

Background: The interplay between dyslipidemia-induced inflammation and angiogenesis remains poorly understood.
Results: Inhibition of caspase-1 improves VEGFR-2 signaling, tube formation, and blood perfusion in ischemic tissues.
Conclusion: The suppression of caspase-1 improves angiogenesis and ischemia prognosis.
Significance: Caspase-1 suppression is a novel therapeutic target for improvement of angiogenesis and ischemia under inflammatory environments.
Keywords: angiogenesis, caspase 1 (CASP1), inflammation, ischemia, vascular endothelial growth factor (VEGF)
Abstract
Deficient angiogenesis may contribute to worsen the prognosis of myocardial ischemia, peripheral arterial disease, ischemic stroke, etc. Dyslipidemic and inflammatory environments attenuate endothelial cell (EC) proliferation and angiogenesis, worsening the prognosis of ischemia. Under these dyslipidemic and inflammatory environments, EC-caspase-1 becomes activated and induces inflammatory cell death that is defined as pyroptosis. However, the underlying mechanism that correlates caspase-1 activation with angiogenic impairment and the prognosis of ischemia remains poorly defined. By using flow cytometric analysis, enzyme and receptor inhibitors, and hind limb ischemia model in caspase-1 knock-out (KO) mice, we examined our novel hypothesis, i.e. inhibition of caspase-1 in ECs under dyslipidemic and inflammatory environments attenuates EC pyroptosis, improves EC survival mediated by vascular endothelial growth factor receptor 2 (VEGFR-2), angiogenesis, and the prognosis of ischemia. We have made the following findings. Proatherogenic lipids induce higher caspase-1 activation in larger sizes of human aortic endothelial cells (HAECs) than in smaller sizes of HAECs. Proatherogenic lipids increase pyroptosis significantly more in smaller sizes of HAECs than in larger sizes of the cells. VEGFR-2 inhibition increases caspase-1 activation in HAECs induced by lysophosphatidylcholine treatment. Caspase-1 activation inhibits VEGFR-2 expression. Caspase-1 inhibition improves the tube formation of lysophosphatidylcholine-treated HAECs. Finally, caspase-1 depletion improves angiogenesis and blood flow in mouse hind limb ischemic tissues. Our results have demonstrated for the first time that inhibition of proatherogenic caspase-1 activation in ECs improves angiogenesis and the prognosis of ischemia.
Introduction
Dyslipidemia, a risk factor for cardiovascular disease and stroke, is defined as pathologically elevated plasma concentrations of cholesterol and other lipids, which are commonly found in patients with atherosclerosis (1). Atherosclerosis and its complications, such as myocardial infarction, peripheral artery disease, and stroke, are the leading causes of morbidity and mortality in the United States (2). Reports from our laboratories and others' support the notion that dyslipidemia, pro-inflammatory mediators, and other risk factors promote endothelial cell activation via several mechanisms, including EC2 activation and injury (2, 3), the increase of monocyte recruitment and differentiation (4, 5), and the decrease of CD4+CD25+ regulatory T cell population (6, 7), etc. EC activation has features of the up-regulation of EC adhesion molecules such as intercellular adhesion molecule-1 and increased secretion of inflammatory cytokines and chemokines, which are defined as an initial event responsible for monocyte recruitment in atherogenesis (8). Moreover, we have recently reported that early hyperlipidemia promotes EC activation in early atherogenesis via the caspase-1–sirtuin-1 pathway (9), suggesting that ECs, similar to other innate immune cells, also have sensor-response functions for elevated metabolites.
Caspase-1, a member of the cysteine protease family of caspases, is initially synthesized in the cell cytosol as pro-caspase-1, an inactive zymogen, and requires the post-translational assembly of a nucleotide-binding and oligomerization domain-like receptor family member-containing protein complex termed “inflammasome” for enzymatic activation. Activated caspase-1 is required for cleaving/processing pro-interleukin-1β (IL-1β) and pro-IL-18 into mature pro-inflammatory cytokines IL-1β and IL-18, respectively, and the activation of other inflammatory pathways (10). Recently, we reported that in early atherosclerosis the caspase-1–inflammasome pathway in ECs can sense elevated lipids as endogenous metabolic danger signal-associated molecular patterns (DAMPs) and activate ECs (9, 10).
ECs that line the inner surface of the vessel wall are the first cells exposed to metabolite-related endogenous danger signals in the circulatory system (1). The issue of whether different EC sizes (11) and EC proliferation status respond differently to dyslipidemic stimuli remains unknown. To address these issues, we searched for an EC sensing-response system for metabolic danger signals and dyslipidemia. The receptors for pathogen-associated molecular patterns have been recently characterized, which bridge the signals generated by the sensory systems for exogenous pathogen-associated molecular patterns and endogenous DAMPs to the initiation of inflammation (12). The Toll-like receptors (TLRs), mostly located on plasma membranes, recognize a variety of conserved microbial pathogen-associated molecular patterns and metabolic DAMPs and promote inflammatory gene transcription and expression. As we pointed out previously in our model of inflammation privilege (13) for the tissues in which inflammasome component genes are not constitutively expressed, Toll-like receptors also work in synergy with cytosolic sensing receptor families, including nucleotide binding and oligomerization domain-like receptors in recognizing endogenous DAMPs in mediating the up-regulation of a range of inflammatory genes (10).
It has been reported that dyslipidemia attenuates EC proliferation in vitro and angiogenesis in vivo (14–16) and that vasculitis is an antiangiogenic state (17). Deficient angiogenesis may contribute to poor prognosis of dyslipidemia-related diseases after ischemic events such as myocardial ischemia, peripheral arterial disease, ischemic stroke, etc. Under ischemic conditions, various types of inflammatory cells are recruited and play an active role in vascular repair and tissue remodeling in the context of myocardial infarction (18). The mechanism underlying the interplay between lipid stimulus-induced EC activation and inflammation and endothelial cell-mediated angiogenesis under ischemic and inflammatory environment is not well defined.
The binding of VEGFR-2 promotes EC survival, angiogenesis, endothelium wound healing, and repair of the damaged existing vasculature (19, 20). Three VEGFRs such as VEGFR-1, VEGFR-2, and VEGFR-3 are expressed exclusively in ECs (21), among which VEGFR-2 (also termed KDR/Flk-1) plays a central role in EC function and proliferation (19, 22). It was reported that matrix metalloproteinase 3 and 9 are responsible for alcohol-induced VEGFR-2 protein degradation in human brain ECs (23). However, an important question remains, whether the transcript levels of VEGFR-2 in ECs are regulated by the activation of caspase-1 in the dyslipidemic and inflammatory environment.
Our novel hypothesis in this study is that the inhibition of caspase-1 attenuates pyroptosis (inflammatory cell death) in ECs, improves EC survival mediated by VEGFR-2 signaling, angiogenesis, and ischemia's prognosis under dyslipidemic and inflammatory environments. To examine this hypothesis, we used the hind limb ischemia model in caspase-1 KO mice and stimulated HAECs with proatherogenic lipids, oxidized low density lipoprotein (oxLDL), carbamylated LDL, oxLDL-derived lipids, lysophosphatidylcholine (LPC), and lysophosphatidic acid (LPA) (24). Our results showed that caspase-1 inhibition improves the tube formation of LPC-treated HAECs and that caspase-1 depletion improves angiogenesis and blood flow in mouse hind limb ischemic tissues. Our results have demonstrated for the first time that inhibition of proatherogenic caspase-1 activation in ECs improves angiogenesis and the prognosis of ischemia.
Materials and Methods
Reagents
The oxLDL and carbamylated LDL were purchased from Biomedical Technologies (Stoughton, MA). LPC and LPA were purchased from Avanti Polar Lipids (Alabaster, AL). Hydrogen peroxide (H2O2) was purchased from Sigma. Vascular endothelial growth factor receptor II inhibitor (SU1498) was purchased from EMD Millipore (Billerica, MA).
Human Aortic Endothelial Cell Culture
Human aortic endothelial cells (HAECs) were purchased at Clonetics Corp. (San Diego). The cells were cultured in a 2% gelatin-coated 75-cm2 flask in M199 (Hyclone Labs., Logan, UT) with 20% fetal bovine serum (FBS), 1% penicillin/streptomycin (Invitrogen), 3 ng/ml EC growth supplement (BD Biosciences), and 5 units/ml heparin (Sigma) at 37 °C under 5% CO2, 95% air until passage 8. For experiments, HAECs (≤ passage 9) were used and treated with the desired stimuli for the indicated time.
Caspase-1 Activity Assay
Active caspase-1 levels were determined with an APO LOGIX kit (Cell Technology, Mountain View, CA). The kit contained a carboxyfluorescein (FAM) (excitation/emission (nm): 490/520)-labeled peptide fluoromethyl ketone (FMK) caspase-1 inhibitor (FAM-YVAD-FMK), which irreversibly binds to active caspase-1. The procedures were performed according to the manufacturer's instructions. HAECs were cultured in 6-well dishes and serum-starved overnight to ensure quiescence of the cells before treatment. The next day, HAECs were treated with the indicated stimuli for 6 h. Cells were harvested and suspended at 1 × 106 cells/ml. The cell suspension (150 μl) was incubated in 37 °C with 1× FAM-YVAD-FMK for 1 h. After being washed with 1× washing buffer, the cells were fixed with fixative buffer and stored in 4 °C for up to 24 h. To determine the cell membrane integrity, the unfixed cells were stained with 7-AAD (Pharmingen) (0.25 μg/test) and incubated for no more than 10 min before analysis.
Flow Cytometer
The FACSCalibur flow cytometer (BD Biosciences) was used to determine FAM+ staining (caspase-1 activity marker, FL-1 channel), 7-AAD+ staining (cell membrane integrity marker, FL-4 channel), and ICAM-1 staining (adhesion molecule marker, FL-4 channel).
Data Analysis Using FlowJo Software
All flow cytometric data were analyzed by using the FlowJo software (Tree Star, Ashland, OR). The uncompensated data were collected from the flow cytometer (either FACSCalibur flow cytometer or LSRII flow cytometer; BD Biosciences). Forward and side scatter gates were used to select live cell populations from clumps and debris. The positive gating was determined by its matched IgG control, and single staining was used to determine the compensation parameter. Different gates were established to analyze the data according to cell size as described in our figures.
Mice
Wild-type mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Caspase-1−/− (Casp-1−/−) mice in a C57BL/6J background were generously provided by Dr. Richard Flavell's laboratory (Yale University School of Medicine, New Haven, CT). Mice were housed in a specific pathogen-free environment. Mice were maintained on a normal chow diet (5% fat, Lab diet 5001). Animal procedures were performed in accordance with the approval of Institutional Animal Care and Use Committee (IACUC) of Temple University.
Murine Hind Limb Ischemia Model
Age-matched 8-week-old male mice were used to create the mouse model of hind limb ischemia as reported previously (25). An incision was made in the skin at the mid-portion of the right hind limb. The femoral artery and vein were then dissected free from nerve. Proximal and distal portions of the femoral artery were ligated followed by complete excision from the hind limb.
Laser Doppler Perfusion Imaging
Blood perfusion in the hind limb was monitored by using laser Doppler perfusion imaging (Lisca, North Brunswick, NJ). The mice were anesthetized before initiating the image scanning. For each time point, the laser Doppler image obtained was analyzed by averaging the perfusion and expressed as the relative unit of flux, as determined by Moor Instruments, over the surface of the ischemic and nonischemic foot.
Tube Formation Assay
The Matrigel matrix (Bedford, MA) was used to coat the 96-well plates. Human aortic endothelial cells were cultured. The cells were treated with caspase-1 inhibitor (Anaspec, Fremont, CA) and proatherogenic stimulus LPC for 24 h. HAECs were harvested and suspended. Cell concentration was determined and added to each Matrigel-coated well and incubated for 16–18 h. Images were collected using the inverted microscope.
Data Analysis
The experiments were performed at least three times, and results were expressed as the means ± S.E. Statistical comparison of single parameters between two groups was performed by paired Student's t test, and multiple group comparisons were performed by one-way analysis of variance. Data were considered statistically significant if p was <0.05.
Results
Proatherogenic Oxidized Lipids Induce Higher Caspase-1 Activation in the Larger Sizes of HAECs than in Smaller Sizes of HAECs
To examine our hypothesis that the size and growth status of endothelial cells regulate caspase-1 activation induced by proatherogenic lipids, we performed the stimulation of HAECs followed by flow cytometric analysis, and we determined the caspase-1 activation in the five groups of HAECs, including untreated control, oxLDL-treated, LPA-treated, LPC-treated, and H2O2-treated control. The results showed that proatherogenic lipids significantly activate caspase-1 (Fig. 1A). To determine whether caspase-1 is activated differently in various cell sizes of HAECs in response to proatherogenic lipid stimulation, we used the forward scatter (FSC) function in the y axis to establish the cell size and caspase-1 activation in the x axis to determine the activation of HAECs during the analysis of the FACS data using the FlowJo software. The results in the dot plot (Fig. 1B, upper panel) showed that caspase-1-positive population spreads in a slightly diagonal manner along the forward scatter (y axis) and caspase-1 activity (x axis), suggesting a possibility that larger sizes of cells have higher caspase-1 activity than smaller sizes of HAECs. After gating out HAECs into three populations, large, medium, and small using FSC in the y axis, we examined caspase-1 activation in the x axis in these three cell size populations as we described previously. The results showed that oxLDL, LPA, and LPC induce caspase-1 activation in a larger size of HAECs by 63.6, 54.4, and 52.8%, respectively, higher than that in the smaller size of HAECs (14.4, 25.2, and 38.2%) (Fig. 1, B and C), suggesting that caspase-1 activation is associated with high cell growth status in larger cell sizes and that various lipid stimuli show no significant differences in activating caspase-1 in various sizes of HAECs. Of note, our previous studies only reported the effect of oxLDL in activating caspase-1 in HAECs. Here, we found the new results on the effects of other proatherogenic lipids in promoting caspase-1 activation (9). Taken together, the results demonstrated that proatherogenic lipids induce caspase-1 activation in HAECs, which may promote EC activation in a larger size of cells and inflammatory cell death in a smaller size of cells.
FIGURE 1.

Proatherogenic oxidized lipids induce higher caspase-1 activation in the larger sizes of HAECs than in smaller sizes of HAECs. A, HAECs were treated with different proatherogenic stimuli as follows: oxLDL (100 μm), LPA (30 μm), LPC (30 μm), and H2O2 (500 μm) for 6 h. Caspase-1(+) cell percentage was measured, and the results showed an increase in caspase-1 activation after proatherogenic lipid treatments. B, three different sizes of cell populations after caspase-1 activation were identified using FSC, which is widely used to define the cell size in flow cytometry. The gates were established as small, medium, and large according to the cell size (FSC, y axis) and caspase-1 activation (x axis) to analyze the differential caspase-1 activation percentage according to the cell size. Analysis of three gates showed an increase in caspase-1 activation percentage in larger HAECs. C, bar graph shows the quantitation data of caspase-1 activation. *, p < 0.05.
Proatherogenic Lipids Significantly Increase Pyroptosis in Smaller Sizes of Human Aortic Endothelial Cells than in Larger Sizes of the Cells
Inflammatory cell death (pyroptosis) is a newly characterized form of cell death with the features of caspase-1 activation, plasma membrane rupture, and release of inflammatory factors (26). In Fig. 1, B and C, we noticed that caspase-1 activities in LPC-treated HAECs and H2O2-treated positive control HAECs are lower in the medium sizes of HAECs than in the smaller sizes of cells, suggesting that increased caspase-1 activities in the smaller sizes of cells result in poorer survival and decreased sizes of cells. To examine the hypothesis that proatherogenic lipids promote caspase-1 activation and promote higher EC pyroptosis in the smaller size of HAECs compared with the larger size of HAECs, we stimulated HAECs with LPC and co-stained them with the caspase-1 fluorescence probe to analyze the activation of caspase-1 and the viable fluorescence dye 7-AAD to determine the plasma membrane integrity, viability, and pyroptosis. We first analyzed the activation of caspase-1 in HAECs, and we established the y axis as FSC, which evaluates the cell size, and the x axis to measure the activation of caspase-1. Using these parameters, we analyzed caspase-1 activation in three sizes of cell populations. After that, we analyzed the 7-AAD staining that determines HAEC pyroptosis in different sizes of caspase-1(+) HAECs. The results showed that in LPC-treated HAECs, the smaller size of caspase-1(+) cells has an increase in pyroptosis (38.77%) compared with that in medium size (20.17%) and that in the large size of caspase-1(+) HAECs (9.93%). The bar graphs results in Fig. 2 showed that LPC stimulation significantly induced 7-AAD+ pyroptosis from 22.50 to 38.77% in smaller size of caspase-1(+) HAECs (p < 0.01). However, LPC stimulation did not induce significant pyroptosis in the medium and larger size of caspase-1(+) HAECs from 10 to 12% (p > 0.05). It should be noted that in unstimulated cells, the percentage of 7-AAD+ cells in the smaller sizes of HAECs (22.50%) was much higher than that in both the medium size (10.71%) and larger size of HAECs (10.49%), suggesting that the membrane integrity in the medium and larger sizes of caspase-1(+) HAECs is significantly better preserved than that of the smaller sizes of caspase-1(+) HAECs. The results demonstrated that caspase-1 activation induced by LPC in the larger sizes of HAECs may promote EC activation but not significant pyroptosis and that as HAECs become smaller in size, LPC-induced caspase-1 activation promotes measurable pyroptosis.
FIGURE 2.
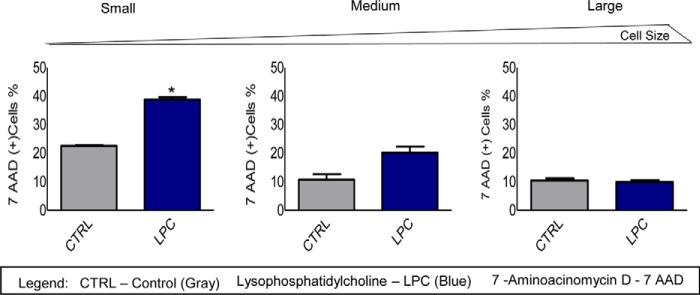
Proatherogenic stimuli significantly increase more pyroptosis in smaller sizes of HAECs than in larger sizes of the cells. The pyroptotic rates in LPC-treated caspase-1(+) HAECs were increased as the cell sizes became smaller. The FACS results demonstrated an increase in 7AAD+ pyroptotic percentage in smaller sizes of caspase-1(+) HAECs when compared with medium size and larger size caspase-1(+) cells. The bar graphs show the quantitation data of 7AAD+ percentages in caspase-1(+) cells. *, p < 0.05.
VEGFR-2 Inhibition Increases Caspase-1 Activation in HAECs Induced with LPC Treatment
Our data suggest that as cell growth status improves, endothelial cell size becomes larger and less pyroptotic than smaller cell sizes. These results lead us to hypothesize that vascular endothelial growth factor signaling may inhibit caspase-1 activation and pyroptosis. To examine this possibility, we searched for supporting evidence with unpublished microarray data deposited in the National Institutes of Health NCBI-Geo-Profile database. The experimental data of a microarray analysis with vegfr2 gene knockdown (KD) samples, compared with wild-type control samples in two different time courses (84 and 95 h after gene knockdown), showed that the expressions of caspase-1 and inflammasome component apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) were increased at a ratio of 1.48 and 2.45, respectively (p = 0.00014–0.06443) (Fig. 3A). The expression of two caspase-1 substrates, such as IL-1β and IL-18, and two housekeeping genes was not significantly changed. To further consolidate the conclusion, we then stimulated HAECs with LPC in the presence or absence of the VEGFR-2 inhibitor SU1498, followed by co-stained HAECs with a caspase-1 activity probe. The results showed that VEGFR-2 inhibition increases caspase-1 activation in HAECs (Fig. 3B). These results suggest that VEGFR-2 cell growth signaling inhibits caspase-1 activation and cell death.
FIGURE 3.

Caspase-1 activation is increased after the inhibition of VEGFR-2 in LPC-treated HAECs. A, VEGFR-2 depletion increases the expressions of caspase-1 and inflammasome adaptor protein ASC. The microarray data-mining analyses were performed using the National Institutes of Health NCBI-Geo-profile database. The microarray dataset IDs were indicated. B, HAECs were pretreated with VEGFR-2 inhibitor (SU1498) and then treated with LPC (30 μm) for 6 h after overnight starvation with serum-free medium. The bar graph shows the quantitation data of caspase-1 activation, which demonstrates an increase in caspase-1 activation after the inhibition of VEGFR-2 in LPC-treated HAECs. *, p < 0.05.
Caspase-1 Activation Inhibits VEGFR-2 Expression
As indicated above, our results give rise to the following question. How can caspase-1 promote HAEC activation (9) in larger cell sizes and simultaneously enhance pyroptosis in the smaller size of HAECs? One possibility is that caspase-1 activation and VEGFR-2 signaling interplay each other in their functions. To examine this possibility, we analyzed the National Institutes of Health NCBI-Geo-Profile database. The experimental data of a microarray analysis showed that the ratio of VEGFR-2 expression levels in the wild-type control samples versus caspase-1 gene-deficient (KO) samples was 0.62 (Fig. 4A), suggesting that VEGFR-2 expression is increased in caspase-1 gene depletion samples and that caspase-1 activation inhibits VEGFR-2 signaling. The caspase-1 expression level was 22.23-fold higher in wild-type samples than in caspase-1 KO samples, justifying the bona fide comparison of wild-type samples and caspase-1 KO samples.
FIGURE 4.

Inhibition of caspase-1 activation and activation of Sirt-1 increase VEGFR-2 expression. A, caspase-1 depletion increases VEGFR-2 expression. A microarray data-mining analysis was performed by analyzing the indicated microarray dataset deposited in the National Institutes of Health NCBI-Geo-profile database. The gene expression value was determined based on the relative threshold cycle on the database. Caspase-1 expression was used as a control to show that caspase-1 expression was absent in the caspase-1 gene depletion samples. In addition, depletion of caspase-1 slightly increases the expressions of the caspase-1-cleaving substrates cytokines IL-1β and IL-18. As the microarray data quality controls, caspase-1 depletion did not significantly change the expressions of three housekeeping genes, Gadph, Nono, and Aldoa. B, caspase-1 inhibition and Sirt-1 activation increase the expression of VEGFR-2 after LPC treatment in HAECs. HAECs pretreated with the caspase-1 inhibitor (10 μm) and Sirt-1 activator (10 μm) and then treated with LPC (30 μm) for 6 h have an increased expression of VEGFR-2. C, three cell size gates were established, small, medium, and large according to FSC (y axis); and VEGFR-2 expression (x axis) was measured in those gated cells to analyze differential VEGFR-2 expressions according to the cell size after treatments with LPC (30 μm), caspase-1 inhibitor (10 μm), and Sirt-1 activator (10 μm) for 6 h. The analysis shows increases in VEGFR-2 expression as the cell sizes became larger. D, bar graphs show the quantitation data of VEGFR-2 expressions. *, p < 0.05.
In addition, the expression levels of three housekeeping genes were within the range of 1.04 to 0.99, suggesting the high quality of the microarray experimental datasets. To gain further support for our conclusion, we then stimulated HAECs with LPC in the presence or absence of caspase-1 inhibitor followed by the analysis of the FACS data, which showed that after the inhibition of caspase-1 in LPC-treated HAECs, VEGFR-2 expression was increased (Fig. 4B). We further determined the expression of VEGFR-2 after caspase-1 inhibition in LPC-treated HAECs in different cell sizes. Three gates were established according to the cell size using FSC in the y axis to analyze the cell sizes and VEGFR-2 expression in the x axis. The results showed that pretreatment with caspase-1 inhibitor in LPC-treated HAECs increased the expression of VEGFR-2 when compared with LPC-treated HAECs from 11.3 to 22.1% in the smaller size of HAECs, from 22.1 to 50.0% in the medium size of HAECs, and from 34.3 to 64.4% in the larger size of HAECs (Fig. 4, C and D). It has been demonstrated in our recent report that caspase-1 deficiency in apolipoprotein E (apoE)−/−/caspase-1−/− mouse aorta and the inhibition of caspase-1 in ECs result in accumulation of anti-inflammatory protein/histone deacetylase sirtuin 1 (Sirt1), which is a proteolytic substrate of caspase-1, suggesting that caspase-1 activation in early atherogenesis promotes endothelial activation via a Sirt1 degradation pathway (9). To determine whether activation of Sirt1 has the same effect as inhibition of caspase-1 in promoting VEGFR-2 expression, we performed the same experiment as previous ones using Sirt1 activator pretreatment in LPC-treated HAECs. We used the same method to analyze the data according to cell sizes and VEGFR-2 expression. The VEGFR-2 expression was increased in LPC-treated HAECs pretreated with Sirt-1 activator compared with LPC-treated HAECs. The VEGFR-2 expression in the above-mentioned groups increased from 11.3 to 21.0% in the smaller size of HAECs, from 22.1 to 52.5% in the medium size of HAECs, and from 34.3 to 70.2% in the larger size of HAECs. Taken together, the results suggest that LPC-induced caspase-1 activation decreases VEGFR-2 expression levels in HAECs, whereas the inhibition of caspase-1 and activation of Sirt-1 rescue VEGFR-2 expression levels in LPC-treated HAECs.
Caspase-1 Inhibition Improves Tube Formation of LPC-treated Human Aortic Endothelial Cells
Because EC tube formation assay has been widely used in determining VEGFR-2-mediated angiogenesis (27), we used the EC tube formation assay in the Matrigel to test the hypothesis that caspase-1 inhibition improves VEGFR-2-mediated HAEC tube formation. To test this hypothesis, we performed tube formation assays with untreated HAECs, LPC-treated HAECs, and LPC plus caspase-1 inhibitor co-treated HAECs. The results showed that the lengths of the total master segment length (in micrometers) of tube formation in LPC-treated HAECs were significantly lower (317 μm) than in untreated HAEC controls (718 μm). In addition, LPC plus caspase-1 inhibitor co-treated HAECs have restored the EC tube formation (827 μm) (Fig. 5). The results suggest that caspase-1 inhibition not only increases VEGFR-2 expression levels but also functionally improves VEGFR-2-mediated EC tube formation.
FIGURE 5.
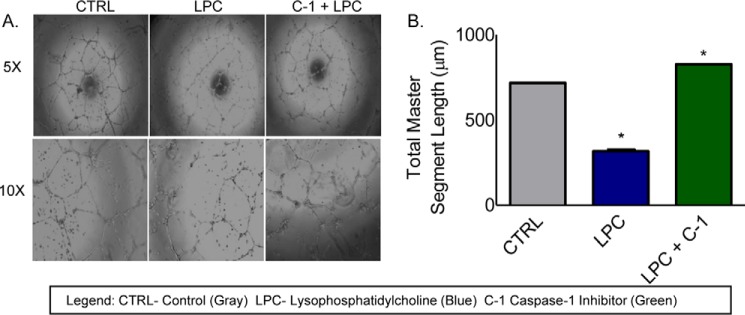
Tube formation, as an in vitro model of angiogenesis, is improved after caspase-1 inhibition in LPC-treated cells. A, HAECs were treated with LPC (30 μm) and caspase-1 inhibitor (10 μm) for 6 h. After the treatment, the cells were incubated in the Matrigel for 16 h for the analysis of tube formation. B, bar graph shows the increases of total master segment lengths after the inhibition of caspase-1 in LPC-treated HAECs.*, p < 0.05.
Caspase-1 Depletion Improves Angiogenesis and Ischemia/Normal Blood Perfusion Ratio in Hind Limb Ischemic Mouse Tissues
Because the mouse model of hind limb ischemia has been widely used in determining the gene effect on hind limb ischemia-triggered angiogenesis (25), we performed the hind limb ischemia model on wild-type mice and caspase-1 KO mice followed by the blood flow measurement with the LDSII Doppler at days 0–3, 7, 10, and 21. The results showed that caspase-1 depletion increased the ischemia/normal leg blood perfusion ratios from 25.5 to 32.7% on day 1, from 37.8 to 44.1% on day 3, and from 57.62 to 66.49% on day 7, respectively, in hind limb ischemia-affected tissues in caspase-1 KO mice in comparison with that in wild-type mice at the same time points (p > X ± 2SD) (Fig. 6). The results suggest that caspase-1 depletion improves angiogenesis and blood supply in hind limb ischemic mouse tissues.
FIGURE 6.

Caspase-1 KO mice have an increase in blood flow after hind limb ischemia compared with wild-type mice. A, hind limb ischemia model was performed in WT and caspase-1 KO mice. The blood flow was recorded with the LDSII Doppler at different time points. The data show that the caspase-1 KO mice have a trend of increased blood flow after hind limb ischemia. B, linear graph shows the quantification of the ischemia/normal leg perfusion ratio. C, an absolute value (y-x) was used to create a confidence interval.
Discussion
The importance of caspase-1 in tissue injury is evidenced by in vivo studies demonstrating that caspase-1 inhibition protects against ischemic injury in brain (28), heart (29), and liver (30). It has been reported that dyslipidemia attenuates EC proliferation in vitro and angiogenesis in vivo (14–16). Although the role of caspase-1 in the dyslipidemic environment and atherogenesis remains controversial (31), the dominant concept supported by our own report (9) is that caspase-1 plays a proatherogenic role, which is supported by results collected from apoE−/−/caspase-1−/− mice (9, 32, 33), inflammasome sensor nlrp3 KO bone marrow cells in ldlr−/− mice (34), apoE−/−/IL-1β−/− mice (35), and apoE−/−/IL-18−/− mice (36). Gage et al. (32) and Usui et al. (33) studied the role of caspase-1 deficiency in full-blown atherosclerosis in apoE−/− mice after high fat feeding for 8 weeks (32) and 12 weeks (33). The differences of endothelial cell sizes were observed previously (37, 38). In addition, endothelial cell sizes were correlated with different proliferation statuses (39). Nonuniformity of important regulatory molecule expression in ECs, such as endothelial nitric-oxide synthase, was also reported (40).
Our results have addressed the issue about the mechanism in the interplay between inflammatory environment, initiation of EC activation, inflammatory cell death (pyroptosis) in ECs, angiogenesis impairment in ECs, and ischemia in animal tissue. We examined caspase-1 activation in ECs, EC pyroptosis, the microarray data, EC tube formation, and hind limb ischemia mouse model, and we investigated our novel hypothesis that the inhibition of caspase-1 in ECs attenuates EC pyroptosis, improves VEGFR-2-mediated ECs survival, angiogenesis, and ischemia's prognosis under inflammatory and dyslipidemic environment. We have made the important finding that dyslipidemic environment induces higher caspase-1 activation in the larger sizes of HAECs than in smaller sizes of HAECs. Moreover, the smaller HAECs have higher pyroptosis rates than larger HAECs after stimulation with proatherogenic lipids. The activation of caspase-1 after stimulation with proatherogenic lipids promotes ECs activation as judged by increased intercellular adhesion molecule-1 expression in HAECs as we demonstrated previously (9). Our results show that the inhibition of VEGFR-2 increases caspase-1 activation in HAECs induced by LPC treatment and that the activation of caspase-1 inhibits VEGFR-2 expression in HAECs. The molecular data were further consolidated with the evidence in functional angiogenesis models that caspase-1 inhibition improves tube formation of LPC-treated HAECs and the depletion of caspase-1 improves angiogenesis and blood supply in the hind limb of ischemic mouse tissues.
Although microvascular endothelial cells are often involved in angiogenesis, aortic endothelial cells are also used as experimental models for angiogenesis such as the aortic ring angiogenesis model (41). In addition, neovessel formation was recently found in the human thoracic aneurysm of the ascending aorta (42) and revascularization in atherosclerotic aorta (43). The use of aortic endothelial cells in the commonly used experimental angiogenesis model and the involvement of aortic endothelial cells in pathological angiogenesis and atherosclerosis justified our use of aortic endothelial cells in this study.
Our recent review summarized that there are 11 endogenous metabolic stress-related danger signals, including oxLDL and cholesterol crystals that induce caspase-1 activation (10). However, the issue of whether several oxLDL-derived lipids, such as LPC and LPA (24), can induce caspase-1 activation was not clear. Our results reported here have demonstrated for the first time that LPC and LPA, small non-crystal lipids, can induce caspase-1 activation, presumably not via particle-lysosome pathway (10). Because LPA acts on the LPC receptors, including LPA1-6 G protein-coupled receptors (38), and LPC acts on the LPC receptor, GPCR-G2A (44), the signals from these receptors to activate caspase-1 have been intensely studied in our investigation.
It was reported that lipopolysaccharide induces a caspase-3/caspase-1-dependent EC apoptosis, which can be inhibited by VEGF (45). However, we showed for the first time that VEGFR-2 signaling inhibits caspase-1-dependent pyroptosis, which is demonstrated by positive 7-AAD staining. Of note, 7-AAD staining is not a feature of apoptosis (26). In addition, the detailed molecular mechanisms underlying caspase-1 inhibition of VEGFR-2 transcript expression remain unknown. It was reported that caspase-1 generated proatherogenic cytokine IL-1β, which counteracts mechanical signals that induced VEGFR-2 signaling and inhibits EC proliferation (46). However, the question of whether other caspase-1 substrates, including IL-18 and sirtuin 1 (9), have direct effects on VEGFR-2-induced EC proliferation remains unknown. Our unpublished results showed that caspase-1 has an IL-1β, IL-18, and sirtuin 1-independent transcription regulatory effect.
In our newly proposed working model (Fig. 7), we summarize our findings and highlight the current understanding that the larger size of HAECs has higher VEGFR-2 expression and less caspase-1(+) induction of pyroptosis than the smaller cells under the dyslipidemic and inflammatory environment, which promotes cell survival and angiogenesis and improves the prognosis of the ischemia. In contrast, under dyslipidemic and inflammatory environments, the smaller size of HAECs has an increase in caspase-1(+) induction of pyroptosis and less VEGFR-2 expression than the larger cells that decrease the angiogenesis and cell survival and worsen ischemia's prognosis. In summary, there is a mutual inhibition between the VEGFR-2 signaling pathway and caspase-1 activation in endothelial cells. Our novel results provide important insight on the future development of a novel therapeutic approach, based on the inhibition of caspase-1 for suppression of vascular inflammation and improvement of angiogenesis and ischemia prognosis under inflammatory environments (47).
FIGURE 7.

Schematic representation of our working model. The larger HAECs have higher vascular endothelial growth factor receptor 2 (VEGFR-2) and less caspase-1(+) pyroptosis induction after dyslipidemic stimuli, which promotes angiogenesis and improves ischemia's prognosis. However, after dyslipidemic stimuli, the smaller HAECs have an increase in caspase-1(+) induction of pyroptosis and less VEGFR-2 expression, which decrease the angiogenesis and worsen ischemia's prognosis. In summary, there is a mutual inhibition between the VEGFR-2 signaling pathway and caspase-1 activation in endothelial cells.
Acknowledgment
We are very grateful to Dr. Richard A. Flavell, Yale University School of Medicine, for generously providing the caspase-1 gene knock-out mice.
This work was supported, in whole or in part, by National Institutes of Health grants (to X. F. Y. and H. W.). The authors declare that they have no conflicts of interest with the contents of this article.
- EC
- endothelial cell
- HAEC
- human aortic endothelial cell
- FAM
- carboxyfluorescein
- FMK
- fluoromethyl ketone
- 7-AAD
- 7-amino-actinomycin D
- DAMP
- danger signal-associated molecular pattern
- oxLDL
- oxidized low density lipoprotein
- LPA
- lysophosphatidic acid
- FSC
- forward scatter.
References
- 1. Libby P., Ridker P. M., Hansson G. K. (2011) Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325 [DOI] [PubMed] [Google Scholar]
- 2. Mestas J., Ley K. (2008) Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 18, 228–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jiang X., Yang F., Tan H., Liao D., Bryan R. M., Jr., Randhawa J. K., Rumbaut R. E., Durante W., Schafer A. I., Yang X., Wang H. (2005) Hyperhomocystinemia impairs endothelial function and eNOS activity via PKC activation. Arterioscler. Thromb. Vasc. Biol. 25, 2515–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang D., Jiang X., Fang P., Yan Y., Song J., Gupta S., Schafer A. I., Durante W., Kruger W. D., Yang X., Wang H. (2009) Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine β-synthase-deficient mice. Circulation 120, 1893–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Combadière C., Potteaux S., Rodero M., Simon T., Pezard A., Esposito B., Merval R., Proudfoot A., Tedgui A., Mallat Z. (2008) Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117, 1649–1657 [DOI] [PubMed] [Google Scholar]
- 6. Xiong Z., Yan Y., Song J., Fang P., Yin Y., Yang Y., Cowan A., Wang H., Yang X. F. (2009) Expression of TCTP antisense in CD25(high) regulatory T cells aggravates cuff-injured vascular inflammation. Atherosclerosis 203, 401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ait-Oufella H., Salomon B. L., Potteaux S., Robertson A. K., Gourdy P., Zoll J., Merval R., Esposito B., Cohen J. L., Fisson S., Flavell R. A., Hansson G. K., Klatzmann D., Tedgui A., Mallat Z. (2006) Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 12, 178–180 [DOI] [PubMed] [Google Scholar]
- 8. Chowienczyk P. J., Watts G. F., Cockcroft J. R., Ritter J. M. (1992) Impaired endothelium-dependent vasodilation of forearm resistance vessels in hypercholesterolaemia. Lancet 340, 1430–1432 [DOI] [PubMed] [Google Scholar]
- 9. Yin Y., Li X., Sha X., Xi H., Li Y. F., Shao Y., Mai J., Virtue A., Lopez-Pastrana J., Meng S., Tilley D. G., Monroy M. A., Choi E. T., Thomas C. J., Jiang X., et al. (2015) Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler. Thromb. Vasc. Biol. 35, 804–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yin Y., Pastrana J. L., Li X., Huang X., Mallilankaraman K., Choi E. T., Madesh M., Wang H., Yang X. F. (2013) Inflammasomes: sensors of metabolic stresses for vascular inflammation. Front. Biosci. 18, 638–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goranov A. I., Gulati A., Dephoure N., Takahara T., Maeda T., Gygi S. P., Manalis S., Amon A. (2013) Changes in cell morphology are coordinated with cell growth through the TORC1 pathway. Curr. Biol. 23, 1269–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang X. F., Yin Y., Wang H. (2008) Vascular inflammation and atherogenesis are activated via receptors for PAMPS and suppressed by regulatory T cells. Drug Discov. Today Ther. Strateg. 5, 125–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin Y., Yan Y., Jiang X., Mai J., Chen N. C., Wang H., Yang X. F. (2009) Inflammasomes are differentially expressed in cardiovascular and other tissues. Int. J. Immunopathol. Pharmacol. 22, 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zechariah A., ElAli A., Hagemann N., Jin F., Doeppner T. R., Helfrich I., Mies G., Hermann D. M. (2013) Hyperlipidemia attenuates vascular endothelial growth factor-induced angiogenesis, impairs cerebral blood flow, and disturbs stroke recovery via decreased pericyte coverage of brain endothelial cells. Arterioscler. Thromb. Vasc. Biol. 33, 1561–1567 [DOI] [PubMed] [Google Scholar]
- 15. Chen C. H., Jiang W., Via D. P., Luo S., Li T. R., Lee Y. T., Henry P. D. (2000) Oxidized low-density lipoproteins inhibit endothelial cell proliferation by suppressing basic fibroblast growth factor expression. Circulation 101, 171–177 [DOI] [PubMed] [Google Scholar]
- 16. Van Belle E., Rivard A., Chen D., Silver M., Bunting S., Ferrara N., Symes J. F., Bauters C., Isner J. M. (1997) Hypercholesterolemia attenuates angiogenesis but does not preclude augmentation by angiogenic cytokines. Circulation 96, 2667–2674 [DOI] [PubMed] [Google Scholar]
- 17. Stillman I. E., Karumanchi S. A. (2012) Vasculitis is an antiangiogenic state. J. Am. Soc. Nephrol. 23, 8–10 [DOI] [PubMed] [Google Scholar]
- 18. Silvestre J. S., Smadja D. M., Lévy B. I. (2013) Postischemic revascularization: from cellular and molecular mechanisms to clinical applications. Physiol. Rev. 93, 1743–1802 [DOI] [PubMed] [Google Scholar]
- 19. Shalaby F., Rossant J., Yamaguchi T. P., Gertsenstein M., Wu X. F., Breitman M. L., Schuh A. C. (1995) Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66 [DOI] [PubMed] [Google Scholar]
- 20. Millauer B., Wizigmann-Voos S., Schnürch H., Martinez R., Møller N. P., Risau W., Ullrich A. (1993) High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 72, 835–846 [DOI] [PubMed] [Google Scholar]
- 21. Neufeld G., Cohen T., Gengrinovitch S., Poltorak Z. (1999) Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 13, 9–22 [PubMed] [Google Scholar]
- 22. Baffert F., Le T., Sennino B., Thurston G., Kuo C. J., Hu-Lowe D., McDonald D. M. (2006) Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am. J. Physiol. Heart Circ. Physiol. 290, H547–H559 [DOI] [PubMed] [Google Scholar]
- 23. Abdul Muneer P. M., Alikunju S., Szlachetka A. M., Haorah J. (2012) The mechanisms of cerebral vascular dysfunction and neuroinflammation by MMP-mediated degradation of VEGFR-2 in alcohol ingestion. Arterioscler. Thromb. Vasc. Biol. 32, 1167–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Z., Subramanian P., Sevilmis G., Globke B., Soehnlein O., Karshovska E., Megens R., Heyll K., Chun J., Saulnier-Blache J. S., Reinholz M., van Zandvoort M., Weber C., Schober A. (2011) Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab. 13, 592–600 [DOI] [PubMed] [Google Scholar]
- 25. Limbourg A., Korff T., Napp L. C., Schaper W., Drexler H., Limbourg F. P. (2009) Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind limb ischemia. Nat. Protoc. 4, 1737–1746 [DOI] [PubMed] [Google Scholar]
- 26. Miao E. A., Leaf I. A., Treuting P. M., Mao D. P., Dors M., Sarkar A., Warren S. E., Wewers M. D., Aderem A. (2010) Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. DeCicco-Skinner K. L., Henry G. H., Cataisson C., Tabib T., Gwilliam J. C., Watson N. J., Bullwinkle E. M., Falkenburg L., O'Neill R. C., Morin A., Wiest J. S. (2014) Endothelial cell tube formation assay for the in vitro study of angiogenesis. J. Vis. Exp. 91, e51312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schielke G. P., Yang G. Y., Shivers B. D., Betz A. L. (1998) Reduced ischemic brain injury in interleukin-1β converting enzyme-deficient mice. J. Cereb. Blood Flow Metab. 18, 180–185 [DOI] [PubMed] [Google Scholar]
- 29. Yaoita H., Ogawa K., Maehara K., Maruyama Y. (1998) Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 97, 276–281 [DOI] [PubMed] [Google Scholar]
- 30. Natori S., Selzner M., Valentino K. L., Fritz L. C., Srinivasan A., Clavien P. A., Gores G. J. (1999) Apoptosis of sinusoidal endothelial cells occurs during liver preservation injury by a caspase-dependent mechanism. Transplantation 68, 89–96 [DOI] [PubMed] [Google Scholar]
- 31. Menu P., Pellegrin M., Aubert J. F., Bouzourene K., Tardivel A., Mazzolai L., Tschopp J. (2011) Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2, e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gage J., Hasu M., Thabet M., Whitman S. C. (2012) Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can. J. Cardiol. 28, 222–229 [DOI] [PubMed] [Google Scholar]
- 33. Usui F., Shirasuna K., Kimura H., Tatsumi K., Kawashima A., Karasawa T., Hida S., Sagara J., Taniguchi S., Takahashi M. (2012) Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 425, 162–168 [DOI] [PubMed] [Google Scholar]
- 34. Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nuñez G., Schnurr M., Espevik T., Lien E., Fitzgerald K. A., Rock K. L., Moore K. J., et al. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kirii H., Niwa T., Yamada Y., Wada H., Saito K., Iwakura Y., Asano M., Moriwaki H., Seishima M. (2003) Lack of interleukin-1β decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 23, 656–660 [DOI] [PubMed] [Google Scholar]
- 36. Elhage R., Jawien J., Rudling M., Ljunggren H. G., Takeda K., Akira S., Bayard F., Hansson G. K. (2003) Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 59, 234–240 [DOI] [PubMed] [Google Scholar]
- 37. Adamson R. H. (1993) Microvascular endothelial cell shape and size in situ. Microvasc. Res. 46, 77–88 [DOI] [PubMed] [Google Scholar]
- 38. Haas T. L., Duling B. R. (1997) Morphology favors an endothelial cell pathway for longitudinal conduction within arterioles. Microvasc. Res. 53, 113–120 [DOI] [PubMed] [Google Scholar]
- 39. Mayhew T. M. (2002) Fetoplacental angiogenesis during gestation is biphasic, longitudinal and occurs by proliferation and remodelling of vascular endothelial cells. Placenta 23, 742–750 [DOI] [PubMed] [Google Scholar]
- 40. Andries L. J., Brutsaert D. L., Sys S. U. (1998) Nonuniformity of endothelial constitutive nitric oxide synthase distribution in cardiac endothelium. Circ. Res. 82, 195–203 [DOI] [PubMed] [Google Scholar]
- 41. Blacher S., Devy L., Burbridge M. F., Roland G., Tucker G., Noël A., Foidart J. M. (2001) Improved quantification of angiogenesis in the rat aortic ring assay. Angiogenesis 4, 133–142 [DOI] [PubMed] [Google Scholar]
- 42. Kessler K., Borges L. F., Ho-Tin-Noé B., Jondeau G., Michel J. B., Vranckx R. (2014) Angiogenesis and remodelling in human thoracic aortic aneurysms. Cardiovasc. Res. 104, 147–159 [DOI] [PubMed] [Google Scholar]
- 43. Luttun A., Tjwa M., Moons L., Wu Y., Angelillo-Scherrer A., Liao F., Nagy J. A., Hooper A., Priller J., De Klerck B., Compernolle V., Daci E., Bohlen P., Dewerchin M., Herbert J. M., et al. (2002) Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat. Med. 8, 831–840 [DOI] [PubMed] [Google Scholar]
- 44. Schmitz G., Ruebsaamen K. (2010) Metabolism and atherogenic disease association of lysophosphatidylcholine. Atherosclerosis 208, 10–18 [DOI] [PubMed] [Google Scholar]
- 45. Munshi N., Fernandis A. Z., Cherla R. P., Park I. W., Ganju R. K. (2002) Lipopolysaccharide-induced apoptosis of endothelial cells and its inhibition by vascular endothelial growth factor. J. Immunol. 168, 5860–5866 [DOI] [PubMed] [Google Scholar]
- 46. Liu J., Agarwal S. (2010) Mechanical signals activate vascular endothelial growth factor receptor-2 to upregulate endothelial cell proliferation during inflammation. J. Immunol. 185, 1215–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Annex B. H. (2013) Therapeutic angiogenesis for critical limb ischaemia. Nat. Rev. Cardiol. 10, 387–396 [DOI] [PubMed] [Google Scholar]