
Relative detection of Aβ oligomer in different assays. Dose-dependent binding of monomeric, F-12 (oligomeric), and globulomer (oligomeric) preparations of synthetic Aβ(1–42) in PLISA (a), 82e1-8243 (b), and 8243-Nu4 (c) assays. Error bars represent S.E. of assay technical replicates. TRF, time-resolved fluorescence.
We therefore performed 82e1-8243 ELISA and PLISA on SEC-fractionated APP/PSEN1 mouse TBS brain extracts to characterize the distribution of oligomeric species. Similar to results with synthetic Aβo, PLISA activity elutes as a sharp peak in early HMW fractions, with an additional smaller peak of activity around 200 kDa indicating the presence of other PrPC-interacting species absent in synthetic preparation (Fig. 1h). The first and major peak of 82e1-8243 oligomer ELISA activity also elutes in HMW fractions, although later than PLISA activity and has two smaller additional peaks. The second peak of 82e1-8243 ELISA activity appears to co-elute with the smaller PLISA activity, suggesting that these peaks were generated by detection of the same Aβo species. Together, this combination of assays distinguishes multiple Aβo forms in the brains of APP/PSEN1 mice after the onset of AD-like symptoms.
Levels of PrPC-interacting Aβo Increase with Age and Predict Behavioral Impairment
Because the presence of PrPC-interacting high molecular weight Aβo was exclusively confined to AD-related samples, we tracked changes in PrPC-interacting Aβo through the life span of APP/PSEN1 mice. These mice are extensively used as an AD model and have been thoroughly characterized in terms of behavioral impairment, and therefore they provided a good system to assess the ability of PrPC-interacting Aβo to predict the severity of behavioral deficit (58, 62). Here, we used the MWM hidden platform task (54) to track the development of learning deficits in APP/PSEN1 mice as a function of age.
Compared with WT mice, APP/PSEN1 mice show a progressive decline in cognitive function from normal spatial learning at 3 months to nearly complete impairment at 18 months (Fig. 3a). In association with this age-dependent disease progression, we performed measurements of PrPC-interacting Aβo in TBS-soluble material, as well as in a subsequent detergent-soluble fraction extracted with 1% Triton X-100 in TBS (TBSX) to assess the amount Aβo in the membrane-associated state. Both TBS and TBSX fractions demonstrated comparable levels of PLISA activity, suggesting equal proportions of free and membrane-associated PrPC-interacting Aβo in both mouse and human brains. The amount of Aβo in both TBS and TBSX extracts increased with mouse age, reaching the mean value observed in human AD patients (13.2 ± 3.2 and 14.4 ± 4.1 ng/g brain tissue for TBS and TBSX, respectively) at about 8–10 months and continuing to rise to 32.3 ± 0.8 ng/g for TBS and 42.2 ± 3.2 ng/g for TBSX at 18 months (Fig. 3a). Interestingly, APP/PSEN1 mice start to develop signs of cognitive impairment (demonstrate learning deficit in MWM hidden platform spatial navigation task) at 8–12 months, approximately the same time that their PrPC-interacting Aβo reaches the average level observed in human AD brains. Moreover, regression analysis of these data demonstrates a very strong connection between PLISA and MWM navigation performance, highlighting the precision of PrPC-interacting Aβo levels in predicting cognitive impairment in APP/PSEN1 mice (Fig. 3b, linear regression curve fit, n = 10; r2 = 0.99, p < 0.0001).
FIGURE 3.

Levels of PrPC-interacting Aβo rise with age and are tightly linked to development of learning deficit in multiple mouse models of AD. a, levels of PrPC-interacting Aβo were measured using PLISA in TBS (black line) and TBSX (red line) brain protein extracts from APP/PSEN mice at 3, 6, 12, and 18 months of age, as well as from human AD-affected (red squares) and control (black squares) individuals. Learning deficits were also assessed in APP/PSEN mice using the Morris water maze hidden platform navigation task at 3, 12, and 18 months (APP/PSEN, dashed blue line; WT, dashed green line). PLISA activity levels increased with the age of APP/PSEN mice and were highly coherent with the progressive decline in spatial learning. Regression analysis using linear growth curve fit further demonstrated the link between PLISA levels and learning deficit (b). Error bars represent S.E. c, levels of PrPC-interacting Aβ oligomers were measured across multiple ages of WT (2, 3, 10, 12, 16, and 18 months), CRND8 (2, 5, and 10 months), 3× Tg (4, 13, and 16 months), Tg2576 (3, 6, 10, 14, and 16 months), 5× FAD (2 and 6 months), and APP/PSEN1 (3, 6, 12, and 18 months) as well as human AD and control individuals (please see Tables 1 and 2 for details on sample sizes for each group). All AD model mice but not WT animals demonstrated an increase in PrPc-interacting Aβ oligomers; however, the rates of accumulation varied greatly between models. To assess interstrain comparison of cognitive decline, behavioral data from in-house experiments (Tg2576, APP/PSEN1, 3× Tg) as well as literature data (5× FAD, CRND8 (65, 69, 70)) were converted into a normalized performance in Morris water maze hidden platform navigation task (normalized spatial learning deficit, nSLD) (d) and Morris water maze probe trials (nSMD) (e). SEC characterization of brain lysates from WT, 5× FAD, Tg2576, and 3× Tg mice coupled to PLISA (f) and Aβ42 ELISA (g) have demonstrated results consistent with ones observed for APP/PSEN1 mice in Fig. 1; PrPC-interacting Aβo specifically detected by PLISA were represented by HMW Aβ assemblies highly conserved across all tested mouse strains, whereas Aβ42 ELISA was largely specific toward monomeric Aβ. Regression analysis using exponential growth curve fit demonstrated that PrPc-interacting Aβ oligomers possess an excellent ability to predict the decline of cognitive performance in mice, both in form of spatial learning (h) and spatial memory (i). Error bars represent S.E.
PrPC-interacting Aβo Linked to Cognitive Impairment in Multiple AD Mouse Models
To determine the predictive potential of PrPC-interacting Aβo measurement for cognitive impairment, we performed PLISA measurements throughout the life span of four widely used AD mouse models (in addition to APP/PSEN1 mice) (63, 64) as follows: CRND8 transgenic mice (65), 3× transgenic mice (53), 5× FAD transgenic mice (67), and Tg2576 transgenic mice (68). Because of the differences in genetic backgrounds and slight variations under “Experimental Procedures” (for instance, the diameter of the maze used in the study), the direct interstrain comparison of cognitive performance between multiple AD mouse models using standard behavioral metrics (like the time the mouse spends in the target quadrant of the maze or the time the mouse takes to find the hidden platform) is extremely challenging. To account for such experimental variations and therefore to make it possible to test the generalizability of our observations on multiple AD mouse strains, we have calculated the normalized cognitive performance coefficients based on animal performance in MWM spatial navigation task (nSLD) and MWM probe trials to assess spatial memory deficit (nSMD). We have then correlated the accumulated PLISA data with nSLD and nSMD. Behavioral experiments on 3×, APP/PSEN1, and Tg2576 mice were performed in-house with the same colony as the Aβo measurements, whereas the behavioral data for CRND8 and 5× FAD mice were extracted from the literature (65, 69, 70). All tested AD models demonstrated a progressive increase in PrPC-interacting Aβo; however, the rates of accumulation as well as maximal Aβo levels dramatically differed from strain to strain (Fig. 3c). Thus, 5× FAD and CRND8 mice demonstrated the most rapid increase in PrPC-interacting Aβo concentration, reaching values 2–3 times higher than APP/PSEN1 mice almost three times faster. Conversely, Tg2576 and particularly 3× transgenic mice were much slower Aβo accumulators, having comparatively low PLISA activity even at 16 months. In terms of cognitive impairment, the trends in both nSLD and nSMD mirrored that observed in PLISA measurements (Fig. 3, d and e) as follows: deterioration of both spatial learning and spatial memory progressed much faster in APP/PSEN1, 5× FAD, and CRND8 mice and slower in Tg2576 and 3× mice. Interestingly, despite dramatic interstrain differences in the rates of PrPC-interacting Aβo accumulation, the size distribution of PLISA activity was similar in all strains and was eluting as a distinct peak at HMW range (Fig. 3, f and g).
Finally, linear regression analysis demonstrated that the capacity of PrPC-interacting Aβo levels to predict cognitive impairment could be extrapolated from APP/PSEN1 to multiple mouse AD models. Moreover, combining data from all transgenic strains provides a strong highly significant correlation of PrPC-interacting Aβo levels with mouse dysfunction of learning and memory (Fig. 3, h and i). Taken together, the examined AD mouse models exhibited a continuous progression of pairs of ln(PLISA)-nSLD and ln(PLISA)-nSMD values, with nSLD showing a better match to PLISA levels than does nSMD (ln(PLISA:nSLD): n = 64, Dfd = 62, r2 = 0.77, p < 0.0001; ln(PLISA): nSMD: n = 62, Dfd = 60 r2 = 0.50, p < 0.0001; where n represents the total number of animals for which both biochemical Aβ measurements and cognitive performance measurements are available). These high correlations are observed with behavioral data derived from our laboratory for three strains and from literature citations for two strains. Any differences between the literature handling and housing would only degrade the correlations. In this setting, the high correlations observed for oligomeric Aβ species are even more striking.
Accumulation of Other Aβ Subsets in Multiple Mouse Strains
To evaluate the association between PrPC-interacting Aβo and cognitive decline, we compared the predictive power of PLISA with an array of metrics commonly used to characterize Aβ burden in biological samples, as well as newly developed 82e1-8243 and 8243-Nu4 oligomer-specific ELISAs. Specifically, we performed three-step sequential protein extraction (TBS followed by TBSX followed by FA) from the brains of APP/PSEN1, 3×, 5× FAD, Tg2576, and CRND8 mice of various ages, and we analyzed the lysates by immunoblotting and ELISAs (see “Experimental Procedures” for details). Additionally, ThT and immunofluorescent staining for Aβ were performed on brain sections of 3×, APP/PSEN1, and Tg2576 mice to calculate the amyloid plaque burden in cerebral cortex. These transgenic mice are extensively studied and have been thoroughly characterized in terms of development of AD-like biochemical, histological, and behavioral pathologies (63, 64). To our knowledge, no investigation of similar scope comparing the pattern of Aβ accumulation across multiple models and its effects on cognitive performance has been performed.
Because WB measurements performed on TBS- and TBSX-extracted material demonstrated similar absolute levels of Aβ in both fractions, these two metrics were averaged into a single one, “Total Soluble Aβ.” The WB samples were denatured by boiling in SDS, so Aβ monomer bands under these conditions include monomeric Aβ and dissociated Aβ from oligomers, thereby estimating total soluble Aβ independently of polymerization status. WB-quantified levels of total soluble Aβ progressively increased with animal age in all model mice, although the absolute values and accumulation rates varied dramatically from strain to strain, resembling the pattern observed in PLISA (Fig. 4, a and b). Similarly to PLISA measurements, 5× FAD and CRND8 mice were quick to accumulate Aβ; at 6 and 10 months, respectively, they demonstrated a 8–10-fold higher amount of total soluble Aβ than average human AD brain (5× FAD at 6 months: 625 ± 35 ng of Aβ/g of brain tissue, n = 3; CRND8 at 10 months: 481 ± 40 ng of Aβ/g of brain tissue, n = 3; human AD: 81 ± 13 ng of Aβ/g of brain tissue). APP/PSEN1 and Tg2576 animals were slower in attaining high levels of total soluble Aβ, reaching 3–4 times the level observed in human AD brains at 16–18 months of age (APP/PSEN1: 230 ± 45 ng of Aβ/g of brain tissue, n = 2; Tg2576: 248 ng of Aβ/g of brain tissue, n = 1). 3× transgenic mice were the slowest to accumulate WB-detectable soluble Aβ only achieving about 20% of human AD average (3× Tg at 16 months: 14 ± 5 ng of Aβ/g of brain tissue, n = 6). Similarly to WB, values from Aβ42 ELISAs on TBS and TBSX extracts were merged into a single variable and referred to as “Soluble Aβ Monomer” due to the high bias of the Aβ42 ELISA kit used toward monomeric Aβ. Consistent with WB results, Aβ42 ELISA showed age-dependent accumulation of soluble Aβ with a large divergence in Aβ levels between models (Fig. 4c). 5× FAD and CRND8 mice were the fastest to accumulate soluble Aβ monomers; APP/PSEN1 and Tg2576 showed intermediate rate of accumulation, and 3× was the slowest (5× FAD at 6 months: 4.44 ± 0.48 ng of Aβ/g of brain tissue, n = 3; CRND8 at 10 months: 4.68 ± 0.70 ng of Aβ/g of brain tissue, n = 2; APP/PSEN1 at 18 months 1.45 ± 0.19 ng of Aβ/g of brain tissue, n = 2; Tg2576 at 16 months: 0.49 ± 0.09 ng of Aβ/g of brain tissue, n = 3; 3× at 16 months: 0.11 ± 0.05 ng of Aβ/g of brain tissue, n = 6). Finally, we used WB and Aβ42 ELISA to measure the levels of FA-extracted Aβ, a fraction that should mostly contain insoluble Aβ that was deposited in brain as amyloid plaques. Consistently with ELISA, PLISA, and WB data, AD mice showed high inter-strain variation in Aβ accumulation rates and in absolute Aβ levels, whereas each of the strains taken separately demonstrated age-dependent increase in FA-soluble Aβ (Fig. 4d; 5× FAD at 6 months: 71,000 ± 2000 ng of Aβ/g of brain tissue, n = 3; CRND8 at 10 months: 79,000 ± 1200 ng of Aβ/g of brain tissue, n = 3; APP/PSEN1 at 18 months: 69,000 ± 2000 ng of Aβ/g of brain tissue, n = 2; Tg2576 at 16 months: 18,000 ± 5000 ng of Aβ/g of brain tissue, n = 3; 3× at 16 months: 1300 ± 400 ng of Aβ/g of brain tissue, n = 6).
FIGURE 4.

Multiple Aβ-related metrics demonstrate coherent increase of multiple fractions of Aβ as a function of mouse age. Total protein was sequentially extracted from the brains of human AD patients and neurologically healthy individuals as well as from brains of several mouse strains of multiple ages (WT: 2, 3, 10, 12, 16, and 18 months; CRND8: 2, 5, and 10 months; 3× Tg: 4, 13, and 16 months; Tg2576: 3, 6, 10, 14, and 16 months; 5× FAD: 2 and 6 months; APP/PSEN1: 3, 6, 12, and 18 months). All the lysates were then assayed for Aβ levels using ELISA and immunoblotting. a, representative images of immunoblots used to assess the level of total Aβ in brain lysates after sequential extraction with TBS, TBSX, and FA. Monomeric preparation of synthetic Aβ at 32, 64, and 128 ng (missing in TBSX blots) were run alongside lysates to aid with absolute quantitation. Averaged results of WB immunoblot quantitation of Aβ in TBS and TBSX fractions (total soluble Aβ) are graphed in b. Levels of total soluble Aβ in increased with animal age in all model mice, although the accumulation rates varied: 5× FAD and CRND8 mice were accumulating Aβ much faster than APP/PSEN mice, whereas Tg2576 and 3× mice were slower Aβ accumulators. A similar discrepancy in Aβ accumulation rates was observed for the levels of soluble Aβ measured by ELISA (c) as well as levels of Aβ in formic acid-extracted material (d), average of quantitation of immunoblots on FA-extracted material and ELISA on FA-extracted material. Overall, levels of multiple subsets of Aβ increase in coherence as a function of mouse age. Error bars represent S.E.
Both ThT and immunofluorescent anti-Aβ staining allowed visualization of the formation of amyloid plaques in cerebral cortex (Fig. 5) of old but not young mice. The area occupied by plaques determined by histological metrics was directly proportional to the levels of FA-soluble Aβ detected by ELISA and WB, confirming similar bias of these methods toward measuring insoluble Aβ deposits (Fig. 5, b and c; % total brain area covered by amyloid plaques, immunostaining: 16-month-old Tg2576, 2.23%; 18-month-old APP/PSEN1, 4.7%; 16-month-old 3×, 0.01%; % total brain area covered by amyloid plaques, ThT staining: 16-month-old Tg2576, 1.00%; 18-month-old APP/PSEN1, 2.4%; 16-month-old 3×, 0.06%).
FIGURE 5.
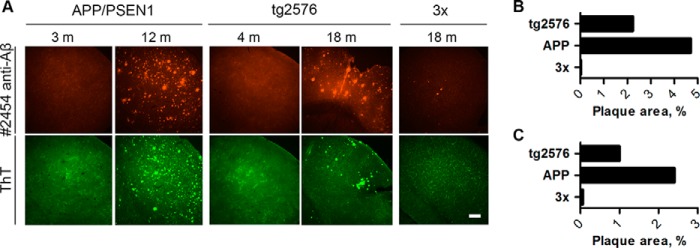
Comparative histological characterization of amyloid plaque deposition in AD mice. a, slices from the brains of old APP/PSEN1 mice (12 months old) and old Tg2576 mice (18 months old) showed considerably higher levels of plaque burden compared with old 3× Tg (18 months old) mice both when stained with anti-Aβ antibody (top row of images) or ThT (bottom row of images). Neither young APP/PSEN1 (3 months) nor young Tg2576 (4 months) mice had any detectable plaques. Scale bar, 200 μm. b, quantification of staining intensities of Aβ immunostaining (b) and ThT staining (c).
Finally, we performed 82e1-8243 and 8243-Nu4 ELISAs on TBS-extracted brain lysates to study the accumulation of all detectable Aβo species in various AD mouse models. Both oligomer-specific ELISAs have revealed similar temporal patterns of Aβo accumulation in AD mice. As for PLISA, the levels of total Aβo increase with animal's age, with 5× FAD and CRND8 being the fastest accumulators and 3× Tg the slowest (Fig. 6, a and b; 82e1-824: 5× FAD at 6 months: 37.84 ± 3.59 ng of Aβ/g of brain tissue, n = 3; CRND8 at 10 months: 41.57 ± 7.93 ng of Aβ/g of brain tissue, n = 3; APP/PSEN1 at 18 months: 17.79 ± 2.93 ng of Aβ/g of brain tissue, n = 2; Tg2576 at 16 months: 13.34 ± 0.89 ng of Aβ/g of brain tissue, n = 5; 3× at 16 months: 1.02 ± 0.22 ng of Aβ/g of brain tissue, n = 6; 8234-Nu4: 5× FAD at 6 months: 116.88 ± 14.27 ng of Aβ/g of brain tissue, n = 3; CRND8 at 10 months: 101.65 ± 5.15 ng of Aβ/g of brain tissue, n = 3; APP/PSEN1 at 18 months: 29.01 ± 7.71 ng of Aβ/g of brain tissue, n = 2; Tg2576 at 16 months: 25.66 ± 1.63 ng of Aβ/g of brain tissue, n = 5; 3× at 16 months: 2.56 ± 0.62 ng of Aβ/g of brain tissue, n = 6).
FIGURE 6.

Characterization of total Aβo accumulation in AD model mice. TBS brain lysates of WT (2, 3, 10, 12, 16, and 18 months), CRND8 (2, 5, and 10) months, 3× Tg (4, 13, and 16 months), Tg2576 (3, 6, 10, 14, and 16 months), 5× FAD (2, 4, and 6 months), and APP/PSEN1 (3, 6, 12, and 18 months) mice were analyzed by 82e1-8243 (a) and 8243-Nu4 (b) assays to quantify total amount of Aβo. Levels of total Aβo increased with animal age in all model mice, although the accumulation rates varied as follows: 5× FAD and CRND8 mice accumulated Aβo much faster than APP/PSEN and Tg2576 mice, whereas 3× mice were slower Aβ accumulators. c, table of correlations between mouse performance in behavioral tests and various Aβ-related biochemical metrics. PLISA is demonstrating the strongest significant correlation with progression of behavioral impairment across the panel of five AD mouse models. Error bars represent S.E.
PrPC-interacting Aβo Level Is an Accurate Predictor of Cognitive Impairment in Mice
To test the comparative potential of various metrics to predict the behavioral impairment, we performed linear regression analysis, receiver-operator characteristic (ROC) curve analysis, and 2 × 2 contingency table analysis (71) allowing us to assess the accuracy of PrPC-interacting Aβo (PLISA), soluble Aβ42 monomer ELISA, total soluble Aβo ELISAs, soluble Aβ WB and FA-extracted Aβ levels as indicators of the onset and progression of cognitive decline (measured as nSLD and nSMD). Linear regression analysis was performed similarly to PLISA-nSMD and PLISA-nSLD (Fig. 3, h and i) and revealed that all Aβ-related metrics demonstrate significant correlations with deficits both in memory and learning across a panel of AD mouse models. However, PrPC-interacting Aβo correlates with the progression of memory and learning deficits more closely than any other Aβ-related metric analyzed (Fig. 6c). For ROC curve and contingency table analysis, we used nSLD = 0.5 and nSMD = 0.15 as cutoffs for disease and classified all the animals showing higher values as behaviorally deficient. In ROC curve analysis, PrP-interacting Aβo had larger area under the curve values (which is a metric used to assess the ability of a test to correctly predict diagnosis) than any other metric (Fig. 7). However, the sample size was not sufficient to demonstrate statistical significance of higher area under the curve values for PLISA as compared with other metrics except Aβ monomer ELISA. For contingency table analysis of Aβ-related metrics, the lowest measurement observed in human AD patients by a corresponding metric was used as a cutoff (2.2 ng/g of tissue for PLISA; 0.01 ng/g of tissue for soluble Aβ monomer ELISA; 26.3 ng/g of tissue for soluble Aβ WB; 725.86 ng/g of tissue for FA-extracted Aβ; 13.15 ng/g of tissue for 82e1-8243; and 24.15 ng/g of tissue for 8243-Nu4), and animals demonstrating Aβ levels above this threshold were considered as testing positive for Aβ. Behaviorally deficient animals testing positive for Aβ were classified as true positives; mice with no cognitive deficit and Aβ-negative were considered true negatives; and Aβ-negative mice with behavioral deficits or cognitively normal Aβ-positive animals were deemed false-negatives or false-positives, respectively (Fig. 8; Table 3). Out of all the metrics analyzed, PrPC-interacting Aβo had the highest accuracy in predicting the cognitive impairment in both the MWM hidden platform task (nSLD, accuracy = 0.97) and probe trials (nSMD, accuracy = 0.79). However, the oligomer-specific 82e1-8243 and 8243-Nu4 ELISAs showed comparable yet lower accuracy values. Although decreasing or increasing the stringency of behavioral deficit cutoffs shifted the calculated accuracy values, PrPC-interacting Aβo demonstrated higher predictive power relative to other metrics regardless of cutoff (data not shown).
FIGURE 7.

ROC curve and contingency table analysis of predictive potential of various Aβ-related metrics. In ROC curve analysis PLISA is trending as the best predictor of deficit onset in both learning (a, quantified in c) and memory (b, quantified in d); however, this trend reaches significance only in comparison with Aβ monomer-specific ELISA. As described in Fig. 3, the normalized behavioral data are from in-house experiments (Tg2576, APP/PSEN1, 3× Tg) as well as literature data (5× FAD and CRND8 (65, 69, 70)).
FIGURE 8.
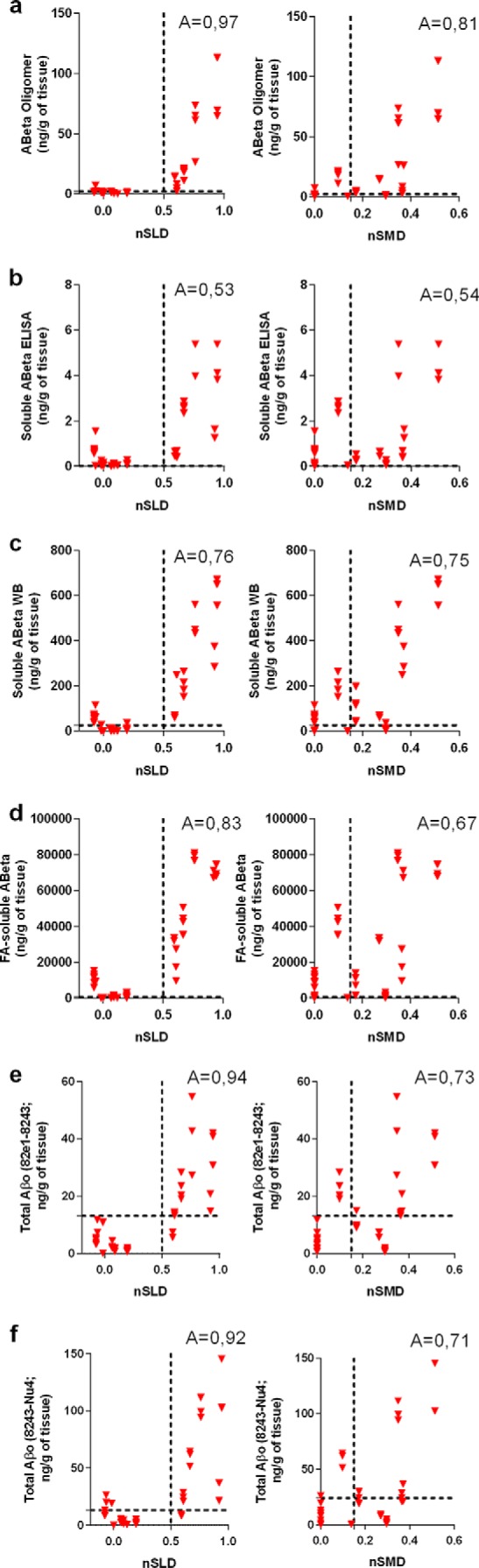
PrPc-interacting Aβo is superior to other Aβ metrics in predicting cognitive impairment in AD model mice. Using the contingency table analysis, we have estimated that PrPC-interacting Aβ oligomers are better at predicting decline cognitive performance in AD mice both in form of spatial learning and spatial memory (a). As in Figs. 3 and 7, the normalized behavioral data are from in-house experiments (Tg2576, APP/PSEN1, 3× Tg) as well as literature data (5× FAD and CRND8 (65, 69, 70)). Soluble Aβ42 ELISA performed the worst in predicting spatial learning and spatial memory decline (b). Total soluble Aβ quantified by WB (c) and Aβ in FA-extracted fraction (d) demonstrated intermediate ability to predict behavioral deficit. Both 82e1-8243 and 8243-Nu4 ELISA (total Aβ) assays were acting as strong cognitive impairment predictors, although still lower than PLISA (e and f). Vertical dashed lines represent cutoffs for considering animals as cognitively impaired (0.5 for normalized spatial learning deficit, left graph in each series; 0.15 for normalized spatial memory deficit, right graph in each series). Horizontal dashed lines represent threshold Aβ concentrations where the animals were considered to be testing positive for the presence of respective Aβ species (minimal values observed in human AD patients were used as a reference here, resulting in the following cutoff values (in ng/g brain tissue): PrPc interacting Aβo, 2,45; soluble Aβ ELISA, 0.01; soluble Aβ WB, 26.26; FA-extracted Aβ, 725.86.
TABLE 3.
Analysis of behavioral impairment, Aβ level contingency table
| PLISA | ELISA | FA | WB | 82e1–8243 | 8243-Nu4 | |
|---|---|---|---|---|---|---|
| Morris water maze hidden platform task (nSLD) | ||||||
| True positive | 18 | 16 | 17 | 15 | 15 | 14 |
| False positive | 2 | 28 | 15 | 9 | 2 | 2 |
| True negative | 43 | 16 | 30 | 28 | 33 | 33 |
| False negative | 0 | 0 | 0 | 0 | 1 | 2 |
| Total | 63 | 60 | 62 | 52 | 51 | 51 |
| Sensitivity | 1.00 | 1.00 | 1.00 | 1.00 | 0.94 | 0.88 |
| Specificity | 0.96 | 0.36 | 0.67 | 0.76 | 0.94 | 0.94 |
| Accuracy | 0.97 | 0.53 | 0.76 | 0.83 | 0.94 | 0.92 |
| Morris water maze probe trials (nSMD) | ||||||
| True positive | 19 | 22 | 23 | 17 | 15 | 14 |
| False positive | 6 | 27 | 14 | 12 | 6 | 6 |
| True negative | 31 | 10 | 23 | 17 | 21 | 21 |
| False negative | 6 | 0 | 1 | 5 | 7 | 8 |
| Total | 62 | 59 | 61 | 51 | 49 | 49 |
| Sensitivity | 0.76 | 1.00 | 0.96 | 0.77 | 0.68 | 0.64 |
| Specificity | 0.84 | 0.27 | 0.62 | 0.59 | 0.78 | 0.78 |
| Accuracy | 0.81 | 0.54 | 0.75 | 0.67 | 0.73 | 0.71 |
Taken together, all the metrics demonstrate a high degree of coherence between multiple populations of Aβ molecular species. Although all AD mouse strains showed progressive increase in all measured Aβ species as a function of age, the Aβo levels (both PrPC-interacting and total Aβo) in behaviorally impaired animals were the closest to the ones observed in human AD patients (Fig. 9a, human/mouse Aβ ratio: PLISA, 0.461 ± 0.108; soluble Aβ ELISA, 0.1 ± 0.006; FA-soluble Aβ, 0.045 ± 0.007; soluble Aβ WB, 0.251 ± 0.052; 82e1-8243, 0.60 ± 0.15; 8243-Nu4, 0.52 ± 0.15). Thus, cognitively impaired mice had PLISA, 82e1-8243, and 8243-Nu4 activity twice that of human AD brain, whereas the same mice exhibited FA-extracted and ELISA-detectable soluble Aβ amounts 50 and 20 times higher than that of humans, respectively. The WB-measured soluble Aβ level was ∼4 times higher in cognitively impaired mice than in human AD patients. From this point of view, the mouse models closely mimic the Aβo burden observed in the human AD samples. These data also suggest that Aβ oligomerization, relative to fibrillization, is more prevalent in human brain than in mouse brain.
FIGURE 9.

PrP-Fc affinity depletion of synaptotoxic activity from AD brain extracts specifically removes HMW but not low molecular weight Aβo species. The average levels of PrPC-interacting Aβo and total Aβo in mice with developed behavioral deficits were only twice higher than the ones observed in human AD patients (a). In contrast, levels of soluble ELISA-detectable Aβ- and FA-extractable Aβ were, respectively, ∼50 and ∼20 times higher in mice than in humans. Total soluble Aβ measured by WB was ∼4 times higher in mice than in humans. The relative fraction of PrPC-interacting Aβo in the total Aβo pool was significantly higher in APP/PSEN1 and CRND8 than in 3× and Tg2576 mice, whereas 5× FAD mice had an intermediate relative amount of PrPC-interacting Aβo (b and c). To investigate the relative potency of PrP-interacting and PrP-inert Aβo, pooled TBS brain extracts from AD patients (n = 6) were affinity-depleted of PrPC-interacting Aβ by incubation with PrP-Fc-Sepharose. Fc-conjugated resin was used for nonspecific pulldown control, whereas pooled extracts from the brains of neurologically healthy patients (n = 5) were used as disease-negative control. PrP-Fc pulldown resulted in complete elimination of PLISA activity from AD samples, whereas incubation with the noncoupled resin did not change PLISA activity level or size distribution by SEC relative to pre-pulldown values (d and e). The levels of total Aβo measured by 82e1-8243 total Aβo assay after PrP-Fc pulldown were decreased but not eliminated (f). Total Aβo was also measured by 82e1-8243 assay after SEC fractionation of affinity chromatography fractions (g). PrP-Fc affinity resin removed the majority of total Aβo from early eluting HMW fractions. Western blot analysis (h, quantified in i) of affinity-depleted and negative control lysates further demonstrates the nonhomogeneity of HMW amyloid oligomers as only a part of HMW immunoreactivity is depleted from AD samples after PrP resin incubation. Because the focus was on oligomeric Aβ, the membrane was not boiled, and the monomeric Aβ band remains faint. Error bars represent S.E.
PrPC-interacting Fraction and Size of Aβo Pool Varies between Mouse Models of AD
Despite the general similarity between the accumulation profiles of total and PrPC-interacting Aβo, the relative speed of accumulation of PrPC-interacting Aβo as a fraction of total Aβo pool varied greatly from model to model. At the time of detectable cognitive impairment PLISA activity in APP/PSEN1 mice is almost six times higher than in Tg2576 mice, whereas 82e1-8243 and 8243-Nu4 assays demonstrate that the levels of total Aβo in these mice are almost the same. Thus, the relative fraction of PrPC-interacting Aβo in the total Aβo pool of cognitively impaired 3× and Tg2576 mice was significantly lower than in CRND8 and APP/PSEN1 mice in both Aβo ELISAs (Fig. 9, b and c, red bars). 5× FAD mice demonstrated intermediate relative levels of PrPC-interacting Aβo, being significantly different from Tg2576 in both assays and from APP/PSEN1 or CRND8 in 82e1-8243 or 8243-Nu4, respectively (Fig. 9, b and c, blue bars). Such segregation of genotypes based on the fraction of PrPC-interacting Aβo in the total Aβo pool may uncover functionally important differences in the patterns of Aβ oligomerization between mouse strains and could dramatically affect the mechanisms involved in triggering the onset of AD-like symptoms in different animals. Interestingly, when we performed regression analysis on various Aβ metrics separately in mice with low (Tg2576 and 3× Tg) and high (APP/PSEN1, 5× FAD, and CRND8) relative amounts of PrP-interacting Aβo in total Aβo pool, there were differences (data not shown). We saw an even stronger significant correlation between behavioral deficit and PLISA activity in mice with high relative fractions of PrP-interacting Aβo compared with the one observed across the mixed sample of mice with low and high relative amounts of PrP-interacting Aβo. In contrast, the correlation between PLISA activity and the severity of behavioral impairment was less in mice with a low relative amount of PrP-interacting Aβo, suggesting that these mice might have alternative and less PrP-dependent mechanisms contributing to the progression of AD-like pathology.
To further assess differences in the Aβo pool between strains, we have compared the size distribution of Aβo in APP/PSEN1 (with a high proportion of PrPC-interacting Aβo) to Tg2576 (with a low proportion of PrPC-interacting Aβo) mice by performing the 82e1-8243 assay on SEC-fractionated brain extracts. Interestingly, when compared with APP/PSEN1, the profile of Aβo in Tg2576 mice is shifted toward lower molecular weight forms (data not shown). This observation further highlights interstrain variation of Aβo accumulation patterns and also explains the lower relative levels of PrPC-interacting Aβo (which is predominantly HMW) in Tg2576 mice. These differences raise the possibility that alternative PrPC-independent mechanisms might contribute to the emergence of AD-like phenotypes in mice with low relative amounts of PrPC-interacting Aβo, whereas phenotypes in mice with high fractions of PrPC-interacting Aβo (like APP/PSEN1) are highly dependent on the presence of PrPC.
Blockade of Aβ Production Reveals a Long Half-life for Soluble Oligomeric Aβ
Earlier studies have demonstrated that various Aβ pools have very different half-lives and respond quite differently to blockade of Aβ production by secretase inhibitors. It is documented that short term treatment with secretase inhibitors rapidly lowers soluble monomeric Aβ40 and Aβ42 within hours (72–75). In contrast, levels of pre-existing plaque Aβ are stable during secretase inhibition such that accumulation is halted but plaques do not regress substantially even over many months of treatment (74–77).
We sought to determine whether soluble Aβ oligomers have a short or long half-life assessed after secretase inhibition in plaque-bearing mice. Therefore, we treated 12-month-old APP/PSEN mice with of the γ-secretase inhibitor LY-411575 (n = 4) or vehicle (n = 5) twice daily by oral gavage for 1 week. We then analyzed Aβ levels in sequentially extracted brain homogenates (TBS, TBSX, and FA) using PLISA, 82e1-8243 oligomer ELISA, Aβ Western blot, Aβ40, and Aβ42 ELISAs (both highly biased toward monomeric Aβ40 or Aβ42). Effective inhibition of γ-secretase in mice treated with LY-411575 is obvious from the pronounced accumulation of APP C99 (a C-terminal fragment of APP produced by β-secretase ACE in the absence of γ-secretase activity) in the treated group (Fig. 10, a and b). Consistent with the short half-life of monomeric Aβ40 and Aβ42, these pools were strongly suppressed in TBS and TBSX fractions, although the change for Aβ42 in the TBSX fraction failed to reach significance. As expected for the very long half-life of Aβ in plaques, levels of Aβ in formic acid extracts of TBSX-insoluble material measured by Aβ40 and Aβ42 ELISA as well as WB demonstrated no significant change after LY-411575 treatment (Fig. 10, a and c). The levels of oligomeric Aβ measured by 82e1-8243 assay and PLISA showed no significant change in the γ-secretase inhibitor-treated group. Thus, soluble Aβ oligomers have a half-life substantially longer than soluble Aβ40 and Aβ42 monomer ELISA. Furthermore, Aβ oligomers and monomers are not in rapid equilibrium.
FIGURE 10.

Short term γ-secretase inhibition does not reduce oligomeric Aβ levels. a, 12-month-old APP/PSEN1 mice were treated with LY-411575 (n = 4, red bars) or vehicle (n = 5, black bars). Neither of the Aβo assays (PLISA or 82e1-8243) detected a significant change in soluble amyloid oligomers. No significant difference was observed in FA-soluble Aβ by Aβ40 and Aβ42 ELISAs; an insignificant increase in FA-soluble Aβ was observed by Western blot (p = 0.083). Levels of monomeric soluble Aβ40 and Aβ42 were reduced significantly in LY-411575-treated group (mAβ40 TBS: p = 0.004; mAβ40 TBSX: p = 0.002; mAβ42 TBS: p < 0.001), and mAβ42 TBSX showed a strong trend toward decrease (p = 0.057). Levels of C99 fragment of APP were increased dramatically in LY-411575-treated mice indicating drug effectiveness. Blots (anti-Aβ 8243) used for quantitation of C99 APP fragment and FA-soluble Aβ are shown in (b and c), respectively. Error bars represent S.E.
PrPC-interacting Aβo Is Required for AD-dependent Fyn Phosphorylation and Ca2+ Mobilization in Vitro
Previously, we showed that the ability of human AD brain soluble extracts to trigger disease-relevant neuronal effects in vitro (particularly, the activation of Fyn kinase (27) and the mobilization of intracellular Ca2+ (37) are abolished by affinity depletion using PrP-Fc conjugated to Sepharose beads). This suggests that PrPC-interacting Aβo species within human AD brain are most critical for these toxicities. Here, we assessed the level of different subsets for Aβo before and after the depletion of activity by affinity chromatography. Specifically, the amounts and size distribution of total Aβo (82e1-8243 assay) and PrPC-interacting Aβo (PLISA) were analyzed after PrP-Fc affinity depletion of pooled TBS brain extracts from AD patients (n = 6). PLISA signal is abolished by PrP-Fc affinity depletion, whereas incubation with negative control resin (human Fc-Sepharose) does not alter PLISA levels (Fig. 9, d and e). In contrast, PrP-Fc affinity resin depletes only 50% of Aβo detected by the 82e1-8243 assay (4216 ± 231 to 2272 ± 402 time-resolved fluorescence counts, Fig. 9f) resulting in a value much higher than observed in extracts from neurologically healthy control patients (−9 ± 173 TRF counts, n = 5). The 82e1-8243 analysis of SEC fractions demonstrates that the reduction in 82e1-8243 activity is largely attributable to removal of the HMW oligomer (Fig. 9g). Western blot analysis of AD and control brain lysates incubated with PrP or noncoupled resins further demonstrates the nonhomogeneity of the Aβo pool in humans (Fig. 9, h and i). Thus, after depletion of PrP-interacting Aβo from the brain lysates, about 20–40% of high molecular weight Aβ immunoreactivity could still be detected and could correspond to PrP-inert Aβo species. Expectedly, boiling the PrP affinity, but not negative control resin, results in recovery of pulled down Aβo immunoreactivity. These data confirm that human AD brain extracts contain distinct separable subsets of Aβo and that PrPC-interacting HMW Aβo but not lower molecular weight PrPC-inert Aβo species account for specific in vitro activity of AD brain extracts.
AD Mice with Low PrPC-interacting Aβo Fractions Are Partially Rescued by PrPC Ablation
It was reported that cognitive performance in some animal models of AD (like APP/PSEN1 mice) is completely rescued by deletion of Prnp expression, whereas some AD mice (like J20 strain) remain cognitively impaired on a Prnp null background. To test whether the relative amount of PrPC-interacting Aβo correlated with the dependence of AD-like phenotype on PrPC, we have assessed the cognitive performance of 4-, 12-, and 18-month-old Tg2576 mice bred on a wild-type or Prnp−/− background. The mice were littermate-matched to avoid background strain differences. At 4 months, animals of all four genotypes tested (WT, Prnp−/−, Tg2576, and Tg2576 Prnp−/−) demonstrated similar performance in spatial learning and spatial memory (Fig. 11, a and b). This is consistent with previous observations that Tg2576 remains cognitively normal until 9–10 months. At 12 months, Tg2576 still performs normally in MWM hidden platform task but demonstrates a marked deficit in the MWM probe trial, indicating a deficit in spatial memory. This early deficit is fully rescued by lack of PrPC expression (Fig. 11, c and d) despite the relatively lower fraction of PrPC-interacting Aβo. By 18 months of age, Tg2576 mice show a trend exhibiting spatial learning deficits, as well as retaining a complete absence of spatial memory in the probe trial (Fig. 11, e and f). Interestingly, the protective effect of ablating the PrPC-encoding gene appears reduced at 18 months. The Tg2576 mice without PrPC learn more slowly than WT (Fig. 11e). In probe trials, although a significant preference for the platform location is retained for Tg2576 mice without PrPC, there is a nonsignificant trend to a reduced preference relative to WT (Fig. 11f). The incompleteness of behavioral preservation during Tg2576 progression supports the hypothesis of the existence of PrPC-independent mechanisms contributing to cognitive impairment. These other mechanisms are likely to be mediated through a fraction of Aβ oligomer that fails to interact with PrPC.
FIGURE 11.

Genetic deletion of PrPC improves learning and memory in Tg2576 transgenic mice. Spatial learning is plotted as the time necessary to find a hidden platform in the Morris water maze at 4 months of age. Data are presented as means ± S.E. WT control n = 32, WT PRNP−/− n = 15, Tg2576 control n = 31, Tg2576 PRNP−/− n = 25. Performance did not differ throughout the experiment (p > 0.05) as determined by a repeated measures ANOVA. (a, p > 0.05). The platform is removed for a probe trial 24 h after the training in the Morris water maze was completed. Time spent in the target quadrant and anti-target quadrant was measured. Random chance is 25%. Data are presented as means ± S.E. for the mice from a. At this age, all groups showed a strong preference for the target quadrant over the anti-target quadrant with there being no difference in performance among the groups (p > 0.05) as determined by an ANOVA (b, Student's t test, *, p < 0.05; ***, p < 0.001; n.s., not significant). The mice were again trained in a Morris water maze paradigm except toward a different hidden platform location. Spatial learning is plotted as latency. WT control n = 28, WT PRNP−/− n = 11, Tg2576 control n = 22, Tg2576 PRNP−/− n = 20. Performance did not differ throughout the experiment (c, p > 0.05). Mice performed a probe trial 24 h after the training in the Morris water maze was completed. Time spent in the target quadrant and anti-target quadrant was measured. Random chance is 25%. Data are presented as mean ± S.E. for the mice from c. Tg2576 mice are impaired as the animals do not show a preference for the target quadrant, although all other groups strongly prefer the target quadrant (*, p < 0.05) as determined by an ANOVA and Fisher's LSD for post hoc analysis. d, *. p < 0.05; **, p < 0.01). Spatial learning is plotted as the time necessary to find a hidden platform in a new third location in the Morris water maze at 18 months of age. WT control n = 24, WT PRNP−/− n = 11, Tg2576 control n = 21,Tg2576 PRNP−/− n = 14. By repeated measures ANOVA for swim blocks 4–6 with post hoc pairwise testing and Tukey correction for multiple tests, both the Tg2576 with and without PrPC differed from control mice as indicated (p < 0.05). Mice performed a probe trial 24 h after the training in the Morris water maze was completed. Time spent in the target quadrant and anti-target quadrant was measured. Random chance is 25%. Data are presented as mean ± S.E. for the mice from e. The Tg2576 control mice continue to have a deficit in the probe trial as they spend significantly less time in the target quadrant than WT mice, although Tg2576 PRNP−/− mice do not perform differently from WT mice as determined by an ANOVA and Fisher's LSD for post hoc analysis (f, p < 0.05).
Discussion
In the field of AD research, the term “Aβ oligomer” is often used as a generic name for any nonmonomeric Aβ species that could be solubilized under nondenaturing conditions (20, 78). Myriad assemblies have been described in vivo and in vitro that fit this definition. However, neither is the biochemical link between these assemblies known nor is their relative contribution toward AD progression certain. The multivalent and unstable nature of Aβo hinders experimental reproducibility and creates controversies in the field. Pinpointing the key AD-mediating Aβo assemblies, resolving their properties, biochemical origin, and mechanism of downstream action are the key challenges of the amyloid hypothesis (10, 20, 79). Another obstacle on the way to the efficient translational AD research is lack of comparative characterization of Aβo accumulation and its impact on cognitive performance across multiple mouse model strains and human AD cases.
A major issue implicit in the amyloid hypothesis of Alzheimer disease is the molecular mechanism of Aβo action on neurons and synapses. Accumulated evidence suggests that PrPC acts as a high affinity receptor for Aβo to mediate signaling (21–43). It has been shown that cellular prion protein is essential for the emergence of behavioral deficits in APP/PSEN1 mice; however, the involvement of particular Aβo species was not investigated previously (29). The levels of PrPC-interacting Aβo were also shown to be correlated with biochemical measures of Aβo-PrPC signaling in vitro, including the assays measuring the mobilization of intracellular calcium and activation of Fyn kinase (27, 31, 34, 37, 38). Moreover, PrPC was demonstrated to be critical for Aβo-dependent decrease in synaptic density, neurotoxicity, and inhibition of long term potentiation (21, 25, 28–33, 36). However, some studies provide evidence for PrPC-independent Aβo-induced phenotypes, thus conflicting with the data supporting the role of PrPC-Aβo interaction in AD (23, 24, 44, 45). To investigate these controversies, we performed comparative analysis of accumulation of Aβ oligomers and other Aβ forms across multiple AD mouse model strains and human AD cases, characterized the size distribution of total and PrPC-interacting fraction of Aβo, and assessed how the relative amount of PrPC-interacting Aβo is influencing the progression of AD-like phenotypes in mice.
The major barrier to characterizing AD-relevant Aβ oligomers and assessing their importance for AD pathophysiology in model mice and in humans is the absence of a reliable detection and quantitation method (20). The majority of immunoassays currently developed to detect Aβ oligomers rely on pairs of antibodies directed against similar or identical epitopes within the Aβ peptide, thus favoring the detection of multimeric assemblies (15, 80, 81). Despite low detection limits, these assays cannot effectively distinguish between various forms of Aβo because the selectivity for oligomers is determined simply by number of monomers >1. Here, we take advantage of multiple Aβ-related metrics, including two highly sensitive oligomer-specific ELISAs (82e1-8243 and 8243-Nu4) to quantify total Aβo as well as PLISA, a recently developed high throughput plate-based assay allowing sensitive and specific detection of PrPC-interacting Aβ oligomers in biological samples (27). We show that PrPC-interacting Aβ oligomers are specifically present only in soluble protein extracts from the brains of AD model mice and post-mortem brain tissue of human patients with AD but not in age-matched control human nor wild-type mouse brain lysates, thus resulting in 100% segregation of disease-relevant and control samples.
By using size-exclusion chromatography coupled to PLISA and 82e1-8243 broad spectrum Aβo-specific ELISA, we determined that PrPC-interacting Aβ oligomers represent a distinct population of high molecular weight Aβ assemblies. The existence of HMW-soluble Aβ oligomers in human and mouse brain extracts has been reported before by several groups by using microdialysis and oligomer-specific ELISAs (14, 15, 19). The size of PLISA-detectable material was similar across multiple mouse models, as well as human AD patients, potentially indicating the existence of a conserved pathway of Aβ aggregation that results in large Aβo assemblies capable of binding to cellular prion protein and thereby inducing AD-related pathology. In contrast, the size distribution profiles of total Aβo varied from strain to strain, with APP/PSEN1 accumulating larger Aβo assemblies than Tg2576 animals. Although it seems that PrP is demonstrating particularly high preference toward HMW Aβo in biological samples and in some synthetic Aβo preparations, there is evidence that lower molecular weight Aβo assemblies like dimers and trimers are also capable of binding to PrP, although it might be attributed to the large proportion of these LMW Aβ species in the Aβo preparations used in the study (82).
Additional evidence for uniform properties and potency of PrPC-binding Aβ oligomers comes from our observation that the threshold amounts of PrPC-interacting Aβ oligomers at which mice and humans begin to demonstrate the signs of prominent cognitive impairment seem to be highly consistent between transgenic animals and human AD patients, averaging about 2–5 ng per g of brain tissue (Fig. 12). At sub-threshold values of PLISA activity, AD mice appear cognitively normal or perform marginally worse than wild-type animals, therefore seeming to be in a diagnostic “gray zone.” When the levels of PrP-interacting Aβ rise as a function of age and eventually hit a critical value, mice demonstrate escalating cognitive decline. Interestingly, the threshold amount of PrPC-interacting Aβo lies close to reported affinity of PrPC toward the Aβ oligomer (KD was determined to be in the low nanomolar range), which could explain why lower concentrations of PLISA-detectable Aβo have little effect on cognitive performance (22, 28). The rates of Aβ oligomer accumulation varied dramatically between transgenic AD model mice. Thus, 5× FAD and CRND8 mice attained the symptomatic cutoff levels of PLISA activity in 4–6 months. It took APP/PSEN1 mice about 10–12 months to reach the same levels of Aβo, and it took even longer for Tg2576 and 3× mice, 14 and 16 months, respectively. Such discrepancy in the rates of PrPC-interacting Aβo accumulation is in close agreement to the rates of behavioral deficit manifestation. Thus, the memory of 5× FAD and CRND8 mice is already substantially impaired at 6 months. At the same time, behavioral deficits in APP/PSEN1 mice are not obvious until 9–11 months; Tg2576s first show signs of cognitive impairment at 14–16 months, and in 3× mice the learning deficit is marginal even at 18 months (Fig. 12). Regression analysis further highlighted the remarkable potential of PrPC-interacting Aβo levels to extrapolate the cognitive impairment throughout the entire panel of mouse models used in the study (r2 = 0.78 for performance in MWM hidden platform navigation task, a value superior to all other Aβ-related metrics tested).
FIGURE 12.
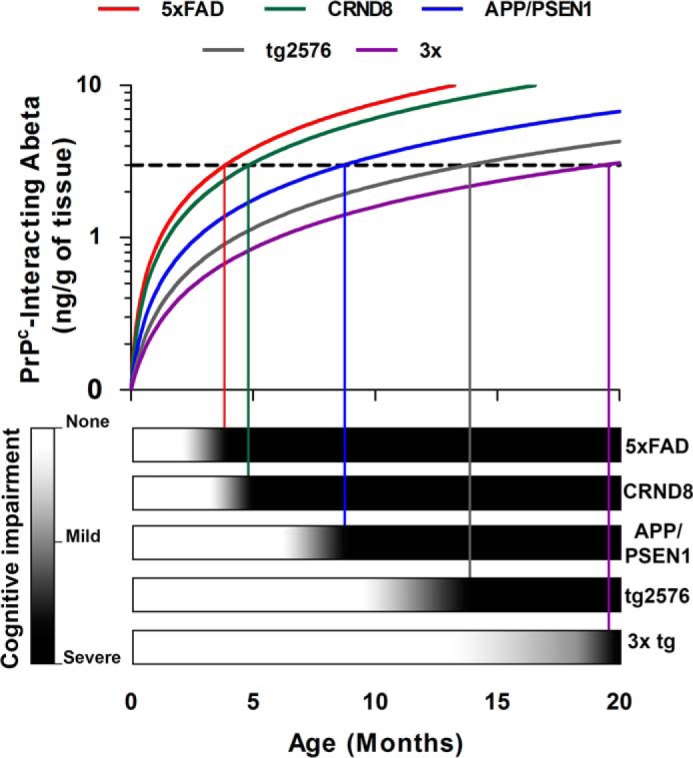
Threshold level of PrPC-interacting Aβo for development of cognitive deficits in AD model mice. The time of onset of symptomatic behavioral impairment varies dramatically between different AD model mouse strains. However, all of the transgenic AD mice as well human AD patients start to show manifestations of behavioral decline when their level of PrPC-interacting Aβo is reaching certain threshold value, ∼2–5 ng/g of brain tissue. The subthreshold values might result in mild forms of cognitive impairment too subtle to measure with usual behavioral tests and thus puts animals with subthreshold amount of PrPc-interacting Aβo in a diagnostic gray zone.
By monitoring various forms of Aβ as a function of animal age, we have observed a large degree of coherence between the rates of Aβ accumulation in different fractions from the animals of the same genotype. The inter-model comparison, however, demonstrated considerable segregation of transgenic strains. Thus, 5× FAD and CRND8 mice accumulate PrPC-interacting Aβo, total oligomerized Aβ (measured by 82e1-8243 or 8243-Nu4 Aβo ELISAs), Aβ monomers (measured by soluble Aβ ELISA), total soluble Aβ (measured by soluble Aβ WB), and insoluble fibrillar Aβ (ELISA and WB on FA-extracted Aβ) much more quickly, and by 6 months have reached higher levels of Aβ (by any metric) than all other model mice used in the study. APP/PSEN1 mice show intermediate rates of Aβ accumulation, whereas Tg2576 and particularly 3× transgenic mice were relatively slow Aβ accumulators. Such concordance between various Aβ metrics could be explained by the existence of dynamic equilibrium between Aβ species, a view widely acknowledged in the field (6, 10, 83, 84).
Despite the general coherence in accumulation of the various forms of Aβ, their correlation with the cognitive decline in mice varies greatly. Using contingency table analysis and regression analysis, we have demonstrated that the ability of PrPC-interacting Aβo to accurately predict the onset and progression of behavioral deficit (measured as decreased performance in MWM hidden platform navigation test and in MWM probe trials) is equal to or superior to all other Aβ measurements used in the study, even though the predictive power of total Aβo levels was statistically similar.
Another important discrepancy in the patterns of Aβo accumulation is the variation of the relative fraction of PrPC-interacting Aβo within the total Aβo pool. Thus, APP/PSEN1, CRND8, and to the lesser extent 5× FAD mice show a higher proportion of PrPC-interacting Aβo then do 3× and Tg2576 mice. The cause of these differences between strains is not obviously based on promotor, APP mutation, or PSEN1 mutation. However, mice with higher total Aβ levels and more rapid progression tend to have higher relative amounts of PrPC-interacting HMW Aβo species.
Treatment with a γ-secretase inhibitor allowed an assessment of the half-life of different pools by halting further Aβ production. It is known that soluble monomers turn over in hours (72–75), whereas Aβ plaques last at least for months (74–77). Both the PrPC-interacting and the total pool of soluble Aβ oligomer are stable over 7 days, showing that they have distinct metabolic kinetics from soluble monomers in mouse brain.
The connection between acute versus chronic secretase inhibition and memory in transgenic mice is not well defined in the literature regardless of Aβ species. In one study with a β-secretase inhibitor, long term treatment improved memory only after several months of treatment (75). In a study of the γ-secretase inhibitor, there were acute effects on memory function in normal rats (85) and pre-plaque Tg2576 (86), suggesting that drug action may or may not be related to APP, Aβ, and AD. It will be of interest to extend our current studies by measuring the progression of memory dysfunction and the levels of multiple Aβ fractions at a range of time points after the initiation of secretase treatment to halt Aβ production.
Development of AD-like symptoms in APP/PSEN1 is critically dependent on the normal expression of the Prnp gene, whereas some AD mice (like the J20 strain) are able to develop symptoms even on a Prnp null background. We have tested whether the relative amount of PrPC-interacting Aβo could determine the dependence of AD-like phenotypes on PrPC by performing behavioral characterization of Tg2576 mice, a strain with a small proportion of PrPc-interacting Aβo. Interestingly, the effect of PrPC ablation in these mice resulted only in full rescue of cognitive performance early during the disease course and to partial rescue at a later time point. This observation indicates the existence of alternative PrPC-independent mechanisms of Aβo action, perhaps mediated by a large fraction of oligomers that poorly interact with PrPC. The effects of these mechanisms might be relatively weak compared with effect of PrPC-Aβo axis and are therefore negligible in mice with a high proportion of PrPC-interacting Aβo (like APP/PSEN1 mice), thus allowing for a dramatic recovery of cognitive performance on Prnp null background. However, in mice with a lower proportion of PrPC-interacting Aβ like Tg2576 and, perhaps, J20, the relative contribution of these PrPC-independent mechanisms is likely to be more significant, thus allowing only for a partial recovery of cognitive function after ablation of PrPC. Thus, studying of D mice with low levels of PrPC-interacting Aβo, such asTg2576, on a Prnp null background may allow isolation and characterization of any such PrPC-independent mechanisms of AD. In this context, it is critical to note that human AD samples exhibit levels of PrPC-interacting Aβo nearly identical to APP/PSEN1 mice at the onset of deficits. Thus, in the clinical situation, PrPC-interacting Aβo, and by extension PrPC, could play a central role.
Certain antibodies have been used to distinguish conformations and populations of Aβo (79, 87), with two major pools having been distinguished. OC and related “fibrillar” antibodies are not specific for oligomers, recognizing insoluble Aβ fibrils as well as soluble Aβo. A11 and related antibodies appear to detect smaller oligomers, related to Aβ*56 (16, 79). Previous work with Aβo binding to PrPC demonstrated preferential interaction with the OC-positive fraction of Aβo, and not with A11-reactive Aβo (36, 38). Similar to PLISA activity, but distinct from A11 immunoreactivity, OC immunoreactivity is elevated in human AD samples (66). However, distinct from OC antibodies, PrPC binding separates a subpopulation of Aβo from fibrils.
The evidence for a critical role of PrPC in AD is growing. In this context, the dramatic coherence between the levels of PrPC-interacting Aβo and cognitive impairment not only provides generalizable evidence for the role of PrPC-interacting Aβo in AD progression but also delivers a further verification for the pivotal role of PrPC in AD pathobiology. A molecular understanding of toxic Aβ oligomers at the atomic level remains elusive but might hold the key to efficient development of AD diagnostics and therapeutics. Here, we demonstrate that the subset of Aβ capable of interacting with cellular prion protein is represented by a distinct population of soluble high molecular weight Aβo assemblies that are conserved across multiple animal models of AD as well as in human AD patients. Moreover, this Aβo species is strongly correlated with cognitive decline (Fig. 12). The proportion of PrPC-interacting Aβo varies from model to model and is likely proportional to the contribution of the PrPC-Aβo axis in driving the AD progression. The combination of tools described here will facilitate further characterization of Aβo assemblies and enable resolution of controversies in the amyloid hypothesis. They also provide a road to development of more sensitive and accurate AD diagnostic tools and novel therapeutic intervention strategies.
Acknowledgments
We thank Todd Golde for samples of CRND8 mouse brain and William Klein for a sample of NU-4 antibody.
This work was supported, in whole or in part, by a National Institutes of Health grant. This work was also supported by the BrightFocus Foundation, Alzheimer's Association, and Falk Medical Research Trust (to S. M. S.). S. M. S. is a co-founder of Axerion Therapeutics seeking to develop PrP-based therapeutics for Alzheimer disease.
- AD
- Alzheimer disease
- PrPC
- cellular prion protein
- Aβo
- amyloid-β oligomer
- PLISA
- PrPC-interacting Aβo species assay
- HMW
- high molecular weight
- Ab
- antibody
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- MWM
- Morris water maze
- ANOVA
- analysis of variance
- nSLD
- normalized spatial learning deficit
- ThT
- thioflavin-T
- WB
- Western blot
- FA
- formic acid
- SEC
- size-exclusion chromatography
- nSMD
- normalized spatial memory deficit
- Tg
- transgenic
- APP
- amyloid precursor protein
- ROC
- receiver-operator characteristic.
References
- 1. Mayeux R. (2003) Epidemiology of neurodegeneration. Annu. Rev. Neurosci. 26, 81–104 [DOI] [PubMed] [Google Scholar]
- 2. Braak H., Braak E. (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 [DOI] [PubMed] [Google Scholar]
- 3. Blennow K., de Leon M. J., Zetterberg H. (2006) Alzheimer's disease. Lancet 368, 387–403 [DOI] [PubMed] [Google Scholar]
- 4. Arendt T. (2009) Synaptic degeneration in Alzheimer's disease. Acta Neuropathol. 118, 167–179 [DOI] [PubMed] [Google Scholar]
- 5. Prince M., Bryce R., Albanese E., Wimo A., Ribeiro W., Ferri C. P. (2013) The global prevalence of dementia: a systematic review and metaanalysis. Alzheimer's Dementia 9, 63–75 [DOI] [PubMed] [Google Scholar]
- 6. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 7. Querfurth H. W., LaFerla F. M. (2010) Alzheimer's disease. N. Engl. J. Med. 362, 329–344 [DOI] [PubMed] [Google Scholar]
- 8. Jonsson T., Atwal J. K., Steinberg S., Snaedal J., Jonsson P. V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., et al. (2012) A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 488, 96–99 [DOI] [PubMed] [Google Scholar]
- 9. Selkoe D. J. (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 [DOI] [PubMed] [Google Scholar]
- 10. Haass C., Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 11. Reiman E. M., Chen K., Liu X., Bandy D., Yu M., Lee W., Ayutyanont N., Keppler J., Reeder S. A., Langbaum J. B., Alexander G. E., Klunk W. E., Mathis C. A., Price J. C., Aizenstein H. J., et al. (2009) Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 106, 6820–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koffie R. M., Meyer-Luehmann M., Hashimoto T., Adams K. W., Mielke M. L., Garcia-Alloza M., Micheva K. D., Smith S. J., Kim M. L., Lee V. M., Hyman B. T., Spires-Jones T. L. (2009) Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U.S.A. 106, 4012–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Esparza T. J., Zhao H., Cirrito J. R., Cairns N. J., Bateman R. J., Holtzman D. M., Brody D. L. (2013) Amyloid-β oligomerization in Alzheimer dementia versus high pathology controls. Ann. Neurol. 73, 104–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Savage M. J., Kalinina J., Wolfe A., Tugusheva K., Korn R., Cash-Mason T., Maxwell J. W., Hatcher N. G., Haugabook S. J., Wu G., Howell B. J., Renger J. J., Shughrue P. J., McCampbell A. (2014) A sensitive Aβ oligomer assay discriminates Alzheimer's and aged control cerebrospinal fluid. J. Neurosci. 34, 2884–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang T., Hong S., O'Malley T., Sperling R. A., Walsh D. M., Selkoe D. J. (2013) New ELISAs with high specificity for soluble oligomers of amyloid β-protein detect natural Aβ oligomers in human brain but not CSF. Alzheimer's Dementia 9, 99–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., Ashe K. H. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 17. Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., Selkoe D. J. (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 [DOI] [PubMed] [Google Scholar]
- 18. Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., Brett F. M., Farrell M. A., Rowan M. J., Lemere C. A., Regan C. M., Walsh D. M., Sabatini B. L., Selkoe D. J. (2008) Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takeda S., Hashimoto T., Roe A. D., Hori Y., Spires-Jones T. L., Hyman B. T. (2013) Brain interstitial oligomeric amyloid β increases with age and is resistant to clearance from brain in a mouse model of Alzheimer's disease. FASEB J. 27, 3239–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benilova I., Karran E., De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 21. Laurén J., Gimbel D. A., Nygaard H. B., Gilbert J. W., Strittmatter S. M. (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen S., Yadav S. P., Surewicz W. K. (2010) Interaction between human prion protein and amyloid-β (Aβ) oligomers: role OF N-terminal residues. J. Biol. Chem. 285, 26377–26383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Calella A. M., Farinelli M., Nuvolone M., Mirante O., Moos R., Falsig J., Mansuy I. M., Aguzzi A. (2010) Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol. Med. 2, 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Balducci C., Beeg M., Stravalaci M., Bastone A., Sclip A., Biasini E., Tapella L., Colombo L., Manzoni C., Borsello T., Chiesa R., Gobbi M., Salmona M., Forloni G. (2010) Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Freir D. B., Nicoll A. J., Klyubin I., Panico S., Mc Donald J. M., Risse E., Asante E. A., Farrow M. A., Sessions R. B., Saibil H. R., Clarke A. R., Rowan M. J., Walsh D. M., Collinge J. (2011) Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zou W. Q., Xiao X., Yuan J., Puoti G., Fujioka H., Wang X., Richardson S., Zhou X., Zou R., Li S., Zhu X., McGeer P. L., McGeehan J., Kneale G., Rincon-Limas, et al. (2011) Amyloid-β42 interacts mainly with insoluble prion protein in the Alzheimer brain. J. Biol. Chem. 286, 15095–15105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Um J. W., Nygaard H. B., Heiss J. K., Kostylev M. A., Stagi M., Vortmeyer A., Wisniewski T., Gunther E. C., Strittmatter S. M. (2012) Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fluharty B. R., Biasini E., Stravalaci M., Sclip A., Diomede L., Balducci C., La Vitola P., Messa M., Colombo L., Forloni G., Borsello T., Gobbi M., Harris D. A. (2013) An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 288, 7857–7866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gimbel D. A., Nygaard H. B., Coffey E. E., Gunther E. C., Laurén J., Gimbel Z. A., Strittmatter S. M. (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chung E., Ji Y., Sun Y., Kascsak R. J., Kascsak R. B., Mehta P. D., Strittmatter S. M., Wisniewski T. (2010) Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 11, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Resenberger U. K., Harmeier A., Woerner A. C., Goodman J. L., Müller V., Krishnan R., Vabulas R. M., Kretzschmar H. A., Lindquist S., Hartl F. U., Multhaup G., Winklhofer K. F., Tatzelt J. (2011) The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J. 30, 2057–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barry A. E., Klyubin I., Mc Donald J. M., Mably A. J., Farrell M. A., Scott M., Walsh D. M., Rowan M. J. (2011) Alzheimer's disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 31, 7259–7263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bate C., Williams A. (2011) Amyloid-β-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J. Biol. Chem. 286, 37955–37963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Larson M., Sherman M. A., Amar F., Nuvolone M., Schneider J. A., Bennett D. A., Aguzzi A., Lesné S. E. (2012) The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer's disease. J. Neurosci. 32, 16857–16871a [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35. Kudo W., Lee H. P., Zou W. Q., Wang X., Perry G., Zhu X., Smith M. A., Petersen R. B., Lee H. G. (2012) Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum. Mol. Genet. 21, 1138–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nicoll A. J., Panico S., Freir D. B., Wright D., Terry C., Risse E., Herron C. E., O'Malley T., Wadsworth J. D., Farrow M. A., Walsh D. M., Saibil H. R., Collinge J. (2013) Amyloid-β nanotubes are associated with prion protein-dependent synaptotoxicity. Nat. Commun. 4, 2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Um J. W., Kaufman A. C., Kostylev M., Heiss J. K., Stagi M., Takahashi H., Kerrisk M. E., Vortmeyer A., Wisniewski T., Koleske A. J., Gunther E. C., Nygaard H. B., Strittmatter S. M. (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer Aβ oligomer bound to cellular prion protein. Neuron 79, 887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rushworth J. V., Griffiths H. H., Watt N. T., Hooper N. M. (2013) Prion protein-mediated toxicity of amyloid-β oligomers requires lipid rafts and the transmembrane LRP1. J. Biol. Chem. 288, 8935–8951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ostapchenko V. G., Beraldo F. H., Mohammad A. H., Xie Y. F., Hirata P. H., Magalhaes A. C., Lamour G., Li H., Maciejewski A., Belrose J. C., Teixeira B. L., Fahnestock M., Ferreira S. T., Cashman N. R., Hajj G. N., et al. (2013) The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-β oligomer toxicity. J. Neurosci. 33, 16552–16564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klyubin I., Nicoll A. J., Khalili-Shirazi A., Farmer M., Canning S., Mably A., Linehan J., Brown A., Wakeling M., Brandner S., Walsh D. M., Rowan M. J., Collinge J. (2014) Peripheral administration of a humanized anti-PrP antibody blocks Alzheimer's disease Aβ synaptotoxicity. J. Neurosci. 34, 6140–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu N. W., Nicoll A. J., Zhang D., Mably A. J., O'Malley T., Purro S. A., Terry C., Collinge J., Walsh D. M., Rowan M. J. (2014) mGlu5 receptors and cellular prion protein mediate amyloid-β-facilitated synaptic long-term depression in vivo. Nat. Commun. 5, 3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Walsh K. P., Minamide L. S., Kane S. J., Shaw A. E., Brown D. R., Pulford B., Zabel M. D., Lambeth J. D., Kuhn T. B., Bamburg J. R. (2014) Amyloid-β and proinflammatory cytokines utilize a prion protein-dependent pathway to activate NADPH oxidase and induce cofilin-actin rods in hippocampal neurons. PLoS One 9, e95995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dohler F., Sepulveda-Falla D., Krasemann S., Altmeppen H., Schlüter H., Hildebrand D., Zerr I., Matschke J., Glatzel M. (2014) High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer's disease. Brain 137, 873–886 [DOI] [PubMed] [Google Scholar]
- 44. Cissé M., Sanchez P. E., Kim D. H., Ho K., Yu G. Q., Mucke L. (2011) Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J. Neurosci. 31, 10427–10431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kessels H. W., Nguyen L. N., Nabavi S., Malinow R. (2010) The prion protein as a receptor for amyloid-β. Nature 466, E3–E4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vonsattel J. P., Del Amaya M. P., Keller C. E. (2008) Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol. 115, 509–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mirra S. S., Heyman A., McKeel D., Sumi S. M., Crain B. J., Brownlee L. M., Vogel F. S., Hughes J. P., van Belle G., Berg L. (1991) The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41, 479–486 [DOI] [PubMed] [Google Scholar]
- 48. Braak H., Alafuzoff I., Arzberger T., Kretzschmar H., Del Tredici K. (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hyman B. T., Phelps C. H., Beach T. G., Bigio E. H., Cairns N. J., Carrillo M. C., Dickson D. W., Duyckaerts C., Frosch M. P., Masliah E., Mirra S. S., Nelson P. T., Schneider J. A., Thal D. R., Thies B., et al. (2012) National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimer's Dementia 8, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Montine T. J., Phelps C. H., Beach T. G., Bigio E. H., Cairns N. J., Dickson D. W., Duyckaerts C., Frosch M. P., Masliah E., Mirra S. S., Nelson P. T., Schneider J. A., Thal D. R., Trojanowski J. Q., Vinters H. V., et al. (2012) National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 123, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hyman B. T., Trojanowski J. Q. (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 56, 1095–1097 [DOI] [PubMed] [Google Scholar]
- 52. National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiol. Aging 18, S1–S2 [PubMed] [Google Scholar]
- 53. Oddo S., Caccamo A., Shepherd J. D., Murphy M. P., Golde T. E., Kayed R., Metherate R., Mattson M. P., Akbari Y., LaFerla F. M. (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 39, 409–421 [DOI] [PubMed] [Google Scholar]
- 54. Morris R. (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 11, 47–60 [DOI] [PubMed] [Google Scholar]
- 55. Haas L. T., Kostylev M. A., Strittmatter S. M. (2014) Therapeutic molecules and endogenous ligands regulate the interaction between brain cellular prion protein (PrPC) and metabotropic glutamate receptor 5 (mGluR5). J. Biol. Chem. 289, 28460–28477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zahn R., Liu A., Lührs T., Riek R., von Schroetter C., López García F., Billeter M., Calzolai L., Wider G., Wüthrich K. (2000) NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. U.S.A. 97, 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shankar G. M., Welzel A. T., McDonald J. M., Selkoe D. J., Walsh D. M. (2011) Isolation of low-n amyloid β-protein oligomers from cultured cells, CSF, and brain. Methods Mol. Biol. 670, 33–44 [DOI] [PubMed] [Google Scholar]
- 58. Lalonde R., Kim H. D., Maxwell J. A., Fukuchi K. (2005) Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neurosci. Lett. 390, 87–92 [DOI] [PubMed] [Google Scholar]
- 59. Garcia-Alloza M., Robbins E. M., Zhang-Nunes S. X., Purcell S. M., Betensky R. A., Raju S., Prada C., Greenberg S. M., Bacskai B. J., Frosch M. P. (2006) Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 24, 516–524 [DOI] [PubMed] [Google Scholar]
- 60. Lambert M. P., Velasco P. T., Chang L., Viola K. L., Fernandez S., Lacor P. N., Khuon D., Gong Y., Bigio E. H., Shaw P., De Felice F. G., Krafft G. A., Klein W. L. (2007) Monoclonal antibodies that target pathological assemblies of Aβ. J. Neurochem. 100, 23–35 [DOI] [PubMed] [Google Scholar]
- 61. Barghorn S., Nimmrich V., Striebinger A., Krantz C., Keller P., Janson B., Bahr M., Schmidt M., Bitner R. S., Harlan J., Barlow E., Ebert U., Hillen H. (2005) Globular amyloid β-peptide oligomer–a homogenous and stable neuropathological protein in Alzheimer's disease. J. Neurochem. 95, 834–847 [DOI] [PubMed] [Google Scholar]
- 62. Jankowsky J. L., Fadale D. J., Anderson J., Xu G. M., Gonzales V., Jenkins N. A., Copeland N. G., Lee M. K., Younkin L. H., Wagner S. L., Younkin S. G., Borchelt D. R. (2004) Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 13, 159–170 [DOI] [PubMed] [Google Scholar]
- 63. Hall A. M., Roberson E. D. (2012) Mouse models of Alzheimer's disease. Brain Res. Bull. 88, 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Czech C., Grueninger F. (2013) Animal models for Alzheimer's disease–the industry perspective. Drug Discov. Today 10, e73–e78 [Google Scholar]
- 65. Hyde L. A., Kazdoba T. M., Grilli M., Lozza G., Brusa R., Brussa R., Zhang Q., Wong G. T., McCool M. F., Zhang L., Parker E. M., Higgins G. A. (2005) Age-progressing cognitive impairments and neuropathology in transgenic CRND8 mice. Behav. Brain Res. 160, 344–355 [DOI] [PubMed] [Google Scholar]
- 66. Tomic J. L., Pensalfini A., Head E., Glabe C. G. (2009) Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol. Dis. 35, 352–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., Berry R., Vassar R. (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 [DOI] [PubMed] [Google Scholar]
- 69. Chen R., Zhang J., Wu Y., Wang D., Feng G., Tang Y.-P., Teng Z., Chen C. (2012) Monoacylglycerol lipase is a therapeutic target for Alzheimer's disease. Cell Rep. 2, 1329–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bouter Y., Kacprowski T., Weissmann R., Dietrich K., Borgers H., Brauss A., Sperling C., Wirths O., Albrecht M., Jensen L. R., Kuss A. W., Bayer T. A. (2014) Deciphering the molecular profile of plaques, memory decline and neuron loss in two mouse models for Alzheimer's disease by deep sequencing. Front. Aging Neurosci. 6, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Loong T. W. (2003) Understanding sensitivity and specificity with the right side of the brain. BMJ 327, 716–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dovey H. F., John V., Anderson J. P., Chen L. Z., de Saint Andrieu P., Fang L. Y., Freedman S. B., Folmer B., Goldbach E., Holsztynska E. J., Hu K. L., Johnson-Wood K. L., Kennedy S. L., Kholodenko D., Knops J. E., et al. (2001) Functional γ-secretase inhibitors reduce β-amyloid peptide levels in brain. J. Neurochem. 76, 173–181 [DOI] [PubMed] [Google Scholar]
- 73. Cirrito J. R., May P. C., O'Dell M. A., Taylor J. W., Parsadanian M., Cramer J. W., Audia J. E., Nissen J. S., Bales K. R., Paul S. M., DeMattos R. B., Holtzman D. M. (2003) In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J. Neurosci. 23, 8844–8853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Abramowski D., Wiederhold K. H., Furrer U., Jaton A. L., Neuenschwander A., Runser M. J., Danner S., Reichwald J., Ammaturo D., Staab D., Stoeckli M., Rueeger H., Neumann U., Staufenbiel M. (2008) Dynamics of Aβ turnover and deposition in different β-amyloid precursor protein transgenic mouse models following γ-secretase inhibition. J. Pharmacol. Exp. Ther. 327, 411–424 [DOI] [PubMed] [Google Scholar]
- 75. Chang W. P., Huang X., Downs D., Cirrito J. R., Koelsch G., Holtzman D. M., Ghosh A. K., Tang J. (2011) β-Secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 25, 775–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Best J. D., Smith D. W., Reilly M. A., O'Donnell R., Lewis H. D., Ellis S., Wilkie N., Rosahl T. W., Laroque P. A., Boussiquet-Leroux C., Churcher I., Atack J. R., Harrison T., Shearman M. S. (2007) The novel γ secretase inhibitor N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1,1-trifluoromethanesulfonamide (MRK-560) reduces amyloid plaque deposition without evidence of notch-related pathology in the Tg2576 mouse. J. Pharmacol. Exp. Ther. 320, 552–558 [DOI] [PubMed] [Google Scholar]
- 77. Garcia-Alloza M., Subramanian M., Thyssen D., Borrelli L. A., Fauq A., Das P., Golde T. E., Hyman B. T., Bacskai B. J. (2009) Existing plaques and neuritic abnormalities in APP:PS1 mice are not affected by administration of the γ-secretase inhibitor LY-411575. Mol. Neurodegener. 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kayed R., Lasagna-Reeves C. A. (2013) Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimer's Dis. 33, S67–S78 [DOI] [PubMed] [Google Scholar]
- 79. Larson M. E., Lesné S. E. (2012) Soluble Aβ oligomer production and toxicity. J. Neurochem. 120, 125–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bruggink K. A., Jongbloed W., Biemans E. A., Veerhuis R., Claassen J. A., Kuiperij H. B., Verbeek M. M. (2013) Amyloid-β oligomer detection by ELISA in cerebrospinal fluid and brain tissue. Anal. Biochem. 433, 112–120 [DOI] [PubMed] [Google Scholar]
- 81. Klaver A. C., Patrias L. M., Finke J. M., Loeffler D. A. (2011) Specificity and sensitivity of the Aβ oligomer ELISA. J. Neurosci. Methods 195, 249–254 [DOI] [PubMed] [Google Scholar]
- 82. Ganzinger K. A., Narayan P., Qamar S. S., Weimann L., Ranasinghe R. T., Aguzzi A., Dobson C. M., McColl J., St George-Hyslop P., Klenerman D. (2014) Single-molecule imaging reveals that small amyloid-β1–42 oligomers interact with the cellular prion protein (PrPC). ChemBioChem 15, 2515–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Karran E., Mercken M., De Strooper B. (2011) The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat. Rev. Drug. Discov. 10, 698–712 [DOI] [PubMed] [Google Scholar]
- 84. Huang Y., Mucke L. (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dash P. K., Moore A. N., Orsi S. A. (2005) Blockade of γsecretase activity within the hippocampus enhances long-term memory. Biochem. Biophys. Res. Commun. 338, 777–782 [DOI] [PubMed] [Google Scholar]
- 86. Comery T. A., Martone R. L., Aschmies S., Atchison K. P., Diamantidis G., Gong X., Zhou H., Kreft A. F., Pangalos M. N., Sonnenberg-Reines J., Jacobsen J. S., Marquis K. L. (2005) Acute γ-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 25, 8898–8902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 [DOI] [PMC free article] [PubMed] [Google Scholar]