

Background: The overexpression of phospholipase A/acyltransferase-3 (PLA/AT-3) in mammalian cells causes specific disappearance of peroxisomes.
Results: PLA/AT-3 bound to Pex19p, one of peroxins, and inhibited the binding of Pex19p to peroxisomal membrane proteins.
Conclusion: The interaction between Pex19p and PLA/AT-3 may be related to the down-regulation of peroxisomes by PLA/AT-3.
Significance: PLA/AT-3 may be involved in a novel regulatory mechanism for peroxisome biogenesis.
Keywords: glycerophospholipid, organelle, peroxisome, phospholipase A, tumor suppressor gene, H-rev107, HRAS-like suppressor, peroxin, phospholipase A/acyltransferase (PLA/AT)
Abstract
Phospholipase A/acyltransferase (PLA/AT)-3 (also known as H-rev107 or AdPLA) was originally isolated as a tumor suppressor and was later shown to have phospholipase A1/A2 activity. We have also found that the overexpression of PLA/AT-3 in mammalian cells results in specific disappearance of peroxisomes. However, its molecular mechanism remained unclear. In the present study, we first established a HEK293 cell line, which stably expresses a fluorescent peroxisome marker protein (DsRed2-Peroxi) and expresses PLA/AT-3 in a tetracycline-dependent manner. The treatment with tetracycline, as expected, caused disappearance of peroxisomes within 24 h, as revealed by diffuse signals of DsRed2-Peroxi and a remarkable decrease in a peroxisomal membrane protein, PMP70. A time-dependent decrease in ether-type lipid levels was also seen. Because the activation of LC3, a marker of autophagy, was not observed, the involvement of autophagy was unlikely. Among various peroxins responsible for peroxisome biogenesis, Pex19p functions as a chaperone protein for the transportation of peroxisomal membrane proteins. Immunoprecipitation analysis showed that PLA/AT-3 binds to Pex19p through its N-terminal proline-rich and C-terminal hydrophobic domains. The protein level and enzyme activity of PLA/AT-3 were increased by its coexpression with Pex19p. Moreover, PLA/AT-3 inhibited the binding of Pex19 to peroxisomal membrane proteins, such as Pex3p and Pex11βp. A catalytically inactive point mutant of PLA/AT-3 could bind to Pex19p but did not inhibit the chaperone activity of Pex19p. Altogether, these results suggest a novel regulatory mechanism for peroxisome biogenesis through the interaction between Pex19p and PLA/AT-3.
Introduction
Peroxisomes are single membrane-surrounded organelles present in virtually all eukaryotic cells and are involved in a wide variety of metabolic events, including scavenging of peroxides and reactive oxygen species, the catabolism of very long chain fatty acids and branched chain fatty acids, and the biosynthesis of all ether-type lipid precursors (1–5). The physiological importance of peroxisomes has been confirmed by the analysis of a number of inherited diseases exhibiting peroxisome dysfunctions (6, 7). Peroxins, encoded by the PEX genes, are a series of proteins responsible for the biogenesis of peroxisomes, and their defects lead to the dysfunction of peroxisomes. So far, 31 peroxins have been reported, and they are involved in the generation and division of peroxisomes as well as the import of peroxisomal proteins (4, 8). In humans, 14 peroxins have been identified and shown to link to peroxisome biogenesis disorders.
As a machinery of peroxisome biogenesis, it is well known that nascent peroxisomal matrix proteins are transported into peroxisomes with the aid of several peroxins, which recognize peroxisomal targeting signals (PTSs),2 PTS1 and PTS2, of peroxisomal matrix proteins (2, 9). However, it is poorly understood how peroxisome membrane structure is formed and how peroxisomal membrane proteins (PMPs) are transported into peroxisomes. In addition, because peroxisomes lack the phospholipid-synthesizing enzymes necessary for the formation of peroxisome membrane structure, phospholipids must be trafficked and supplied to peroxisomes from other organelles, such as the ER. Pex3p, Pex16p, and Pex19p have been identified as peroxins indispensable for peroxisome membrane assembly and PMP transport, and the cells deficient in these proteins are devoid of peroxisome structure itself (8, 9). Pex19p is predominantly localized to cytoplasm and binds to various PMPs, whereas Pex3p and Pex16p are associated with peroxisomal membrane and function as the membrane-anchoring site for Pex19p·PMP complexes and as the receptor for Pex3p·Pex19p complex, respectively (10).
The abundance of peroxisomes is remarkably affected by the nutritional environment and specific conditions (11). For example, peroxisomes can be induced and proliferated in yeast cultured in methanol as a sole carbon source and in rodents treated with peroxisome proliferators. Increased peroxisomes are rapidly decreased and degraded by altering the environment or by withdrawing peroxisome proliferators (12). These results indicate that peroxisome levels are reciprocally regulated by the balance between their biogenesis and degradation. Recently, it has been reported that the selective autophagy of peroxisomes, pexophagy, contributes to the maintenance of quality and quantity of peroxisomes (11, 13).
The HRAS-like suppressor (HRASLS) family, consisting of five members (HRASLS1 to 5), was originally isolated as tumor suppressors negatively regulating the oncogene H-Ras (14, 15). It has been reported that these proteins are related to various diseases, such as cancers (16, 17), obesity (18, 19), and Poland syndrome, a rare disorder characterized by hypoplasia/aplasia of the pectoralis major muscle (20). We and others have demonstrated that all of these members function as enzymes with phospholipase A1/2 (PLA1/2) and phospholipid acyltransferase activities (21–26). Thus, we proposed to rename HRASLS1 to 5 as phospholipase/acyltransferase-1 to -5 (PLA/AT-1 to -5), respectively (26). Unexpectedly, we found that the overexpression of PLA/AT-3 (HRASLS3, H-rev107, or AdPLA) or PLA/AT-2 (HRASLS2) in mammalian cells results in the disappearance of peroxisome membrane structure and the dysfunction of peroxisomes, as revealed by a remarkable decrease in the intracellular levels of ether-type lipids (27, 28). The disappearance of peroxisomes was dependent on the enzyme activity of PLA/AT-3 because the mutant devoid of enzyme activity failed to affect peroxisome functions (27). However, the mechanism by which PLA/AT-3 and -2 regulate the abundance of peroxisomes remained unclear.
Because the phenotypes appearing in cells by the overexpression of PLA/AT-3 or -2 were similar to those in Pex3p-, Pex16p-, or Pex19p-deficient cells, we presumed that the negative regulation of peroxisomes by these PLA/AT proteins is mediated by these peroxins. In this study, we show that PLA/AT-3 is a novel Pex19p-binding protein and suppresses the binding of Pex19p to PMPs. The enzyme activity of PLA/AT-3 was unrelated to its binding to Pex19p but was required to suppress the interaction of Pex19p with PMPs. These results suggest the presence of a novel regulatory mechanism for peroxisome biogenesis through the interaction between Pex19p and PLA/AT-3.
Experimental Procedures
Materials
1,2-[1-14C]Dipalmitoyl-phosphatidylcholine (PC) was purchased from PerkinElmer Life Sciences. [1-14C]Hexadecanol was from Moravek Biochemicals (Brea, CA). Horseradish peroxidase-linked anti-mouse IgG, horseradish peroxidase-linked anti-rabbit IgG, Hybond P, an ECL Plus kit, and Protein A-Sepharose CL-4B were from GE Healthcare. 1,2-Dipalmitoyl-PC, 1-O-palmityl-2,3-dipalmitoyl-rac-glycerol, 1-hexadecanol, anti-70-kDa peroxisomal membrane protein (PMP70) monoclonal antibody, anti-FLAG monoclonal antibody M2, and anti-FLAG M2-conjugated agarose were from Sigma. Dulbecco's modified Eagle's medium, Alexa 488-conjugated anti-mouse IgG, Alexa 594-conjugated anti-rabbit IgG, Lipofectamine 2000, Lipofectamine RNAiMAX, fetal calf serum, hygromycin B, blasticidin, Geneticin, pcDNA3.1(+), pcDNA3.1/Myc-His, pEF1/Myc-His, pEF6/Myc-His, pcDNA5/TO, pcDNA6/TR, pCMV/myc/nuc, small interfering RNAs (siRNAs) directed against human Pex19p, and a control siRNA were from Invitrogen. Antibodies against lamin A/C, HA tag, Myc tag, and DYKDDDDK tag were from Cell Signaling Technology (Danvers, MA). Antibodies against catalase and Pex19p were from Abcam Inc. (Cambridge, MA). pEGFP-C1, pEGFP-N1, pDsRed2-Peroxi vectors, and human MTC panel I were from Clontech. Anti-V5 antibody and anti-LC3 antibody were from Medical and Biological Laboratories (Nagoya, Japan). Permafluor was from Immunotech (Marseille, France). Triton X-100 and Nonidet P-40 were from Nacalai Tesque, Inc. (Kyoto, Japan). Ex Taq DNA polymerase and PrimeScript RT reagent kit were from TaKaRa Bio Inc. (Ohtsu, Japan). The RNeasy Mini Kit was from Qiagen. KOD-Plus-Neo DNA polymerase was from TOYOBO (Osaka, Japan). Normal goat serum was from Vector Laboratories (Burlingame, CA). Protease inhibitor mixture set III and precoated silica gel 60 F254 aluminum sheets (20 × 20 cm, 0.2 mm thick) for TLC were from Merck (Darmstadt, Germany). Protein assay dye reagent concentrate was from Bio-Rad. Mouse Hepa1-6 cells were purchased from the RIKEN BRC Cell Bank (Tsukuba, Japan). pEGFP-LC3 was obtained from Addgene (Cambridge, MA). pmCitrine-C1 is a kind gift from Dr. Joel A. Swanson (University of Michigan).
Construction of Expression Vectors
All of the primers used for this purpose are shown in supplemental Table 1. PCR was carried out with KOD-Plus-Neo DNA polymerase. The cDNAs encoding C-terminally HA-tagged Pex3p (Pex3p-HA), C-terminally V5-tagged Pex16p (Pex16p-V5), N-terminally Myc-tagged Pex19p (Myc-Pex19p), N-terminally HA-tagged Pex19p (HA-Pex19p), N-terminally Myc-tagged Pex11βp (Myc-Pex11βp), and C-terminally Myc-tagged alkyl-dihydroxyacetonephosphate synthase (ADHAPS-Myc) were amplified by PCR with the human brain cDNA (human MTC panel I) as a template, and the obtained DNA fragments were subcloned into the corresponding sites of pcDNA3.1/Myc-His (for Myc-Pex11βp), pcDNA3.1(+) (for HA-Pex19p and ADHAPS-Myc), or pEF1/Myc-His (for others). The cDNA encoding C-terminally Myc-tagged peroxisomal 3-ketoacyl-CoA thiolase (thiolase-Myc) was amplified by PCR with the cDNA prepared from mouse Hepa1-6 cells, and the obtained DNA fragment was subcloned into pcDNA3.1(+). In order to construct Pex19p containing two nuclear localization signals (NLSs) and a Myc tag at the C terminus (Pex19p-NLS-Myc), PCR was performed using pEF1/Myc-His vector harboring human Myc-Pex19p, and the obtained DNA fragment was subcloned into pCMV/myc/nuc. In order to construct Pex3p or Pex16p fused to the N terminus of EGFP (Pex3p-EGFP or Pex16p-EGFP), PCR was performed using pEF1/Myc-His vector harboring human Pex3p-HA or human Pex16p-V5, and the obtained DNA fragments were subcloned into pEGFP-N1. To construct Pex11βp or Pex19p fused to the C terminus of Citrine (Citrine-Pex11βp or Citrine-Pex19p), PCR was performed using pcDNA3.1/Myc-His vector harboring human Myc-Pex11βp or pEF1/Myc-His vector harboring human HA-Pex19p, and the obtained DNA fragments were subcloned into pmCitrine-C1.
In order to construct PLA/AT-3 (PLA/AT-3-FL) and PLA/AT-3-C113S (PLA/AT-3-C113S-FL), which have a C-terminal FLAG tag, PCR was performed using pEF1/Myc-His vector harboring N-terminally FLAG-tagged mouse PLA/AT-3 (FL-PLA/AT-3) or PLA/AT-3-C113S (FL-PLA/AT-3-C113S), respectively (27). The deletion mutants (PLA/AT-3-ΔN10-FL and PLA/AT-3-ΔC27-FL) were also constructed using the expression vector harboring mouse PLA/AT-3-FL. C-terminally FLAG-tagged PLA/AT-2 (PLA/AT-2-FL) was constructed using pEF1/Myc-His vector harboring N-terminally FLAG-tagged human PLA/AT-2 (28), and the obtained DNA fragment was subcloned into pEF6/Myc-His. The expression vector pEF6/Myc-His harboring C-terminally FLAG-tagged human PLA/AT-1 (PLA/AT-1-FL) was constructed as reported previously (26). To construct pcDNA5/TO vector harboring FL-PLA/AT-3, pEF1/Myc-His vector harboring FL-PLA/AT-3 (27) was digested with BamHI and NotI, and the resultant DNA fragment was subcloned into the corresponding sites of pcDNA5/TO. All constructs were sequenced in both directions using an ABI 3130 Genetic Analyzer (Invitrogen).
Cell Culture, Transfection, and RNA Interference
HEK293, HEK293T, and COS-7 cells were maintained in Dulbecco's modified Eagle's medium with 10% fetal calf serum at 37 °C in humidified air containing 5% CO2. Expression vectors or siRNAs were introduced into cells with Lipofectamine 2000 or Lipofectamine RNAiMAX according to the manufacturer's instructions, respectively. The final concentration of siRNA was 20 nm. Forty-eight hours after transfection, the cells were used for RT-PCR, PLA1/2 assay, and metabolic labeling with [14C]hexadecanol.
Establishment of FL-PLA/AT-3-Tet-on Cells Expressing DsRed2-Peroxi
HEK293 cells were transfected with pcDNA6/TR vector, expressing tetracycline repressor, by the use of Lipofectamine 2000. Cells were selected in the medium containing 10 μg/ml blasticidin. Clonal cell lines were isolated by colony lifting and maintained in the blasticidin-containing medium. The cell line expressing the highest level of tetracycline repressor was transfected with DsRed2-Peroxi vector by the use of Lipofectamine 2000. Cells were selected in the medium containing 1 mg/ml Geneticin and 6 μg/ml blasticidin. Clonal cell lines were isolated by colony lifting and maintained in the medium containing Geneticin and blasticidin. One of the obtained clones, named DsRed2-PTS1 cell, was transfected with pcDNA5/TO vector, expressing FL-PLA/AT-3, by the use of Lipofectamine 2000. Cells were selected in the medium containing 200 μg/ml hygromycin, 600 μg/ml Geneticin, and 6 μg/ml blasticidin. Clonal cell lines were isolated by colony lifting and maintained in the medium containing hygromycin, Geneticin, and blasticidin. One of the obtained cell lines was named FL-PLA/AT-3-Tet-on and used throughout this study.
To measure PLA1/2 activity, the harvested cells were suspended in homogenization buffer (0.25 m sucrose, 1 mm EDTA, and 20 mm Tris-HCl, pH 7.4) and sonicated three times each for 3 s. The cell homogenates (30 μg of protein) were incubated with 200 μm 1,2-[1-14C]dipalmitoyl-PC (45,000 cpm) in 100 μl of 50 mm Tris-HCl (pH 8.0), 2 mm DTT, and 0.1% Nonidet P-40 at 37 °C for 30 min. The reaction was terminated with the addition of 320 μl of a mixture of chloroform/methanol (2:1, v/v) containing 5 mm 3(2)-t-butyl-4-hydroxyanisole. After centrifugation, 100 μl of the lower fraction was spotted on a silica gel thin layer plate (10-cm height) and developed at 4 °C for 25 min in chloroform/methanol/H2O (65:25:4, v/v/v). The distribution of radioactivity on the plate was quantified using a BAS1500 bioimaging analyzer (FUJIX Ltd., Tokyo, Japan). The protein concentration was determined by the method of Bradford with bovine serum albumin as a standard.
Western Blotting and Immunoprecipitation
For Western blotting unrelated to the immunoprecipitation assay, cells were homogenized in homogenization buffer by being passed through a 25-gauge syringe (for COS-7 cells) or a 27-gauge syringe (for HEK293 cells) (27), and nuclear fractions were prepared from the homogenates by centrifugation at 800 × g for 10 min at 4 °C. The resultant postnuclear supernatant (PNS) fractions were then centrifuged at 105,000 × g for 30 min at 4 °C to separate the cytosol (supernatant fractions) from cellular organelles (particulate fractions). For the immunoprecipitation assay, cells were lysed in ice-cold lysis buffer (0.25 m sucrose, 1 mm EDTA, 0.5% Triton X-100, and 20 mm Tris-HCl, pH 7.4) containing protease inhibitors for 1 h at 4 °C. After centrifugation at 18,000 × g for 10 min at 4 °C, the supernatant was used as cell lysate. The cell lysates were immunoprecipitated with the aid of anti-FLAG M2 antibody-conjugated agarose or antibody-conjugated Protein A-Sepharose CL-4B. Samples were separated by SDS-PAGE and electrotransferred to a hydrophobic polyvinylidene difluoride membrane (Hybond P). The membrane was blocked with PBS containing 5% dried milk and 0.1% Tween 20 (buffer A) and then incubated with primary antibodies in buffer A at room temperature for 1 h, followed by incubation with horseradish peroxidase-labeled secondary antibodies in buffer A at room temperature for 1 h. Proteins were finally treated with an ECL Plus kit and visualized with the aid of a LAS1000plus lumino-imaging analyzer (FUJIX Ltd.).
RT-PCR
Total RNAs were isolated from cells using an RNeasy Mini Kit. cDNAs were prepared from total RNAs using a PrimeScript RT reagent kit and subjected to PCR amplification by Ex Taq DNA polymerase. The primers used are shown in supplemental Table 2. The PCR conditions used were as follows: denaturation at 96 °C for 20 s, annealing at 60 °C for 20 s, and extension at 72 °C for 20 s (24 cycles for GAPDH, 28 cycles for others).
Metabolic Labeling
Cells were grown at 37 °C to 80% confluence in a 6-well plate containing Dulbecco's modified Eagle's medium with 10% fetal calf serum and were labeled with [1-14C]hexadecanol (1.6 μCi) for 18 h. Cells were then harvested and washed twice with PBS. The total lipids were extracted by the method of Bligh and Dyer (29), spotted on a silica gel thin layer plate (20-cm height), and developed at 4 °C for 1 h in hexane/diethyl ether/acetic acid (75:25:1, v/v/v). The distribution of radioactivity on the plate was quantified using a BAS1500 bioimaging analyzer.
LC-ESI/MS Analysis
The lipids of cultured cells were extracted by the method of Bligh and Dyer. Mass spectrometry (MS) analysis was performed as described previously by using a Quattro Micro tandem quadrupole mass spectrometer (Waters, Milford, MA) equipped with an electrospray ionization (ESI) source (27). Phospholipid molecular species were separated and analyzed using normal phase capillary-liquid chromatography (LC)-ESI/MS with a Deverosil Si60 silica column (150 × 0.3-mm inner diameter, 5-μm particle size; Nomura Chemicals, Nagoya, Japan). The samples were provided by an UltiMate high performance liquid chromatography system (Dionex Corp.) into the electrospray interface at a flow rate of 4 μl/min. For both positive and negative ionization, the mobile phase consisted of acetonitrile/methanol (2:1) containing 0.1% ammonium formate (pH 6.4) (solvent A) and methanol/water (2:1) containing 0.1% ammonium formate (pH 6.4) (solvent B). The mobile phase was started with 100% acetonitrile. After 5 min, the mobile phase was changed from 100% acetonitrile to a mixture of solvent A and B (90:10). The linear gradient reached 40% solvent A at 20 min. The mobile phase was then held constant for 15 min. Nitrogen was used as drying gas. Desolvation gas flow was 523 liters/h, and cone gas flow was maintained at 42 liters/h. Desolvation temperature was 150 °C, and source temperature was 90 °C. The capillary and cone voltages were set at 3.4–3.7 kV and 36–40 V, respectively. Argon was used as collision gas for collision-associated dissociation experiments at a pressure of 5 × 10−4 millibars. The collision energy was set to 40 eV.
LC-ESI-MS/MS Analysis
The LC-ESI-MS/MS analysis was performed on a Shimadzu Nexera ultrahigh performance liquid chromatography system (Shimadzu, Kyoto, Japan) coupled with a QTRAP 4500 hybrid triple quadrupole linear ion trap mass spectrometer (AB SCIEX, Framingham, MA). Chromatographic separation was performed on an Acquity UPLC HSS T3 column (100 mm × 2.1 mm, 1.8 μm; Waters) maintained at 40 °C using mobile phase A (water/acetonitrile (50:50, v/v) containing 10 mm ammonium acetate and 0.2% acetic acid) and mobile phase B (isopropanol/acetone (50:50, v/v)) in a gradient program (0–3 min, 30% B to 50% B; 3–24 min, 50% B to 90%; 24–28 min, 30% B) with a flow of 0.3 ml/min. The instrument parameters were as follows (arbitrary units if not specified): curtain gas = 10 p.s.i.; collision gas = 7; ion spray voltage = −4500 V; temperature = 700 °C; ion source gas 1 = 40 p.s.i.; ion source gas 2 = 60 p.s.i.; declustering potential = −60 V; entrance potential = −10 V; collision energy = −40 V. Product ion analysis in the negative ion mode was performed to determine the fatty acid composition of each ether-type ethanolamine phospholipid species.
Microscopy
For live cell imaging, cells were plated onto glass bottom plates (Greiner Bio-one) and transiently transfected with the expression vectors harboring Pex3p-EGFP, Pex16p-EGFP, Citrine-Pex11βp, Citrine-Pex19p, or EGFP-LC3. Twenty-four hours after transfection, the cells were placed in prewarmed Ringer's buffer (10 mm Hepes-NaOH (pH 7.2), 155 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 2 mm NaH2PO4, 10 mm d-glucose, and 0.5 mg/ml bovine serum albumin) on the thermo-controlled stage at 37 °C and observed with an LSM700 confocal laser microscope (Carl Zeiss).
For immunocytochemistry, cells were cultured on 18-mm glass coverslips containing Dulbecco's modified Eagle's medium with 10% fetal calf serum. The cells were then fixed with 4% paraformaldehyde in 0.1 m phosphate buffer (pH 7.4) for 15 min. The fixed cells were rinsed in PBS and treated with 0.25% NH4Cl in PBS for 10 min to quench free aldehyde groups. The cells were then washed with PBS and permeabilized with 0.15% Triton X-100 in PBS for 10 min. After treatment with a blocking buffer (5% normal goat serum in PBS) for 1 h, the cells were incubated with anti-FLAG antibody (1:1,000 dilution) or anti-Myc antibody (1:500 dilution) in 1% normal goat serum plus PBS for 1 h at room temperature. The cells were washed with PBS and labeled with Alexa 488-conjugated anti-mouse IgG (1:1,000 dilution) or Alexa 594-conjugated anti-rabbit IgG (1:1,000 dilution) in 1% normal goat serum plus PBS for 1 h. The specimen coverslips were mounted on glass slides using a mounting medium Permafluor and were observed with the LSM 700 confocal laser microscope.
Results
Peroxisomal Proteins Are Abrogated in PLA/AT-3-expressing Cells
We first examined the effect of PLA/AT-3 expression on the expression levels and intracellular localization of a peroxisomal membrane protein (PMP70) and three peroxisomal matrix proteins (catalase, ADHAPS), and peroxisomal 3-ketoacyl-CoA thiolase (thiolase)). As for PTS of these proteins, PMP70 possesses mPTS, catalase possesses PTS1, and ADHAPS and thiolase possess PTS2 (2, 9). In the assays shown in Fig. 1, control HEK293 cells and HEK293 cells stably expressing N-terminally FLAG-tagged PLA/AT-3 (FL-PLA/AT-3 cells) were homogenized and centrifuged at 800 × g. The resultant PNS was then fractionated into particulate and soluble fractions by ultracentrifugation at 105,000 × g. As analyzed by Western blotting, endogenous PMP70 and catalase in the control cells were mostly detected in the particulate fractions, where peroxisomes were expected to exist. By contrast, in FL-PLA/AT-3 cells, faint signals of PMP70 were seen in both particulate and soluble fractions, and catalase was mostly detected in soluble rather than particulate fractions. These effects of PLA/AT-3 expression were in agreement with our previous results (27). Moreover, when Myc-tagged ADHAPS or Myc-tagged thiolase was transiently expressed, the Myc signals of both proteins were detected primarily in the particulate fractions of the control cells and were much fainter in the FL-PLA/AT-3 cells. These results suggested that the overexpression of PLA/AT-3 affects the abundance or localization of the peroxisomal proteins with PTS2 as well as those with mPTS or PTS1.
FIGURE 1.
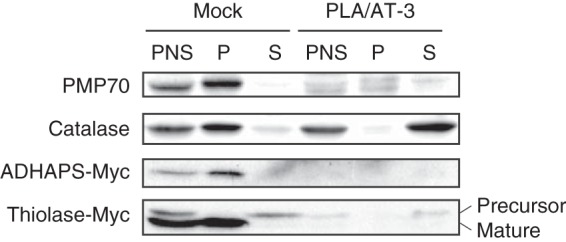
Analysis of peroxisomal proteins in PLA/AT-3-expressing cells by Western blotting. PNS and particulate (P) and soluble (S) fractions of control HEK293 cells (Mock) and HEK293 cells stably expressing FL-PLA/AT-3 (PLA/AT-3) were analyzed for endogenous PMP70 and catalase with their antibodies. Moreover, the cells were transiently transfected with ADHAPS-Myc-expressing vector or thiolase-Myc-expressing vector. Forty-eight hours after transfection, the cells were analyzed for recombinant ADHAPS and thiolase with anti-Myc antibody. The positions of the precursor and mature forms of thiolase are indicated.
Expression of PLA/AT-3 Rapidly Causes Disappearance of Peroxisomes
To cytochemically monitor peroxisomes, we established a HEK293 cell line named DsRed2-PTS1. In these cells, peroxisomes were fluorescently labeled with DsRed2-Peroxi (a fluorescent peroxisome marker protein possessing a PTS1 sequence at the C terminus) (30). Using DsRed2-PTS1 cells, we further established a Tet-on HEK293 cell line (FL-PLA/AT-3-Tet-on cells), which can express FL-PLA/AT-3 in a tetracycline-dependent manner (31). When FL-PLA/AT-3-Tet-on cells were observed by fluorescent confocal microscopy, peroxisomes, visualized by DsRed2-Peroxi, were discernible as punctate signals (Fig. 2A). Before the addition of the tetracycline analog doxycycline (DOX) to the cells, FL-PLA/AT-3 signals were not detected. At 12 h after the addition of DOX, FL-PLA/AT-3 signals appeared in cytoplasm, and at 24 h, the signals were further enhanced. This localization was consistent with the previous observation that PLA/AT-3 has a hydrophobic domain at the C terminus, yet the overexpressed PLA/AT-3 is distributed throughout cells, including cytoplasm (22). Consistently, as analyzed by RT-PCR and Western blotting, DOX induced mRNA and protein of FL-PLA/AT-3 (Fig. 2, B and C). The PLA1/2 activity of PLA/AT-3 in the cell homogenates was also enhanced from 0.07 to 1.4 nmol/min/mg protein at 24 h, and this level was maintained at least until 96 h (Fig. 2D). Regarding time-dependent changes of peroxisomal proteins, DsRed2-Peroxi signals became diffuse at 24 h (Fig. 2A), and the level of endogenous PMP70 protein began to decrease at 12 h and became hardly detectable at 48 h (Fig. 2C). To determine whether the peroxisomal loss effect of PLA/AT-3 is reversible, the medium was returned to DOX-free medium after FL-PLA/AT-3-Tet-on cells were cultured in the presence of DOX for 48 h. As analyzed by Western blotting, endogenous PMP70 protein became detectable 7 days after the removal of DOX. This result suggests that the effect of PLA/AT-3 on peroxisomes is reversible (data not shown).
FIGURE 2.

Characterization of FL-PLA/AT-3-Tet-on cells. A, FL-PLA/AT-3-Tet-on cells cultured on glass slips were incubated with DOX for the indicated time, and the fixed and permeabilized cells were immunostained with anti-FLAG antibody (green, PLA/AT-3) and observed with a confocal laser-scanning microscope. Native fluorescence of DsRed2-Peroxi protein (red) was also observed with the same cells. Scale bars, 10 μm. B, after the cells were treated with DOX for the indicated time, total RNAs were isolated and analyzed by RT-PCR using the primers specific for PLA/AT-3 and GAPDH (control). C and D, cell homogenates (40 μg of protein) were analyzed for FL-PLA/AT-3, PMP70, and LC3 by Western blotting (C) and for PLA1/2 activity with 200 μm 1,2-[14C]dipalmitoyl-PC as a substrate (D). The reaction products were separated by TLC, and PLA1/2 activity was quantified (mean values ± S.D. (error bars), n = 3). E, cells were radiolabeled with [14C]hexadecanol, and their total lipids were extracted and separated by TLC. Relative radioactivities of MADAG (top) and free fatty alcohol (bottom) are shown (mean values ± S.D., n = 3). F, FL-PLA/AT-3-Tet-on cells transiently transfected with the EGFP-LC3-expressing vector (green) were cultured on glass slips for 24 h in the presence or absence of DOX and were observed with a confocal laser-scanning microscope. The cells were also cultured under starvation for 3 h. DsRed2-Peroxi signals are shown in red, and the green and red images are merged (Merge). Scale bars, 10 μm.
Peroxisomal enzymes, such as dihydroxyacetone phosphate acyltransferase and ADHAPS, play essential roles in the biosynthesis of ether-type lipids, including monoalkyldiacylglycerol (MADAG), plasmenylethanolamine, and plasmanylcholine (2). When FL-PLA/AT-3-Tet-on cells were treated with DOX for different time periods, followed by metabolic labeling with [14C]hexadecanol as a precursor of MADAG, the intracellular levels of [14C]MADAG were time-dependently decreased concomitant with the accumulation of [14C]hexadecanol in the cells (Fig. 2E). Similarly, LC-ESI/MS analyses showed that the levels of major plasmenylethanolamine species (e32:2, e34:2, e36:5, e38:5, and e40:7) were decreased in a DOX-dependent manner without obvious changes in the levels of diacyl-type phosphatidylethanolamine species (Fig. 3). To establish the structural assignments of plasmenylethanolamine species, we also performed LC-ESI-MS/MS for [M − H]− ions (supplemental Fig. 1). Plasmanylcholine levels were also reduced (data not shown). These results indicated that the dysfunction of peroxisomes rapidly occurs, according to the increasing expression level of PLA/AT-3.
FIGURE 3.

Analysis of ethanolamine phospholipids by LC-ESI/MS. After the cells were treated with DOX for the indicated time, total lipids were extracted and analyzed by LC-ESI/MS. Mass spectra of [M + H]+ ions of ethanolamine phospholipids were detected. Arrowheads, major plasmenylethanolamine species.
Because peroxins, such as Pex3p, Pex11βp, Pex16p, and Pex19p, are indispensable for the biogenesis of peroxisome, we analyzed a possible DOX-dependent translocation of these proteins in FL-PLA/AT-3-Tet-on cells. In the absence of DOX, Pex3p-EGFP and Pex16p-EGFP were clearly detected as punctate signals and mainly colocalized with DsRed2-Peroxi signals (Fig. 4, A and B). However, after the treatment with DOX for 24 h, the EGFP signals of both proteins were detected in non-peroxisomal structures. These results were similar to previous reports that the deficiency of peroxisomes resulted in the mislocalization of Pex3p and Pex16p to non-peroxisomal structures (32–34). Another peroxisomal membrane protein, Pex11βp, was similarly translocated (Fig. 4C). By contrast, Pex19p signals were detected throughout the cytoplasm irrespective of the DOX treatment (Fig. 4D). Importantly, the diffuse localization of DsRed2-Peroxi signals induced by DOX was not restored to their original localization by the overexpression of these peroxins. However, we could not rule out the possibility that the expression levels of these proteins were not high enough to overcome the effect of PLA/AT-3 on peroxisomal structure.
FIGURE 4.

Intracellular localization of peroxisomal proteins in FL-PLA/AT-3-Tet-on cells. FL-PLA/AT-3-Tet-on cells transiently transfected with the vectors expressing Pex3p-EGFP (A), Pex16p-EGFP (B), Citrine-Pex11βp (C), or Citrine-Pex19p (D) were cultured on glass slips for 24 h in the presence (+) or absence (−) of DOX and were observed with a confocal laser-scanning microscope (green). DsRed2-Peroxi signals are also shown in red, and both images are merged (Merge). Scale bars, 10 μm.
Our finding might result from the enhanced clearance of peroxisomes by the selective autophagy “pexophagy” (11, 13, 35). We therefore examined whether PLA/AT-3 stimulates the conversion of LC3-I protein to its phosphatidylethanolamine-conjugated form, LC3-II, which is known to occur during autophagosome formation (36). However, the DOX-dependent expression of PLA/AT-3 did not stimulate LC3-II formation (Fig. 2C). Furthermore, we analyzed a possible translocation of EGFP-LC3 in FL-PLA/AT-3-Tet-on cells in a DOX-dependent manner. As reported previously (37), the diffuse distribution of EGFP-LC3 was changed to punctate autophagosome signals by culturing the cells under the starved conditions (Fig. 2F). However, such translocation of EGFP-LC3 did not occur by the treatment with DOX, suggesting that autophagy is not involved in the peroxisome disappearance caused by PLA/AT-3.
Interaction of PLA/AT-3 with Pex19p
Because the functional abnormality of Pex3p, Pex16p, or Pex19p results in the deficiency of peroxisomes due to the defects in peroxisome membrane biogenesis (1, 6, 38), we examined whether PLA/AT-3 interacts with these peroxins and regulates their functions. To this end, we used COS-7 cells, which were more suitable for transient expression, instead of HEK293 cells, which were suitable for stable expression and were used in the experiments of Figs. 1–4, and carried out the immunoprecipitation assay between PLA/AT-3 and these peroxins. The C-terminally FLAG-tagged PLA/AT-3 (PLA/AT-3-FL) was transiently co-expressed with either HA-tagged Pex3p (Pex3p-HA), V5-tagged Pex16p (Pex16p-V5), or Myc-tagged Pex19p (Myc-Pex19p) in COS-7 cells, and cell lysates were subjected to immunoprecipitation using anti-FLAG antibody-conjugated agarose. The immunoprecipitate, which we used for Western blotting, corresponded to 13 times the volume of the lysate. Because the remaining FLAG-tagged PLA/AT-3 was not detected in the lysate after the immunoprecipitation with anti-FLAG antibody, the efficiency of the immunoprecipitation was thought to be high enough.
When analyzed by Western blotting, Pex3p-HA and Myc-Pex19p, but not Pex16p-V5, were coimmunoprecipitated with PLA/AT-3-FL (Fig. 5A). The doublet bands of Myc-Pex19p were presumably attributed to its partial farnesylation (39). We also examined whether the immunoprecipitation occurs in the other directions. The cell lysates were subjected to immunoprecipitation using anti-HA antibody for Pex3p-HA, anti-V5 antibody for Pex16p-V5, and anti-Myc antibody for Myc-Pex19p. Consistent with the aforementioned results, Western blot analysis with anti-FLAG antibody showed that PLA/AT-3-FL was coimmunoprecipitated with Pex3p-HA and Myc-Pex19p but not with Pex16p-V5 (data not shown). The amount of PLA/AT-3 immunoprecipitated decreased when Pex3p-HA or Pex16-V5 was coexpressed (Fig. 5A). Because the expression level of PLA/AT-3 in the lysates was not affected by the coexpression with Pex3p, the accessibility of anti-FLAG antibody to a complex consisting of PLA/AT-3-FL and Pex3p-HA might be affected. On the other hand, the expression level of PLA/AT-3 in the lysates was remarkably decreased by the coexpression with Pex16p-V5. Although this result is reproduced, the reason remains unclear.
FIGURE 5.

Interaction of PLA/AT-3 with peroxins in COS-7 cells. A, COS-7 cells were transiently cotransfected with PLA/AT-3-FL and either Pex3p-HA, Pex16p-V5, or Myc-Pex19p, and the obtained cell lysates were subjected to immunoprecipitation using anti-FLAG antibody-conjugated agarose. The immunoprecipitates (IP-αFLAG) and cell lysates were then analyzed by Western blotting with the indicated antibodies. The positions of the farnesylated (F) and nonfarnesylated (N) Pex19p are indicated. B, schematic primary structures of the constructed PLA/AT-3 mutants are shown. Wt, wild type; PRD, proline-rich domain; NCD, NC domain; HD, hydrophobic domain; FLAG, FLAG tag. C, COS-7 cells were transiently cotransfected with Myc-Pex19p and either wild-type PLA/AT-3-FL or one of its mutants, and the obtained cell lysates were subjected to immunoprecipitation using anti-FLAG antibody-conjugated agarose. The immunoprecipitates and cell lysates were then analyzed by Western blotting with the indicated antibodies. D and E, COS-7 cells were transiently cotransfected with Myc-Pex19p and either Pex3p-HA (D) or PLA/AT-3-FL (E), and the obtained cell lysates were analyzed by Western blotting with the indicated antibodies. Total RNAs were also isolated and analyzed for the indicated mRNAs by RT-PCR. E, cell homogenates (30 μg of protein) were allowed to react with 200 μm 1,2-[14C]dipalmitoyl-PC for the PLA1/2 assay. The products were separated by TLC, and PLA1/2 activity was quantified (mean values ± S.D. (error bars), n = 3). Mix, Myc-Pex19p-expressing cell homogenate (15 μg of protein) and PLA/AT-3-FL-expressing cell homogenate (15 μg of protein) were mixed immediately before the PLA1/2 assay. *, significant difference from PLA/AT-3-FL-expressing cells (p < 0.001). F, Myc-Pex19p-expressing cell lysate and PLA/AT-3-FL-expressing cell lysate were mixed in the same volume and incubated for 12 h, followed by immunoprecipitation using anti-FLAG antibody-conjugated agarose. The immunoprecipitates and cell lysates were then analyzed by Western blotting with the indicated antibodies.
Because Pex19p is a chaperone protein binding to and regulating multiple PMPs (33, 40) and because both Pex19p (Fig. 4D) (33) and PLA/AT-3 (Fig. 2A) are mainly present in the cytoplasm, it was likely that the interaction between PLA/AT-3 and Pex19p is related to the effect of PLA/AT-3 on peroxisomes. We therefore examined which domain of PLA/AT-3 is required for this interaction. Because the primary structure of PLA/AT-3 comprises the NC domain, which contains the catalytic center cysteine 113, N-terminal proline-rich domain, and C-terminal hydrophobic domain (24), we constructed three mutants: 1) PLA/AT-3-C113S-FL, in which cysteine 113 was replaced with serine (C113S); 2) PLA/AT-3-ΔN10-FL, which lacks the N-terminal 10 amino acids contained in the proline-rich domain (ΔN10); and 3) PLA/AT-3-ΔC27-FL, which lacks the C-terminal 27 amino acids contained in the hydrophobic domain (ΔC27) (Fig. 5B). These mutants were previously reported to be catalytically inactive (24). When these PLA/AT-3 mutants were co-expressed with Myc-Pex19p and treated with anti-FLAG antibody-conjugated agarose gel, Myc-Pex19p was coimmunoprecipitated with PLA/AT-3-C113S-FL as well as the wild-type PLA/AT-3, whereas the intensity of the Myc-Pex19p signal was weak or hardly detectable in the presence of PLA/AT-3-ΔN10-FL or PLA/AT-3-ΔC27-FL (Fig. 5C). These results suggested that the proline-rich domain and hydrophobic domain of PLA/AT-3 play important roles in its binding to Pex19p. Judging from relative intensities of the doublet Myc-Pex19p bands in the cell lysate, the expression of PLA/AT-3 did not appear to affect the farnesylation of Pex19p (Fig. 5C). It was previously reported that Pex19p increases the level of Pex3p by stabilizing this protein (34). We could reproduce this result by co-expressing Pex3p and Pex19p (Fig. 5D). Similarly, the expression of Pex19p increased the amount of PLA/AT-3 protein in the cell lysate, whereas the PLA/AT-3 mRNA level was unaltered (Fig. 5E). Consistent with this observation, the PLA1/2 activity derived from PLA/AT-3 in the cell lysate was 1.5-fold increased by co-expression of Pex19p (Fig. 5E). Pex19p itself did not have PLA1/2 activity. Furthermore, Pex19p did not stimulate the PLA1/2 activity of PLA/AT-3 when both proteins were separately prepared and mixed upon enzyme assay (Fig. 5E). These results suggest that Pex19p binds to PLA/AT-3 and enhances the intracellular level of the catalytically active PLA/AT-3.
We next examined whether the interaction between PLA/AT-3 and Pex19p occurs in a cell-free system. The lysate expressing PLA/AT-3-FL and that expressing Myc-Pex19p were prepared separately and mixed together. When the mixture was subjected to immunoprecipitation using anti-FLAG antibody, Myc-Pex19p was coimmunoprecipitated with PLA/AT-3-FL (Fig. 5F). Thus, the interaction between PLA/AT-3 and Pex19p was reproduced in a cell-free system.
To confirm the results so far obtained with COS-7 cells, we next used HEK293T as host cells. When co-expressed in HEK293T cells, Myc-Pex19p was coimmunoprecipitated with wild-type PLA/AT-3-FL and PLA/AT-3-C113S-FL but not with PLA/AT-3-ΔC27-FL (Fig. 6A). In this assay, the expression of Myc-Pex19p increased the amounts of wild-type PLA/AT-3 and C113S mutant, but not ΔC27 mutant, in the cell lysates. In contrast, mRNA levels of these PLA/AT-3s were hardly affected by the co-expression of Pex19p (Fig. 6B). The PLA1/2 activity in the homogenate of wild-type PLA/AT-3-expressing cells was significantly increased by co-expression of Pex19p (Fig. 6C). The C113S and ΔC27 mutants were catalytically inactive as expected.
FIGURE 6.

Interaction of PLA/AT-3 with Pex19p in HEK293T cells. HEK293T cells were cotransfected with Myc-Pex19p and either wild-type PLA/AT-3-FL (Wt) or one of its mutants, and the obtained cell lysates were subjected to immunoprecipitation using anti-FLAG antibody-conjugated agarose. A, immunoprecipitates (IP-αFLAG) and cell lysates were analyzed by Western blotting with the indicated antibodies. B, total RNAs were isolated and analyzed for mRNAs of PLA/AT-3, Pex19p, and GAPDH (control) by RT-PCR. C, cell homogenates (30 μg of protein) were allowed to react with 200 μm 1,2-[14C]dipalmitoyl-PC for the PLA1/2 assay. The products were separated by TLC, and PLA1/2 activity was quantified (mean values ± S.D. (error bars), n = 3). Mix, Myc-Pex19p-expressing cell homogenate (15 μg of protein) and PLA/AT-3-FL-expressing cell homogenates (15 μg of protein) were mixed immediately before the PLA1/2 assay. *, significant difference from PLA/AT-3-FL-expressing cells (p < 0.001).
In the preceding and following experiments concerning Pex19p, we detected recombinant Pex19p with a tag. Here, we also examined the effect of endogenous Pex19p on recombinant FL-PLA/AT-3 in HEK293 cells by using FL-PLA/AT-3-Tet-on cells. The expression of endogenous Pex19p was successfully suppressed by two different siRNAs against Pex19p mRNA, as examined by RT-PCR (Fig. 7A). This suppression significantly reduced the PLA1/2 activity in the cell homogenates (Fig. 7B). In another assay, after FL-PLA/AT-3 was DOX-dependently expressed in FL-PLA/AT-3-Tet-on cells, FL-PLA/AT-3 was immunoprecipitated with anti-FLAG antibody from the cell lysates and visualized as a ∼18.9 kDa band by silver staining (Fig. 7C). When these immunoprecipitates were immunostained with anti-Pex19p antibody, endogenous Pex19p was detected only in the DOX-treated cells (Fig. 7D), indicating that PLA/AT-3 interacts with endogenous Pex19p as well as recombinant Pex19p.
FIGURE 7.
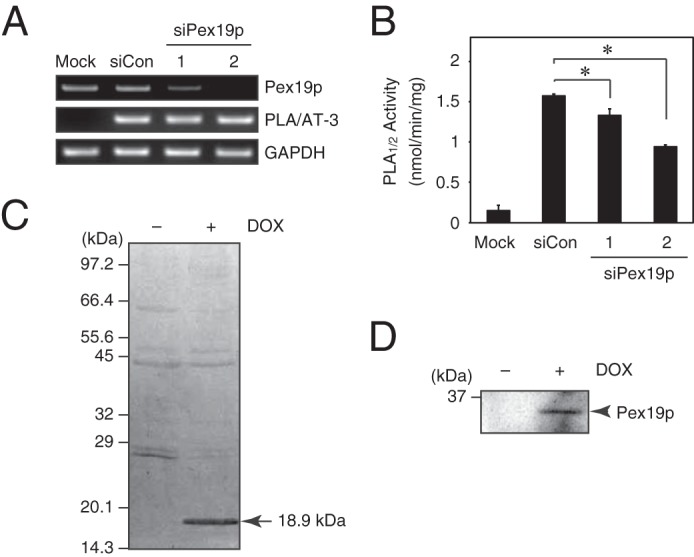
Endogenous Pex19p binds to PLA/AT-3. A, FL-PLA/AT-3-Tet-on cells were transiently transfected with a control siRNA (siCon) or Pex19p siRNAs (siPex19p-1 and -2). After 48 h, total RNAs were isolated and analyzed by RT-PCR using specific primers for Pex19p, PLA/AT-3, and GAPDH (a control). Mock, control HEK293 cells. B, the cell homogenates (20 μg of protein) were also prepared and allowed to react with 200 μm 1,2-[14C]dipalmitoyl-PC for the PLA1/2 assay (mean values ± S.D. (error bars), n = 3). *, significant differences from FL-PLA/AT-3-Tet-on cells transfected with a control siRNA (p < 0.007). C and D, FL-PLA/AT-3 was immunoprecipitated from FL-PLA/AT-3-Tet-on cells cultured for 48 h in the presence (+) or absence (−) of DOX. The precipitates were analyzed by SDS-PAGE, followed by staining with silver nitrate (C) or by Western blotting with anti-Pex19p antibody (D). Error bars, S.D.
PLA/AT-3 Interacts with Pex19p in Vivo
To further confirm that PLA/AT-3 binds to Pex19p in living cells, we employed the nuclear mislocalization assay (32). For this purpose, we constructed Pex19p-NLS-Myc, which has two tandem copies of NLS and a Myc tag. PLA/AT-3-FL was co-expressed with Myc-Pex19p or Pex19p-NLS-Myc in COS-7 cells, and the cell homogenates were fractionated into nuclear and PNS fractions. When analyzed by Western blotting, Myc-Pex19p was mostly localized in the PNS fraction, whereas Pex19p-NLS-Myc was seen in both nuclear and PNS fractions (Fig. 8A). As for the localization of PLA/AT-3-FL, its signal in the nuclear fraction of Pex19p-NLS-Myc-expressing cells was more intense than that in the nuclear fraction of Myc-Pex19p-expressing cells (Fig. 8B). Immunofluorescence microscopy also revealed that PLA/AT-3-FL accumulates in the nucleoplasm when Pex19p-NLS-Myc, but not Myc-Pex19p, was co-expressed (Fig. 8C). These results showed that PLA/AT-3 at least partly binds to Pex19p in living cells.
FIGURE 8.

Targeting of Pex19p to nucleus leads to nuclear accumulation of PLA/AT-3. COS-7 cells were cotransfected with PLA/AT-3-FL and either Myc-Pex19p or Pex19p-NLS-Myc, and nuclear (Nu) and PNS fractions were analyzed by Western blotting with antibodies against lamin A/C (a nuclear marker), Myc, and FLAG (A and B). The positions of the farnesylated (F) and nonfarnesylated (N) Myc-Pex19p and Pex19p-NLS-Myc (arrowhead) are indicated. C, the cells were immunostained with anti-FLAG antibody (green) and anti-Myc antibody (red) and observed with a confocal laser-scanning microscope. Both images are merged (Merge). Scale bars, 10 μm.
PLA/AT-3 Inhibits the Interaction of Pex19p with PMPs in a Catalytic Activity-dependent Manner
We examined whether the coexistence of PLA/AT-3 affects the ability of Pex19p to bind to PMPs. When Myc-Pex19p and Pex3p-HA were co-expressed in COS-7 cells, both proteins in the cell lysate were precipitated by anti-HA antibody (Fig. 9A), as reported previously (34, 41). The additional expression of wild-type PLA/AT-3-FL abolished the binding of Pex19p to Pex3p despite the unaltered protein levels of Pex19p and Pex3p in the cell lysate. Interestingly, the C113S mutant of PLA/AT-3 did not interfere with the binding of Pex19p to Pex3p. Similarly, when HA-Pex19p and Myc-Pex11βp were co-expressed, the additional expression of PLA/AT-3-FL, but not its C113S mutant, potently inhibited the co-precipitation of Pex19p and Pex11βp (Fig. 9B). Notably, the protein level of Pex11βp in the lysate, but not its mRNA level, was remarkably increased by the simultaneous expression of Pex19p (Fig. 9B). This result suggested that Pex19p stabilized Pex11βp. Moreover, the overexpression of PLA/AT-3 reduced the levels of recombinant PMP70 in an enzyme activity-dependent manner (Fig. 9C). Similar results were obtained with the cells in which Pex19p was additionally expressed (Fig. 9C). These results suggested that PLA/AT-3 interferes with the binding of both Pex3p and Pex11βp to Pex19p and destabilizes PMP70. Ineffectiveness of the catalytically inactive C113S mutant suggested an important role of the enzyme activity in this action of PLA/AT-3.
FIGURE 9.

PLA/AT-3 inhibits the binding of Pex19p to PMPs in an enzyme activity-dependent manner. A, COS-7 cells were cotransfected with three cDNAs (Myc-Pex19p, Pex3-HA, and PLA/AT-3-FL), and the cell lysates were subjected to immunoprecipitation using anti-HA antibody. The immunoprecipitates (IP-αHA) and cell lysates were then analyzed by Western blotting with the indicated antibodies. Wt and C113S, wild-type PLA/AT-3-FL and its C113S mutant, respectively. B, COS-7 cells were cotransfected with three cDNAs (HA-Pex19p; Pex11βp-Myc; and either PLA/AT-1-FL (1), PLA/AT-2-FL (2), or PLA/AT-3-FL (Wt or C113S)), and the cell lysates were subjected to immunoprecipitation using anti-HA antibody. The immunoprecipitates and cell lysates were then analyzed by Western blotting with the indicated antibodies. Total RNAs were also isolated and analyzed by RT-PCR using the indicated primers. C, COS-7 cells were cotransfected with three cDNAs (HA-Pex19p, DsRed-PMP70, and either PLA/AT-1-FL (1), PLA/AT-2-FL (2), or PLA/AT-3-FL (Wt or C113S)), and the cell lysates were analyzed by Western blotting with the indicated antibodies. Total RNAs were also isolated and analyzed by RT-PCR using the indicated primers.
Effects of PLA/AT-1 and -2
We also examined whether other members of the PLA/AT family show this inhibitory effect. The FLAG-tagged PLA/AT-2 (PLA/AT-2-FL) inhibited the binding of Pex19p to Pex11βp potently, whereas PLA/AT-1-FL hardly inhibited this binding (Fig. 9B). Both PLA/AT-1 and -2 reduced the PMP70 levels in the presence or absence of recombinant Pex19p (Fig. 9C).
Interaction of PLA/AT-3 with Pex3p
As shown in Fig. 4A, Pex3p was also coimmunoprecipitated with PLA/AT-3. In order to clarify which domain of PLA/AT-3 is necessary for this interaction, we examined a possible co-immunoprecipitation of Pex3p-HA with PLA/AT-3-C113S-FL, PLA/AT-3-ΔN10-FL, and PLA/AT-3-ΔC27-FL (Fig. 10). The results showed that Pex3p was immunoprecipitated with the C113S and ΔN10 mutants as well as wild-type PLA/AT-3, whereas the ΔC27 mutant was ineffective. These results suggested that PLA/AT-3 binds to Pex3p through the C-terminal hydrophobic domain in an enzyme activity-independent manner.
FIGURE 10.
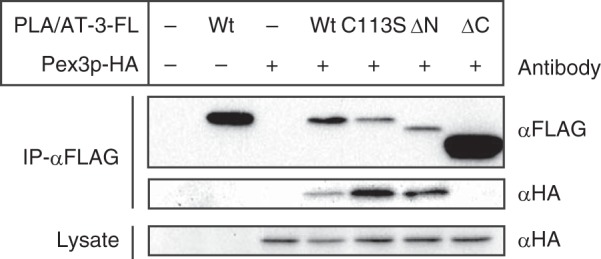
C-terminal hydrophobic domain of PLA/AT-3 is required for its binding to Pex3p. COS-7 cells were cotransfected with Pex3p-HA and either wild-type PLA/AT-3-FL (Wt) or one of its mutants, and the cell lysates were subjected to immunoprecipitation using anti-FLAG antibody-conjugated agarose. The immunoprecipitates (IP-αFLAG) and cell lysates were then analyzed by Western blotting with the indicated antibodies.
Discussion
We previously reported that the overexpression of recombinant PLA/AT-3 (also referred to as HRASLS3, H-rev107, or AdPLA) causes specific disappearance of peroxisomes in mammalian cells (27). In the present study, we first confirmed this finding (Figs. 1–4). In FL-PLA/AT-3-Tet-on cells, the induction of PLA/AT-3 with DOX readily decreased the abundance of peroxisomes labeled with DsRed2-Peroxi, and the peroxisomes almost completely disappeared within 24 h. The dysfunction of peroxisomes was also revealed by the decreased levels or abnormal distribution of PMPs (PMP70, Pex3p, Pex11βp, and Pex16) and peroxisomal matrix proteins (catalase, ADHAPS, and thiolase) as well as large reductions in the levels of ether-type lipids, such as MADAG, plasmenylethanolamine, and plasmanylcholine. An obvious decrease in PMP70 level was observed even with a low concentration (0.01 μg/ml) of DOX, and in the presence of this concentration of DOX, the PLA1/A2 activity was 10-fold higher than that of DOX-free control cells (data not shown). Thus, it appeared that the expression level of PLA/AT-3, increasing PLA1/A2 activity 10-fold over the control cells, is sufficient to see the loss of peroxisomes.
It has been reported that the quality and quantity of peroxisomes are maintained by peroxisome-specific autophagy, pexophagy (11, 13). However, during the DOX-dependent reduction of peroxisomes, the activation of LC3, a marker of autophagy, was not observed. Thus, we could not show evidence that PLA/AT-3 stimulates the degradation of peroxisomes. Because it is thought that the level of peroxisomes is regulated by the balance between their biogenesis and degradation (11), we next focused on a possible inhibition of peroxisome biogenesis by PLA/AT-3.
Among peroxins responsible for peroxisome membrane biogenesis, Pex19p is mainly localized in cytoplasm, whereas Pex3p and Pex16p are mainly associated with peroxisomes (1). In the present study, immunoprecipitation assays revealed that PLA/AT-3 binds to both Pex19p and Pex3p but not to Pex16p. Compared with the expression of PLA/AT-3 alone, co-expression of PLA/AT-3 with Pex19p caused an increase in the protein level and PLA1/2 activity of PLA/AT-3. Using different PLA/AT-3 mutants, we showed that PLA/AT-3 binds to Pex19p through its N-terminal proline-rich and C-terminal hydrophobic domains. PLA/AT-3-C113S, a single point mutant devoid of the enzyme activity, was still capable of binding to Pex19p, suggesting that the binding is independent of its enzyme activity. The nuclear mislocalization assay using Pex19p-NLS also revealed an interaction between PLA/AT-3 and Pex19p. Taken together, these findings strongly suggested that PLA/AT-3 is a novel Pex19p-binding protein. PLA/AT-3 also bound to Pex3p through its C-terminal hydrophobic domain. Because Pex3p is one of the Pex19p-binding proteins (41), it remains unclear whether this binding occurs directly or indirectly through the binding of PLA/AT-3 to Pex19p.
One of the most important functions of Pex19p is to capture PMPs in the cytoplasm or on the ER membrane and transport them to peroxisomes (9, 42, 43). Although it is still controversial where Pex19p binds to PMPs, a growing body of evidence has suggested that nascent PMPs existing on the ER membrane are assembled to form nascent peroxisomes (preperoxisomal vesicles) and that the nascent peroxisomes bud from ER and are transported to preexisting peroxisomes in a Pex19p-dependent manner (42, 43) (Fig. 11A). Moreover, a recent report demonstrated that Pex16p is transported to peroxisomes from a specialized domain in ER membrane in a Sec16p-dependent manner (44). Therefore, if PLA/AT-3 attaches to the ER membrane as the PLA/AT-3·Pex19p complex, PLA/AT-3 may act as a PLA1/2 and acyltransferase enzyme toward glycerophospholipids of the membrane and disrupt the membrane structure required for the formation and budding of nascent peroxisomes (Fig. 11B). Alternatively, PLA/AT-3 may be transported to peroxisomes by Pex19p and enzymatically decompose membrane phospholipids of existing peroxisomes (Fig. 11C). These two possibilities are supported by the previous reports that a certain population of PLA/AT-3 localizes to the ER (22) or to peroxisomes (27) and may explain why the C113S mutant fails to disturb peroxisomal functions despite its ability to bind to Pex19p. However, there is no direct evidence for these possibilities, and further experiments will be needed to clarify where and how PLA/AT-3 as an enzyme decomposes the peroxisome membrane structure.
FIGURE 11.

Possible mechanisms of the disappearance of peroxisomes caused by PLA/AT-3. A, generally accepted roles of Pex19p in peroxisome biogenesis. Shown are possible functions of PLA/AT-3 on the ER membrane (B) and peroxisome membrane (C).
Our results also indicated that PLA/AT-3 inhibits the binding of Pex19p to PMPs, such as Pex3p and Pex11βp. This finding suggests that PLA/AT-3 causes mislocalization of PMPs. Interestingly, despite the ability to bind to Pex19p, the C113S mutant failed to inhibit the binding of Pex19p to Pex3p and Pex11βp. However, it remains unclear why the enzyme activity of PLA/AT-3 is necessary for this inhibitory effect.
Previously, we suggested that the overexpression of PLA/AT-2 (HRASLS2) also causes the dysfunction of peroxisomes (28), whereas PLA/AT-1 (HRASLS1, A-C1) did not appear to affect peroxisomal functions (45). In the present study, we observed that PLA/AT-2 inhibited the binding of Pex19p to Pex3p and Pex11βp and decreased the protein level of PMP70. As for PLA/AT-1, this inhibitory effect was not clearly seen, and a moderate effect on the PMP70 level was observed. These results may be related to the fact that PLA/AT-3 is closer to PLA/AT-2 than PLA/AT-1 in terms of primary structures (26).
Peroxisomes are responsible for many metabolic events, including the synthesis of precursors of ether-type lipids, including MADAG. PLA/AT-3 is abundant in adipose tissues of mice, rats, and humans (22, 24, 25), and its expression is further induced in obesity (19). Furthermore, the expression level of PLA/AT-3 was highly enhanced through the differentiation of mouse adipogenic cell line 3T3-L1 cells into adipocyte-like cells (18, 22). Interestingly, MADAG levels rapidly decrease as the adipogenesis proceeds in 3T3-L1 cells (46). Given our present results, the negative regulation of peroxisome biogenesis by the increasing PLA/AT-3 may be one of the reasons for the decrease in MADAG levels. Jaworski et al. (19) reported that the mice deficient in PLA/AT-3 (AdPLA) have a defect in the growth of lipid droplets in adipocytes, causing a lean phenotype. This finding was explained by high rate of lipolysis caused by the marked reduction in the signaling pathway of the prostaglandin E2-EP3 axis because the deficiency in PLA/AT-3, acting as arachidonic acid-releasing PLA2, results in a decrease of prostaglandin E2 levels. The elimination of adipocyte peroxisomes by the adipocyte-specific disruption of the Pex5p gene in mice was reported to lead to increased fat mass and reduced lipolysis in white adipose tissue (47), suggesting the inverse correlation between peroxisome level and lipid droplet production in adipose tissues. Thus, the dysregulation of peroxisomes in adipose tissues may be another reason for the thinness of PLA/AT-3-deficient mice.
Because PLA/AT-3 was originally isolated as a tumor suppressor negatively regulating the oncogene H-Ras (14), it is of particular interest to examine how PLA/AT-3 regulates the oncogenic activity of H-Ras. H-Ras is modified by prenylation at the C-terminal cysteine residue in the CAAX motif and palmitoylation at two cysteine residues (48). These lipid modifications are required for the intracellular trafficking of H-Ras from the endomembrane system to the plasma membrane (49–51). Recently, Wang et al. (52) reported that PLA/AT-3 binds to H-Ras and decreases the level of the GTP-loaded active form of H-Ras by inhibiting its palmitoylation in an enzyme activity-dependent manner. However, it remains unclear how PLA/AT-3 enzymatically inhibits the palmitoylation. Based on our present results, PLA/AT-3 may affect the oncogenic activity and/or intracellular localization of H-Ras by altering the membrane structure through the reduction in the content of ether-type lipids. It was also reported that aggressive cancer cells possess significantly higher levels of ether-type lipids than less aggressive counterparts (53). Aggressive cancer cells highly expressed ADHAPS, an enzyme involved in the biosynthesis of the ether-type lipids, and the transformation of less aggressive cancer cells with H-Ras up-regulated ADHAPS expression, leading to an increase in the levels of ether-type lipids. This change also increased the levels of oncogenic signaling lipids, such as ether-type and ester-type lysophosphatidic acids and eicosanoids. Similarly, the tumor suppressing activity of PLA/AT-3 may be related to the decreased levels of ether-type lipids resulting from the dysfunction of peroxisomes. Further studies on PLA/AT-3 are required to elucidate its role in obesity and cancers.
In conclusion, our results showed for the first time that PLA/AT-3 is a Pex19p-binding protein and inhibits the chaperone functions of Pex19p. This action may be responsible for the negative regulation of PLA/AT-3 on peroxisome biogenesis in mammalian cells.
Author Contributions
T. U. and N. U. conceived and coordinated the study and wrote the paper. T. U., M. W., T. I., and K. T. designed, performed, and analyzed the experiments shown in Figs. 1, 2, and 5–11. K. K. and N. A. designed, performed, and analyzed the experiments shown in Figs. 2, 4, and 8. N. K. and H. A. designed, performed, and analyzed the experiments shown in Fig. 3 and supplemental Fig. 1. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Yumi Tani and Ami Yamada for technical assistance. We also acknowledge technical assistance from the Divisions of Research Instrument and Equipment and Radioisotope Research, the Life Science Research Center, Kagawa University.
This work was supported by Grants-in-Aid for Scientific Research (C) 24590355 (to T. U.) and 25460370 (to N. U.) from the Japan Society for the Promotion of Science; the Fund for Kagawa University Young Scientists 2014 (to T. U.); the Naito Foundation (to T. U.); and the Japan Foundation for Applied Enzymology (to N. U.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables 1 and 2 and Fig. 1.
- PTS
- peroxisome targeting signal
- ADHAPS
- alkyl-dihydroxyacetone phosphate synthase
- DOX
- doxycycline
- EGFP
- enhanced green fluorescence protein
- ESI
- electrospray ionization
- HRASLS
- HRAS-like suppressor
- MADAG
- monoalkyldiacylglycerol
- NLS
- nuclear localization signal
- PC
- phosphatidylcholine
- PLA
- phospholipase A
- PLA/AT
- phospholipase A/acyltransferase
- PMP
- peroxisomal membrane protein
- PNS
- postnuclear supernatant
- ER
- endoplasmic reticulum.
References
- 1. Purdue P. E., Lazarow P. B. (2001) Peroxisome biogenesis. Annu. Rev. Cell Dev. Biol. 17, 701–752 [DOI] [PubMed] [Google Scholar]
- 2. Wanders R. J. A., Waterham H. R. (2006) Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 75, 295–332 [DOI] [PubMed] [Google Scholar]
- 3. Van Veldhoven P. P. (2010) Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 51, 2863–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith J. J., Aitchison J. D. (2013) Peroxisomes take shape. Nat. Rev. Mol. Cell Biol. 14, 803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lodhi I. J., Semenkovich C. F. (2014) Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. 19, 380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weller S., Gould S. J., Valle D. (2003) Peroxisome biogenesis disorders. Annu. Rev. Genomics Hum. Genet. 4, 165–211 [DOI] [PubMed] [Google Scholar]
- 7. Steinberg S. J., Dodt G., Raymond G. V., Braverman N. E., Moser A. B., Moser H. W. (2006) Peroxisome biogenesis disorders. Biochim. Biophys. Acta 1763, 1733–1748 [DOI] [PubMed] [Google Scholar]
- 8. Fujiki Y., Yagita Y., Matsuzaki T. (2012) Peroxisome biogenesis disorders: Molecular basis for impaired peroxisomal membrane assembly: in metabolic functions and biogenesis of peroxisomes in health and disease. Biochim. Biophys. Acta 1822, 1337–1342 [DOI] [PubMed] [Google Scholar]
- 9. Theodoulou F. L., Bernhardt K., Linka N., Baker A. (2013) Peroxisome membrane proteins: multiple trafficking routes and multiple functions? Biochem. J. 451, 345–352 [DOI] [PubMed] [Google Scholar]
- 10. Fujiki Y., Okumoto K., Mukai S., Honsho M., Tamura S. (2014) Peroxisome biogenesis in mammalian cells. Front. Physiol. 5, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nordgren M., Wang B., Apanasets O., Fransen M. (2013) Peroxisome degradation in mammals: mechanisms of action, recent advances, and perspectives. Front. Physiol. 4, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iwata J., Ezaki J., Komatsu M., Yokota S., Ueno T., Tanida I., Chiba T., Tanaka K., Kominami E. (2006) Excess peroxisomes are degraded by autophagic machinery in mammals. J. Biol. Chem. 281, 4035–4041 [DOI] [PubMed] [Google Scholar]
- 13. Manjithaya R., Nazarko T. Y., Farré J. C., Subramani S. (2010) Molecular mechanism and physiological role of pexophagy. FEBS Lett. 584, 1367–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hajnal A., Klemenz R., Schäfer R. (1994) Subtraction cloning of H-rev107, a gene specifically expressed in H-ras resistant fibroblasts. Oncogene 9, 479–490 [PubMed] [Google Scholar]
- 15. Ueda N., Tsuboi K., Uyama T. (2013) Metabolism of endocannabinoids and related N-acylethanolamines: canonical and alternative pathways. FEBS J. 280, 1874–1894 [DOI] [PubMed] [Google Scholar]
- 16. Morales M., Arenas E. J., Urosevic J., Guiu M., Fernández E., Planet E., Fenwick R. B., Fernández-Ruiz S., Salvatella X., Reverter D., Carracedo A., Massagué J., Gomis R. R. (2014) RARRES3 suppresses breast cancer lung metastasis by regulating adhesion and differentiation. EMBO Mol. Med. 6, 865–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiong S., Tu H., Kollareddy M., Pant V., Li Q., Zhang Y., Jackson J. G., Suh Y. A., Elizondo-Fraire A. C., Yang P., Chau G., Tashakori M., Wasylishen A. R., Ju Z., Solomon H., Rotter V., Liu B., El-Naggar A. K., Donehower L. A., Martinez L. A., Lozano G. (2014) Pla2g16 phospholipase mediates gain-of-function activities of mutant p53. Proc. Natl. Acad. Sci. U.S.A. 111, 11145–11150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hummasti S., Hong C., Bensinger S. J., Tontonoz P. (2008) HRASLS3 is a PPARγ-selective target gene that promotes adipocyte differentiation. J. Lipid Res. 49, 2535–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jaworski K., Ahmadian M., Duncan R. E., Sarkadi-Nagy E., Varady K. A., Hellerstein M. K., Lee H.-Y., Samuel V. T., Shulman G. I., Kim K.-H., de Val S., Kang C., Sul H. S. (2009) AdPLA ablation increases lipolysis and prevents obesity induced by high-fat feeding or leptin deficiency. Nat. Med. 15, 159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vaccari C. M., Romanini M. V., Musante I., Tassano E., Gimelli S., Divizia M. T., Torre M., Morovic C. G., Lerone M., Ravazzolo R., Puliti A. (2014) De novo deletion of chromosome 11q12.3 in monozygotic twins affected by Poland syndrome. BMC Med. Genet. 15, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jin X.-H., Okamoto Y., Morishita J., Tsuboi K., Tonai T., Ueda N. (2007) Discovery and characterization of a Ca2+-independent phosphatidylethanolamine N-acyltransferase generating the anandamide precursor and its congeners. J. Biol. Chem. 282, 3614–3623 [DOI] [PubMed] [Google Scholar]
- 22. Duncan R. E., Sarkadi-Nagy E., Jaworski K., Ahmadian M., Sul H. S. (2008) Identification and functional characterization of adipose-specific phospholipase A2 (AdPLA). J. Biol. Chem. 283, 25428–25436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin X.-H., Uyama T., Wang J., Okamoto Y., Tonai T., Ueda N. (2009) cDNA cloning and characterization of human and mouse Ca2+-independent phosphatidylethanolamine N-acyltransferases. Biochim. Biophys. Acta 1791, 32–38 [DOI] [PubMed] [Google Scholar]
- 24. Uyama T., Morishita J., Jin X.-H., Okamoto Y., Tsuboi K., Tonai T., Ueda N. (2009) The tumor suppressor gene H-Rev107 functions as a novel Ca2+-independent cytosolic phospholipase A1/2 of the thiol hydrolase-type. J. Lipid Res. 50, 685–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uyama T., Jin X.-H., Tsuboi K., Tonai T., Ueda N. (2009) Characterization of the human tumor suppressors TIG3 and HRASLS2 as phospholipid-metabolizing enzymes. Biochim. Biophys. Acta 1791, 1114–1124 [DOI] [PubMed] [Google Scholar]
- 26. Shinohara N., Uyama T., Jin X.-H., Tsuboi K., Tonai T., Houchi H., Ueda N. (2011) Enzymological analysis of the tumor suppressor A-C1 reveals a novel group of phospholipid-metabolizing enzymes. J. Lipid Res. 52, 1927–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uyama T., Ichi I., Kono N., Inoue A., Tsuboi K., Jin X.-H., Araki N., Aoki J., Arai H., Ueda N. (2012) Regulation of peroxisomal lipid metabolism by catalytic activity of tumor suppressor H-rev107. J. Biol. Chem. 287, 2706–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Uyama T., Ikematsu N., Inoue M., Shinohara N., Jin X.-H., Tsuboi K., Tonai T., Tokumura A., Ueda N. (2012) Generation of N-acylphosphatidylethanolamine by members of the phospholipase A/acyltransferase (PLA/AT) family. J. Biol. Chem. 287, 31905–31919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 30. Kurochkin I. V., Mizuno Y., Konagaya A., Sakaki Y., Schönbach C., Okazaki Y. (2007) Novel peroxisomal protease Tysnd1 processes PTS1- and PTS2-containing enzymes involved in β-oxidation of fatty acids. EMBO J. 26, 835–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yao F., Svensjö T., Winkler T., Lu M., Eriksson C., Eriksson E. (1998) Tetracycline repressor, tetR, rather than the tetR-mammalian cell transcription factor fusion derivatives, regulates inducible gene expression in mammalian cells. Hum. Gene Ther. 9, 1939–1950 [DOI] [PubMed] [Google Scholar]
- 32. Sacksteder K. A., Jones J. M., South S. T., Li X., Liu Y., Gould S. J. (2000) PEX19 binds multiple peroxisomal membrane proteins, is predominantly cytoplasmic, and is required for peroxisome membrane synthesis. J. Cell Biol. 148, 931–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones J. M., Morrell J. C., Gould S. J. (2004) PEX19 is a predominantly cytosolic chaperone and import receptor for class 1 peroxisomal membrane proteins. J. Cell Biol. 164, 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matsuzaki T., Fujiki Y. (2008) The peroxisomal membrane protein import receptor Pex3p is directly transported to peroxisomes by a novel Pex19p- and Pex16p-dependent pathway. J. Cell Biol. 183, 1275–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hara-Kuge S., Fujiki Y. (2008) The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes. Exp. Cell Res. 314, 3531–3541 [DOI] [PubMed] [Google Scholar]
- 36. Kabeya Y., Mizushima N., Yamamoto A., Oshitani-Okamoto S., Ohsumi Y., Yoshimori T. (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117, 2805–2812 [DOI] [PubMed] [Google Scholar]
- 37. Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Islinger M., Cardoso M. J., Schrader M. (2010) Be different: the diversity of peroxisomes in the animal kingdom. Biochim. Biophys. Acta 1803, 881–897 [DOI] [PubMed] [Google Scholar]
- 39. Matsuzono Y., Kinoshita N., Tamura S., Shimozawa N., Hamasaki M., Ghaedi K., Wanders R. J., Suzuki Y., Kondo N., Fujiki Y. (1999) Human PEX19: cDNA cloning by functional complementation, mutation analysis in a patient with Zellweger syndrome, and potential role in peroxisomal membrane assembly. Proc. Natl. Acad. Sci. U.S.A. 96, 2116–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matsuzono Y., Fujiki Y. (2006) In vitro transport of membrane proteins to peroxisomes by shuttling receptor Pex19p. J. Biol. Chem. 281, 36–42 [DOI] [PubMed] [Google Scholar]
- 41. Matsuzono Y., Matsuzaki T., Fujiki Y. (2006) Functional domain mapping of peroxin Pex19p: interaction with Pex3p is essential for function and translocation. J. Cell Sci. 119, 3539–3550 [DOI] [PubMed] [Google Scholar]
- 42. Ma C., Agrawal G., Subramani S. (2011) Peroxisome assembly: matrix and membrane protein biogenesis. J. Cell Biol. 193, 7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tabak H. F., Braakman I., van der Zand A. (2013) Peroxisome formation and maintenance are dependent on the endoplasmic reticulum. Annu. Rev. Biochem. 82, 723–744 [DOI] [PubMed] [Google Scholar]
- 44. Yonekawa S., Furuno A., Baba T., Fujiki Y., Ogasawara Y., Yamamoto A., Tagaya M., Tani K. (2011) Sec16B is involved in the endoplasmic reticulum export of the peroxisomal membrane biogenesis factor peroxin 16 (Pex16) in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 108, 12746–12751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Uyama T., Inoue M., Okamoto Y., Shinohara N., Tai T., Tsuboi K., Inoue T., Tokumura A., Ueda N. (2013) Involvement of phospholipase A/acyltransferase-1 in N-acylphosphatidylethanolamine generation. Biochim. Biophys. Acta 1831, 1690–1701 [DOI] [PubMed] [Google Scholar]
- 46. Bartz R., Li W. H., Venables B., Zehmer J. K., Roth M. R., Welti R., Anderson R. G., Liu P., Chapman K. D. (2007) Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J. Lipid Res. 48, 837–847 [DOI] [PubMed] [Google Scholar]
- 47. Martens K., Bottelbergs A., Peeters A., Jacobs F., Espeel M., Carmeliet P., Van Veldhoven P. P., Baes M. (2012) Peroxisome deficient aP2-Pex5 knockout mice display impaired white adipocyte and muscle function concomitant with reduced adrenergic tone. Mol. Genet. Metab. 107, 735–747 [DOI] [PubMed] [Google Scholar]
- 48. Hancock J. F. (2003) Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 4, 373–384 [DOI] [PubMed] [Google Scholar]
- 49. Hancock J. F., Magee A. I., Childs J. E., Marshall C. J. (1989) All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57, 1167–1177 [DOI] [PubMed] [Google Scholar]
- 50. Hancock J. F., Paterson H., Marshall C. J. (1990) A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63, 133–139 [DOI] [PubMed] [Google Scholar]
- 51. Misaki R., Morimatsu M., Uemura T., Waguri S., Miyoshi E., Taniguchi N., Matsuda M., Taguchi T. (2010) Palmitoylated Ras proteins traffic through recycling endosomes to the plasma membrane during exocytosis. J. Cell Biol. 191, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang C. H., Shyu R. Y., Wu C. C., Tsai T. C., Wang L. K., Chen M. L., Jiang S. Y., Tsai F. M. (2014) Phospholipase A/Acyltransferase enzyme activity of H-rev107 inhibits the H-RAS signaling pathway. J. Biomed. Sci. 21, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Benjamin D. I., Cozzo A., Ji X., Roberts L. S., Louie S. M., Mulvihill M. M., Luo K., Nomura D. K. (2013) Ether lipid generating enzyme AGPS alters the balance of structural and signaling lipids to fuel cancer pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 110, 14912–14917 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.