

Background: Nitric oxide (•NO) inhibits neutrophil β2 integrin adhesion by an unknown mechanism.
Results: Exposure to •NO causes actin S-nitrosylation, which elicits actin turnover and an interdependent process whereby nitric-oxide synthase-2 and NADPH oxidase activation generate reactive species to maintain cytoskeletal changes that inhibit β2 integrin adhesion.
Conclusion: By concurrent perturbation of several enzymes •NO impedes neutrophil adherence.
Significance: This mechanism offers physiological insight into vascular biology.
Keywords: actin, guanylate cyclase (guanylyl cyclase), NADPH oxidase, nitric-oxide synthase, S-nitrosylation, focal adhesion kinase
Abstract
This investigation explored the mechanism for inhibition of β2 integrin adhesion molecules when neutrophils are exposed to nitric oxide (•NO). Roles for specific proteins were elucidated using chemical inhibitors, depletion with small inhibitory RNA, and cells from knock-out mice. Optimal inhibition occurs with exposures to a •NO flux of ∼28 nmol/min for 2 min or more, which sets up an autocatalytic cascade triggered by activating type 2 nitric-oxide synthase (NOS-2) and NADPH oxidase (NOX). Integrin inhibition does not occur with neutrophils exposed to a NOX inhibitor (Nox2ds), a NOS-2 inhibitor (1400W), or with cells from mice lacking NOS-2 or the gp91phox component of NOX. Reactive species cause S-nitrosylation of cytosolic actin that enhances actin polymerization. Protein cross-linking and actin filament formation assays indicate that increased polymerization occurs because of associations involving vasodilator-stimulated phosphoprotein, focal adhesion kinase, and protein-disulfide isomerase in proximity to actin filaments. These effects were inhibited in cells exposed to ultraviolet light which photo-reverses S-nitrosylated cysteine residues and by co-incubations with cytochalasin D. The autocatalytic cycle can be arrested by protein kinase G activated with 8-bromo-cyclic GMP and by a high •NO flux (∼112 nmol/min) that inactivates NOX.
Introduction
The project goal was to identify the mechanism for neutrophil β2 integrin adhesion molecule inhibition by nitric oxide (•NO). This phenomenon has relevance to basic physiology, pathophysiology, and therapeutic interventions. β2 integrins play a central role in regulating neutrophil activation and endothelial adhesion (1). Inhibition of β2 integrins by •NO diminishes potentially harmful neutrophil interactions with the microvasculature (2–5). Administration of •NO donors has been shown to have benefit in abrogating β2 integrin function and neutrophil adhesion after ischemia reperfusion (6–10). Dose response is difficult to predict, however, and •NO administration can have the opposite effect under some conditions, thus exacerbating inflammatory injuries (5, 11–15).
The flux of •NO encountered by neutrophils under normal physiological conditions is difficult to predict. Production by the vascular endothelium has been estimated to be ∼13 pmol/min/cm2 (16). In a 25-μm diameter blood vessel, 1 cm2 of endothelium surface would be contained by a 12.7-cm length, with a volume of 0.06 ml. Hence, the local intravascular •NO concentration could be as high as 210 nm. Some •NO will, of course, be taken up by erythrocyte hemoglobin, although there are physical barriers that diminish this process (17).
Previous work has shown that when neutrophils are exposed to •NO or to chemical agents that spontaneously generate •NO at a flux of ∼20 nmol/min or more, synthesis of cGMP by the particulate or membrane-bound form of guanylate cyclase (mGC)2 is inhibited (18). The inhibitory effect on mGC was thought possibly relevant to •NO-dependent inhibition of β2 integrins because incubating cells with the membrane-permeable agent, 8-bromo-cyclic GMP (8-br-cGMP), reverses β2 integrin inhibition (18). However, the role of mGC and how cGMP impacts •NO-mediated β2 integrin inhibition are not known.
Integrins are normally poorly adhesive, and their activation involves modifications of the actin cytoskeleton that lead to alterations in the conformation of extracellular integrin components to increase the affinity for binding and a coordinated grouping of integrins in the plane of the membrane to increase their avidity (19, 20). Actin binding via an assortment of linking proteins provides a platform that brings integrins and activated enzymes in close proximity (21, 22).
Nitric oxide and related nitrogen oxides can influence cell signaling by S-nitrosylation of labile cysteine residues (23). With regard to β2 integrin regulation, S-nitrosylation of cytoplasmic actin (SNO-actin) was shown to increase formation of Triton-soluble actin filaments, leading to alterations in the cytoskeletal network that inhibit β2 integrin-dependent adherence-associated with exposure to high pressure oxygen (24). Reactive nitrogen species are generated because nitric-oxide synthase type 2 (NOS-2 or iNOS) activation occurs (25). Agents capable of causing S-nitrosylation are generated when •NO synthesized by iNOS reacts with reactive oxygen species (ROS) that are synthesized by myeloperoxidase that is concurrently activated by the hyperoxic exposure (24). An autocatalytic cycle occurs because vasodilator stimulated phosphoprotein (VASP) exhibits high affinity for S-nitrosylated Triton-soluble filamentous actin (sF-actin) (26). VASP bundles Rac1, Rac2, PKA, and PKG in close proximity to sF-actin, and subsequent Rac activation increases free actin incorporation into filamentous actin (26). Focal adhesion kinase links to the sF-actin and secondary association of iNOS enhances •NO synthesis (25). The disordered actin polymerization process is reversed by 8-br-cGMP, which activates PKG outside of the sF-actin pool and phosphorylates VASP. Phosphorylated VASP does not exhibit heightened binding to SNO-actin, which abrogates the accelerated actin turnover and restores normal β2 integrin function (26).
We reasoned that a similar cascade of protein activation as occurs with exposure to high pressure oxygen may occur in response to exogenous •NO. How reactive species capable of S-nitrosylation reactions would be generated is unclear, however. In this regard, it is important to recognize that mGC exhibits complex biological effects; some involve activation of NADPH oxidase (NOX), although this has not been reported in neutrophils (27–30). As this investigation developed and consistent with previous work pertaining to SNO-actin, an interdependent complex of enzyme activation was found to be influenced by exogenous •NO. We found that production of reactive species by NOX is required for •NO-mediated inhibition of β2 integrins. Bundling several proteins with sF-actin leads to Rac activation, increased actin turnover, and increased iNOS activity. Thus, S-nitrosylation occurs because of concurrent NOX and iNOS activation.
Experimental Procedures
Materials
The majority of chemicals and their sources used in this investigation have been listed in prior publications (24–26, 31); therefore only new reagents not used previously will be listed. Diethylamine NONOate (DENO) and fluorescein bis(benzyl boronic ester) (FBBBE) were purchased from Cayman Chemical Inc. (Ann Arbor, MI). The siRNA sequences (5′ to 3′ orientation) to cause degradation of mRNA for mGC is a pool of three different siRNA duplexes: (a) sense: GGAUUGUCCUGUGACUAUAtt and antisense: UAUAGUCACAGGACAAUCCtt; (b) sense: CCAAGUGGCUGAAGAUACUtt and antisense: AGUAUCUUCAGCCACUUGGtt; and (c) sense: GUAGCAACCUCCUAUAUCAtt and antisense: UGAUAUAGGAGGUUGCUACtt. The siRNA sequences to cause degradation of mRNA for soluble GC is a pool of three different siRNA duplexes: (a) sense: CAAGGGUUAUGGAUCUCAAtt and antisense: UUGAGAUCCAUAACCCUUGtt; (b) sense: GGAUGCUUAUCACCAUCAAtt and antisense: UUGAUGGUGAUAAGCAUCCtt; and (c) sense: GUGAUAGUGUCGCCUGUAAtt and antisense: UUACAGGCGACACUAUCACtt. The siRNA sequences to cause degradation of mRNA for HSP90 is a pool of four different siRNA duplexes: (a) sense: CUGAUGACAUCACUAAUGAtt and antisense: UCAUUAGUGAUGUCAUCAGtt; (b) sense: GCAUGGAAGAAGUAGACUAtt and antisense: UAGUCUACUUCUUCCAUGCtt; (c) sense: CAAGCACUUCUCUGUAGAAtt and antisense: UUCUACAGAGAAGUGCUUGtt; and (d) sense: GCAAGAACAUCGUCAAGAAtt and antisense: UUCUUGACGAUGUUCUUGCtt. The siRNA sequences to cause degradation of mRNA for gp91phox is a pool of three different siRNA duplexes: (a) sense: CAAGGUAUCCAAGUUAGAAtt andntisense: UUCUAACUUGGAUACCUUGtt; (b) sense: GUACAGCCAGUGAAGAUGUtt and antisense: ACAUCUUCACUGGCUGUACtt; and (c) sense: CACCAGUCUGAAACUCAAAtt and antisense: UUUGAGUUUCAGACUGGUGtt.
Studies with Human Blood
All procedures were reviewed and approved by the University Committee on Studies Involving Human Beings. Subjects were healthy adults who had taken no medications for at least 4 days prior to phlebotomy. Blood was acquired in heparinized tubes, and neutrophils were isolated as previously described (24, 31).
Animals
Mice (Mus musculus) were purchased (Jackson Laboratories, Bar Harbor, ME), fed a standard rodent diet and water ad libitum, and housed in the university animal facility. A colony of iNOS and gp91phox knock-out mice was maintained from breading pairs purchased from Jackson Laboratories. Mice were anesthetized (intraperitoneal administration of ketamine (100 mg/kg) and xylazine (10 mg/kg)), skin was prepared by swabbing with Betadine, and blood was obtained into heparinized syringes by aortic puncture. Neutrophils were isolated from heparinized blood as previously described (26).
Isolation of Neutrophils and Exposure to Various Agents
Studies with mouse and human neutrophils were carried out in an identical fashion. A concentration of 5 × 105 neutrophils/ml of PBS + 5.5 mm glucose was exposed to •NO-donating agents for 2 min. Where indicated, prior to some mouse cell exposures, suspensions were incubated for 24 h at room temperature with 0.08 nm siRNA following the manufacturer's instructions using control siRNA or siRNA specific for a mouse protein. The magnitude of protein concentration reduction was evaluated from Western blots by comparing the ratio of band density for each protein to β-actin using lysates from cells incubated with each specific siRNA compared against measurements made with lysates from cells incubated with the control siRNA. Expressed as a percentage of the protein concentration compared with control siRNA incubated cells, the FAK presence was 34.8 ± 4.5 (S.E., n = 5), the gp91phox presence was 24.8 ± 1.1 (n = 4), the HSP90 presence was 21.0 ± 1.8 (n = 4), the mGC presence was 19.7 ± 1.6 (n = 4), the soluble GC presence was 26.4 ± 0.1 (n = 3), the PDI presence was 19.2 ± 4.9 (n = 5), and the VASP presence was 29.1 ± 5.3 (n = 5).
Exposure to Nitric Oxide
The majority of studies were performed with DENO. Solutions of 200–500 mm DENO were prepared daily in 50 mm potassium phosphate, pH 8.5, and the concentration was verified by measuring ultraviolet absorbance (E250 = 6,500). Neutrophils were exposed to concentrations of DENO that were prepared by making appropriate dilutions of the stock solution directly into cell suspensions, and cells were incubated for at least 2 min before performing assays. The half-life of DENO in buffer at pH 7.4 was 2 min, based on its characteristic ultraviolet absorption spectrum and liberation of •NO measured directly with a •NO-selective electrochemical sensor. The concentrations of DENO used in studies, 10–1000 nm, generated •NO at rates of ∼2.8 to 276 nmol/min (18).
Fibrinogen-coated Plate Adherence
Preparation and use of fibrinogen-coated plates to measure β2 integrin-specific neutrophil adherence in calcein AM-loaded cells were as previously described (24, 26). Suspensions of 25,000 cells in 100 μl of PBS were added to plate wells, and at the end of a 10-min incubation, wells were washed twice with 4 ml of PBS to remove nonadherent cells. Cell adherence calculated as in Refs. 24 and 26.
Cell Extract Preparation and Biotin Switch Assay
Isolated neutrophils were suspended in 2 ml of HEN buffer (250 mm Hepes, pH 7.7, 1 mm EDTA, 0.1 mm neocuproine), sonicated on ice for 30 s and then passed through a 28-gauge needle five times. Lysates were centrifuged at 2000 × g for 10 min, and supernatant was recovered and made 0.4% CHAPS using the 10% stock solution. The biotin switch assay was carried out following published methods (24).
Cytoskeletal Protein Analysis Based on Triton Solubility
Neutrophils were processed following a published protocol to separate G-actin, sF-actin, and Triton-insoluble F-actin (26). For some studies, neutrophils were suspended in a solution of 0.5 mm dithiobis (succinimidyl proprionate) (DTSP) to cross-link sulfhydryl-containing proteins within a proximity of ∼12 Å following published procedures (26, 32). Cell lysates were partitioned into the three fractions and subjected to Western blotting as described (26).
NOS Activity Assay in Permeabilized Neutrophils
Isolated neutrophils were subjected to permeabilization using 0.2% n-octyl-β-glucopyranoside (OG), and NOS activity was assessed exactly as described (26).
NOS Dimer/Monomer Differences
Differences in presence of NOS dimers versus monomers were assayed following published methods (33, 34).
Confocal Microscopy
Isolated neutrophils suspended in PBS were exposed to 100 nm DENO for 2 min and then placed on slides coated with fibrinogen. Cells were allowed to settle for 1 h to provide a more uniform focal plane, because cells were only poorly adherent to the slide surface and then fixed with 4% paraformaldehyde following published methods (26). Cells were permeabilized by incubation for 1 h at room temperature with PBS containing 0.2% (v/v) Triton X-100, 5% (v/v) fetal bovine serum, and 5% (v/v) normal goat serum and then incubated overnight with 1:200 dilutions of Alexa 488-conjugated phalloidin plus primary antibodies to either gp91phox (1:500), FAK (1:500), iNOS (1:500), or HSP90 (1:250). The next morning, slides were rinsed three times with PBS and counterstained with a 1:200 dilution of Alexa 647 or R-phycoerythrin-conjugated secondary antibodies. Images of neutrophils were acquired using a Zeiss Meta510 confocal microscope equipped with a Plan-Apochromat 63×/1.4 NA oil objective. Fluorophore excitation was provided by 488, 560-, and 633-nm laser lines, and the resulting fluorescence was separated using 505-nm-long pass for Alexa 488, a 575-nm-long pass for R-phycoerythrin, and a 650-nm-long pass filter for Alexa 647.
Actin Polymerization in Permeabilized Cells
Neutrophil suspensions permeabilized with 0.2% OG were exposed to PBS or DENO as outlined above, and actin polymerization was assayed exactly as described (26).
Total F-actin
Total F-actin was assessed following the procedure described by Chan et al. (35). Briefly, cells were exposed to PBS or 100 nm DENO, fixed with 11% formaldehyde, and permeabilized with 0.1% saponin, and fluorescence was measured after cells were incubated with 1 μm N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) phallacidin.
Reactive Species Generation
Neutrophil suspensions were prepared with 10 μm 2,7-dihydrodichlorofluorescein-diacetate (DCF-DA) or 50 μm FBBBE, and fluorescence was monitored (DCF at 492-nm excitation, 530-nm emission; FBBBE at 480-nm excitation, 512-nm emission) after incubations in PBS or PBS + 100 nm DENO following the procedures described previously (36).
Rac Activation
Neutrophil suspensions were assayed for Rac activity using a commercial kit (Cytoskeleton, Inc., Denver, CO) exactly as described (26).
Cyclic GMP Assay
Neutrophils were suspended in PBS + 5.5 mm glucose and exposed to a range of DENO concentrations for 2 min. Suspensions were then made 1 n HCl, cells were lysed, and cGMP was assayed using a commercial competitive enzyme-linked immunosorbent assay following the manufacturer's instructions (Cayman Chemical).
Statistical Analysis
The results are expressed as means ± S.E. for three or more independent experiments. The data were compared by ANOVA using SigmaStat (Jandel Scientific, San Jose, CA) and Newman-Keuls post-hoc test. The level of statistical significance was defined as p < 0.05.
Results
Neutrophil β2 Integrin Adhesion
Using DENO as the •NO-generating agent, a biphasic dose-response effect on β2 integrin-specific adherence to fibrinogen-coated plates was found using isolated murine neutrophils (Fig. 1). Inhibition of adherence was due to •NO and not diethylamine because when DENO was left on the bench top for 24 h to deplete its •NO and then added to neutrophil suspensions, adherence was 20.2 ± 0.2% (n = 3, NS versus PBS/control). We also found that when cells were incubated with 1 mm isosorbide dinitrate, which yields a comparable concentration of •NO as 50 nm DENO during the assay time (37), adherence decreased to 5.4 ± 0.5% (n = 4, p < 0.05 versus PBS/control).
FIGURE 1.
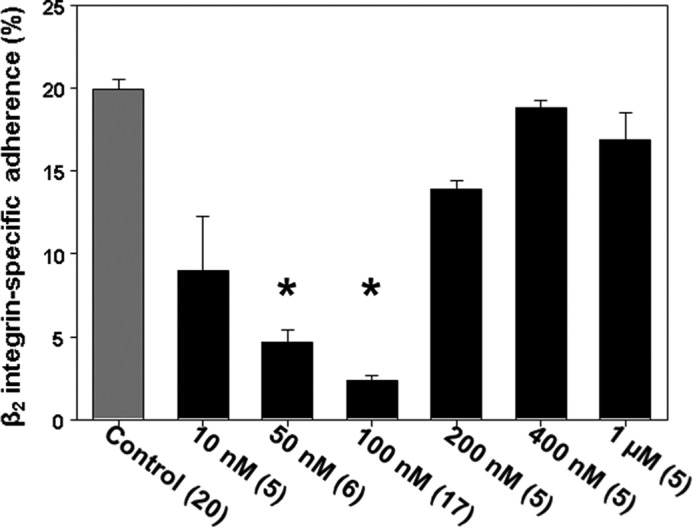
Neutrophil β2 integrin-dependent adherence. Murine neutrophils suspended in PBS + 5.5 mm glucose were exposed to the indicated concentrations of DENO for 2 min and then adherence to fibrinogen-coated plates was assessed. The values are means ± S.E. The numbers in parentheses indicate the numbers of replicate experiments performed with fresh neutrophil isolates from different mice. *, p < 0.05 versus PBS/control.
A majority of studies to elucidate mechanisms were performed with murine cells to take advantage of knock-out mouse strains and because of the ease with depleting specific proteins using small inhibitory RNA (siRNA). Human neutrophils, however, exhibit similar effects as murine cells. Following the same techniques as with murine cells, β2 integrin-specific adherence of human neutrophils to fibrinogen-coated plates was 14.7 ± 0.8% (n = 6), but after exposure to 100 nm DENO for 2 min, adherence was just 0.3 ± 0.6% (n = 6, p < 0.001). As will be discussed below, protein S-nitrosylation is involved with the mechanism for β2 integrin inhibition. If human neutrophils are exposed to 100 nm DENO and then to UV light for 5 min (which photo-reverses S-nitrosylated cysteine residues) adherence was 12.9 ± 1.4% (n = 3, NS versus control). There was no effect of UV exposure on baseline/control adherence, which was found to be 13.8 ± 1.1% (n = 3).
A series of studies was performed using cells from knock-out mice lacking iNOS or the gp91phox component of NOX. As is shown in Table 1, 100 nm DENO did not inhibit β2 integrin adhesion of these cells. Similarly, when wild-type cells were incubated with the iNOS inhibitor, 1400W, or with a membrane-permeable NOX inhibitor Nox2ds (but not a control, scrambled sequence Nox2ds peptide) prior to DENO exposure, no inhibition of adherence occurred. The inhibitory effect of DENO was also abrogated in cells incubated with siRNA to deplete the gp91phox component of NOX. One pathway for NOX activation involves enzyme complex assembly facilitated by protein-disulfide isomerase (PDI) (38). If cells were depleted of PDI by siRNA incubation or if cells were incubated with quercetin-3-rutinoside, a PDI inhibitor, DENO did not inhibit adherence.
TABLE 1.
Impact of various agents on .NO-mediated inhibition of β2 integrin-dependent murine neutrophil adherence, iNOS activation, DCF-DA fluorescence, and actin polymerization
Neutrophils were exposed to PBS or 100 nm DENO for 2 min before assays were performed. Cell adherence reflects β2 integrin-specific adherence to fibrinogen-coated plates. NOS activity reflects pmol [H3]citrulline produced when [H3]arginine was added to cells that were then incubated for 15 min. DCF fluorescence was assessed when 10 μm DCF-DA was added to cell suspensions and monitored for up to 10 min. Actin polymerization reflect pyrene actin fluorescence in arbitrary units/min. All values are means ± S.E. (n = number of independent trials). Where studies were done with small chemical inhibitors, cells were incubated with the agent for 10 min prior to adding DENO or an equal volume of PBS (control) before performing the assay. Where studies were done with siRNA, cells were incubated with control, scrambled sequence, or siRNA sequences specific for the protein indicated for 24 h prior to performing the study. iNOS KO, neutrophils from NOS-2 knock-out mice; gp91phox KO, neutrophils from knock-out mice lacking the gp91phox subunit of NADPH oxidase; 1400W, incubation with 1 mm 1400W; UV, cells exposed to UV light for 5 min after PBS or DENO incubation for 2 min; Cyto D, incubation with 5 μm cytochalasin D; Nox2ds, incubation with 10 μm Nox-2ds; Scrmb-Nox2ds, incubations performed with 10 μm control, scrambled sequence peptide to Nox-2ds; Rac-i, incubation with 50 μm NSC 23766; Cont-si, cells incubated with control scrambled sequence siRNA; gp91phox-si, cells incubated with siRNA to gp91phox; VASP-si, cells incubated with siRNA to vasodilator stimulated phosphoprotein; FAK-i, cells incubated with 20 μm PT 573228; FAK-si, cells incubated siRNA to focal adhesion kinase; Geld, cells incubated with 3 μm geldanamycin; HSP90-si, cells incubated with siRNA to HSP90; PDI-i, cells incubated with 30 μm quercetin-3-rutinoside, PDI-si, cells incubated with siRNA to protein disulfide isomerase; mGC-si, cells incubated with siRNA to membrane guanylate cyclase; ND, no experiments performed.
| Agent | Neutrophil adherence |
iNOS |
DCF |
Actin polymerization |
||||
|---|---|---|---|---|---|---|---|---|
| PBS | DENO | PBS | DENO | PBS | DENO | PBS | DENO | |
| % | pmol [H3] citrulline/h | fluorescence/min | fluor/min × 103 | |||||
| PBS | 20.6 ± 0.4 (21) | 2.4 ± 0.4 (20)a | 0.20 ± 0.05 (15) | 2.38 ± 0.05 (12)a | 0.88 ± 0.09 (16) | 2.2 ± 0.10 (7)a | 1.00 ± 0.06 (8) | 3.63 ± 0.26 (8)a |
| iNOS KO | 19.7 ± 0.2 (5) | 18.3 ± 0.3 (5) | 0 ± 0 (5) | 0 ± 0 (5) | ND | ND | 0.80 ± 0.20 (3) | 1.10 ± 0.06 (6) |
| gp91phox KO | 20.4 ± 0.8 (3) | 20.0 ± 0.7 (4) | ND | ND | ND | ND | 0.93 ± 0.23 (3) | 1.00 ± 0.12 (4) |
| 1400W | 21.3 ± 0.5 (5) | 20.8 ± 0.7 (5) | 0 ± 0 (8) | 0 ± 0 (8) | 0.98 ± 0.07 (3) | 1.08 ± 0.11 (3) | 0.87 ± 0.13 (5) | 1.00 ± 0.02 (3) |
| Nox2ds | 20.1 ± 0.1 (5) | 19.7 ± 0.5 (5) | 0.25 ± 0.05 (6) | 0.32 ± 0.05 (4) | 1.00 ± 0.01 (3) | 0.67 ± 0.33 (3) | 1.05 ± 0.13 (4) | 0.76 ± 0.11 (5) |
| Scrmb- Nox2ds | 22.2 ± 0.9 (4) | 3.4 ± 0.8 (4)a | 0.21 ± 0.09 (3) | 2.0 ± 0.18 (4)a | 1.17 ± 0.17 (3) | 1.99 ± 0.03 (3)a | 1.1 ± 0.15 (7) | 3.11 ± 0.25 (3)a |
| Cont-si | 17.1 ± 1.2 (12) | 2.7 ± 0.4 (10)a | 0.20 ± 0.02 (9) | 2.1 ± 0.16 (7)a | 1.10 ± 0.16 (9) | 2.51 ± 0.22 (8)a | 0.94 ± 0.11 (5) | 4.04 ± 0.44 (5)a |
| gp91phox-si | 20.6 ± 0.9 (3) | 20.3 ± 0.1 (3) | 0.23 ± 0.13 (3) | 0.40 ± 0.13 (3) | 0.98 ± 0.33 (3) | 1.00 ± 0.27 (3) | ND | ND |
| PDI-i | 22.6 ± 0.2 (3) | 20.8 ± 0.9 (3) | 0.21 ± 0.08 (3) | 0. 61 ± 0.06 (3) | 1.00 ± 0.08 (3) | 0.67 ± 0.14 (3) | 0.53 ± 0.24 (3) | 0.57 ± 0.07 (3) |
| PDI-si | 22.6 ± 0.3 (3) | 20.3 ± 0.2 (3) | 0.21 ± 0.10 (6) | 0.33 ± 0.10 (6) | 1.00 ± 0.33 (3) | 1.00 ± 0.38 (3) | 1.00 ± 0.03 (3) | 1.03 ± 0.03 (3) |
| UV | 21.4 ± 0.6 (6) | 20.7 ± 0.7 (6) | 0.13 ± 0.03 (7) | 0.25 ± 0.01 (4) | 1.00 ± 0.11 (3) | 1.1 ± 0.10 (3) | 0.90 ± 0.10 (3) | 0.90 ± 0.08 (6) |
| Cyto D | 22.4 ± 0.4 (3) | 21.1 ± 0.3 (3) | 0.15 ± 0.04 (6) | 0.20 ± 0.08 (3) | 0.98 ± 0.17 (3) | 1.1 ± 0.17 (3) | 0.78 ± 0.13 (4) | 0.85 ± 0.13 (6) |
| Rac-i | 20.7 ± 1.1 (5) | 19.5 ± 0.7 (6) | 0.21 ± 0.01 (5) | 0.32 ± 0.04 (4) | 1.05 ± 0.07 (3) | 0.70 ± 0.02 (3) | 0.87 ± 0.08 (3) | 0.85 ± 0.13 (6) |
| VASP-si | 14.5 ± 1.2 (3) | 14.7 ± 0.9 (5) | 0.20 ± 0.03 (4) | 0.19 ± 0.01 (4) | 0.80 ± 0.20 (3) | 0.22 ± 0.15 (3)a | 1.10 ± 0.10 (3) | 0.90 ± 0.10 (3) |
| FAK-i | 21.6 ± 1.5 (3) | 20.6 ± 1.3 (3) | 0.23 ± 0.03 (3) | 0.35 ± 0.06 (3) | 1.00 ± 0.08 (3) | 0.28 ± 0.03 (3) | 0.97 ± 0.03 (3) | 0.10 ± 0.01 (3) |
| FAK-si | 15.8 ± 1.7 (3) | 14.1 ± 1.9 (4) | 0.19 ± 0.01 (4) | 0.20 ± 0.02 (4) | 0.92 ± 0.08 (3) | 0.27 ± 0.03 (3) | 1.10 ± 0.09 (3) | 1.12 ± 0.10 (3) |
| Geld | 21.1 ± 1.2 (5) | 20.7 ± 0.6 (5) | 0.31 ± 0.0 (3) | 0.39 ± 0.05 (3) | 0.83 ± 0.17 (3) | 1.10 ± 0.33 (3) | 1.17 ± 0.17 (5) | 1.23 ± 0.15 (3) |
| HSP90-si | 20.1 ± 0.9 (5) | 18.7 ± 0.7 (5) | 0.24 ± 0.02 (3) | 0.39 ± 0.08 (3) | 0.95 ± 0.23 (6) | 0.97 ± 0.19 (6) | 1.10 ± 0.07 (3) | 1.33 ± 0.17 (3) |
| 8-br-cGMP | 20.7 ± 0.2 (4) | 20.3 ± 0.1 (4) | 0.24 ± 0.04 (3) | 0.32 ± 0.07 (3) | 1.10 ± 0.10 (3) | 1.11 ± 0.08 (3) | 1.10 ± 0.10 (3) | 1.17 ± 0.09 (3) |
| mGC-si | 21.8 ± 0.7 (5) | 20.0 ± 0.2 (5) | 0.23 ± 0.07 (5) | 0.33 ± 0.10 (5) | 0.68 ± 0.10 (3) | 0.67 ± 0.08 (3) | 1.00 ± 0.01 (3) | 1.01 ± 0.03 (3) |
a p < 0.05 versus control/PBS value
If neutrophils were exposed to UV light for 5 min after 100 nm DENO exposure and prior to plating, impaired adherence was not identified (Table 1). Alternatively, if cells were incubated with cytochalasin D, with small chemical inhibitors to Rac or FAK prior to DENO exposure, or if cells were depleted of VASP or FAK by prior incubations with siRNA, no discernible •NO-mediated inhibition of β2 integrin adherence occurred.
The effect of 1400W and observations with cells from iNOS knock-out mice support a role of iNOS activation in β2 integrin inhibition. Because an association with heat shock protein 90 (HSP90) is one pathway for iNOS activation (39), we investigated whether siRNA to HSP90 and a HSP90 inhibitor, geldanamycin, had effects. As shown in Table 1, no inhibition of β2 integrin adherence occurred with DENO after these interventions.
Inhibition of β2 integrin function is abrogated by incubating cells with 8-br-cGMP (Table 1), which has been reported previously (18). Exposure to •NO has also been shown to inhibit the catalytic activity of mGC (18). To investigate the role of guanylate cyclase enzymes in •NO-mediated β2 integrin inhibition, cells were incubated with siRNA to deplete either mGC or the soluble GC enzyme. Whereas cells depleted of mGC do not exhibit impaired β2 integrin function after DENO exposure (Table 1), depletion of soluble GC had no significant effect on the DENO-mediated response. β2 integrin-specific adherence in soluble GC-depleted cells exposed just to PBS was 21.9 ± 0.6% (n = 6), and adherence was reduced by 100 nm DENO to 3.9 ± 1.4% (n = 6, p < 0.001). Similarly, when cells were exposed to 0.1 mm LY 83583, a guanylate cyclase inhibitor, for 15 min no abrogation in the β2 integrin inhibition by 100 nm DENO was observed (data not shown).
Actin S-Nitrosylation
S-Nitrosylation of murine neutrophil proteins was surveyed by the biotin switch assay, which covalently adds a disulfide-linked biotin to the labile S-nitrosylation sites on proteins. A Western blot probing for biotin-containing proteins in neutrophils is shown in Fig. 2. In separate trials, the prominent band at ∼43 kDa was cut from nitrocellulose paper, subjected to amino acid sequencing, and identified as β-actin. If the analysis was performed on cell lysates treated with just N-[6-(biotinamido) hexyl]-3′-(2′-pyridyldithio) propionamide-biotin or with ascorbate (e.g. an incomplete biotin-switch formulation) or with 1 mm HgCl2 (which cleaves S-NO bonds), the bands are not visualized (data not shown).
FIGURE 2.
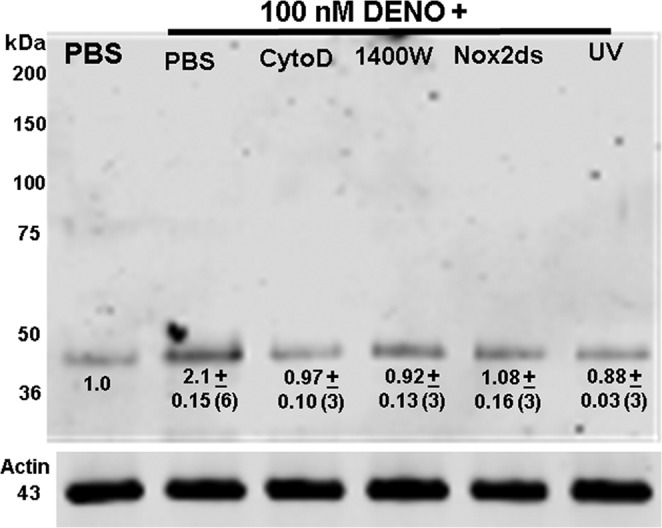
Western blot showing biotinylated proteins. Lysates of neutrophils exposed to PBS + 5.5 mm glucose or to PBS/glucose + 100 nm DENO for 2 min were prepared according to the biotin switch assay. The entire gel is shown. Where indicated cells were concurrently incubation with DENO and 5 μm cytochalasin D, 1 mm 1400W, or 10 μm Nox-2ds. UV indicates cells exposed to UV light for 5 min after DENO incubation. Although not shown, cytochalasin D, 1400W, Nox-2ds, and UV had no effect on biotin switch results in cells incubated with PBS rather than DENO. The numbers beneath the band at ∼43 kDa reflect means ± S.E. (n = replicate experiments) for the fold difference in 43-kDa band density/actin band density normalized to the ratio of PBS-only exposed control cells for each experiment. *, p < 0.05 versus control.
For serial studies, the magnitude of S-nitrosylated actin (SNO-actin) was evaluated by measuring the biotin band density normalized to blots assessing actin band density in the same cell lysates. The numbers shown in Fig. 2 indicate fold band density versus control for DENO-exposed cells and for cells incubated with DENO along with cytochalasin D, 1400W, and Nox2ds and when exposed to UV light for 5 min after DENO incubation. These interventions all reversed actin S-nitrosylation. Although not shown, these interventions had no significant effect on biotin switch analysis of control (PBS suspended) neutrophils. Among cells incubated with 8-br-cGMP, the SNO-actin band density was 0.99 ± 0.16 (n = 3, NS) fold that of control cells. When cells were co-incubated with 8-br-cGMP plus DENO the SNO-actin band density was 1.20 ± 0.06 (n = 3, NS versus control), thus showing that 8-br-cGMP also reversed actin S-nitrosylation.
NOS Activation
The activity of iNOS was monitored as [3H]citrulline production that could be inhibited by co-incubation with 1400W in cells permeabilized with OG to remove the need for active [3H]arginine transport (prior studies have shown that OG treatment does not impede β2 integrin function (25)). Table 1 shows [3H]citrulline production when [3H]arginine was added to cells that were then either exposed to only PBS or to 100 nm DENO. As shown, iNOS activation occurred in response to DENO, and the same interventions that abrogated the effect of •NO on β2 integrin adherence also inhibited iNOS activation.
Activation of iNOS occurs with formation of dimers. Total intracellular content of iNOS normalized to actin content was not altered by 100 nm DENO (data not shown). Analysis of iNOS dimers/actin in cell lysate Western blots exhibited over a 2-fold elevation with DENO-exposed cells. A representative blot is shown in Fig. 3. No significant increase in iNOS dimers occurred when cells were exposed to UV light after DENO incubation or when cells were depleted of HSP90 by prior incubation with siRNA.
FIGURE 3.
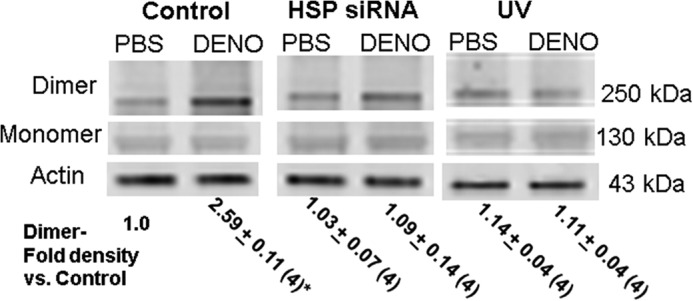
Western blot showing iNOS dimer and monomers. Neutrophil suspensions were exposed to PBS/glucose (control) ± 100 nm DENO for 2 min. Some had been incubated with siRNA to HSP 90 for 24 h before the study, and others were irradiated with UV for 5 min after incubation and before lysis. Western blots were probed for iNOS and β-actin. The figure shows a representative experiment, and the numbers beneath the figure are mean data for four replicate studies performed with fresh neutrophil isolates from different mice normalized to the control iNOS dimer/actin band density ratio for each experiment. The data are means ± S.E. *, p < 0.05 versus control.
As the study progressed, it became important to evaluate whether NOS activation may accelerate with time after cells were exposed to •NO. Therefore, a series of studies was performed by incubating cells with DENO, except rather than adding [3H]arginine concurrently with DENO and measuring [3H]citrulline 30 min later, [3H]arginine was added 30, 60, or 90 min after DENO addition, and [3H]citrulline was measured 30 min later. In this way, NOS activity could be compared at times up to 2 h following DENO addition. We found NOS activity to be essentially the same at each time interval. For example, in cells assayed by incubating with [3H]arginine between 1.5 and 2 h after DENO addition, the rate of [3H]citrulline formation was 1.85 ± 0.02 (n = 4) pmol/h, significantly greater than control but not significantly different from the standard assay when [3H]arginine was added to cell preparations concurrently with DENO (Table 1).
Actin Polymerization in Permeabilized Neutrophils
The data generated thus far demonstrate that •NO-mediated β2 integrin inhibition involves a complex interaction that includes actin polymerization, given the inhibitory effects of cytochalasin D, and perturbations of proteins such as FAK and VASP. Therefore, we next examined whether exposure to 100 nm DENO altered the dynamics of intracellular actin polymerization assessed as free actin incorporation sites. This was done with OG-permeabilized murine neutrophils incubated with pyrene-actin immediately following 2-min incubations with DENO. As shown in Table 1, DENO exposure increased actin polymerization by 3.6-fold, and the same manipulations that abrogated •NO-mediated β2 integrin inhibition and iNOS activation also inhibited augmented actin polymerization.
Total F-actin was measured by NBD-phalloidin binding and expressed as fluorescence/μg cell protein. In seven replicate trials, fluorescence of PBS-exposed cells (control) was 1.4 ± 0.2, and in PBS cells exposed to UV light fluorescence was 0.8 ± 0.5 (NS versus control), whereas in cells exposed for 2 min to 100 nm DENO, fluorescence was 131.1 ± 9.9 (p < 0.05 versus control, ANOVA). Consistent with a role for SNO-actin, if cells were exposed to UV light after DENO addition, fluorescence was 8.0 ± 1.3 (NS versus control).
The elevation in total F-actin caused by DENO diminished with time. If cells were fixed at 1 h after DENO addition, rather than at 2 min, fluorescence was 62.0 ± 11.5 (n = 6), 32.5 ± 3.1 at 2 h and at 3 h, and 17.6 ± 2.5—values significantly different from control and also different from the 2 min DENO value.
The distribution of Triton-soluble G-actin, Triton-soluble sF-actin, and Triton-insoluble F-actin before and after •NO treatment is shown in Fig. 4. The numbers below the representative Western blot are the percentages of total actin in the cell preparations. The results show the increase in sF-actin at the expense of G- and Triton-insoluble F-actin caused by exposure to 100 nm DENO. Reversal of this effect by UV light is also shown.
FIGURE 4.
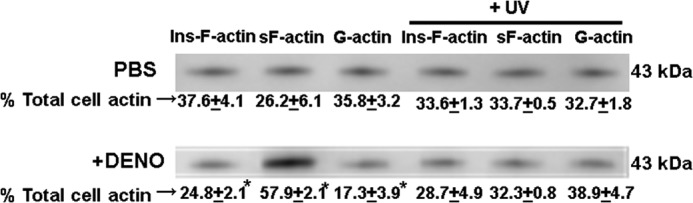
Distribution of G-actin, sF-actin, and Triton-insoluble F-actin. Murine neutrophils were exposed to PBS/glucose ± 100 nm DENO for 2 min, and where indicated, cells were then exposed to UV light for 5 min prior to lysis and fractionation (see “Experimental Procedures”). Representative Western blots are shown for air-exposed, control cells (top), and cells exposed to DENO. The percentage of Triton-insoluble F-actin (Ins-F-actin), Triton-soluble F-actin (sF-actin), and G-actin are shown as means ± S.E. for four replicate studies. *, p < 0.05 versus PBS exposed (control) cells.
Reactive Species Generation
Because of the complex pattern of protein involvement in •NO-mediated β2 integrin inhibition, we were interested in a closer evaluation of reactive species produced in response to DENO exposure. First, a series of studies was conducted by adding membrane permeable DCF-DA to murine neutrophil suspensions. Table 1 data demonstrate that DCF fluorescence was increased by exposure to DENO, and fluorescence was impeded by all of the same interventions blocking •NO-mediated β2 integrin inhibition.
DCF fluorescence enhancement by DENO did not occur when cells were exposed to Nox2ds or in cells depleted of gp91phox, indicating that NOX activation was occurring. Because NOX activity would result in elevations of H2O2, we also performed studies with 50 μm FBBBE, an agent viewed as highly specific for H2O2 versus DCF-DA, which reacts with a relatively wide variety of reactive species (40). As shown in Table 2, exposure to DENO resulted in a near 3-fold elevation in the rate of FBBBE fluorescence, and the response was inhibited by the NOX inhibitor, Nox2ds, the iNOS inhibitor 1400W, and the HSP90 inhibitor, geldanamycin.
TABLE 2.
FBBBE fluorescence
Fluorescence in arbitrary units/min when 1.8 × 105 murine neutrophils were incubated for 10 min with agents listed in the left column prior to adding 50 μm FBBBE + PBS or 100 nm DENO and monitoring for 5 min, which was the duration of linear rise in fluorescence readings. The data are mean rates ± S.E., n = 6 replicate experiments performed with fresh neutrophil isolates from different mice for all groups.
| Agent | Control | 100 nm DENO |
|---|---|---|
| PBS | 1.29 ± 0.07 | 3.07 ± 0.15a |
| Nox2ds | 1.00 ± 0.17 | 0.97 ± 0.11 |
| 1400W | 0.87 ± 0.07 | 1.20 ± 0.12 |
| Geldanamycin | 1.33 ± 0.17 | 1.40 ± 0.38 |
a p < 0.05 versus control.
Rac Activation
We next wanted to evaluate the activity of Rac proteins because the pathways that regulate cytoskeletal actin polymerization formation in neutrophils involve the Rac GTPase proteins (41). Neutrophils were exposed to PBS (control) or DENO, and activity of Rac-1/2/3 was assayed as described under “Experimental Procedures” using a commercial kit. Cells incubated with DENO exhibited a marked elevation in Rac activity (Table 3). A role for S-nitrosylated proteins was shown because elevated Rac activity did not occur in cells exposed to UV light after DENO. Similarly, Rac activation did not occur if cells had been incubated with Nox2ds, 1400W, or cytochalasin D.
TABLE 3.
Rac activation (ng/10 μg of protein)
Neutrophil was incubated for 10 min with agents listed in the left column prior to being exposed to PBS (control) or 100 nM DENO for 2 min and lysed, and Rac activity was assayed. Prior to lysis, some cells were exposed for 5 min to UV light. The values are the means ± S.E. The numbers in parentheses indicate the numbers of replicate experiments performed with fresh neutrophil isolates from different mice.
| Agent | Control | 100 nm DENO |
|---|---|---|
| PBS | 0.99 ± 0.07 (6) | 3.72 ± 0.16 (6)a |
| Nox2ds | 1.08 ± 0.05 (3) | 1.03 ± 0.06 (3) |
| 1400W | 0.94 ± 0.09 (3) | 1.05 ± 0.07 (3) |
| Cytochalasin D | 1.33 ± 0.17 (3) | 1.40 ± 0.38 (3) |
| UV | 1.01 ± 0.09 (3) | 1.10 ± 0.09 (3) |
a p < 0.05 versus air (control).
Protein Cross-linking to Actin Fractions
The small chemical inhibitors and siRNA-mediated protein depletion effects on •NO-mediated actin polymerization outlined in Table 1 and the biotin switch assay that identified S-nitrosylated actin (Fig. 2) implicate actin turnover as essential for inhibiting β2 integrin adherence. Therefore, we were interested in evaluating associations of cytoplasmic actin with proteins exhibiting a role in •NO-mediated β2 integrin inhibition. After cells were exposed for 2 min to either PBS (control) or 100 nm DENO, they were treated with the membrane-permeable protein cross-linker DTSP and lysed 30 min later. Cell lysates were separated into G-actin, sF-actin, and Triton-insoluble F-actin fractions. Blots were analyzed looking for differences in protein band densities relative to actin and whether changes seen in the DENO-exposed cells were abrogated by exposure to UV light for 5 min before DTSP incubations. A representative Western blot of the sF-actin cell fractions is shown in Fig. 5, and quantitative changes for all three actin fractions are outlined in Table 4. There were marked elevations in protein associations in the sF-actin fraction of DENO-exposed cells. Rather than elevated ratios, we found reductions in the same protein associations of DENO-exposed cells in G-actin and Triton-insoluble F-actin fractions, and they were once again abrogated by exposure to UV light (Table 4).
FIGURE 5.
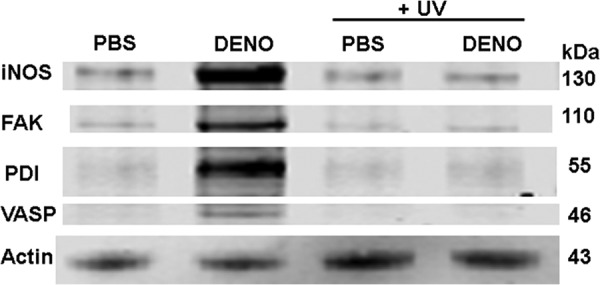
Protein associations in the Triton-soluble F-actin fraction. Murine neutrophils were exposed to PBS/glucose ± 100 nm DENO for 2 min. Where indicated, samples were then exposed to UV light for 5 min prior to addition of DTSP to cross-link proteins, then fractioned based on Triton solubility (see “Experimental Procedures”), and subjected to Western blotting. This is a representative blot among four replicate experiments. Data based on the blots are shown in Table 4.
TABLE 4.
Protein associations assessed by DTSP cross-linking in sF-actin, G-actin, and Triton-insoluble actin fractions of neutrophils incubated with PBS (control) or 100 nm DENO for 2 min
The values are ratios calculated based on the band densities of the identified protein relative to the actin band density in each sample. In each table, the values are normalized to air-exposed control protein band ratios of individual experiments; thus, values greater than 1.0 reflect increases in protein associations with actin, whereas values less than 1.0 indicate lower protein associations. Where indicated cells were exposed to UV light for 5 min prior to addition of DTSP to cross-link proteins and then fractioned based on Triton solubility (see “Experimental Procedures”). The data are the means ± S.E. (n = 4 for all samples).
| VASP/actin | FAK/actin | iNOS/actin | PDI/actin | |
|---|---|---|---|---|
| sF-actin | ||||
| DENO | 3.85 ± 1.02a | 2.08 ± 0.17a | 2.31 ± 0.17a | 5.07 ± 1.25a |
| PBS + UV | 1.37 ± 0.6 | 1.14 ± 0.24 | 1.10 ± 0.23 | 1.51 ± 0.33 |
| DENO +UV | 1.13 ± 0.19 | 0.97 ± 0.19 | 1.21 ± 0.47 | 1.26 ± 0.24 |
| G-actin | ||||
| DENO | 0.16 ± 0.09a | 0.15 ± 0.08a | 0.39 ± 0.16a | 0.21 + 0.10a |
| PBS + UV | 0.82 ± 0.22 | 0.81 ± 0.12 | 0.73 ± 0.12 | 0.74 ± 0.13 |
| DENO +UV | 1.21 ± 0.32 | 1.02 ± 0.18 | 0.65 ± 0.08 | 0.81 ± 0.25 |
| Triton-insoluble | ||||
| DENO | 0.14 ± 0.12a | 0.22 ± 0.08a | 0.51 ± 0.13a | 0.17 ± 0.08a |
| PBS + UV | 1.06 ± 0.18 | 0.93 ± 0.07 | 0.93 ± 0.08 | 1.07 ± 0.14 |
| DENO +UV | 0.88 ± 0.16 | 0.62 ± 0.11 | 0.87 ± 0.07 | 0.95 ± 0.16 |
a Values significantly different from normalized air-exposed control values (p < 0.05, ANOVA).
Confocal Images
Confocal microscope images were obtained to evaluate the appearance of actin filaments and several proteins that impact •NO-mediated β2 integrin inhibition. Fig. 6 shows representative images of neutrophils exposed to either PBS (control) or 100 nm DENO and processed as described under “Experimental Procedures.” Phalloidin shows filamentous actin; iNOS, HSP90, FAK, and gp91phox all appeared in a more aggregated state in •NO-exposed cells.
FIGURE 6.
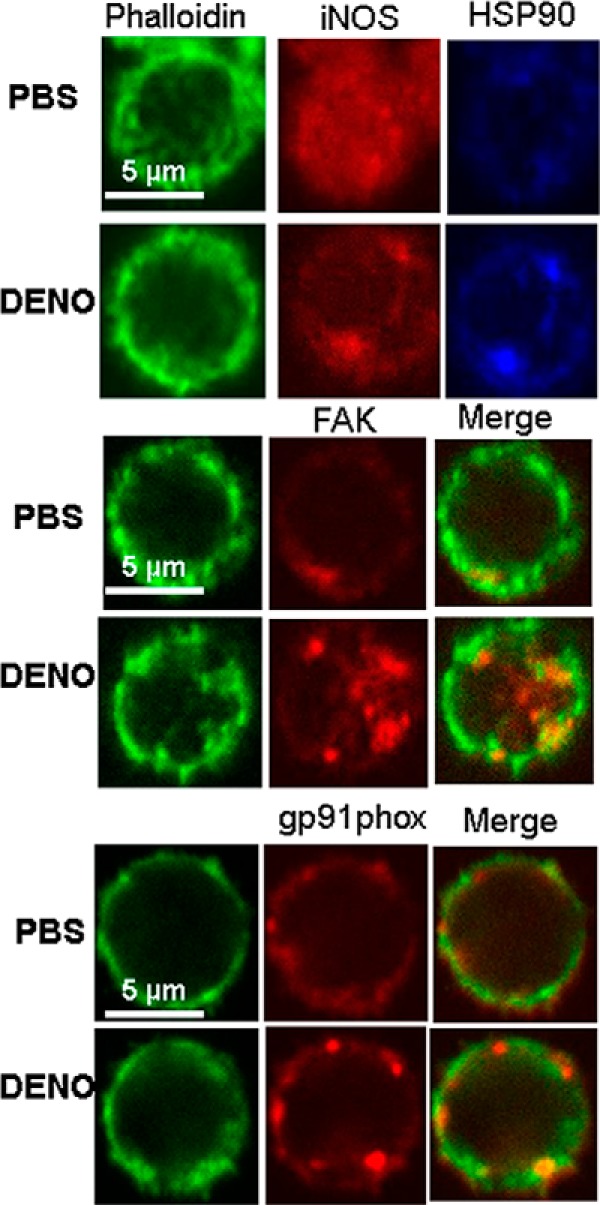
Confocal microscope images of murine neutrophils exposed to PBS/glucose ± 100 nm DENO for 2 min.
The fluorescence intensity of phalloidin was found to differ between PBS and DENO-exposed samples. Using the same laser settings in five studies conducted with neutrophils from different mice, fluorescence in arbitrary units was 933 ± 29 (n = 244 cells) for PBS-exposed (control) neutrophils and 1114 ± 19 (n = 265 cells, p < 0.05) for DENO-exposed cells. Images suggest that although co-fractionation among the various proteins is a response to •NO exposure (Fig. 5), it does not appear to be due to direct protein-protein binding.
Reversal of •NO-dependent β2 Integrin Inhibition by 8-br-cGMP
The data in Table 1 document that incubating neutrophils with 8-br-cGMP abrogates the inhibitory effect of DENO on β2 integrin adhesion. Further, membrane guanylate cyclase, but not soluble guanylate cyclase, is required for •NO-dependent β2 integrin inhibition. Finding a role for 8-br-cGMP as a reversal agent suggests that one or more protein kinases, perhaps PKG or PKA, which can also be activated by cGMP (42), have roles in reversing the DENO effect.
Involvement of PKG and PKA was investigated by first incubating cells with either 0.1 mm Rp-8-br-PET-cGMPS, a selective PKG inhibitor, or 1 mm Rp-cAMPS, a selective inhibitor of PKA. In four trials, neither the PKG nor PKA inhibitors had an effect on β2 integrin-dependent adhesion to fibrinogen-coated plates; adherence was, respectively, 20.6 ± 1.4% and 19.8 ± 1.4%. Incubation with 100 nm DENO significantly reduced β2 integrin adherence. With cells that had been incubated with the PKG inhibitor and then with DENO, adherence was just 1.8 ± 0.3%, with the PKA inhibitor, 2.8 ± 0.5% (for both, p < 0.05 versus PBS-exposed cells). As with normal, control cells shown in Table 1, reversal of •NO-mediated β2 integrin inhibition occurred when cells previously exposed to the PKA inhibitor were incubated with DENO and 100 μm 8-br-cGMP (adherence 18.6 ± 1.1%, NS versus PBS alone). However, 8-br-cGMP did not reverse DENO-mediated inhibition when cells had been incubated with the PKG inhibitor. Adherence after cells were incubated with DENO and 100 μm 8-br-cGMP was 2.9 ± 0.4% (p < 0.05 versus PBS alone). We conclude that 8-br-cGMP abrogates •NO-dependent β2 integrin inhibition by activating PKG.
Biphasic Effect of DENO on β2 Integrin Function
Finally, we investigated the mechanism for the biphasic impact of DENO on neutrophil β2 integrin-dependent adhesion. Fig. 1 demonstrates that 50–100 nm DENO inhibits β2 integrin adherence, and the effect is fully reversed by 400 nm DENO or more. Given that 8-br-cGMP abrogates the inhibitory effect of DENO on β2 integrin adhesion, and •NO can activate soluble guanylate cyclase to generate cGMP, it was possible that high concentrations of DENO might reinstate β2 integrin-dependent adhesion by increasing the intracellular concentration of cGMP.
Fig. 7 shows cGMP concentrations after neutrophils were incubated for 2 min with DENO. Exposure to 100 nm DENO or more resulted in statistically significant elevations of intracellular cGMP concentrations, but there were no notable differences among exposures to 100 nm or more. We conclude, therefore, that reversal of •NO-mediated β2 integrin inhibition at high (∼400 nm DENO) was not be due to elevations of intracellular cGMP concentrations.
FIGURE 7.
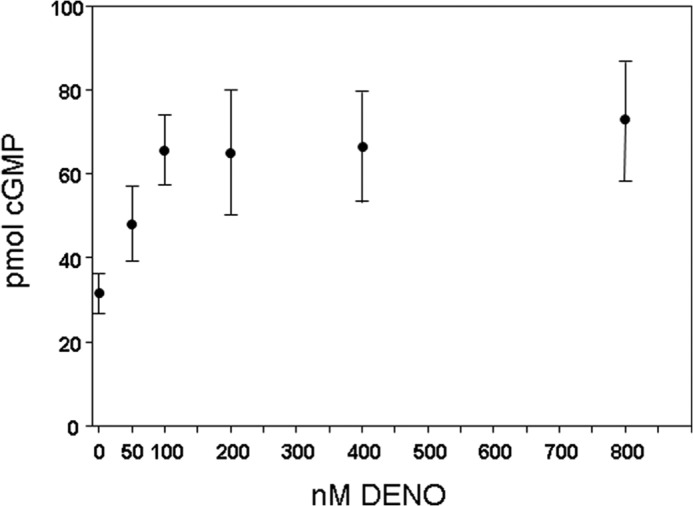
Cyclic GMP concentration in neutrophils. Murine neutrophils were exposed for 2 min to PBS/glucose and the DENO concentrations shown, then lysed for assays of cGMP. The values (pmol cGMP/5 × 105 neutrophils) are means ± S.E., n = 5 for each measurement. The values for 100–800 nm DENO are significantly different from the concentration in cells exposed only to PBS/glucose (p < 0.05, ANOVA) but not significantly different from each other.
Whereas 100 nm DENO increases fluorescence when cells were exposed to 50 μm FBBBE (specific probe for H2O2) nearly 3-fold (Table 2), exposure to 400 nm DENO diminished the rate of fluorescence change by approximately half that seen in normal, control cells to 0.72 ± 0.15 arbitrary units/min (n = 6, p < 0.05 versus control). When 10 μm DCF-DA was used as probe, both 400 nm and 1 μm DENO rendered fluorescence change over 10 min undetectable in six trials. We conclude that high concentrations of DENO inhibit NOX.
As shown in Table 1, iNOS activity in control cells was 0.20 ± 0.05 (n = 15) pmol [H3]citrulline/h, and 100 nm DENO increased iNOS activity by ∼ 12-fold. When cells were incubated with 400 nm DENO, iNOS activity was 0.82 ± 0.15 (n = 3, NS versus control) and with 1 μm DENO 0.90 ± 0.32 (n = 3, NS versus control) pmol [H3]citrulline/h.
Discussion
Study results demonstrate that •NO inhibits neutrophil β2 integrin adherence by a complex series of perturbations related to actin S-nitrosylation. The array of inhibitory interventions shown in Table 1 suggests an interdependency among cell processes. For example, DENO exposure increases actin polymerization, and inhibiting polymerization with cytochalasin D also prevents enhanced iNOS activation and increased DCF fluorescence that involves NOX activation. This pattern leads us to conclude that maintaining adherence inhibition occurs because of an autocatalytic process. Fig. 8 illustrates the proteins involved. Each was included within the gray circle to indicate the interdependence of the interactions, and all can be perturbed by reversing actin nitrosylation with UV light.
FIGURE 8.
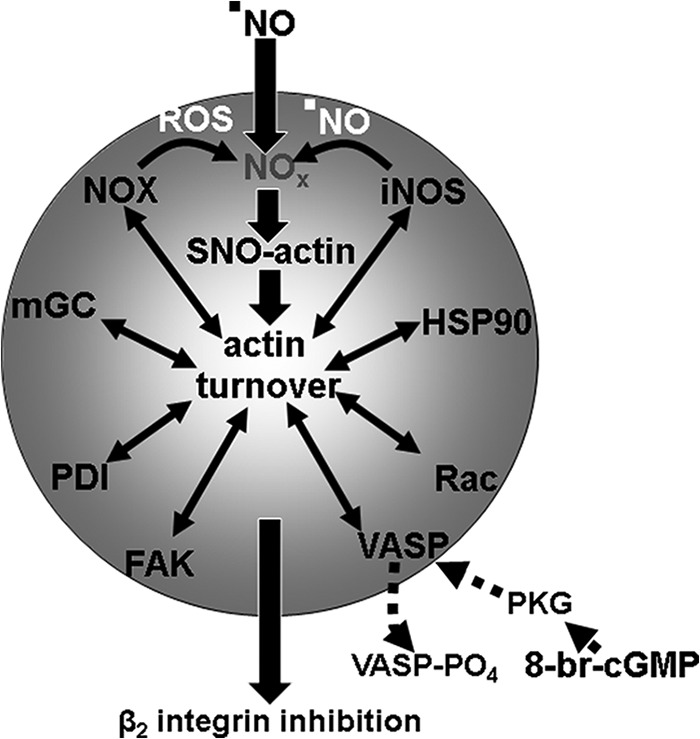
Schematic of proposed mechanism for ·NO-mediated inhibition of β2 integrin dependent neutrophil adherence. See “Discussion” for a detailed explanation. NOx, higher order reactive nitrogen species such as nitrogen dioxide or peroxynitrite; VASP-PO4, phosphorylated VASP mediated by PKG; Rac, Rac1 and Rac2 GTPases. The interdependent nature of the various proteins for maintenance of actin turnover leading to β2 integrin inhibition is shown by their placement within the gray circle.
Exposure to 50–100 nm DENO (a •NO flux of ∼14–28 nmol/min) results in SNO-actin formation (Fig. 2). Reactive nitrogen species such as NO2 or peroxynitrite are necessary for this to occur. NOX is a logical source for the requisite initial ROS needed to react with DENO-derived •NO to produce agents capable of S-nitrosylation. The on-going process appears to be centered on actin polymerization/turnover and inhibited by cytochalasin D. This is similar to a mechanism we reported in other studies, where S-nitrosylation drives a series of steps leading to excessive actin turnover (24, 31). VASP exhibits higher affinity for S-nitrosylated sF-actin filaments (26); VASP also bundles Rac proteins in close proximity to sF-actin, and subsequent Rac activation increases actin polymerization, which leads to enhanced binding of FAK and then association of iNOS (25). Activation of iNOS involves linkage to actin via FAK, and results from this investigation indicate that HSP90 is also required. The process appears to be perpetuated by production of •NO from iNOS and ROS by NOX. In this regard, PDI is known to facilitate NOX activation (38).
The role for mGC in the interdependent process depicted in Fig. 8 is supported by siRNA knockdown studies. This could involve promotion of NOX activity as reported by others (27–30). Synthesis of cGMP by mGC does not play a role because DENO concentrations that inhibit integrin function significantly increase the intracellular cGMP concentration (Fig. 7). To the contrary, inhibition of mGC-mediated cGMP synthesis as reported with exposures to •NO that inhibit β2 integrins could be important (18). Cytoskeletal localizations play an important role in functional consequences of protein kinases, and cGMP synthesized by mGC can activate PKG and cause VASP phosphorylation (26, 43). Results in this study show that addition of 8-br-cGMP abrogates •NO-dependent β2 integrin inhibition via PKG. This action is likely because of the higher affinity 8-br-cGMP has for PKG versus merely an increase in intracellular cGMP concentration, based on results shown in Fig. 7 (44). In a prior publication, we demonstrated that PKG-mediated VASP phosphorylation will shut down iNOS activation mediated by the SNO-actin-driven process because phospho-VASP does not exhibit the same heightened affinity for SNO-actin as does nonphosphorylated VASP (26).
Absence of β2 integrin inhibition by high DENO concentrations (Fig. 1) appears to be related to inhibition of NOX based on studies with FBBBE and DCF-DA, possibility via a •NO reaction with the p47phox subunit (45). Activity of iNOS was also diminished with exposures to 400 nm DENO or more, relative to that seen with 100 nm DENO. This decrease could be due to NOX inhibition and/or inhibition of iNOS directly. High •NO concentrations can inhibit iNOS by competing with O2 at the heme active site or by inhibiting dimer formation (37, 46).
Given the appearance of an autocatalytic process related to NOX and iNOS, we evaluated whether iNOS activity might accelerate over time after addition of DENO. We found, however, that for as long as 2 h after addition of 100 nm DENO, iNOS activity remained elevated at the same level as when assays were done immediately after DENO addition. We believe that persistent, rather than increasing, enzyme activity occurs because the iNOS-FAK-sF-actin association is unstable. Elevated iNOS activity involves linkage to FAK and FAK linkage to sF-actin. Elevated iNOS activity requires an association with filamentous actin (25). Whereas VASP exhibits high affinity for SNO-actin, FAK binding is diminished as more actin is S-nitrosylated (25). These relationships underscore the need for an ongoing actin polymerization process and likely a denitrosylation mechanism. This could include thioredoxin reductase, as shown in other studies, but further investigation with regard to DENO will be required (47). In short, ongoing SNO-actin formation, VASP linkage followed by enhanced actin polymerization, and FAK-mediated iNOS association can explain the apparent prolongation, versus acceleration, of iNOS activation and with it, persistent •NO-mediated β2 integrin inhibition.
There is an interesting discrepancy regarding filamentous actin in our results. Total F-actin in neutrophils appears to increase nearly 94-fold based on NBD-phalloidin binding with addition of 100 nm DENO (fluorescence increase from 1.4 to 131). However, phalloidin fluorescence intensity on confocal images indicates only a 19% increase (mean fluorescence of 1114 for DENO-exposed cells versus 933 for PBS-exposed cells). Similarly, Western blotting of actin fractions (Fig. 4) demonstrates a 19% increase in the total amount of F-actin (Triton-insoluble ± sF-actin).
This discrepancy can be explained as being due to differences in time of cell fixation. NBD-phalloidin binding was reduced by ∼53% if cells were fixed at 1 h after DENO addition and by 75% at 2 h. Because cells only slowly settled onto slides for the imaging studies, they were fixed over 1 h after DENO addition, and the cells used in the Western blot experiments were not fixed, just lysed after centrifugation steps.
Fig. 5 and Table 4 demonstrate that DENO addition led to an augmentation of regulatory protein associations with sF-actin between 2- and 5-fold. There was a increase of ∼2.2-fold in the sF-actin pool, as shown in Fig. 4 (the sF-actin fraction represents ∼26.2% of total actin in control cells, 57.9% in DENO-exposed cells). From this we conclude that the increased protein associations depicted in Fig. 5 and Table 4 are not due solely to the increase in sF-actin. The role of protein scaffolds bundling multiple proteins together with secondary association to sF-actin, as occurs with FAK and VASP, is likely to explain this difference. This interpretation also provides an explanation for the lack of a precise co-localization of proteins with F-actin in the confocal images (Fig. 6).
In the introduction, we discuss what is likely to be a physiological level of •NO generated by endothelium. The biphasic effect of •NO shown in Fig. 1, with the absence of β2 integrin inhibition when cells are exposed to high •NO concentrations, could also have physiological relevance to immune surveillance. Although it is difficult to predict precise •NO concentrations, marked elevations of •NO synthesis occur in association with infection and in wound exudates when surgical complications arise (48, 49). These are instances where effective neutrophil adherence would be advantageous.
In summary, this study adds to our knowledge of how β2 integrin function is regulated in neutrophils. The results demonstrate the complex role that the cytoskeleton plays in this important cell function. We believe that the concentrations of •NO we used to inhibit β2 integrins are in the physiological range, and thus events as described occur within the microvasculature. An important caveat to this consideration is that NOX activity is required for impaired β2 integrin adherence. Further work is required to evaluate the balance between •NO entry to the neutrophil and intracellular production of ROS to inhibit β2 integrins. With regard to possible clinical utility of these findings, the results underscore the need for careful selection of •NO-donor dosage if attempting to impede neutrophil adherence in the setting of, say, reperfusion injury.
Author Contributions
V. M. B., M. Y., and K. Y. performed laboratory studies, interpreted data, reviewed the manuscript, provided advice, and approved final version of the article. S. R. T. conceived ideas, oversaw the research program, performed laboratory studies, analyzed data, interpreted data, and wrote the manuscript.
This work was supported by Grant N00014–14-10094 from the Office of Naval Research.
- mGC
- membrane or particulate guanylate cyclase
- DCF-DA
- dihydrodichlorofluorescein-diacetate
- DENO
- diethylamine NONOate
- DTSB
- dithiobis (succinimidyl proprionate)
- FAK
- focal adhesion kinase
- FBBBE
- fluorescein bis(benzyl boronic ester)
- HSP90
- heat shock protein 90
- iNOS
- type 2 nitric-oxide synthase
- NOS
- nitric-oxide synthase
- NOX
- NADPH oxidase
- OG
- n-octyl-β-glucopyranoside
- PKA
- cAMP-dependent protein kinase
- sF-actin
- Triton-soluble filamentous actin
- SNO-actin
- S-nitrosylation actin
- VASP
- vasodilator-stimulated phosphoprotein
- ANOVA
- analysis of variance
- ROS
- reactive oxygen species
- DTSP
- dithiobis (succinimidyl proprionate)
- PDI
- protein-disulfide isomerase
- NS
- not significant.
References
- 1. Brown E. J., Lindberg F. P. (1996) Leucocyte adhesion molecules in host defence against infection. Ann. Med. 28, 201–208 [DOI] [PubMed] [Google Scholar]
- 2. Niu X. F., Smith C. W., Kubes P. (1994) Intracellular oxidative stress induced by nitric oxide synthesis inhibition increases endothelial cell adhesion to neutrophils. Circ. Res. 74, 1133–1140 [DOI] [PubMed] [Google Scholar]
- 3. Radomski M. W., Palmer R. M., Moncada S. (1987) Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 2, 1057–1058 [DOI] [PubMed] [Google Scholar]
- 4. Arndt H., Russell J. B., Kurose I., Kubes P., Granger D. N. (1993) Mediators of leukocyte adhesion in rat mesenteric venules elicited by inhibition of nitric oxide synthesis. Gastroenterology 105, 675–680 [DOI] [PubMed] [Google Scholar]
- 5. Kubes P., Suzuki M., Granger D. N. (1991) Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. U.S.A. 88, 4651–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kubes P., Jutila M., Payne D. (1995) Therapeutic potential of inhibiting leukocyte rolling in ischemia/reperfusion. J. Clin. Invest. 95, 2510–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kubes P., Kurose I., Granger D. N. (1994) NO donors prevent integrin-induced leukocyte adhesion but not P-selectin-dependent rolling in postischemic venules. Am. J. Physiol. 267, H931–H937 [DOI] [PubMed] [Google Scholar]
- 8. Gaboury J., Woodman R. C., Granger D. N., Reinhardt P., Kubes P. (1993) Nitric oxide prevents leukocyte adherence role of superoxide. Am. J. Physiol. 265, H862–H867 [DOI] [PubMed] [Google Scholar]
- 9. Tsao P. S., Ma X. L., Lefer A. M. (1992) Activated neutrophils aggravate endothelial dysfunction after reperfusion of the ischemic feline myocardium. Am. Heart J. 123, 1464–1471 [DOI] [PubMed] [Google Scholar]
- 10. Johnson G., 3rd, Tsao P. S., Lefer A. M. (1991) Cardioprotective effects of authentic nitric oxide in myocardial ischemia with reperfusion. Crit. Care Med. 19, 244–252 [DOI] [PubMed] [Google Scholar]
- 11. Leite A. C., Cunha F. Q., Dal-Secco D., Fukada S. Y., Girão V. C., Rocha F. A. (2009) Effects of nitric oxide on neutrophil influx depends on the tissue: role of leukotriene B4 and adhesion molecules. Br. J. Pharmacol. 156, 818–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leeuwenburgh C., Hardy M. M., Hazen S. L., Wagner P., Oh-ishi S., Steinbrecher U. P., Heinecke J. W. (1997) Reactive nitrogen intermediates promote low density lipoprotein oxidation in human atherosclerotic intima. J. Biol. Chem. 272, 1433–1436 [DOI] [PubMed] [Google Scholar]
- 13. Münzel T., Sayegh H., Freeman B. A., Tarpey M. M., Harrison D. G. (1995) Evidence for enhanced vascular superoxide anion production in nitrate tolerance. J. Clin. Invest. 95, 187–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lopez-Belmonte J., Whittle B. J., Moncada S. (1993) The actions of nitric oxide donors in the prevention or induction of injury to the rat gastric mucosa. Br. J. Pharmacol. 108, 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pieper G. M., Clarke G. A., Gross G. J. (1994) Stimulatory and inhibitory action of nitric oxide donor agents vs. nitrovasodilators on reactive oxygen production by isolated polymorphonuclear leukocytes. J. Pharmacol. Exp. Ther. 269, 451–456 [PubMed] [Google Scholar]
- 16. Schmidt H. H., Nau H., Wittfoht W., Gerlach J., Prescher K. E., Klein M. M., Niroomand F., Böhme E. (1988) Arginine is a physiological precursor of endothelium-derived nitric oxide. Eur. J. Pharmacol. 154, 213–216 [DOI] [PubMed] [Google Scholar]
- 17. Vaughn M. W., Huang K. T., Kuo L., Liao J. C. (2000) Erythrocytes possess an intrinsic barrier to nitric oxide consumption. J. Biol. Chem. 275, 2342–2348 [DOI] [PubMed] [Google Scholar]
- 18. Banick P. D., Chen Q., Xu Y. A., Thom S. R. (1997) Nitric oxide inhibits neutrophil B2 integrin function by inhibiting membrane-associated cyclic GMP synthesis. J. Cell Physiol. 172, 12–24 [DOI] [PubMed] [Google Scholar]
- 19. Cambi A., Joosten B., Koopman M., de Lange F., Beeren I., Torensma R., Fransen J. A., Garcia-Parajó M., van Leeuwen F. N., Figdor C. G. (2006) Organization of the integrin LFA-1 in nanoclusters regulates its activity. Mol. Biol. Cell 17, 4270–4281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hato T., Pampori N., Shattil S. (1998) Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin αIIb β3. J. Cell Biol. 141, 1685–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Calderwood D. A., Shattil S. J., Ginsberg M. H. (2000) Integrins and actin filaments: Reciprocal regulation of cell adhesion and signaling. J. Biol. Chem. 275, 22607–22610 [DOI] [PubMed] [Google Scholar]
- 22. Blystone S. (2004) Integrating an integrin: a direct route to actin. Biochim. Biophys. Acta 1692, 47–54 [DOI] [PubMed] [Google Scholar]
- 23. Stamler J. S. (1994) Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell 78, 931–936 [DOI] [PubMed] [Google Scholar]
- 24. Thom S. R., Bhopale V. M., Mancini D. J., Milovanova T. N. (2008) Actin S-nitrosylation inhibits neutrophil β2 integrin function. J. Biol. Chem. 283, 10822–10834 [DOI] [PubMed] [Google Scholar]
- 25. Thom S. R., Bhopale V. M., Milovanova T. N., Yang M., Bogush M., Buerk D. G. (2013) Nitric oxide synthase-2 linkage to focal adhesion kinase in neutrophils influences enzyme activity and β2 integrin function. J. Biol. Chem. 288, 4810–4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thom S. R., Bhopale V. M., Yang M., Bogush M., Huang S., Milovanova T. (2011) Neutrophil β2 integrin inhibition by enhanced interactions of vasodilator stimulated phosphoprotein with S-nitrosylated actin. J. Biol. Chem. 286, 32854–32865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vollmar A. M., Förster R., Schulz R. (1997) Effects of atrial natriuretic peptide on phagocytosis and respiratory burst in murine macrophages. Eur. J. Pharmacol. 319, 279–285 [DOI] [PubMed] [Google Scholar]
- 28. Kiemer A. K., Vollmar A. M. (2001) The atrial natriuretic peptide regulates the production of inflammatory mediators in macrophages. Ann. Rheum. Dis. 60, iii68–iii70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baldini P. M., De Vito P., Martino A., Fraziano M., Grimaldi C., Luly P., Zalfa F., Colizzi V. (2003) Differential sensitivity of human monocytes and macrophages to ANP: a role of intracellular pH on reactive oxygen species production through the phospholipase involvement. J. Leukoc. Biol. 73, 502–510 [DOI] [PubMed] [Google Scholar]
- 30. Fürst R., Brueckl C., Kuebler W. M., Zahler S., Krötz F., Görlach A., Vollmar A. M., Kiemer A. K. (2005) Atrial natriuretic peptide induces mitogen-activated protein kinase phosphatase-1 in human endothelial cells via Rac1 and NAD(P)H oxidase/Nox2-activation. Circ. Res. 96, 43–53 [DOI] [PubMed] [Google Scholar]
- 31. Thom S. R., Bhopale V. M., Yang M. (2014) Neutrophils generate microparticles during exposure to inert gases due to cytoskeletal oxidative stress. J. Biol. Chem. 289, 18831–18845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hüttelmaier S., Mayboroda O., Harbeck B., Jarchau T., Jockusch B. M., Rüdiger M. (1998) The interaction of the cell-contact proteins VASP and vinculin is regulated by phosphatidylinositol-4,5-bisphosphate. Curr. Biol. 8, 479–488 [DOI] [PubMed] [Google Scholar]
- 33. Daniliuc S., Bitterman H., Rahat M. A., Kinarty A., Rosenzweig D., Lahat N., Nitza L. (2003) Hypoxia inactivates inducible nitric oxide synthase in mouse macrophages by disrupting its interaction with α-actinin 4. J. Immunol. 171, 3225–3232 [DOI] [PubMed] [Google Scholar]
- 34. Klatt P., Schmidt K., Lehner D., Glatter O., Bächinger H. P., Mayer B. (1995) Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and l-arginine in the formation of an SDS-resistant dimer. EMBO J. 14, 3687–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chan A. Y., Raft S., Bailly M., Wyckoff J. B., Segall J. E., Condeelis J. S. (1998) EGF stimulates an increase in actin nucleation and filamnet number at the leading edge of the lamellipod in mammary adenocarcinoma cells. J. Cell Sci. 111, 199–211 [DOI] [PubMed] [Google Scholar]
- 36. Thom S. R., Yang M., Bhopale V. M., Milovanova T. N., Bogush M., Buerk D. G. (2013) Intra-microparticle nitrogen dioxide is a bubble nucleation site leading to decompression-induced neutrophil activation and vascular injury. J. Appl. Physiol. 114, 550–558 [DOI] [PubMed] [Google Scholar]
- 37. Jiang H. B., Yoneyama H., Furukawa A., Hamamoto T., Takahara J., Ichikawa Y. (2001) Effect of isosorbide dinitrate on nitric oxide synthase under hypoxia. Pharmacology 62, 10–16 [DOI] [PubMed] [Google Scholar]
- 38. de A Paes A. M., Veríssimo-Filho S., Guimarães L. L., Silva A. C., Takiuti J. T., Santos C. X., Janiszewski M., Laurindo F. R., Lopes L. R. (2011) Protein disulfide isomerase redox-dependent association with p47phox: evidence for an organizer role in leukocyte NADPH oxidase activation. J. Leukoc. Biol. 90, 799–810 [DOI] [PubMed] [Google Scholar]
- 39. Yoshida M., Xia Y. (2003) Heat shock protein 90 as an endogenous protein enhancer of inducible nitric-oxide synthase. J. Biol. Chem. 278, 36953–36958 [DOI] [PubMed] [Google Scholar]
- 40. Daniel K. B., Agrawal A., Manchester M., Cohen S. M. (2013) Readily accessible fluorescent probes for sensitive biological imaging of hydrogen peroxide. ChemBioChem 14, 593–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun C. X., Magalhães M. A., Glogauer M. (2007) Rac1 and Rac2 differentially regulate actin free barbed end formation downstream of the fMLP receptor. J. Cell Biol. 179, 239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vila-Petroff M. G., Younes A., Egan J., Lakatta E. G., Sollott S. J. (1999) Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ. Res. 84, 1020–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen H., Levine Y. C., Golan D. E., Michel T., Lin A. J. (2008) Atrial natriuretic peptide-initiated cGMP pathways regulate vasodilator-stimulated phosphoprotein phosphorylation and angiogenesis in vascular endothelium. J. Biol. Chem. 283, 4439–4447 [DOI] [PubMed] [Google Scholar]
- 44. Lincoln T. M., Corbin J. D. (1983) Characterization and biological role of the cGMP-dependent protein kinase. In Advances in Cyclic Nucleotide Research (Greengard P., Robison G. A., eds) pp. 139–192, Raven Press, New York [Google Scholar]
- 45. Selemidis S., Dusting G. J., Peshavariya H., Kemp-Harper B. K., Drummond G. R. (2007) Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc. Res. 75, 349–358 [DOI] [PubMed] [Google Scholar]
- 46. Chen Y., Panda K., Stuehr D. J. (2002) Control of nitric oxide synthase dimer assembly by a heme-NO-dependent mechanism. Biochemistry 41, 4618–4625 [DOI] [PubMed] [Google Scholar]
- 47. Thom S. R., Bhopale V. M., Milovanova T. N., Yang M., Bogush M. (2012) Thioredoxin reductase linked to cytoskeleton by focal adhesion kinase reverses actin S-nitrosylation and restores neutrophil β2 integrin function. J. Biol. Chem. 287, 30346–30357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hirabayashi N., Tanimura H., Yamaue H. (2005) Nitrite/nitrate and cytokine changes in patients with surgical stress. Dig. Dis. Sci. 50, 893–897 [DOI] [PubMed] [Google Scholar]
- 49. Endo S., Inada K., Nakae H., Arakawa N., Takakuwa T., Yamada Y., Shimamura T., Suzuki T., Taniguchi S., Yoshida M. (1996) Nitrite/nitrate oxide (NOX) and cytokine levels in patients with septic shock. Res. Commun. Mol. Pathol. Pharmacol. 91, 347–356 [PubMed] [Google Scholar]