

Background: The mechanism underlying thioredoxin-induced expression of manganese superoxide dismutase (MnSOD) is unknown.
Results: Reduced thioredoxin activates the MKK4-NFκB pathway to repress MKK7-AP-1-mediated inhibition of MnSOD expression.
Conclusion: Cellular redox status regulates gene expression via redox control of MKK4, an MAPK.
Significance: This study reveals a novel mechanism underlying transcriptional regulation of MnSOD encoding a critical antioxidant enzyme.
Keywords: AP-1 transcription factor (AP-1), endothelial cell, mitogen-activated protein kinase (MAPK), NF-κB transcription factor, redox regulation, redox signaling, thioredoxin, Sod2, manganese superoxide dismutase
Abstract
The mitogen-activated protein kinase kinase 4 (MKK4) is activated via phosphorylation of Ser-257 and Thr-261 by upstream MAP3Ks and activates JNK and p38 MAPKs in response to cellular stress. We show that thioredoxin (Trx), a cellular redox protein, activates MKK4 via Cys-246 and Cys-266 residues as mutation of these residues renders MKK4 insensitive to phosphorylation by MAP3Ks, TNFα, or Trx. MKK4 is activated in vitro by reduced Trx but not oxidized Trx in the absence of an upstream kinase, suggesting that autophosphorylation of this protein occurs due to reduction of Cys-246 and Cys-266 by Trx. Additionally, mutation of Cys-246 and Cys-266 resulted in loss of kinase activity suggesting that the redox state of Cys-246 and Cys-266 is a critical determinant of MKK4 activation. Trx induces manganese superoxide dismutase (MnSOD) gene transcription by activating MKK4 via redox control of Cys-246 and Cys-266, as mutation of these residues abrogates MKK4 activation and MnSOD expression. We further show that MKK4 activates NFκB for its binding to the MnSOD promoter, which leads to AP-1 dissociation followed by MnSOD transcription. Taken together, our studies show that the redox status of Cys-246 and Cys-266 in MKK4 controls its activities independent of MAP3K, demonstrating integration of the endothelial redox environment to MAPK signaling.
Introduction
Endothelial dysfunction is a precursor to several cardiovascular diseases such as hypertension, inflammation, atherosclerosis, diabetes, myocardial infarction, and other cardiovascular diseases. The most common implicating factor in these diseases is the overwhelming production of superoxide anion (O2˙̄). In addition to NADPH oxidase, endothelial mitochondria are a significant source of O2˙̄ (1). Superoxide dismutase-2 (MnSOD)2 is a mitochondrial antioxidant enzyme that protects the mitochondria against excessive oxidative burden by converting the superoxide radical to cell-permeable hydrogen peroxide (H2O2), thereby preventing mitochondrial dysfunction (2–4). The physiological importance of MnSOD was demonstrated when mice with homozygous null mutations in the MnSOD gene were produced. Mice lacking MnSOD die within 1–18 days from dilated cardiomyopathy (5). MnSOD also plays an important role as an anti-apoptotic protein by decreasing the level of mitochondrial O2˙̄ and the release of cytochrome c from mitochondria. A wide array of substances induce the activation of MnSOD, including cytokines, LPS, chemotherapeutic agents, and other substances (3, 4, 6–14).
Thioredoxin (Trx) is a small ubiquitous protein originally identified in Escherichia coli as a hydrogen donor for ribonucleotide reductase, the essential enzyme that provides deoxyribonucleotides for DNA replication (15). Trx reduces several proteins and enzymes inactivated by oxidation by converting disulfides to active thiols using reducing equivalents from NADPH via thioredoxin reductase (TrxR). The active site of Trx, Trp-Cys-Gly-Pro-Cys, is highly conserved across species (16, 17). Although Trx induces MnSOD transcription, the signaling mechanisms remain unclear (4). We have previously demonstrated that Trx activates c-Jun N-terminal kinase (JNK) in adenocarcinoma A549 cells and induces phosphorylation of mitogen-activated protein kinases (MAPKs) (18). However, the mechanism of activation of MAPKs and the induction of MnSOD by Trx remain incompletely understood in vascular endothelial cells.
MAPKs respond to myriad cellular and extracellular cues by modulating the activation or repression of gene expression (20). There are three distinct MAPK signaling pathways such as extracellular signal-regulated kinase (ERKs), the p38 MAPK, and JNK. MAPKs are activated by MAP2Ks, which in turn are activated by MAP3Ks. MKK4 (also known as SEK1 and JNKK1) is a MAP2K that is activated by dual phosphorylation of Ser-257 and Thr-261 (21). Although the specific kinase that activates MKK4 in vivo remains unclear (20, 21), MKK4 is activated by upstream MAP3Ks such as MEKK1–4, ASK1, mixed lineage kinases, and TGFβ-activated kinase 1 (TAK1) in transfection studies (22). Furthermore, MKK4 and MKK7 phosphorylate JNK and share similar upstream kinases, but their functions are nonredundant and diverse with respect to different stimuli (22). It is still unclear how MAPK kinases carryout diverse functions by phosphorylating their JNK substrate (22–25). Studies have shown that although MKK7 specifically activates JNKs, MKK4 can activate both JNK and p38 MAPK. MKK4 has also been shown to activate NFκB via activation of IκB kinase (1, 20, 22). Our studies provide an improved understanding of MnSOD gene transcription due to cellular redox perturbations in the endothelium leading to simultaneous MKK4 and MKK7 signaling and its predicted outcome on cell survival or cell death.
In this report, we present evidence that reduced Trx directly activates MKK4 via Cys-246 and Cys-266 residues without involvement of MAP3K. Treatment of wild-type MKK4 with oxidants or mutation of Cys-246 and Cys-266 inactivates MKK4. Our in vitro kinase assay shows that oxidized or reduced Trx differentially regulates MKK4 signaling by autophosphorylation without an upstream MAP3K activation. Additionally, TNFα, a physiological activator of MKK4, fails to activate MKK4 with Cys-246 or Cys-266 mutation, and also following treatment with H2O2. Our data further show that whereas MKK4-NFκB positively regulates MnSOD gene expression, MKK7-AP-1 negatively regulates its expression. Association of NFκB with the MnSOD promoter induces AP-1 displacement from the MnSOD promoter in a time-dependent manner. Taken together, this study provides a novel mechanism of MKK4 activation, which is independent of upstream MAP3Ks that integrate cellular redox state with MAPK activation or suppression leading to the induction or inhibition of gene expression.
Experimental Procedures
Reagents
E. coli thioredoxin was obtained from Promega Corp. (Madison, WI) and American Diagnostica. Human Trx (hTrx) was obtained from Sigma and American Diagnostica. Trx was reduced before use as described previously (4). Unless otherwise stated, reduced Trx was used throughout this study. MEKK, JNK, Fra-1, JunB, c-Jun, MnSOD, Trx, and β-actin antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals were obtained in the highest available grade. SP600125 was obtained from Tocris (Minneapolis, MN). JNK-IN-8 and SB203580 were purchased from Sigma. pJNK, p38 MAPK, MKK4, pMKK4(Thr-261), pMKK4(Ser-257), MKK7, pMKK7(Ser-271/Thr-275), and MEKK1 antibodies were purchased from Cell Signaling Technologies (Beverly, MA). pMEKK1(Thr-1381) was purchased from Abcam (Cambridge, MA).
Cell Culture, Transfections, and Mutagenesis
Human microvascular endothelial cells (HMVEC) from lung were obtained from Clonetics Corp. and propagated in endothelial growth medium (Clonetics). phCMV-Trx has been described in our previously published report (18). The dominant-negative JNKK1 expression plasmid (pSRα-K116R) and dominant-negative JNK (pSRα-AFP-JNK) expression plasmid were generous gifts of Dr. Gary L. Johnson (University of North Carolina, Chapel Hill), and have been described previously (26, 27). pcDNA3-MEKK1 and pcDNA3-MEKK1 (kinase-inactive MEKK1, K→M) are generous gifts from Dr. Thomas Maniatis, Boston, MA, and Gary L. Johnson (28, 29), respectively. pcDNA3-FLAG-MKK7 and pcDNA3-FLAG-MKK4 were generous gifts of Dr. Roger Davis (University of Massachusetts, Worcester, MA). Transfection of various expression plasmids into HMVEC was carried out using FuGENE 6 (Roche Applied Science) or Lipofectamine 2000 (Life Technologies, Inc.) as per manufacturer's protocol. The expression construct pcDNA3-MKK4 was mutagenized using the site-directed mutagenesis kit (Clontech) with primers synthesized by IDT (San Diego). To study the MKK4 mutants, endogenous MKK4 in HMVECs was down-regulated by MKK4 DsiRNA, Dicer substrate siRNA (30), using RNAi design tool (Integrated DNA Technologies) that targeted the noncoding 3′ region of MKK4 mRNA (GenBankTM accession number NM_001281435.1).
RNA Interference
Short interfering RNA (siRNA) duplexes and smart pool siRNA for MKK4 (L-003574) and MKK7 (L-004016) were obtained from Dharmacon (Lafayette, CO) or were synthesized by Integrated DNA Technologies, Coralville, IA (IDT), as per our sequence provided below. A nontargeting (NT) sequence was used as control for off-target effects of siRNA approach. Duplex siRNAs for c-Jun, Fra1, JunB, and DsiRNA for MKK4 (5′-GUAGAAUUCCAAUGCCAAGAAAUGG-3′) were synthesized by IDT. The JNK siRNA was obtained from Dharmacon (Lafayette, CO, ON-TARGETplus SMARTpool L-003514-00). For transfection, HMVEC were seeded in 60-mm2 dishes to obtain 60–70% confluency at the time of transfection. XtremeGENE transfection reagent (Roche Applied Science), SureSilencing kit from Genlantis, or Lipofectamine 2000 (Life Technologies, Inc.) was used to transfect siRNA to a final concentration of 100 nm. Inhibition of gene expression by siRNA was determined after 48–72 h by Western analysis.
The siRNA sequences used are as follows: c-Jun, 5′-GGA ACA GGU GGC ACA GCU UAA ACA G-3′; Fra-1, 5′-GCA GUG GAU GGU ACA GCC UCA UUT C-3′; JunB, 5′-CCA CCA UCA GCU ACC UCC CAC ACG C-3′; MEKK1, 5′-UGGCAAUUGCUAAAGCUU-3′; MKK7, 5′-CAGGAAGAGACCAAAGUAUAAUAAG-3′; MKK4, 5′-GGACGAGGAGCUUAUGGUUCUGUCA-3′; and JNK, 5′-UGGCAAUUGCUAAAGCUU-3′.
Northern Analysis of MnSOD
Total RNA was isolated from endothelial cells using the RNA Minieasy RNA isolation kit obtained from Qiagen Inc. (Valencia, CA) or TRIzol reagent (Invitrogen). In some experiments, RNA was isolated using CsCl2 density gradient centrifugation as reported earlier (3, 11). Ten micrograms of RNA was resolved by electrophoresis in 1% agarose, 2.5 m formaldehyde gel in a buffer containing 20 mm MOPS and 1 mm EDTA, pH 7.4. RNA was transferred to nitrocellulose, and blots were prehybridized for 2–12 h in 50% formamide, 0.75 m sodium chloride, 0.075 m sodium citrate, pH 7.0, 5× Denhardt's solution, 50 μg/ml salmon sperm DNA, and 0.1% SDS at 42 °C. Blots were hybridized with cDNA for human MnSOD (4) or β-actin, labeled to a specific activity of 2–7 × 107 cpm using [γ-32P]CTP (ICN) in hybridization solution at 42 °C overnight, and then were washed in 0.3 m sodium chloride, 0.03 m sodium citrate, 0.1% SDS at 42 °C. Autoradiographs were made by exposing blots to x-ray film (Eastman Kodak) at −70 °C with intensifying screens. MnSOD was detected as 4-kb transcripts as previously done (3, 11). To quantify MnSOD expression, the blots were exposed to phosphor screens (GE Healthcare), and densitometry was performed on phosphor screens using MD ImageQuant (Version 3.3) software.
Nuclear Extract Preparation
Nuclear extracts were prepared as described previously (18). Briefly, 107 cells were washed in 10 ml of phosphate-buffered saline (PBS) and centrifuged (1,500 × g for 5 min). The pellet was resuspended in PBS (1 ml), transferred into an Eppendorf tube, and centrifuged again (16,000 × g for 15 s). PBS was removed, and the cell pellet was resuspended in 400 μl of buffer A (10 mm HEPES, pH 7.8, 10 mm KCl, 0.1 mm EDTA, 2 mm DTT, 1 mm PMSF, leupeptin (0.5 mg/ml), and antipain (0.3 mg/ml)) by gentle pipetting. The cells were allowed to swell on ice for 15 min after which 25 μl of 10% Nonidet-P40 (IgePal, Sigma) was added, and the tube was vortexed vigorously for 10 s. The homogenate was centrifuged for 30 s in a microcentrifuge. The nuclear pellet was resuspended in buffer C (20 mm HEPES, pH 7.8, 0.42 m NaCl, 5 mm EDTA, 5 mm DTT, 1 mm PMSF, 10% (v/v) glycerol)], and the tube was rocked gently at 4 °C for 30 min on a shaking platform. The nuclear extract was centrifuged for 10 min in a microcentrifuge at 4 °C, and the supernatant was frozen at −70 °C in aliquots until electrophoretic mobility shift assay (EMSA) was performed. Protein was quantified by Bradford protein assay (Bio-Rad).
Electrophoretic Mobility Shift Assay (EMSA) of AP-1 and NFκB
The NFκB and AP-1-specific oligonucleotides were obtained from Promega Corp. (Madison, WI). Oligonucleotide was end-labeled using T4 polynucleotide kinase (Promega, Madison, WI) and [γ-32P]ATP (PerkinElmer Life Sciences) in 10× kinase buffer (0.5 m Tris-HCl, pH 7.5, 0.1 m MgCl2, 50 mm DTT, 1 mm spermidine, and 1 mm EDTA). For competition studies, 3.5 pmol of unlabeled oligonucleotide was used. Nuclear extract without labeled oligonucleotide was preincubated for 15 min at 4 °C followed by 20 min of incubation at room temperature, after the addition of labeled oligonucleotide. The binding reaction contained 10 μg of sample proteins and 5 μl of 5× incubation buffer (20% glycerol, 5 mm MgCl2, 5 mm EDTA, 5 mm DTT, 500 mm NaCl, 50 mm Tris-HCl, pH 7.5, 0.4 mg/ml calf thymus DNA). In some of the binding reactions poly(dIdC) (Amersham Biosciences and Sigma) was added to a final concentration of 2 μg. The nuclear protein-γ-32P-labeled oligonucleotide complex was separated from free γ-32P-labeled oligonucleotide by electrophoresis through a 6% native polyacrylamide gel in a running buffer of 0.25× TBE (5× TBE = 500 mm Tris, pH 8.0, 450 mm borate, 5 mm EDTA).
Chromatin Immunoprecipitation (ChIP) Assay
Chromatin was prepared using Chip-IT express kit (Active Motif, Carlsbad, CA kit, catalog no. 53008) as recommended by the manufacturer. HMVECs were equally seeded and treated with reduced Trx for 2 or 4 h, washed twice in cold PBS, and then fixed in 1% formaldehyde at room temperature for 10 min. The fixing reaction was stopped by addition of 1× glycine. The cells were collected in 1 ml of ice-cold PBS, and cell pellets were resuspended in ChIP lysis buffer with protease inhibitors. Samples were gently homogenized using a Dounce homogenizer and were then centrifuged at low speed (5000 rpm at 4 °C for 10 min) to separate the cytosolic fraction from the nuclear pellet. Shearing buffer was added to the nuclear pellet, and the pellet was further sonicated for about 20 pulses using sonic dismembrator model 100 (Fisher). Samples were centrifuged in a microcentrifuge to get sheared chromatin and were immunoprecipitated with p65 (sc-109, Santa Cruz Biotechnology), c-Jun (sc-1694, Santa Cruz Biotechnology), and FraI (sc-605, Santa Cruz Biotechnology). Rabbit IgG was used as a negative control. Immunoprecipitates were washed in ChIP assay buffer and further reverse cross-linked followed by digestion with proteinase K to remove any protein impurities. PCR primers were designed based on the MnSOD promoter sequence to approximately produce 210- and 201-bp PCR fragments covering the putative AP-1 forward primer 5′-CCC TGG GAG GGT ATG AAT GTC TTT-3′ and reverse primer 5-TGC CTG TCT GCC GTA CTT GAGTG-3) and the NFκB forward primer 5-AAA GTC CCT GTC CT-3 and reverse primer 5-AAA GAC ATT CAT ACC CTC CCA GGG C-3) sites, respectively. For each PCR (Life Technologies, Inc.), a 1–2-μl DNA template was prepared from immunoprecipitated chromatin, and the appropriate primer set was used. The whole-cell DNA served as the positive control (data not shown) and the IgG-immunoprecipitated samples as the negative control. The PCR products were separated on 2% agarose gel.
MKK4 in Vitro Kinase Assay
HEK293T cells were trypsinized and seeded at 25% confluency and allowed to grow overnight. Cells were transfected with pcDNA3-MKK4-HA, pcDNA3-MKK4 (C246A)-HA, or pcDNA3-MKK4 (C266A)-HA vectors using 2× HBS and CaCl2. After 6 h of incubation with transfection mix, complete medium (DMEM plus 10% FBS) was replaced. After 48 h of transfection, cells were lysed in lysis buffer (20 mm HEPES, pH 7.4, 150 mm NaCl, 0.1% Nonidet P-40, and 10 mg/ml aprotinin), and MKK4 was immunoprecipitated using anti-HA antibodies. Protein from the immunocomplex was eluted using 100 mm sodium citrate buffer, pH 3.5. The elute was concentrated and buffer exchanged with the kinase assay buffer (20 mm HEPES, pH 7.4, 20 mm MgCl2, 0.1 mm Na3VO4, 1 mm EGTA) using Amicon ultracentrifugal filters (Ultracell 10K, catalog no. UFC501096, Millipore). Recombinant Trx (T8690, Sigma) was reduced using 2 mm DTT or oxidized using 20 μm diamide in kinase assay buffer for 30 min at 30 °C and purified using Microspin G-25 columns (catalog no. 27-5325-01, GE Healthcare). In vitro phosphorylation assay was performed using 5 μg of MKK4 or its mutant in 50 μl of kinase assay buffer containing 20 μm ATP with or without 2 μg of oxidized or reduced Trx at 32 °C for 30 min. Phosphorylation reaction was stopped by adding Laemmli SDS-PAGE sample buffer. Phosphorylation of MKK4 or its mutants at Ser-257 and Thr-261 was analyzed by Western blotting using its phospho-specific antibodies. To estimate the basal phosphorylation level of MKK4 or its mutant, 2 μg of purified substrate was analyzed for its phosphorylation at Ser-257 and Thr-261 before the kinase reaction.
MKK4 kinase activity was assayed using the method described by Deacon and Blank (31). Briefly, HMVECs were treated with or without hTrx (2 μg/ml) for 2–8 h and lysed in lysis buffer, and MKK4 was immunoprecipitated using anti-MKK4 antibodies at 4 °C. To study the effect of C266A and C266A mutation on its kinase activity, HMVECs were transfected with pcDNA-MKK4-HA, pcDNA3-MKK4(C246A)-HA, or pcDNA3-MKK4(C266A)-HA using Lipofectamine 2000. After 48 h of transfection, cells were treated with or without hTrx (2 μg/ml) for 4 h and lysed. Ectopically expressed WT or mutant MKK4 was immunoprecipitated using anti-HA antibodies. Immunoprecipitates were washed twice with lysis buffer and once with kinase assay buffer. Immunoprecipitates were suspended in 25 μl of kinase assay buffer containing 2 mm DTT and 100 μm ATP, and the kinase reaction was initiated by adding 5 μg of inactive MAPK8 protein (PV3320, Invitrogen) and 5 μCi of [γ-32P]ATP. The kinase reaction was allowed for 20 min at 32 °C and terminated by adding Laemmli sample buffer followed by incubation in a boiling water bath for 7 min. Kinase reaction products were separated by SDS, and the 32P-labeled MKK4 substrate, MAPK8, was visualized by autoradiography.
Immunofluorescence and Proximity Ligation Assay (PLA)
HMVECs were cultured on glass coverslips and incubated with E. coli Trx (2 μg/ml) for 12 h. The cells were then washed, fixed, permeabilized, and probed with sheep anti-E. coli Trx antibody followed by Alexa Fluor 488-conjugated donkey anti-sheep secondary antibodies and Alexa Fluor 568-conjugated phalloidin. Fluorescence images were obtained via 20×/0.8 NA objective using Zeiss Axio Imager Z2 upright fluorescent microscope. To observe the internalization of the Trx, a stack of fluorescent images were obtained along z axis at 60-nm intervals via a ×63/1.40 NA objective and deconvolved using AxioVision 4.9 software. xz and yz cut view of the stacks presented to show location of Trx.
In Situ PLA
HMVECs were seeded onto glass coverslips and allowed to grow to 90% confluence. After appropriate treatments, the cells were fixed with 3% paraformaldehyde prepared in PBS for 10 min at 37 °C, permeabilized with 0.1% Triton X-100 for 10 min at room temperature, blocked with 5% donkey serum and 3% BSA in PBS for 1 h at room temperature, and incubated with primary antibodies in 50% Da Vinci Green antibody diluent (Abcam, Cambridge, MA). PLA was performed following the supplier's instruction using Duolink Anti-Rabbit PLUS and anti-mouse MINUS PLA probes and Duolink green detection reagent (Duolink, Sigma). To mark the border of the cells, they were stained with goat anti-VE-cadherin antibodies followed by Alexa Fluor 594-conjugated secondary antibodies. 30–35 fluorescent images along the z axis with 80 nm intervals (optical sections) were obtained using Zeiss Axio Imager Z2 upright fluorescent microscope via a 63×/1.40 NA objective and deconvolved using AxioVision 4.9 software. PLA reaction products appear as green foci, which originate from the location of Trx1 interaction with its partners. The number of green foci was counted using Adobe Photoshop.
Western Analysis
Cells were harvested after treatments as indicated in the figures using radioimmunoprecipitation assay (RIPA) buffer (0.5% sodium deoxycholate, 1% SDS, and 1% IgePal in PBS with protease inhibitors) and was quantified with Bradford assay (Bio-Rad). Western analysis was performed using the Bio-Rad mini protean system. Equal amounts of protein were resolved on a 6–15% SDS-PAGE. Following electrophoresis, protein was transferred to a nitrocellulose (Hybond-ECL, GE Healthcare) or PVDF membrane (Bio-Rad), immunoblotted with respective primary antibodies, and visualized by the ECL system (GE Healthcare) using appropriate secondary antibodies conjugated with HRP IgG (Santa Cruz Biotechnology). Some of the images were acquired with G:BOX Chemi XL1.4 (Syngene, Frederick, MD) using West Femto reagent from Pierce.
MnSOD Activity Assay
MnSOD activity was determined as mentioned in our previous publication (11)
Statistical Analysis
The experiments were performed in replicate and repeated for a minimum of 2 times. Northern and Western data densitometry was performed by ImageQuant or ImageJ software. Data were statistically analyzed (ANOVA) or Student's t test using GraphPad Prism software (Version 6.0).
Results
Inhibition of JNK Potentiates MnSOD Induction by Trx in Endothelial Cells
We determined whether JNK signaling is involved in Trx-induced MnSOD expression in endothelial cells (ECs) as JNK has been shown to be activated in the lung adenocarcinoma cell line A549 (18).
Surprisingly, as demonstrated in Fig. 1A, SP600125, a JNK inhibitor in combination with externally added E. coli Trx, potentiated MnSOD gene expression (Fig. 1, A and B). We also confirmed that externally added hTrx potentiates MnSOD induction in response to JNK inhibition (Fig. 1, C and D). These data indicate that JNK signaling is inhibitory in MnSOD induction by Trx. We further tested whether the expression of MnSOD protein is regulated by JNK. As shown in Fig. 1, E and F, the expression of MnSOD protein was increased in response to Trx, due to JNK inhibition and both in combination. Because MnSOD activity is ultimately responsible for detoxification of the superoxide radical, we determined the MnSOD enzymatic activity in response to Trx, SP600125, or both in combination. As shown in Fig. 1G, MnSOD activity was significantly increased in HMVEC treated with E. coli Trx, SP600125, or Trx + SP600125, indicating that overexpressed MnSOD remains functional in the ECs. We used another specific inhibitor of JNK, JNK-IN-8, to confirm our findings with SP600125. As shown in Fig. 1H, JNK-IN-8 treatment also induced MnSOD expression alone or in combination with Trx. Inhibition of p38 MAPK by SB203580, a specific inhibitor of p38, did not affect MnSOD expression by Trx (Fig. 1, I and J), demonstrating the specificity of JNK inhibition on MnSOD expression. Chemical inhibitors are known to have nonspecific effects on various cellular functions. Therefore, we adopted a genetic approach to determine whether inhibition of JNK induces MnSOD expression by Trx. As shown in Fig. 1K down-regulation of JNK by RNAi significantly induced the expression of MnSOD mRNA (Fig. 1K, top panel). Collectively, these data established that JNK is a negative regulator of MnSOD in endothelial cells. Because we have previously reported JNK activation in A549 cell by Trx (18), we evaluated whether there is a fundamental difference in the activation of JNK in A549 and endothelial cells. As shown in Fig. 1L, although Trx activated JNK in A549 cells, it failed to do so in HMVEC. We determined whether externally added Trx is internalized by ECs because our experiments using ectopic expression of Trx could be reproduced with addition of recombinant Trx (E. coli Trx or human Trx) to the culture medium. Our data (Fig. 1M) show that Trx is internalized by HMVEC as indicated by bright green fluorescence and is localized primarily within the cytoplasm (Fig. 1M, left and right panel s(one HMVEC with actin (red) and Trx (green) immunofluorescence). We further used PLA (32–34) to determine colocalization of externally added human recombinant Trx with TrxR using specific antibodies. Because TrxR is a cytosolic protein and Trx interacts with TrxR for reduction, colocalization of externally added Trx with TrxR would suggest the internalization of externally added hTrx. As shown in Fig. 1N (upper left panel) endogenous Trx colocalized with cytosolic TrxR indicating that Trx and TrxR interact in the cytosol (>1000 PLA signals/cell). When external Trx was added in the culture medium, the number of PLA signals increased to more than 3000 (Fig. 1N, upper right panel), indicating that externally added Trx is able to interact with cytosolic TrxR. Furthermore, we used VE-cadherin as an endothelial membrane marker protein, and we show that externally added Trx does not interact with membrane-bound VE-cadherin (no difference in PLA signal in control or treated) indicating that Trx does not localize with the EC membrane.
FIGURE 1.

JNK inhibition induces MnSOD expression and potentiates MnSOD expression by Trx. A, JNK inhibition potentiates E. coli Trx-mediated MnSOD expression. HMVECs were treated with SP600125 (20 μm) followed by treatment with reduced E. coli Trx (3 μm). Northern blotting was performed as described under “Experimental Procedures.” MnSOD mRNA is detected as 4-kb transcripts (3, 11). B, densitometry of autoradiograph (ratio of MnSOD to β-actin) of A. The experiment was run in triplicate and repeated at least twice. *, significantly higher than the control (p < 0.05); ** significantly higher than the Trx- or SP600125-treated cells, (p < 0.05, ANOVA). C, JNK inhibition potentiates hTrx-mediated MnSOD induction. HMVECs were treated with 3 μm human reduced Trx alone or with indicated concentration of SP600125. MnSOD expression was analyzed by Northern blot. β-Actin was used as a loading and transfer control. D, densitometry of Northern blot of C and the data expressed as ratio of MnSOD to β-actin. *, significantly higher than control at (p < 0.05, ANOVA); **, significantly higher than Trx or SP600125 alone (p < 0.05); E, Trx alone or in combination with JNK inhibitor increases MnSOD protein expression. HMVEC were treated with hTrx or SP600125, alone or in combination as indicated, and the Western analysis was performed as described under “Experimental Procedures.” F, densitometry of blot in E, and the data expressed as the ratio of MnSOD to β-actin. **, significantly higher compared with untreated cells (p < 0.05, ANOVA); *, significantly higher than untreated cells (p < 0.05, Student's t test). G, MnSOD activity is increased in response to Trx alone or in combination with SP600125. MnSOD activity is expressed as units of MnSOD/mg of protein; *, significantly higher than controls (p < 0.05); #, significantly higher compared with E. coli Trx alone (p < 0.05); ##, significantly higher compared with SP600125 (p < 0.05). H, JNK-IN-8, a specific JNK inhibitor, potentiated Trx-mediated MnSOD expression. HMVECs were treated with 5 μm JNK-IN-8 with or without hTrx, and the MnSOD expression was analyzed by Western blotting as described under “Experimental Procedures.” I, p38 MAPK inhibitor does not increase MnSOD expression in response to Trx. Cells were treated with indicated concentration of SP600125 or p38 MAPK inhibitor SB203580 for 2 h followed by treatment with hTrx for 16 h. Northern analysis was performed as described under “Experimental Procedures.” J, densitometry of blot mentioned in Fig. 1I, and data expressed as ratio of MnSOD and β-actin. *, significantly higher than control (p < 0.05); **, significantly higher than Trx alone (p < 0.05, ANOVA). K, depletion of JNK induces MnSOD expression. ECs were treated with JNK siRNA, and after 48 h of MnSOD expression was detected by Western analysis. L, Trx treatment induces JNK activation in A549 cells but not in HMVEC. HMVEC or A549 cells were treated with hTrx (3 μm) for 24 h followed by Western analysis of phospho-JNK. M, internalization of E. coli Trx by ECs. ECs were treated with 2 μm E. coli Trx. Immunofluorescence was performed as described under “Experimental Procedures.” Green fluorescence indicates Trx, and red fluorescence indicates f-actin; N, exogenously added Trx colocalizes with endogenous TrxR but not VE-cadherin. HMVECs were cultured on glass coverslips and incubated with recombinant human Trx (2 μg/ml) for 4 h. Then the cells were fixed and permeabilized, and PLA was performed using anti-Trx antibodies in combination with anti-TrxR or anti-VE-cadherin antibodies. Cellular nuclei were stained with DAPI. PLA reaction products that appear as green foci and were counted using Adobe Photoshop and plotted as a bar graph. *, p < 0.05 versus control.
Down-regulation of MKK4 Decreases but Down-regulation of MKK7 Up-regulates MnSOD Expression in ECs
Because JNK is activated by two upstream kinases, MKK4 and MKK7, we determined the specific upstream kinase that may be involved in the negative regulation of MnSOD expression. We down-regulated MKK4 or MKK7 expression by RNAi as shown in Fig. 2, A and B. Trx-induced MnSOD mRNA expression was decreased in ECs depleted of MKK4 but not depleted of MKK7 (Fig. 2, C and D). Interestingly, MnSOD mRNA expression was increased in ECs depleted of MKK7. Furthermore, MKK7 down-regulation by siRNA increased the level of MnSOD mRNA in response to Trx (Fig. 2, C and D). The MnSOD protein expression was decreased in MKK4-depleted cells but was increased in MKK7-depleted ECs (Fig. 2E). Because MnSOD induction is directly linked to NFκB activation (3, 9, 11) and JNK activates AP-1 transcription factors (35), we determined whether activation of NFκB and AP-1 by Trx is modulated by MKK4 or MKK7. As demonstrated in Fig. 2F, depletion of MKK4, but not MKK7, decreased the activation of NFκB. Surprisingly, AP-1-DNA binding was down-regulated by Trx and in response to MKK7 depletion. However, MKK4 down-regulation did not diminish AP-1-DNA binding (Fig. 2G). Collectively, these data show that Trx-induced MnSOD expression is mediated by MKK4, and MKK7-JNK-AP-1 signaling functions as an endogenous repressor of MnSOD in endothelial cells.
FIGURE 2.

Down-regulation of MKK7 increases MnSOD expression, but down-regulation of MKK4 decreases MnSOD expression in HMVECs. A and B, down-regulation of MKK4 or MKK7 by RNAi. HMVECs were transfected with indicated concentrations of siRNA (MKK7, MKK4, and NT controls). After 48 h, cell lysate was prepared using RIPA lysis buffer, and levels of MKK7 and MKK4 were detected by Western analysis as described under “Experimental Procedures.” C, down-regulation of MKK7 potentiates MnSOD expression, but down-regulation of MKK4 decreases MnSOD expression in response to Trx. HMVECs were transfected with NT, MKK4 siRNA, or MKK7 siRNA as described under “Experimental Procedures.” Following 48 h of transfection, cells were treated with reduced hTrx (3 μm) for 16–18 h. MnSOD and β-actin Northern analysis was performed as described under “Experimental Procedures.” D, densitometry of Northern blot in C. *, significantly higher than control (p < 0.05, ANOVA); #, significantly lower than Trx only (p < 0.05); ##, significantly higher than NT siRNA-treated cells (p < 0.05); **, significantly higher than Trx-treated cells (p < 0.05, ANOVA). E, decreased levels of MKK7 increase, and decreased levels of MKK4 decreases MnSOD protein expression in HMVEC treated with Trx. HMVECs were transfected with NT, MKK4, or MKK7 siRNA as described for A and B. Following 48 h of transfection, cells were treated with reduced hTrx (3 μm) for 16–18 h. MnSOD Western analysis was performed as described under “Experimental Procedures.” F, down-regulation of MKK7 potentiates NFκB activation, but down-regulation of MKK4 decreases NFκB activation by Trx. HMVECs were transfected with NT, MKK4, or MKK7 siRNA as described under “Experimental Procedures.” Following transfection, cells were treated with hTrx for 16 h, and NFκB EMSA was performed as described under “Experimental Procedures.” G, down-regulation of MKK7 decreases AP-1 activation. MKK7 or MKK4 levels in HMVECs were down-regulated, and AP-1 EMSA was performed as described under “Experimental Procedures.”
Trx Directly Activates MKK4 without the Involvement of MEKK1 in ECs
MEKK1 is an upstream kinase that phosphorylates MKK4/MKK7 leading to their activation (31). Studies have shown that Cys-1238 in MEKK1 is redox-sensitive, and the glutathionylation of this residue inhibits MEKK1 activation (36). Given that Trx is a protein-disulfide reductase, we determined whether Trx activates MEKK1 via reduction of Cys-1238. As shown in Fig. 3A, transfection of increasing concentrations of phCMV1-Trx induced the expression of MnSOD but did not increase the phosphorylation of MEKK1. However, MKK4 phosphorylation was increased due to increased expression of Trx. In contrast, there was no increase in MKK7 protein expression or phosphorylation (Fig. 3A). These experiments show that Trx does not induce the activation of either MEKK1 or MKK7 in ECs. To further determine the role of MEKK1 in MKK7 or MKK4 activation and MnSOD expression, we down-regulated MEKK1 by RNAi and evaluated its effect on MKK7 or MKK4 phosphorylation and MnSOD induction by Trx. As shown in Fig. 3B, loss of MEKK1 decreased the baseline phosphorylation of MKK7 but not the phosphorylation of endogenous or ectopically expressed MKK4. Interestingly, loss of MEKK1 increased the expression of MnSOD. These data further confirm our finding that the MEKK1-MKK7 pathway is an endogenous negative regulator of MnSOD induction.
FIGURE 3.

Trx activates MKK4 independent of MEKK1. A, Trx activates MKK4 and induces MnSOD expression in a dose-dependent manner, but it does not activate MKK7 or MEKK1. HMVECs were transfected with 10 μg of phCMV1 or different amounts of phCMV1-Trx (1–20 μg), and samples were analyzed by Western blotting for the expression level of MnSOD using anti-MnSOD antibodies. The blot was reprobed with anti-Trx and anti-β-actin antibodies. Cell lysates were also analyzed by immunoblotting for MEKK1, MKK4, and MKK7 phosphorylation using their phospho-specific antibodies. The blots were reprobed with anti-MEKK1, anti-MKK7, and anti-MKK7 antibodies, respectively. B, down-regulation of MEKK1 does not decrease the expression of MnSOD induced by Trx. HMVECs were transfected with NT siRNA or MEKK1 siRNA (100 nm) in combination with phCMV1, phCMV1-Trx, or pcDNA3-MKK4, and after 48 h, cell lysates were prepared and analyzed for MnSOD expression and activation of MKK4, MKK7, and MEKK1 as described in A. C, overexpression of MEKK1 does not increase MnSOD expression, but overexpression of kinase-inactive MEKK1 increases MnSOD expression. HMVECs were transfected with pcDNA3-MEKK1 or pcDNA3-MEKK1 (K→M) in combination with pcDNA3 or phCMV1-Trx, and after 48 h samples were analyzed for expression levels of MnSOD and phosphorylation of MKK4 by Western blotting as described in A. D, overexpression of MKK7 alone or in combination with MEKK1 did not increase MnSOD expression. HMVECs were transfected with pcDNA3-MKK4, pcDNA3-MKK7, pcDNA3-dnMKK7, or psc-IκB(S32A/S36A) alone or in combination with pcDNA3-MEKK1, and after 48 h, the lysates were analyzed for MnSOD expression and activation of MKK7 and MKK4 as described in A. The blot was reprobed with anti-Trx and anti-β-actin antibodies.
Because the activation of MKK4 independent of upstream kinases is a novel observation, we further evaluated whether cotransfection of MEKK1 and Trx in ECs would result in increased MnSOD expression. As expected, coexpression of MEKK1 and Trx did not increase MnSOD induction compared with Trx alone (Fig. 3C). Additionally, inhibition of MEKK1 kinase activity by dominant-negative expression of kinase-inactive mutant MEKK1 (K→M) had a stimulatory effect on Trx-dependent MnSOD induction (Fig. 3C), demonstrating that MEKK1 activation represses MnSOD expression. Overexpression of MKK4 alone increased the phosphorylation of MKK4 and the expression of MnSOD (Fig. 3D). Coexpression of MKK4 and MEKK1 did increase MnSOD expression and MKK4 phosphorylation. Phosphorylation of MKK4 was stronger in MEKK1 and MKK4 cotransfection compared with MKK transfection alone. However, there was also strong activation of MKK7 in response to MEKK1 transfection as shown by phosphorylation of MKK7 (Fig. 3D).
Given that direct phosphorylation of IκB by MEKK1 activates NFκB (37), we determined whether MEKK1 could induce MnSOD expression via MEKK1-dependent NFκB activation. As shown in Fig. 3D, overexpression of MEKK1 did not result in the induction of MnSOD in HMVECs. Furthermore, ectopic expression of IκB (S32A/S36A, super-repressor of NFκB) in combination with MEKK1 did not show any effect on MnSOD expression (Fig. 3D). Transfection of MKK7 alone or in combination with MEKK1 did not increase MnSOD expression (Fig. 3D). In contrast, transfection of dnMKK7 alone or in combination with MEKK1 induced the expression of MnSOD, suggesting again that suppression of MKK7 promotes MnSOD activation.
Cys-246 and Cys-266 in MKK4 Are Specific Targets of Trx and Are Required for MKK4 Activation by Physiological Activators such as TNFα
Trx is not a protein kinase, and it does not induce MEKK1-dependent MnSOD expression in ECs. Because MKK4 has been shown to have a low level of autocatalytic activity in unstimulated cells (31), we reasoned that Trx might modulate a thiol-disulfide exchange reaction in MKK4, which could facilitate MKK4 autophosphorylation. We first analyzed the MKK4 coding region for possible cysteine residues, which could be targets of Trx for reduction. We found eight cysteine residues in the MKK4 protein sequence as follows: Cys-158, Cys-174, Cys-177, Cys-246, Cys-266, Cys-347, Cys-379, and Cys-382. We mutagenized each of these cysteine residues in the MKK4 sequence (C158A, C174A/C177A, C246A, C266A, C347A, and C379A/C382A) to determine whether any of these cysteine residues is a target of Trx. To reduce interference from endogenous active MKK4 from endothelial cells, we depleted endogenous MKK4 using DsiRNA designed to target the 3′-noncoding region of MKK4 (Fig. 4A). DsiRNA specifically depleted endogenous MKK4 and did not affect ectopically expressed MKK4 and also did not affect levels of other cellular proteins like MKK7 or β-actin (Fig. 4A). As demonstrated in Fig. 4B, the expression of all mutants except the Cys-246 or Cys-266, in ECs depleted of endogenous MKK4 by DsiRNA, resulted in the induction of MnSOD when transfected alone or in combination with Trx. Furthermore, transfection of C246A or C266A mutant constructs did not induce the phosphorylation of either Ser-257 or Thr-261 in MKK4. However, transfection of other mutants induced the phosphorylation of MKK4 on Thr-261 and to a lesser extent on Ser-257. These data from ECs depleted of endogenous MKK4 suggest that Cys-246 and Cys-266 are specific residues on MKK4 that are redox-sensitive and are required for the autophosphorylation of MKK4 on Thr-261 and MnSOD induction.
FIGURE 4.

Cys-246 and Cys-266 of MKK4 are essential for Trx-mediated MKK4 activation and MnSOD expression. A, MKK4 DsiRNA that targets the 3′-noncoding region of MKK4 down-regulates endogenous but not ectopically expressed MKK4. HMVECs were transfected with NT or MKK4 DsiRNA in combination with pcDNA3 empty vector or pcDNA3-MKK4, and after 48 h, cell lysates were prepared and analyzed for levels of MKK4, MKK7, and β-actin as described under “Experimental Procedures.” B, C246A and C266A mutations in MKK4 abrogates Trx-induced phosphorylation of Thr-261 and Ser-257. HMVECs were transfected with wild-type MKK4 construct (pcDNA3-MKK4) or constructs with various cysteine mutants in combinations with MKK4 DsiRNA and with or without pCMV1-hTrx, and after 48 h, cell lysates were prepared using RIPA buffer. Equal amounts of protein from each sample were analyzed for expression levels of MnSOD, Trx, and phosphorylation levels of MKK4 (Thr-261/Ser-257) by Western blotting using their specific antibodies. The blots were reprobed with anti-MKK4, anti-GAPDH, and anti-β-actin antibodies. C, TNFα fails to activate mutant MKK4 (C246A and C266A). HMVECs were transfected with pcDNA3-MKK4, pcDNA3-MKK4 (C246A), or pcDNA3-MKK4(C266A) and after 48 h treated with or without TNFα in presence of absence of H2O2. Cell lysates were prepared and analyzed for MnSOD expression and activation of MKK4 as described in B. D, treatment of HMVEC with Trx results in a reduced redox state of Trx, but treatment with H2O2 promotes an oxidized redox environment. HMVECs were treated with 2 μm hTrx or treated with 0.5 mm H2O2 for 30 min followed by a Trx redox state assay. E, Cys-246 and Cys-266 are required for MKK4 phosphorylation by Trx. MKK4 or its mutants (C246A and C266A) were phosphorylated in vitro in the presence or absence of reduced or oxidized Trx, and the products were analyzed for phosphorylation at Thr-261 or Ser-257 by Western blotting using anti-phospho-MKK4 (Thr-261) or anti-phospho-MKK4 (Ser-257) antibodies. The reaction mix was also immunoblotted to determine Trx levels. To determine basal phosphorylation level of purified MKK4 or its mutant, 2 μg of purified substrate was analyzed by Western blotting using anti-phospho-MKK4 (Thr-261) or anti-phospho-MKK4 (Ser-257) antibodies. F, Trx induces MKK4 kinase activity. HMVECs were treated with hTrx for various times, and cell lysates were prepared. Equal amounts of cell lysates from control and treatments were immunoprecipitated using anti-MKK4 antibodies, and in vitro kinase activity was detected as described under “Experimental Procedures.” G, Cys-246 and Cys-266 of MKK4 are essential for its kinase activity. HMVECs were transfected with pcDNA3-MKK4, pcDNA3-MKK4 (C246A), or pcDNA3-MKK4(C266A) for 48 h followed by 4 h of treatment with hTrx (2 μm). Cell lysates were prepared, and equal amounts of cell lysates from control and treatments were immunoprecipitated with anti-HA antibodies, and in vitro kinase activity was assayed as described under “Experimental Procedures.” H, treatment of ECs with hTrx induces cytosolic interaction of Trx with MKK4. PLA was performed using anti-hTrx and anti-MKK4 as described under “Experimental Procedures.” Green foci-proximity signals of MKK4 and Trx were counted and plotted as a bar graph. *, significantly higher than untreated HMVEC (p < 0.05, Student's t test).
TNFα is a potent inducer of MnSOD (3, 11, 38). TNFα is also an activator of MKK4 (39). To understand how MKK4 activation is modulated by the cellular redox state, we employed TNFα as a physiological activator of MKK4. We also determined whether pretreatment of cells with H2O2, an oxidizing agent, would modulate MKK4 phosphorylation (Ser-257/Thr-261) by TNFα in ECs. As demonstrated in Fig. 4C, there was no effect of H2O2 alone on the basal level of MKK4 phosphorylation. However, treatment with H2O2 decreased MKK4 phosphorylation and MnSOD induction in response to TNFα (Fig. 4C). Furthermore, TNFα failed to induce the phosphorylation of mutant MKK4 (C246A and C266A), demonstrating that Cys-246 and Cys-266 are required for physiological activation of MKK4. We also determined the redox state of ECs treated with either Trx or H2O2. As shown in Fig. 4D, increasing the intracellular Trx resulted in a more reducing Trx redox state (untreated compared with Trx transfection). However, treatment of cells with H2O2 resulted in marked increase in the oxidized Trx level. Taken together, these data further support the conclusion that Cys-246 or Cys-266 in MKK4 must remain in the reduced state for its activation by upstream MAP3Ks induced by TNFα, as treatment of cells with oxidizer H2O2 inhibited MKK4 phosphorylation.
Because MKK4 displays a low level of autophosphorylation in vitro (31), we determined whether this autophosphorylation could be increased in the presence of reduced Trx. It is plausible that Trx might directly reduce the Cys-246 and Cys-266 due to its thiol-reducing activity, and it could thus induce autophosphorylation of MKK4 on Thr-261 or Ser-257. To evaluate this hypothesis, we determined whether MKK4 activation could be modulated in vitro using oxidized or reduced Trx and whether Cys-246 or Cys-266 is required for redox-mediated autophosphorylation of MKK4. When oxidized Trx was used in a kinase assay with wild-type MKK4 as substrate, there was no phosphorylation of Thr-261 (Fig. 4E). In contrast, when reduced Trx was included in the in vitro kinase assay, MKK4 was phosphorylated on Thr-261 and to a lesser extent on Ser-257. However, the mutant MKK4 protein (C246A or C266A) did not respond to oxidized or reduced Trx in the kinase assay and remained unphosphorylated. These data suggest that Trx alone could activate MKK4 due to reduction of Cys-246/Cys-266. Treating ECs with Trx induced MKK4 kinase activity (Fig. 4E) as determined by a kinase assay using MAPK8 as the substrate for MKK4. We further determined the effect of C246A and C266A mutations on Trx-induced MKK4 activation. As shown in Fig. 4F, the mutant MKK4 (C246A and C266A) failed to show basal as well as Trx-induced kinase activity in contrast to WT MKK4. Collectively, these data further confirm that Cys-246 and Cys-266 are critical residues required for the activation of MKK4 via autophosphorylation of Thr-261. The reduction of Cys-246/Cys-266 could only occur if Trx would make physical contact with MKK4 as the disulfide-thiol exchange reaction would occur when both proteins were in close proximity. To determine whether both Trx and MKK4 were in close proximity in response to Trx treatment, we performed PLA using anti-Trx and anti-MKK4 antibodies. As shown in Fig. 4H, addition of hTrx induced a significant PLA signal (>1200 foci compared with ∼100 in untreated cells). These data suggest that MKK4 and externally added Trx associate in the cytosol.
Redox Control of MKK4 Activation and MnSOD Expression
Because MKK4 positively regulates Trx-mediated MnSOD, we determined whether MnSOD expression would follow a pattern similar to MKK4 phosphorylation by either TNFα or IL-1β, as TNFα and IL-1β are potent inducers of MnSOD and MAP3Ks. As shown in Fig. 5, A and B, TNFα, IL-1β, or Trx induced the expression of MnSOD in ECs. The combination of Trx with TNFα or IL-1β potentiated MnSOD expression. The activation of NFκB also followed a similar pattern of induction by these agents, alone or in combination with Trx (Fig. 5C). Given that Trx is a reducing agent, we evaluated the effect of an oxidizing agent such as H2O2 on NFκB activation and MnSOD induction by TNFα or IL-1β. As shown in Fig. 5D, NFκB activation induced by TNFα or IL-1β was decreased in the presence of H2O2. H2O2 alone did not activate NFκB in ECs. Furthermore, H2O2 at 0.1 to 1 mm concentrations did not induce MnSOD expression in ECs (Fig. 5, E and F). In contrast to Trx, H2O2 decreased the expression of MnSOD induced by TNFα or IL-1β (Fig. 5, E and F). Collectively, these studies support the conclusion that MKK4 activation is controlled by the cellular redox environment involving Cys-246 and Cys-266. We further determined the MKK4 activation in response to TNFα or IL-1β in the presence of Trx or H2O2. As shown in Fig. 5F, MKK4 phosphorylation by TNFα or IL-1β was decreased in the presence of H2O2, but it increased in the presence of Trx. Additionally, TNFα- or IL-1β-dependent MKK4 activation was significantly higher in the presence of Trx.
FIGURE 5.

TNFα or IL-1β-mediated NFκB activation and MnSOD induction is potentiated by Trx but decreased in presence of H2O2. A, potentiation of TNFα- or IL-1β-induced MnSOD expression by Trx. HMVECs were treated with E. coli Trx (7 μm) for 2 h, followed by 16 h of treatment with TNFα (1 ng/ml) or IL-1β (0.02 ng/ml). Following incubation, Northern analysis was performed for MnSOD and β-actin gene expression. B, ratio of MnSOD to β-actin mRNA densitometry. *, significantly higher than untreated cells (p < 0.05, ANOVA); **, significantly higher than TNFα- or IL-1β-treated cells (p < 0.05, ANOVA). C, potentiation of TNFα- or IL-1β-induced NFκB activation by Trx. HMVECs were treated with E. coli (7 μm) for 2 h followed by treatment with TNFα (1 ng/ml) or IL-1β (0.02 ng/ml) for 4 h. Following incubation, NFκB EMSA was performed as described earlier. D, TNFα- or IL-1β-induced NFκB activation is decreased by treatment of cells with H2O2. HMVECs were pretreated with indicated concentrations of H2O2 for 1 h, followed by addition of TNFα (1 ng/ml) or IL-1β (0.2 ng/ml) for 1 h, following which NFκB EMSA was performed as described under “Experimental Procedures.” E, TNFα- or IL-1β-induced MnSOD expression is decreased by H2O2. Cells were treated with the indicated concentrations of H2O2 for 1 h followed by treatment with TNFα (1 ng/ml) or IL-1β (1 ng/ml) for 6 h. Northern analysis was performed to detect the levels of MnSOD and β-actin transcripts as described earlier. F, densitometry of 4-kb band of MnSOD of E. G, TNFα- or IL-1β-mediated MKK4 activation is inhibited by H2O2 and potentiated by Trx. Cells were pretreated with H2O2 (500 μm) or hTrx (2 μg/ml) followed by treatment with TNFα (2 ng/ml) or IL-1β (0.05 ng/ml). Western analysis for phospho-MKK4/MKK4 and MnSOD was performed as described under “Experimental Procedures.”‘
Temporal Occupation of MnSOD Promoter by p65, Fra-1, and c-Jun in Response to Trx
Because MnSOD expression was induced by depletion of MKK7 alone or in combination with Trx, we considered the possibility that MKK7 is a constitutive repressor of MnSOD expression. MKK7 is an upstream kinase, which phosphorylates JNK resulting in its activation. JNK phosphorylates Jun and Fra proteins that constitute the AP-1 transcription factor (20). Because either inhibition of JNK or silencing of MKK7 induced MnSOD expression, we sought to determine whether AP-1 complex proteins are endogenous repressors of MnSOD. In addition, because NFκB is required for MnSOD induction, we determined whether Fra-1, c-Jun, or p65 proteins occupy the MnSOD promoter in a temporal manner that would control MnSOD transcription by a specific stimulus. To evaluate these possibilities, we first performed the kinetics of NFκB and AP-1 activation by Trx. As shown in Fig. 6A, NFκB was activated due to the treatment with Trx within 1–2 h and was maximally activated at 4 h. In contrast, AP-1 was found to be associated with the DNA in resting HMVECs, and treatment with Trx caused a progressive dissociation of AP-1 from the MnSOD promoter (Fig. 6B). NFκB activation was not affected by the JNK inhibitor JNK-IN-8 (Fig. 6C). Interestingly, treatment of ECs with JNK-IN-8 caused AP-1 dissociation from DNA, and this dissociation was potentiated in the presence of Trx (Fig. 6D). We determined temporal occupation of MnSOD promoter by AP-1 and NFκB proteins using ChIP assay. We performed the assay with a primer specific for the AP-1 region in the MnSOD promoter to determine whether AP-1 remains bound to the MnSOD promoter in unstimulated ECs. As shown in Fig. 6E, chromatin immunoprecipitates from the resting ECs with either Fra-1 or c-Jun antibodies generated a PCR product specific to the AP-1 region. When cells were incubated with E. coli Trx for 2 or 4 h, we observed a gradual decrease in the abundance of PCR product suggesting that the removal of AP-1 proteins from the MnSOD promoter occurs due to the treatment of ECs with Trx. Additionally, we used p65 antibody to immunoprecipitate the p65-specific protein-DNA complex from the chromatin of ECs treated with or without Trx. As shown in Fig. 6E, untreated ECs did not show any PCR product from p65 chromatin immunoprecipitates, demonstrating the absence of basal NFκB binding to the MnSOD promoter. However, incubation of cells with Trx for 2 or 4 h strongly increased the level of NFκB-specific PCR product, suggesting the enhanced binding of NFκB. These data suggest a sequential trafficking of NFκB and AP-1 proteins to and from the MnSOD promoter that balances the activation of MnSOD in response to Trx.
FIGURE 6.

Temporal occupation of MnSOD promoter by Fra-1, c-Jun, and p65 in response to Trx. A and B, Trx activates NFκB and inactivates AP-1 in a time-dependent manner. HMVECs were treated with hTrx (3 μm) for 1, 2, and 4 h. At the end of the treatments, nuclear extract was prepared and NFκB (A) or AP-1 (B) activation was determined by EMSA as described under “Experimental Procedures.” C and D, effect of JNK Inhibition on NFκB and AP-1 activation. HMVECs were treated with 5 μm JNK-IN-8 in combination with or without 3 μm hTrx for 2 or 4 h. At the end of the incubation, nuclear extract was prepared and EMSA was performed to determine NFκB (C) or AP-1 (D) activation as described under “Experimental Procedures.” E, Trx treatment induces removal of Fra-1 and c-Jun but allows p65 to occupy the MnSOD promoter. ChIP assay was performed using antibodies to Fra-1, c-Jun, and p65 as described under “Experimental Procedures.” F, down-regulation of Fra-1, c-Jun, or JunB up-regulates MnSOD expression. ECs were transfected with siRNA for Fra-1, c-Jun, or JunB as described under “Experimental Procedures,” and MnSOD, β-actin, JunB, Fra-1, and c-Jun Western analysis was performed as described under “Experimental Procedures.” G, redox regulation of MnSOD by Trx. In the resting ECs, MnSOD is maintained at low levels due to the binding of AP-1 proteins to MnSOD promoter. When cells are treated with Trx, AP-1 proteins from the −391 and −260 site in the promoter are dislodged, and p65 is allowed to bind to its consensus sequence in the MnSOD promoter. Thus, binding of NFκB with concomitant removal of AP-1 induces MnSOD gene expression by Trx.
Given the fact that Fra-1, JunB, and c-Jun proteins form homo- or heterodimers of the AP-1 transcription factor, down-regulation of these proteins should increase the expression of MnSOD in unstimulated cells if AP-1 acts as a repressor of MnSOD expression. As demonstrated in Fig. 6F, down-regulation of each of the AP-1 component proteins up-regulated the MnSOD expression in unstimulated ECs, demonstrating that constitutive levels of Fra-1, JunB, or c-Jun are required for repression of MnSOD expression in unstimulated ECs. Because these proteins constitute the AP-1 transcription factor in various combinations, AP-1 transcription factor functions as a repressor of MnSOD in unstimulated ECs. Based on our experimental findings, we propose a model of MnSOD induction by sequential occupation of AP-1 and NFκB in MnSOD promoter as shown in Fig. 6G.
Discussion
Although it is well established that cells respond to internal or external cues by signal transduction via MAPKs, direct activation of MAPKs by modulation of the cellular redox state has not been demonstrated. Our report shows a direct connection between cellular redox state and MAPK activation resulting in the expression of a gene relevant to detoxification of superoxide radicals in the mitochondria of endothelial cells. Mitochondrial oxidative stress is a key mechanism in the onset and propagation of cardiovascular diseases as loss of MnSOD results in dilated cardiomyopathy and limits the survival of mice to 2 weeks (5). Increased mitochondrial oxidative stress also occurs due to an activated renin-angiotensin system that impacts heart and the vascular system, and it has also been implicated in hypertension (41, 42). This study shows a redox regulatory mechanism in MKK4 activation resulting in MnSOD activation in ECs. Trx directly activates MKK4 via Cys-246 and Cys-266 residues without the activation of an upstream MAP3K. Furthermore, the oxidation state of MKK4 is critically important for its activation by upstream kinases. We propose this mechanism of Trx-mediated MKK4 activation based on our following findings. 1) Overexpression of hTrx in ECs induces MKK4 activation (Fig. 3A). 2) MKK4 activation by Trx is independent of upstream kinase MEKK1 (Fig. 3, B and C). 3) Mutation of Cys-246 or Cys-266 abolished Trx-induced MKK4 activation (Fig. 4B), 4) Reduced Trx, but not its oxidized form, induced MKK4 autophosphorylation in vitro, and this activation was abrogated in C246A or C246A mutants (Fig. 4E). 5) Trx-induced MKK4 kinase activity was abolished due to C246A or C246A mutation (Fig. 4G). Furthermore, physiological activators of MAP3Ks such as TNFα failed to activate mutant MKK4 (C246A and C266A; Fig. 4C), suggesting that these residues control MKK4 activation by upstream kinases. Thus, even if the upstream kinases of MKK4 are activated, they may fail to activate MKK4 if Cys-246 and Cys-266 are in an oxidized state. In support of this notion, we showed that TNFα or IL-1 failed to activate MKK4 when ECs are treated with oxidant H2O2. This inactivation of MKK4 was also reflected in decreased MnSOD expression by TNFα or IL-1β in the presence of H2O2.
MKK4 is known to undergo low levels of autophosphorylation in the absence of MEKK due to phosphorylation on Ser-221 because mutation of this residue greatly attenuated MKK4 phosphorylation (31). Furthermore, the Thr-225 residue is required for phosphorylation of MKK4 by MEKK2 and MEKK3 (31). Mutation of both of these residues abrogates MKK4 phosphorylation by MEKK2 or MEKK3. Furthermore, Thr-261 and Ser-257 are phosphorylated by MEKK1. Our studies show that MKK4 mutants (C246A or C266A) are unable to undergo phosphorylation on Thr-261/Ser-257 in the presence of Trx or TNFα, demonstrating that Cys-246 and Cys-266 are necessary for phosphorylation of MKK4 on Thr-261. Furthermore, oxidizing agents such as H2O2 inhibited MKK4 activation induced by TNFα. On the contrary, Trx potentiated MKK4 phosphorylation by TNFα. These data demonstrate that the cellular redox state could directly control a specific MAPKK signaling mechanism resulting in the induction or inhibition of gene expression (Fig. 7).
FIGURE 7.
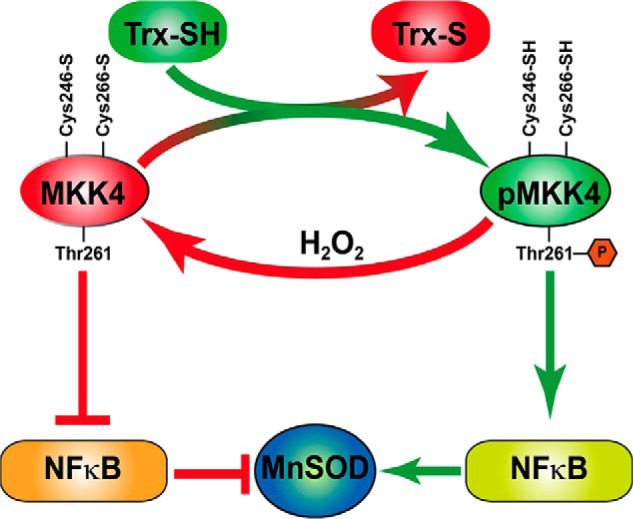
Redox regulation of MKK4 and MnSOD expression in endothelial cells. H2O2 oxidizes MKK4 on Cys-246 and Cys-266 resulting in its inactivation in HMVEC. This inactivation inhibits NFκB and MnSOD expression. Treatment of HMVEC with Trx-SH reduces Cys-246 and Cys-266 that activate MKK4 due to phosphorylation of Thr-261 resulting in NFκB activation and subsequent induction of MnSOD expression.
Given that a variety of unrelated agents induce MnSOD (3, 4, 7–14, 43), it remains enigmatic as to how such diverse agents induce a protein with the specific function of keeping the mitochondria functional by removal of excess superoxide radical. This study provides a mechanism that may explain the control of this gene by MAPKs. Based on the findings in this study, we propose that MnSOD is activated by either removal of AP-1 component proteins due to inhibition of JNK signaling or by activation of MKK4 signaling inducing NFκB activation (Fig. 6G). Additionally, substances that induce both MKK7 and MKK4 signaling, a balance of activation leading to increased MKK4 would result in higher expression of MnSOD, and the balance shifting to higher MKK7 signaling would favor suppression of MnSOD induction in endothelial cells. In our previous report, we have demonstrated that Trx induces both MKK4 and MKK7 activation in A549 adenocarcinoma cells and AP-1 transcription factors after prolonged incubation (24–48 h) of cells with Trx (44). Although adenocarcinoma cells and endothelial cells do significantly differ in their physiology and signal transduction mechanisms, it is likely that sequential removal of NFκB and entry of AP-1 might occur in the MnSOD promoter at prolonged incubations with Trx. Simultaneous activation of JNK and NFκB has been reported (45). Activation of NFκB induces the expression of Gadd45β, which binds to MKK7 and inhibits its activity (46, 47) resulting in inhibition of the JNK cascade (46). Inhibition of MKK7 would inhibit JNK in our endothelial system. In addition to Gadd45β, studies have demonstrated that X chromosome-linked inhibitor of apoptosis is also an NFκB-responsive (48, 49) gene that inactivates JNK via MKK7 inhibition (50). Therefore, NFκB-dependent down-regulation of JNK activity could decrease AP-1 DNA binding. Based on this experimental evidence, Trx-mediated NFκB activation could inhibit JNK, resulting in down-regulation of AP-1 activation. Thus, inhibition of Trx-induced MKK4 activation would decrease ΝFκΒ activation resulting in increased AP-1 DNA binding.
Induction of a powerful mitochondrial antioxidant enzyme such as MnSOD by the reducing agent Trx raises the logical question as to why a reducing agent would induce an antioxidant enzyme, as it may be more advantageous for the cell to induce MnSOD due to oxidative stress imposed by H2O2. MnSOD cannot neutralize H2O2; rather it produces H2O2 in the mitochondria due to dismutation of superoxide anion. We and others have shown that that H2O2 does not induce MnSOD in a variety of cell types (3, 4, 11, 51, 52). However, one explanation for induction of MnSOD by a reducing agent could be the redox compartmentalization of the cellular environment. For example, the redox state of mitochondria may be oxidized, but the redox state of cytosol may remain in a reduced state. Mitochondrially produced H2O2 acts as a signaling molecule in the normal physiological state of the cell, and excess H2O2 produced by abnormal oxidative activity in the mitochondria is detrimental, as it may induce apoptosis. Thus, physiological levels of H2O2 may not oxidize MKK4, but pathological levels of H2O2 would oxidize MKK4 cysteines and render it nonfunctional. This is important because in overwhelming oxidative stress conditions, cells would die by apoptosis, as it cannot repair itself. Therefore, in the presence of pathological levels of H2O2 in the cytosol, it will be less useful for the cell to produce more MnSOD by keeping MKK4 functional (see Fig. 8).
FIGURE 8.
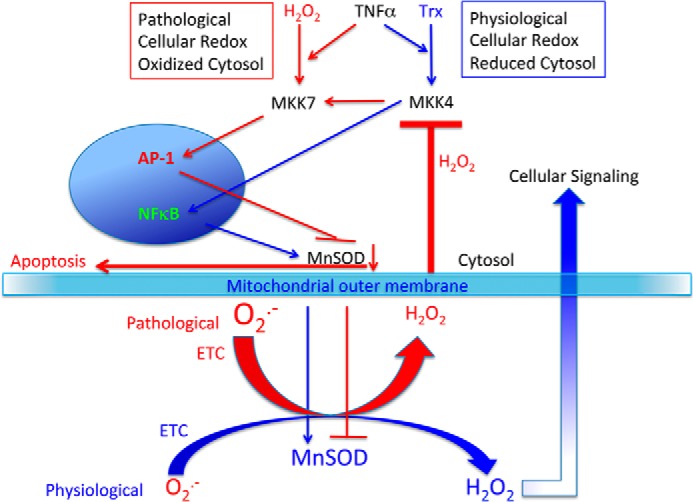
Redox signaling in ECs and role of MnSOD. The figure shows a role of cellular redox modulation by relative levels of physiological H2O2 and pathological H2O2 in the cell and the pivotal role of MnSOD in the cell survival via MKK4 redox control in ECs. Blue arrows, survival pathways; red arrows, apoptotic pathways.
In our studies, we used pharmacological levels of Trx to activate MnSOD via activation of MKK4. High levels of reduced Trx are expected to outcompete the oxidized Trx and would shift the balance to a reduced state, as recently reported in our publication (53). Thus, Trx could be used as a pharmacological agent to decrease oxidant-related diseases by improving cellular redox conditions and induction of pro-survival genes such as MnSOD. The ability of Trx to be internalized by cells is specifically useful for therapeutic application of Trx. We have previously reported that Trx is internalized into A549 and MCF-7 cells (4, 54). Furthermore, injected Trx has been shown to be internalized by cardiomyocytes and brain cells (40, 55, 56). As of today, no cell surface receptor has been discovered for Trx. Thus, it appears that Trx is permeable to cells by a simple diffusion process or due to some assisted transport. Further studies are required to conclusively establish the mechanism of Trx transport across the cell membrane. Trx has been shown to be secreted by a leaderless pathway bypassing the usual endoplasmic reticulum-Golgi-mediated secretion (5). Thus, it is plausible that this secretory protein could move in and out of the cells due to unrestricted diffusion.
Because MKK4 catalytic domain contains 11 subdomains that are found in other protein kinases (21), it is likely that redox-sensitive cysteine residues in their catalytic domains may control their activity. Thus, identification of these cysteine residues in the kinase domains may provide critical understanding of specific MAPK activation in the context of gene expression. The signaling events leading to positive and negative regulation of MnSOD resulting in the final decision of the fate of the cell either to undergo apoptosis or to survive are physiologically significant. For example, a net increase in MnSOD expression with activation of both MKK4 or MKK7 could possibly favor survival over cell death. On the contrary, a net decrease in MnSOD expression due to positive or negative signaling could induce apoptosis.
Acknowledgments
We gratefully acknowledge the generous contribution of expression plasmids by Dr. Rodger Davis (University of Massachusetts, Worcester, MA), Dr. Thomas Maniatis (Harvard University, Cambridge, MA), and Dr. Gary L. Johnson (University of North Carolina, Chapel Hills, NC). Technical assistance of Harish Muniyappa is gratefully acknowledged.
This work was supported, in whole or in part, by National Institutes of Health Grants HL 1R01HL107885 and 1R01HL109397 (to K. C. D.). The authors declare that they have no conflicts of interest with the contents of this article.
- MnSOD
- manganese superoxide dismutase
- Trx
- thioredoxin
- ANOVA
- analysis of variance
- HMVEC
- human microvascular endothelial cell
- hTrx
- human Trx
- PLA
- proximity ligation assay
- EC
- endothelial cell
- TrxR
- thioredoxin reductase
- NT
- nontargeting.
References
- 1. Davidson S. M. (2010) Endothelial mitochondria and heart disease. Cardiovasc. Res. 88, 58–66 [DOI] [PubMed] [Google Scholar]
- 2. Suzuki K., Tatsumi H., Satoh S., Senda T., Nakata T., Fujii J., Taniguchi N. (1993) Manganese-superoxide dismutase in endothelial cells: localization and mechanism of induction. Am. J. Physiol. 265, H1173–H1178 [DOI] [PubMed] [Google Scholar]
- 3. Das K. C., Lewis-Molock Y., White C. W. (1995) Thiol modulation of TNFα and IL-1 induced MnSOD gene expression and activation of NF-κB. Mol. Cell. Biochem. 148, 45–57 [DOI] [PubMed] [Google Scholar]
- 4. Das K. C., Lewis-Molock Y., White C. W. (1997) Elevation of manganese superoxide dismutase gene expression by thioredoxin. Am. J. Respir. Cell Mol. Biol. 17, 713–726 [DOI] [PubMed] [Google Scholar]
- 5. Li Y., Huang T. T., Carlson E. J., Melov S., Ursell P. C., Olson J. L., Noble L. J., Yoshimura M. P., Berger C., Chan P. H., Wallace D. C., Epstein C. J. (1995) Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 11, 376–381 [DOI] [PubMed] [Google Scholar]
- 6. Abid M. R., Tsai J. C., Spokes K. C., Deshpande S. S., Irani K., Aird W. C. (2001) Vascular endothelial growth factor induces manganese-superoxide dismutase expression in endothelial cells by a Rac1-regulated NADPH oxidase-dependent mechanism. FASEB J. 15, 2548–2550 [DOI] [PubMed] [Google Scholar]
- 7. Bianchi A., Bécuwe P., Franck P., Dauça M. (2002) Induction of MnSOD gene by arachidonic acid is mediated by reactive oxygen species and p38 MAPK signaling pathway in human HepG2 hepatoma cells. Free Radic. Biol. Med. 32, 1132–1142 [DOI] [PubMed] [Google Scholar]
- 8. Clerch L. B., Wright A. E., Coalson J. J. (1996) Lung manganese superoxide dismutase protein expression increases in the baboon model of bronchopulmonary dysplasia and is regulated at a posttranscriptional level. Pediatr. Res. 39, 253–258 [DOI] [PubMed] [Google Scholar]
- 9. Darville M. I., Ho Y. S., Eizirik D. L. (2000) NF-κB is required for cytokine-induced manganese superoxide dismutase expression in insulin-producing cells. Endocrinology 141, 153–162 [DOI] [PubMed] [Google Scholar]
- 10. Das K. C., Guo X. L., White C. W. (1998) Protein kinase Cδ-dependent induction of manganese superoxide dismutase gene expression by microtubule-active anticancer drugs. J. Biol. Chem. 273, 34639–34645 [DOI] [PubMed] [Google Scholar]
- 11. Das K. C., Lewis-Molock Y., White C. W. (1995) Activation of NF-κB and elevation of MnSOD gene expression by thiol reducing agents in lung adenocarcinoma (A549) cells. Am. J. Physiol. 269, L588–L602 [DOI] [PubMed] [Google Scholar]
- 12. Delhalle S., Deregowski V., Benoit V., Merville M. P., Bours V. (2002) NF-κB-dependent MnSOD expression protects adenocarcinoma cells from TNF-α-induced apoptosis. Oncogene 21, 3917–3924 [DOI] [PubMed] [Google Scholar]
- 13. Kinscherf R., Deigner H. P., Usinger C., Pill J., Wagner M., Kamencic H., Hou D., Chen M., Schmiedt W., Schrader M., Kovacs G., Kato K., Metz J. (1997) Induction of mitochondrial manganese superoxide dismutase in macrophages by oxidized LDL: its relevance in atherosclerosis of humans and heritable hyperlipidemic rabbits. FASEB J. 11, 1317–1328 [DOI] [PubMed] [Google Scholar]
- 14. Xu Y., Kiningham K. K., Devalaraja M. N., Yeh C. C., Majima H., Kasarskis E. J., St Clair D. K. (1999) An intronic NF-κB element is essential for induction of the human manganese superoxide dismutase gene by tumor necrosis factor-α and interleukin-1β. DNA Cell Biol. 18, 709–722 [DOI] [PubMed] [Google Scholar]
- 15. Laurent T. C., Moore E. C., Reichard P. (1964) Enzymatic synthesis of deoxyribonucleotides. Iv. isolation and characterization of thioredoxin, the hydrogen donor from Escherichia coli B. J. Biol. Chem. 239, 3436–3444 [PubMed] [Google Scholar]
- 16. Holmgren A. (1985) Thioredoxin. Annu. Rev. Biochem. 54, 237–271 [DOI] [PubMed] [Google Scholar]
- 17. Holmgren A., Björnstedt M. (1995) Thioredoxin and thioredoxin reductase. Methods Enzymol. 252, 199–208 [DOI] [PubMed] [Google Scholar]
- 18. Das K. C. (2001) c-Jun NH2-terminal kinase-mediated redox-dependent degradation of IκB: role of thioredoxin in NF-κB activation. J. Biol. Chem. 276, 4662–4670 [DOI] [PubMed] [Google Scholar]
- 19. Muniyappa H., Song S., Mathews C. K., Das K. C. (2009) Reactive oxygen species-independent oxidation of thioredoxin in hypoxia: inactivation of ribonucleotide reductase and redox-mediated checkpoint control. J. Biol. Chem. 284, 17069–17081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davis R. J. (2000) Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 [DOI] [PubMed] [Google Scholar]
- 21. Cuenda A. (2000) Mitogen-activated protein kinase kinase 4 (MKK4). Int. J. Biochem. Cell Biol. 32, 581–587 [DOI] [PubMed] [Google Scholar]
- 22. Haeusgen W., Herdegen T., Waetzig V. (2011) The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur. J. Cell Biol. 90, 536–544 [DOI] [PubMed] [Google Scholar]
- 23. Davies C., Tournier C. (2012) Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem. Soc. Trans. 40, 85–89 [DOI] [PubMed] [Google Scholar]
- 24. Wada T., Penninger J. M. (2004) Mitogen-activated protein kinases in apoptosis regulation. Oncogene 23, 2838–2849 [DOI] [PubMed] [Google Scholar]
- 25. Wang X., Destrument A., Tournier C. (2007) Physiological roles of MKK4 and MKK7: insights from animal models. Biochim. Biophys. Acta 1773, 1349–1357 [DOI] [PubMed] [Google Scholar]
- 26. Wang T. H., Wang H. S., Ichijo H., Giannakakou P., Foster J. S., Fojo T., Wimalasena J. (1998) Microtubule-interfering agents activate c-Jun N-terminal kinase/stress-activated protein kinase through both Ras and apoptosis signal-regulating kinase pathways. J. Biol. Chem. 273, 4928–4936 [DOI] [PubMed] [Google Scholar]
- 27. Johnson N. L., Gardner A. M., Diener K. M., Lange-Carter C. A., Gleavy J., Jarpe M. B., Minden A., Karin M., Zon L. I., Johnson G. L. (1996) Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase kinase induce cell death. J. Biol. Chem. 271, 3229–3237 [DOI] [PubMed] [Google Scholar]
- 28. Widmann C., Johnson N. L., Gardner A. M., Smith R. J., Johnson G. L. (1997) Potentiation of apoptosis by low dose stress stimuli in cells expressing activated MEK kinase 1. Oncogene 15, 2439–2447 [DOI] [PubMed] [Google Scholar]
- 29. Widmann C., Gerwins P., Johnson N. L., Jarpe M. B., Johnson G. L. (1998) MEK kinase 1, a substrate for DEVD-directed caspases, is involved in genotoxin-induced apoptosis. Mol. Cell. Biol. 18, 2416–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim D. H., Behlke M. A., Rose S. D., Chang M. S., Choi S., Rossi J. J. (2005) Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotechnol. 23, 222–226 [DOI] [PubMed] [Google Scholar]
- 31. Deacon K., Blank J. L. (1997) Characterization of the mitogen-activated protein kinase kinase 4 (MKK4)/c-Jun NH2-terminal kinase 1 and MKK3/p38 pathways regulated by MEK kinases 2 and 3. MEK kinase 3 activates MKK3 but does not cause activation of p38 kinase in vivo. J. Biol. Chem. 272, 14489–14496 [DOI] [PubMed] [Google Scholar]
- 32. Weibrecht I., Leuchowius K. J., Clausson C. M., Conze T., Jarvius M., Howell W. M., Kamali-Moghaddam M., Söderberg O. (2010) Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev. Proteomics 7, 401–409 [DOI] [PubMed] [Google Scholar]
- 33. Bunse L., Schumacher T., Sahm F., Pusch S., Oezen I., Rauschenbach K., Gonzalez M., Solecki G., Osswald M., Capper D., Wiestler B., Winkler F., Herold-Mende C., von Deimling A., Wick W., Platten M. (2015) Proximity ligation assay evaluates IDH1R132H presentation in gliomas. J. Clin. Invest. 125, 593–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pacchiana R., Abbate M., Armato U., Dal Prà I., Chiarini A. (2014) Combining immunofluorescence with in situ proximity ligation assay: a novel imaging approach to monitor protein-protein interactions in relation to subcellular localization. Histochem. Cell Biol. 142, 593–600 [DOI] [PubMed] [Google Scholar]
- 35. Brandt B., Abou-Eladab E. F., Tiedge M., Walzel H. (2010) Role of the JNK/c-Jun/AP-1 signaling pathway in galectin-1-induced T-cell death. Cell Death Dis. 1, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cross J. V., Templeton D. J. (2004) Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem. J. 381, 675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee F. S., Peters R. T., Dang L. C., Maniatis T. (1998) MEKK1 activates both IκB kinase α and IκB kinase β. Proc. Natl. Acad. Sci. U.S.A. 95, 9319–9324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Visner G. A., Chesrown S. E., Monnier J., Ryan U. S., Nick H. S. (1992) Regulation of manganese superoxide dismutase: IL-1 and TNF induction in pulmonary artery and microvascular endothelial cells. Biochem. Biophys. Res. Commun. 188, 453–462 [DOI] [PubMed] [Google Scholar]
- 39. Xia D., Srinivas H., Ahn Y. H., Sethi G., Sheng X., Yung W. K., Xia Q., Chiao P. J., Kim H., Brown P. H., Wistuba I. I., Aggarwal B. B., Kurie J. M. (2007) Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NFκB-dependent pathway. J. Biol. Chem. 282, 3507–3519 [DOI] [PubMed] [Google Scholar]
- 40. Wang B., Tian S., Wang J., Han F., Zhao L., Wang R., Ning W., Chen W., Qu Y. (2015) Intraperitoneal administration of thioredoxin decreases brain damage from ischemic stroke. Brain Res. 10.1016/j.brainres.2015.04.033 [DOI] [PubMed] [Google Scholar]
- 41. Wen H., Gwathmey J. K., Xie L. H. (2012) Oxidative stress-mediated effects of angiotensin II in the cardiovascular system. World J. Hypertens. 2, 34–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee D. Y., Wauquier F., Eid A. A., Roman L. J., Ghosh-Choudhury G., Khazim K., Block K., Gorin Y. (2013) Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: role of mitochondrial reactive oxygen species. J. Biol. Chem. 288, 28668–28686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Abid M. R., Schoots I. G., Spokes K. C., Wu S. Q., Mawhinney C., Aird W. C. (2004) Vascular endothelial growth factor-mediated induction of manganese superoxide dismutase occurs through redox-dependent regulation of forkhead and IκB/NF-κB. J. Biol. Chem. 279, 44030–44038 [DOI] [PubMed] [Google Scholar]
- 44. Das K. C., Muniyappa H. (2010) c-Jun-NH2 terminal kinase (JNK)-mediates AP-1 activation by thioredoxin: phosphorylation of c-Jun, JunB, and Fra-1. Mol. Cell. Biochem. 337, 53–63 [DOI] [PubMed] [Google Scholar]
- 45. Luo J. L., Kamata H., Karin M. (2005) IKK/NF-κB signaling: balancing life and death–a new approach to cancer therapy. J. Clin. Invest. 115, 2625–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Papa S., Zazzeroni F., Bubici C., Jayawardena S., Alvarez K., Matsuda S., Nguyen D. U., Pham C. G., Nelsbach A. H., Melis T., De Smaele E., Tang W. J., D'Adamio L., Franzoso G. (2004) Gadd45β mediates the NF-κB suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 6, 146–153 [DOI] [PubMed] [Google Scholar]
- 47. Papa S., Zazzeroni F., Pham C. G., Bubici C., Franzoso G. (2004) Linking JNK signaling to NF-κB: a key to survival. J. Cell Sci. 117, 5197–5208 [DOI] [PubMed] [Google Scholar]
- 48. Sanna M. G., da Silva Correia J., Ducrey O., Lee J., Nomoto K., Schrantz N., Deveraux Q. L., Ulevitch R. J. (2002) IAP suppression of apoptosis involves distinct mechanisms: the TAK1/JNK1 signaling cascade and caspase inhibition. Mol. Cell. Biol. 22, 1754–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stehlik C., de Martin R., Kumabashiri I., Schmid J. A., Binder B. R., Lipp J. (1998) Nuclear factor (NF)-κB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor α-induced apoptosis. J. Exp. Med. 188, 211–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaur S., Wang F., Venkatraman M., Arsura M. (2005) X-linked inhibitor of apoptosis (XIAP) inhibits c-Jun N-terminal kinase 1 (JNK1) activation by transforming growth factor β1 (TGF-β1) through ubiquitin-mediated proteosomal degradation of the TGF-β1-activated kinase 1 (TAK1). J. Biol. Chem. 280, 38599–38608 [DOI] [PubMed] [Google Scholar]
- 51. Wong G. H., Elwell J. H., Oberley L. W., Goeddel D. V. (1989) Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell 58, 923–931 [DOI] [PubMed] [Google Scholar]
- 52. Wong G. H., Goeddel D. V. (1988) Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science 242, 941–944 [DOI] [PubMed] [Google Scholar]
- 53. Das K. C. (2015) Thioredoxin-deficient mice, a novel phenotype sensitive to ambient air and hypersensitive to hyperoxia-induced lung injury. Am. J. Physiol. Lung. Cell. Mol. Physiol. 308, L429–L442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ravi D., Das K. C. (2004) Redox-cycling of anthracyclines by thioredoxin system: increased superoxide generation and DNA damage. Cancer Chemother. Pharmacol. 54, 449–458 [DOI] [PubMed] [Google Scholar]
- 55. Tao L., Gao E., Bryan N. S., Qu Y., Liu H. R., Hu A., Christopher T. A., Lopez B. L., Yodoi J., Koch W. J., Feelisch M., Ma X. L. (2004) Cardioprotective effects of thioredoxin in myocardial ischemia and reperfusion: role of S-nitrosation [corrected]. Proc. Natl. Acad. Sci. U.S.A. 101, 11471–11476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tao L., Gao E., Hu A., Coletti C., Wang Y., Christopher T. A., Lopez B. L., Koch W., Ma X. L. (2006) Thioredoxin reduces post-ischemic myocardial apoptosis by reducing oxidative/nitrative stress. Br. J. Pharmacol. 149, 311–318 [DOI] [PMC free article] [PubMed] [Google Scholar]