

Background: In heart failure, the release of calcium becomes erratic leading to the generation of arrhythmias. Dysregulated Zn2+ homeostasis occurs in chronic heart failure.
Results: Zn2+ can directly activate RyR2, removing the dependence of Ca2+ for channel activation.
Conclusion: Zn2+ shapes Ca2+ dynamics by directly interacting with and modulating RyR2 function.
Significance: This highlights a new role for Zn2+ in cardiac excitation-contraction coupling.
Keywords: calcium, excitation-contraction coupling (E-C coupling), heart failure, ryanodine receptor, zinc
Abstract
Aberrant Zn2+ homeostasis is a hallmark of certain cardiomyopathies associated with altered contractile force. In this study, we addressed whether Zn2+ modulates cardiac ryanodine receptor gating and Ca2+ dynamics in isolated cardiomyocytes. We reveal that Zn2+ is a high affinity regulator of RyR2 displaying three modes of operation. Picomolar free Zn2+ concentrations potentiate RyR2 responses, but channel activation is still dependent on the presence of cytosolic Ca2+. At concentrations of free Zn2+ >1 nm, Zn2+ is the main activating ligand, and the dependence on Ca2+ is removed. Zn2+ is therefore a higher affinity activator of RyR2 than Ca2+. Millimolar levels of free Zn2+ were found to inhibit channel openings. In cardiomyocytes, consistent with our single channel results, we show that Zn2+ modulates both the frequency and amplitude of Ca2+ waves in a concentration-dependent manner and that physiological levels of Zn2+ elicit Ca2+ release in the absence of activating levels of cytosolic Ca2+. This highlights a new role for intracellular Zn2+ in shaping Ca2+ dynamics in cardiomyocytes through modulation of RyR2 gating.
Introduction
In cardiac muscle, the intracellular signal that triggers muscle contraction is thought to be a transient rise in intracellular Ca2+ that leads to the opening of Ca2+ release channels called type 2 ryanodine receptors (RyR2)2 on the sarcoplasmic reticulum (SR). The resulting release of Ca2+ into the cytosol causes movement of contractile myofibrils leading to cell contraction. Damaging changes in Ca2+ homeostasis are associated with heart failure, conduction abnormalities, and contractile dysfunction. Abnormal RyR2 function is recognized as an important component in the etiology of such disease states (1–3).
Recently, there has been much interest in the role of Zn2+ as an intracellular signaling molecule (4–6). Like Ca2+, intracellular Zn2+ is heavily buffered (7), and cellular Zn2+ homeostasis requires mechanisms that tightly control the uptake, storage, and distribution of Zn2+. This is achieved through the coordinated actions of metallothionein proteins and Zn2+ transporters (8–10).
In cardiomyocytes, the resting intracellular Zn2+ concentration is reported to be at picomolar levels (∼100 pm) (11, 12). During cardiac excitation-contraction coupling, intracellular Zn2+ concentrations are altered, and spatiotemporal fluctuations in free Zn2+ levels, including both Zn2+ transients and Zn2+ sparks, are suggested to be remarkably similar to those previously shown for Ca2+ (13). Other groups suggest that Zn2+ can permeate through the L-type channel with a greater affinity than Ca2+ but with a much lower permeability leading to a reduction in the inward current (14, 15). It has also been suggested that the SR can function as an intracellular store for Zn2+ alongside Ca2+ (6, 13).
Importantly aberrant Zn2+ homeostasis has been shown to be associated with cardiomyopathy including chronic heart failure, and myocardial damage as a result of dysregulated intracellular Ca2+ release, reduced cardiac contractility, and significantly prolonged rises of systolic Ca2+ (16–19). The potential role of Zn2+ in shaping intracellular Ca2+ release and regulating intracellular Ca2+ dynamics in heart however, is poorly characterized. Here we show that Zn2+ is a potent regulator of SR Ca2+ release through modulation of RyR2 channel function and that Zn2+ plays a key role in shaping intracellular Ca2+ dynamics important in cardiac excitation-contraction coupling.
Experimental Procedures
Reagents
Chemicals were AnalaR or the best equivalent grade from BDH Chemicals (Poole, UK) or Sigma-Aldrich. All solutions were made in deionized water, and those for use in bilayer experiments were filtered through a Millipore membrane filter (0.45-μm pore). 2,2′-(Ethylenedioxy)dianiline-N,N,N′,N′-tetraacetic acid (BAPTA) (Dorset, UK) and N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) were from Sigma-Aldrich.
SR Vesicle Preparation and Planar Phospholipid Bilayer Techniques
Sheep hearts were obtained from a local abattoir, and heavy SR membrane vesicles were prepared and fused with planar phosphatidylethanolamine lipid bilayers, as described previously (20). Briefly, heavy SR vesicles were prepared as follows. Homogenized ventricular cardiac tissue from sheep was subjected to centrifugation at 6,500 × g followed by ultracentrifugation of the supernatant at 100,000 × g. The heavy SR membrane fraction was obtained from loading mixed membranes onto a discontinuous sucrose density gradient. Heavy SR was collected, snap frozen in liquid N2, and stored at −80 °C. As reported previously, SR vesicles fused in a fixed orientation such that the cis-chamber corresponded to the cytosolic face of the channel and the trans-chamber to the SR lumen (21, 22). The trans-chamber was held at ground and the cis-chamber at potentials relative to ground. After fusion, the cis-chamber was perfused with 250 mm HEPES, 80 mm Tris, and 10 μm free Ca2+ (pH 7.2). The trans-chamber was perfused with 250 mm glutamic acid and 10 mm HEPES (pH to 7.2) with Ca(OH)2 (concentration of free Ca2+ ∼50 mm). The identity of RyR2 was confirmed by the single-channel conductance, and the number of channels gating in the bilayer was assessed by the application of caffeine at the end of the experiment. Zn2+ was added as ZnCl2 to the cis-chamber at the required concentration from an appropriate stock solution. 100 mm stock solution of ZnCl2 was prepared in 0.029 m HCl, and serial ZnCl2 dilutions were made in a Tris/HEPES buffer which contained, 250 mm HEPES, 80 mm Tris, and 10 μm free Ca2+ (pH 7.2) to make appropriate stock solutions. The concentration of each Zn2+ stock was further assessed using a pZn meter (Neurobiotex.com, Galveston, TX) as previously described (23) to confirm the accuracy of our serial dilutions. The lowest concentration Zn2+ stock made was 10 nm. The addition of 100 μm ZnCl2 to our Tris/HEPES buffer had no significant effect on the pH (pH was 7.22 ± 0.014 before and 7.21 ± 0.016 after the addition of 100 μm ZnCl2; n = 3). Special care was taken when preparing Zn2+ stock solutions. All glassware was prewashed with 100 nm TPEN and then rinsed carefully with MilliQ water passed through a Chelex Resin (Bio-Rad). The use of colored pipette tips was also avoided.
Experiments were carried out at room temperature (22 ± 2 °C). The concentration of free Ca2+ and pH of all of our solutions were determined using a Ca2+ electrode and a pH electrode (Hanna Instruments, Bedfordshire, UK) as previously described (20). Our measurements showed that the addition of Zn2+ ≤ 100 μm had no significant effect on the concentration of free Ca2+ ([Ca2+] was 10 ± 3.8 μm in the absence of Zn2+ and 10 ± 3.2 μm in the presence of 100 μm Zn2+ (n = 3). The addition of 1 mm BAPTA reduced the free Ca2+ concentration from 10 ± 2 μm to 10 ± 3.8 nm (n = 3). Subsequent addition of ZnCl2 (≤100 μm) had no significant effect on the measured free Ca2+ concentration (10 ± 3.2 nm; n = 3) under these conditions. The total concentration of Zn2+ in all of our recording solutions was assessed using inductively coupled plasma optical emission spectrometry (ICP-OES) (University of Edinburgh, Grant Institute, commercial facility) and shown to be below the detectable limit (1 ppb). Free Zn2+ was also assessed using a pZn meter and calculated to be ∼9 pm. The addition of concentrations of Ca2+ ≤ 100 μm had no significant effect on the concentration of free Zn2+ ([Zn2+] was 9 ± 7 pm in the presence of 10 μm Ca2+ and 10 ± 6 pm when Ca2+ was raised to 100 μm; n = 3). The calculated free Zn2+ concentration following addition of 1 mm cytosolic BAPTA was first estimated using the MaxChelator program and then assessed by actual measurements of free Zn2+ using a pZn meter. When the starting free Ca2+ concentration was 10 μm, the addition of 1 mm BAPTA had no significant effect on free Zn2+ levels.
Single Channel Recording and Analysis
Single-channel currents were monitored under voltage-clamp conditions using a BC-525C amplifier (Warner Instruments, Harvard Instruments). Channel recordings were low pass filtered at 10 kHz with a 4-pole Bessel filter, digitized at 100 kHz using a National Instruments acquisition interface (NIDAQ-MX; National Instruments, Austin, TX), and recorded on a computer hard drive using WinEDR 3.05 software (John Dempster, University of Strathclyde, Glasgow, UK). The recordings were subsequently filtered at 800 Hz (−3 dB) using a low pass digital filter implemented in WinEDR 3.05. Channel events were detected by the 50% threshold method (24) using TAC 4.2.0 software (Bruxton Corporation, Seattle, WA). Open probability (Po) and lifetime distributions were calculated from 3 min of continuous recording using TACfit 4.2.0 software (Bruxton Corporation). Lifetime analysis was carried out only when a single channel was incorporated into the bilayer. Individual lifetimes were fitted to a probability density function using the method of maximum likelihood (24). Lifetimes of <1 ms were not fully resolved under the conditions of data acquisition described here and were therefore excluded from the fitting procedure. A missed events correction was applied as previously described (25).
Ca2+ Waves
Adult male Wistar rats (300–400 g) were killed by concussion followed by cervical dislocation. The care and sacrifice of the animals conformed to the requirements of Directive 2010/63/EU of the European Parliament.
Isolation of Cardiomyocytes
All extracellular solutions were based on a modified Tyrode's solution. The basic solution contained, 135 mm NaCl, 5 mm KCl, 330 μm NaH2PO4, 5 mm glucose, 5 mm sodium pyruvate, 10 mm HEPES, 1 mm MgCl2, and 2 mm CaCl2 (pH 7.4). Nominally Ca2+-free Tyrode's solution used during cardiomyocyte isolation was as outlined above with no added Ca2+.
The protocol for isolation of cardiomyocytes was as described previously (26). Briefly, the whole heart was rapidly excised and placed into cold, nominally Ca2+-free Tyrode's solution (NT). The heart was then cannulated via the aorta on a Langendorff type apparatus and warmed NT buffer at 37 °C was perfused through the heart in a retrograde fashion for 6 min to clear residual blood. The solution was then exchanged for a Ca2+-free NT buffer with enzyme mix (containing 15 mg of collagenase type II (Worthington), with 50 mg of bovine serum albumin prepared from Cohn fraction V albumin and 18 mg of protease (type XIV 15% Ca2+) in 30 ml) for 8–15 min. Identification of rod-shaped cardiomyocytes in the perfusate was used as an indication of digestion being complete. The solution was then exchanged for a 2 mm Ca2+ NT solution, the heart was cut down, and cardiomyocytes were mechanically dispersed from the tissue in a shaking water bath. Typically this method yielded 70–90% rod-shaped cardiomyocytes, which were stored in NT solution at room temperature and used within 18 h of isolating. In intact myocytes, cells were exposed to ZnCl2 in the presence of the zinc ionophore, zinc pyrithione (ZnPy) to enable the entry of Zn2+ into the cell because ZnCl2 is cell-impermeable.
Cell Permeabilization
Isolated cardiomyocytes were permeabilized using saponin. Briefly, cardiomyocytes were perfused with 0.5 mm Ca2+ Tyrode's solution for 2 min prior to perfusion with a solution containing 100 μm EGTA, 5 mm ATP, 10 mm HEPES, 150 mm potassium gluconate, 25 μm MgCl2 at 20 °C. The sarcolemmal membrane was permeabilized with saponin (50 μg/ml) for 30 s. After permeabilization, cardiomyocytes were maintained in recording solution containing 1 mm EGTA, 10 mm HEPES, 120 mm potassium gluconate, 5 mm ATP, 1 mm pyruvate, 1 mm free MgCl2, and 100 nm free Ca2+ (calculated using the MaxChelator program), and 30 μm Fluo-4, pH 7.2. 1 mm BAPTA was added as indicated in the text, and additional Zn2+ was added to maintain as required.
Imaging
Images from permeabilized cardiomyoyctes were recorded using an Olympus 1X81 confocal microscope system using Olympus Fluoview software (version 4.0). Line scans were recorded using a 60× oil immersion lens using 2 × 2 pixel binning where each pixel was 0.43 μm in diameter. Fluo-4 indicator was excited using a 488-nm laser, and line scans were acquired at a rate of 333 Hz. The data were analyzed offline using ImageJ software. In our Tyrode's solution alone, in the presence of 100 nm Ca2+, there was no significant change in Fluo-4 fluorescence with increasing concentrations of Zn2+ (ΔFluo-4 F/F0 0.026 ± 0.01 units in 10 nm Zn2+ compared with control Tyrode's solution with no ZnCl2 added). For the cardiomyocyte experiments, ZnCl2 stocks were made directly in Tyrode's solution because the maximum concentration of Zn2+ used was 100 nm. For measurements of Ca2+ or Zn2+ in intact cardiomyocytes using Fluo-4-AM or Zinpyr-1, cells were illuminated with 488 nm using a monochromator (Photon Technology International, Birmingham, NJ) with fluorescence emissions captured above 520 nm using a Cascade 512B CCD camera (Photometrics, Tuscan, AZ) and continuously perfused at 32 ± 2 °C. The data were acquired using EasyRatioPro software (Photon Technology International, Birmingham, NJ) and analyzed using Winfluor v3.6.8 software (John Dempster, University of Strathclyde, Glasgow, UK).
Contractile Measurements
Spontaneous contractions were measured from cardiomyocytes perfused with Tyrode's solution at 32 ± 1 °C where videos were recorded to DVD. Analysis of contractions was carried out using edge detection measurements in Winfluor version 3.6.8 software.
Statistics
The data were expressed as means ± S.E. Where appropriate, a Student's t test was used to assess the difference between mean values. A p value of 0.05 was taken as significant. Where multiple treatments were compared, ANOVA followed by a Bonferroni post hoc test was used to assess the difference between treatments. A p value of 0.05 was taken as significant.
Results
Effect of Zn2+ on RyR2 Function
To determine whether Zn2+ can alter RyR2 gating, single RyR2 channels were incorporated into phospholipid bilayers enabling the membrane environment to be carefully controlled, and the direct action of Zn2+ at the cytosolic face of the channel was studied. Using Ca2+ as a permeant ion and holding at a command potential of 0 mV, which is considered to be the resting membrane potential of the sarcoplasmic reticulum (27), the addition of 100 pm Zn2+ to the cytosolic face of RyR2 channels significantly increased channel Po from 0.10 ± 0.03 to 0.45 ± 0.04 (Fig. 1), indicating that RyR2 has high affinity for Zn2+. The potentiation of RyR2 gating by physiological concentrations of free Zn2+ suggests that the amount of Ca2+ released through a single RyR2 channel following activation by cytosolic Ca2+ is significantly greater than previously thought. When Zn2+ levels were incremented in a cumulative fashion, channel activation plateaued at 1 nm Zn2+, and higher concentrations of Zn2+ (>10 nm) had no further significant effect on channel Po (Fig. 1B). Physiological concentrations of Zn2+ will therefore play a key role in regulating graded Ca2+ release events via RyR2 channels, enabling flexibility in the force of cardiac contraction, permitting the heart to adjust to altered diastolic filling (28). The addition of 1 mm Zn2+ to the cytosolic face of RyR2 completely abolished all channel openings (Fig. 1, A and B), possibly a consequence of Zn2+ binding to the low affinity divalent inhibitory site of RyR2 (29). Control HCl buffer solution containing no Zn2+ had no significant effect on RyR2 gating (Fig. 2A). To confirm that the level of free Zn2+ in our recording solutions, which was measured at ∼9 pm, was insufficient to influence channel gating, we exposed RyR2 channels to the potent Zn2+ chelator TPEN. In the absence of exogenously added Zn2+, the addition of 100 nm TPEN to the cis-chamber caused no significant change in channel Po (Fig. 2B).
FIGURE 1.

The effect of Zn2+ on RyR2 gating. A, a typical single RyR2 channel showing the effect of sequential additions of Zn2+ to the cis-chamber (cytosolic face of the channel) in the presence of 10 μm activating Ca2+ with Ca2+ as permeant ion at a holding potential of 0 mV. Open and closed states are shown by O and C, respectively. B, the relationship between Zn2+ concentration and RyR2 Po. Error bars show the mean values ± S.E. (n = 5; *, p < 0.05). C, the effects of Zn2+ on open and closed lifetime distributions of RyR2. Open (left) and closed (right) lifetime distributions and probability density functions of a typical RyR2 channel in the presence of 10 μm cytosolic Ca2+ alone (control) and after sequential addition of 100 pm, 1 nm, 100 nm, and 100 μm Zn2+ to the cytosolic face of the channel. The best fits to the data were obtained by maximum likelihood fitting. Time constants and percentage areas are shown for each distribution.
FIGURE 2.

HCl buffer and TPEN have no effect on RyR2 gating. The top traces in A and B show representative single RyR2 channel current fluctuations using 10 μm Ca2+ as the sole activating ligand. The bilayer was voltage-clamped at 0 mV. The lower traces in A and B show that addition of the HCl buffer (A) or the addition of 100 nm TPEN (B) to the cis-chamber had no significant effect on channel Po. The open and closed channel levels are denoted by O and C, respectively. The bar charts in A and B show both the average Po and the individual data points following the addition of the HCl buffer (A, n = 6) or 100 nm TPEN (B, n = 7). The data are expressed as means ± S.E.
Mechanisms by Which Zn2+ Regulates RyR2 Gating
To investigate the mechanisms underlying RyR2 activation by Zn2+, we performed lifetime analysis on experiments where only a single RyR2 channel was gating in the bilayer. Dwell time histograms of the apparent open and apparent closed states uncovered three modes of Zn2+ operation at the cytosolic face of RyR2 (Fig. 1C). Control channel-gating was best characterized by short openings and long closing as described previously (30, 31). The addition of Zn2+ in the concentration range 100 pm to 10 nm appeared to sensitize RyR2 to cytosolic Ca2+, revealed by a reduction in the duration of all the closed states with little effect on the duration of channel openings (32). When the free Zn2+ concentration was elevated to >1 nm, RyR2 dwelled for longer sojourns in the open state and displayed briefer sojourns in the closed state (see Table 1 for average lifetime data). This type of gating is characteristic of Ca2+-independent channel openings (21).
TABLE 1.
Comparison of the changes in RyR2 channel gating in response to Zn2+
The table shows the open and closed lifetime parameters in the presence of 10 μm cytosolic Ca2+ and following the addition of Zn2+ (100 pm to 100 μm) to the cytosolic face of the channel. The values are means ± S.E. (n = 3).
| [Zn2+] | T1 |
T2 |
T3 |
T4 |
||||
|---|---|---|---|---|---|---|---|---|
| Time | Area | Time | Area | Time | Area | Time | Area | |
| ms | % | ms | % | ms | % | ms | % | |
| Open lifetime parameters | ||||||||
| Control | 1.0 ± 0.05 | 89.0 ± 3.0 | 5.0 ± 0.9 | 11.0 ± 3 | ||||
| 100 pm | 1.0 ± 0.1 | 88.0 ± 1.2 | 4.6 ± 0.6 | 12.0 ± 1.0 | 15.0 ± 8.0 | 2.0 ± 1.0 | ||
| 1 nm | 1.0 ± 0.1 | 62.9 ± 9.0 | 4.5 ± 0.78 | 32.0 ± 8.0 | 27.0 ± 4.0 | 4.6 ± 2.5 | ||
| 100 nm | 1.2 ± 0.3 | 55.0 ± 14.0 | 5.2 ± 0.84 | 34.0 ± 6.0 | 21.2 ± 4.5 | 10.6 ± 9.0 | ||
| 100 μm | 2.2 ± 0.4 | 46.5 ± 0.9 | 10.2 ± 3.6 | 43.0 ± 8.0 | 60.6 ± 20 | 10.2 ± 6.8 | ||
| Closed lifetime parameters | ||||||||
| Control | 1.1 ± 0.1 | 85.8 ± 3.9 | 5.9 ± 0.9 | 39.3 ± 0.5 | 25.3 ± 1.8 | 24.6 ± 6.3 | 170.0 ± 12.7 | 6.8 ± 3.1 |
| 100 pm | 1.0 ± 0.1 | 72.0 ± 2.3 | 5.3 ± 0.6 | 26.0 ± 2.6 | 46.0 ± 15.0 | 2.3 ± 0.3 | ||
| 1 nm | 1.0 ± 0.0 | 75.0 ± 5.3 | 6.0 ± 1.7 | 21.6 ± 5.5 | 42.5 ± 13.4 | 3.0 ± 1.0 | 184.0 ± 27.0 | 1.3 ± 0.6 |
| 100 nm | 1.1 ± 0.1 | 48.0 ± 19.8 | 4.7 ± 1.2 | 36.5 ± 3.7 | 31.1 ± 1.3 | 15.0 ± 18.0 | ||
| 100 μm | 1.0 ± 0.1 | 62.3 ± 11.1 | 6.4 ± 2.4 | 30.3 ± 6.5 | 41.7 ± 17.2 | 7.3 ± 5.1 | ||
It is clear from the presented data that activating Zn2+-binding sites exist on the cytosolic face of RyR2. To assess whether Zn2+ binds to the same cytosolic activation sites on RyR2 as Ca2+, we first exposed RyR2 to a concentration of cytosolic Ca2+ known to elevate Po to peak levels (100 μm) (30) and then added subsequent cumulative doses of Zn2+ to the cytosolic face of the channel (Fig. 3). In the presence of 100 μm cytosolic Ca2+ and using Ca2+ as permeant ion, subsequent addition of cytosolic Zn2+ in the range 1 nm to 100 μm had no significant effect on RyR2 Po. Lifetime analysis, however, showed that at concentrations of Zn2+ > 1 nm channel gating was altered, and the channel dwelled in longer-lived open states, consistent with Ca2+-independent channel openings (21). These data suggest that at least some of the Zn2+-binding sites are distinct from the Ca2+-binding sites. Irrespective of whether the concentration of cytosolic Ca2+ was 10 or 100 μm, the addition of 1 mm cytosolic Zn2+ was always found to abolish channel openings. This suggests that in addition to high affinity Zn2+ activation sites, RyR2 also has low affinity Zn2+ inhibition sites.
FIGURE 3.

The effect of Zn2+ on RyR2 gating in the presence of a peak levels of Ca2+. A, subsequent effect of Zn2+ on RyR2 activation following exposure of the channel to 100 μm Ca2+ with Ca2+ as permeant ion, at a holding potential of 0 mV. O is the open state, and C is the closed state. B, the relationship between RyR2 Po and the concentration of Zn2+ in the continued presence of 10 μm Ca2+ (open squares) or 100 μm Ca2+ (open circles) is shown. The average Po using 10 μm (black dotted line) or 100 μm Ca2+ (gray dotted line) as the sole ligand is shown for comparison. Error bars show the mean Po value ± S.E. (n = 5). C, open (left) and closed (right) lifetime distributions for the channel shown in A in the presence of 10 μm cytosolic Ca2+ alone (control), after raising the cytosolic Ca2+ to a peak concentration of 100 μm and following the sequential addition of 1 and 100 nm Zn2+ to the cytosolic face of the channel in the continued presence of 100 μm Ca2+. The best fits to the data were obtained by maximum likelihood fitting. Time constants and percentage areas are shown for each distribution.
We next examined the interplay between Zn2+ and Ca2+ at the cytoplasmic face of RyR2 (Fig. 4). Using Ca2+ as permeant ion and holding at a command potential of 0 mV, when the free Ca2+ was reduced to a subactivating concentration (<10 nm) by the addition of 1 mm BAPTA, as expected, the Po value for RyR2 reduced to zero (31). The subsequent addition of 1 nm Zn2+ to the cytosolic face of the channel had no effect on Po, and the channel remained closed because under these conditions, channel gating is dependent on activation by cytosolic Ca2+. The subsequent addition of Zn2+ in the range 100 nm to 100 μm, however, resulted in channel activations. This is important because RyR2 is now gating in a Ca2+-independent manner, whereby Zn2+ is the sole activating ligand and the need for Ca2+ to activate the channel is removed.
FIGURE 4.
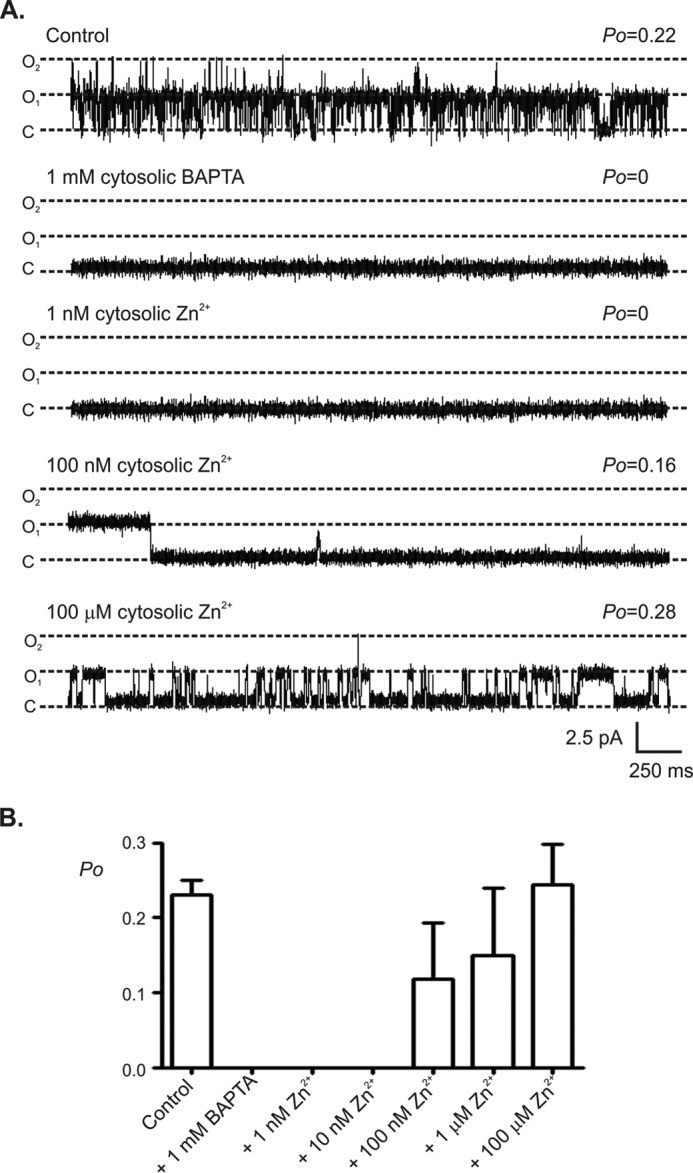
The effect of Zn2+ on RyR2 gating at a subactivating concentration of Ca2+. The top trace in A shows a typical single RyR2 channel with cytosolic Ca2+ as the sole activating ligand and using Ca2+ as the permeant ion. The bilayer was voltage-clamped at 0 mV. Subsequent addition of 1 mm BAPTA lowers the Ca2+ concentration to <10 nm, which is subactivating, and as expected the channel closes. Subsequent addition of 1 nm Zn2+ to the cytosolic face of the channel has no effect, and the channel remains closed. Subsequent addition of 100 nm Zn2+ to the cytosolic face of the channel causes the channel to open. These openings are independent of Ca2+. Further elevation in the Zn2+ concentration causes further channel activation. The open and closed states are denoted by O and C, respectively. The mean data in B show that there is a critical concentration of Zn2+ (10 nm), above which RyR2 openings are independent of the cytosolic Ca2+ concentration and channel openings are regulated solely by Zn2+. The data show the mean values ± S.E. (n = 5).
Effects of Cytosolic Zn2+ on Calcium Waves in Cardiac Cells
We then measured the effect of Zn2+ on Ca2+ waves recorded in isolated cardiomyocytes to provide evidence for the modulation of RyR2 function by Zn2+ in a cellular environment (Fig. 5). In these experiments, cardiomyocytes were permeabilized to allow the intracellular concentration of Zn2+ to be controlled, and Ca2+ fluorescence measurements were made using Fluo-4. In control recording conditions, where cardiomyocytes were perfused with an intracellular solution containing 100 nm free Ca2+, there were no appreciable change in Ca2+ release from the intracellular stores. Perfusion with increasing concentrations of Zn2+ (between 100 pm and 10 nm Zn2+), however, caused a marked increase in both the incidence of spontaneous Ca2+ waves and the amplitude of these events in a Zn2+ concentration-dependent manner (Fig. 5C). Increasing the Zn2+ concentration to >100 nm caused a marked decrease in the amplitude and frequency of waves, and the cardiomyocytes entered an irreversible series of Ca2+ waves often culminating in hypercontracture resulting in a loss of membrane integrity. This concentration of Zn2+ is close to the pathophysiological concentration of Zn2+ previously reported (∼30 nm) (33–36).
FIGURE 5.

The effect of intracellular Zn2+ on the frequency and amplitude of spontaneous Ca2+ waves in isolated cardiomyocytes. A, panels i and ii show a representative image of Fluo-4 fluorescence in a cardiomyocyte in control and 1 nm Zn2+ solution. B, a confocal line scan recording taken from a 5-pixel region of interest of Fluo-4 fluorescence from a single cardiomyocyte in control (panel i), 0.1 nm Zn2+ (panel ii), and 1 nm Zn2+ (panel iii). Confocal line scan recorded from the dotted line as indicated in A. C, the bar charts show the mean number of Ca2+ waves (panel i) and the mean amplitude of the waves from cardiomyocytes with increasing concentrations of Zn2+ (panel ii). (*, p < 0.05; ***, p < 0.001; one-way ANOVA with Bonferroni's post-hoc test, n ≥ 6 cardiomyocytes per data set).
Interestingly, lowering the concentration of cytosolic Ca2+ to subactivating levels while maintaining the concentration of Zn2+ did not diminish the Zn2+-induced frequency of the Ca2+ waves seen in the permeabilized cells. In the presence of 100 nm Zn2+, removal of Ca2+ by chelation with 1 mm BAPTA markedly reduced the wave amplitude (Fig. 6), presumably as a result of substantial Ca2+ buffering. We next examined the caffeine-induced release of SR Ca2+ in intact cardiomyocytes in the presence and absence of the ZnPy in Tyrode's solution containing 100 pm Zn2+ (Fig. 7). The increase in Fluo-4 fluorescence on application of 5 mm caffeine was markedly potentiated by treatment with 10 μm ZnPy, suggesting that the increase in intracellular Zn2+ enhanced the caffeine-induced Ca2+ release via RyR2. Consistent with our single channel studies, this shows that Zn2+ can directly influence cellular Ca2+ dynamics through direct modulation of RyR2. The finding that Zn2+ can act as the sole activator of RyR2 highlights a potential pathophysiological consequence of dysregulated Zn2+ homeostasis. Such a deleterious cycling of intracellular Ca2+ with pathophysiological concentrations of Zn2+ could be arrhythmogenic in the intact myocardium.
FIGURE 6.

The effect of buffering intracellular Ca2+ with BAPTA on wave amplitude and frequency in isolated cardiomyocytes. A, an example image of an isolated cardiomyocyte in control (panel i), in 10 nm Zn2+ solution (panel ii), and in 10 nm Zn2+ solution containing 1 mm BAPTA (panel iii). B, an example confocal line scan and associated fluorescence changes from a 5-pixel region of interest in control (panel i), in 10 nm Zn2+ solution (panel ii), and in 10 nm Zn2+ solution containing 1 mm BAPTA (panel iii). C, bar charts compare the mean number of Ca2+ waves (panel i) and the mean wave amplitude in control and after treatment with 1 mm BAPTA with increasing concentrations of Zn2+ (panel ii) (***, p < 0.001; two-way ANOVA with Bonferroni's post-hoc test, n ≥ 6 cardiomyocytes per data set).
FIGURE 7.
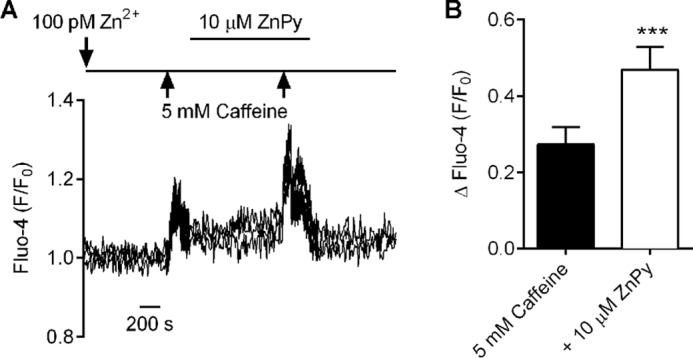
Elevated intracellular Zn2+ levels increase the caffeine-induced Ca2+ release. A, example trace showing Fluo-4 fluorescence from three cardiomyocytes perfused with 100 pm Zn2+-Tyrode's solution with applications of 5 mm caffeine in the absence and presence of 10 μm ZnPy. B, mean change in Fluo-4 fluorescence on application of 5 mm caffeine in 100 pm Zn2+-Tyrode's control solution and after application of 10 μm ZnPy (n = 4 experiments (66 cardiomyocytes) from 2 animals; ***, p < 0.001, paired t test).
Further evidence to suggest that changes in intracellular Zn2+ levels have a marked effect on cellular cardiac contractions comes from raising intracellular Zn2+ levels by pretreating intact cardiomyocytes with 10 μm ZnPy and exposing these cells to 100 pm extracellular ZnCl2. Under these conditions, there was a significant increase in the inward current carried by Zn2+ (Fig. 8, A and B), and a concomitant increase in intracellular Zn2+ measured using the fluorescent indicator Zinpyr-1 (Fig. 8, C and D). As shown by edge detection measurements, this resulted in spontaneous contractions compared with control cells (Fig. 9, A and B). The observed spontaneous contractions were reversed by chelating intracellular Zn2+ by the addition of 10 μm TPEN (Fig. 9, C and D), confirming that these cellular effects are attributed to elevated intracellular Zn2+ levels.
FIGURE 8.
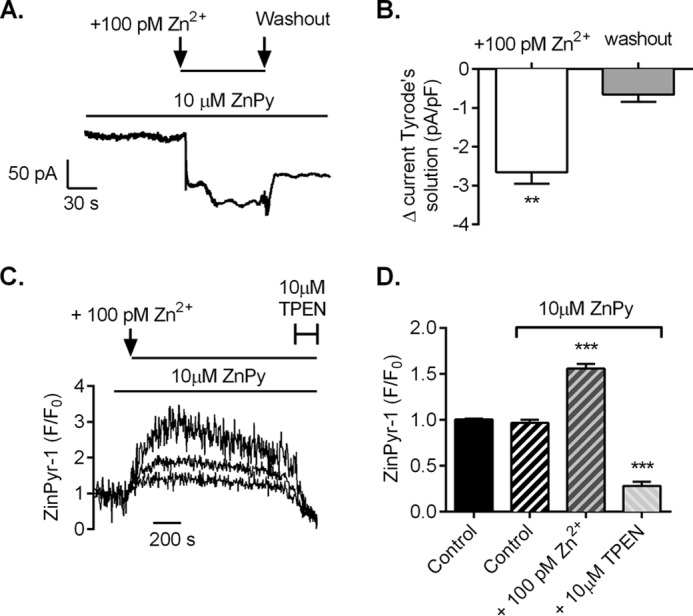
Elevated extracellular zinc levels result in an increase in the concentration of intracellular zinc. A, example whole cell recording showing the development of an inward current on application of a Tyrode's solution containing an additional 100 pm Zn2+ to an isolated cardiomyocyte voltage-clamped at 0 mV. B, mean data showing the increase in current from basal in 100 pm Zn2+ and washout (n = 4 cardiomyocytes from 2 animals; **, p < 0.01, repeated-measured ANOVA with Bonferroni's post-test). C, example fluorescence recordings from three cardiomyocytes loaded with 10 μm Zinpyr-1 showing that application of 10 μm ZnPy facilitates an increase in intracellular Zn2+ when extracellular Zn2+ is increased. D, mean change in intracellular Zinpyr-1 fluorescence in control, in the presence of ZnPy and after application of TPEN (n = 4 experiments (156 cardiomyocytes) from 2 animals; ***, p < 0.001; one-way ANOVA with Bonferroni's post-test.).
FIGURE 9.

Elevated intracellular Zn2+ can evoke spontaneous contractions in intact cardiomyocytes. A, edge detection measurements from a single cardiomyocyte treated with 10 μm ZnPy in control (panel i), with an additional 10 pm ZnCl2 (panel ii), and with an additional 100 pm ZnCl2 showing increasing spontaneous contractions with increasing [Zn2+] (panel iii). B, mean percentage of cardiomyocytes showing spontaneous contractions with increasing [Zn2+] in the presence (black circles) and absence (white circles) of 10 μm ZnPy (n = 4 experiments (>100 cardiomyocytes) from 2 animals for each data set). C, mean percentage of cardiomyocytes showing spontaneous contractions before (dashed line) and after (gray circles) pretreatment with 10 μm TPEN. Cardiomyocytes were perfused either with control-Tyrode's solution alone or with an additional 10 or 100 pm Zn2+-Tyrode's solution with 10 μm ZnPy. D, mean percentage of cells showing spontaneous contractions in 100 pm Zn2+ in the absence or presence of 10 μm ZnPy and with 10 μm ZnPy plus 10 μm TPEN (n = 4 experiments (>100 cardiomyocytes) from 2 animals for each data set; **, p < 0.01; one-way ANOVA with Bonferroni's post-test).
Discussion
In this study, we reveal that cytosolic Zn2+ is a high affinity activator of RyR2, effective at low picomolar concentrations. Lifetime analysis of RyR2 reveals unique and distinctive gating characteristics dependent on the concentration of cytosolic Zn2+. When Zn2+ levels are in the range 100 pm to 1 nm, RyR2 activation occurs by sensitization of the channel to cytosolic Ca2+. When the concentration of Zn2+ is elevated above 1 nm, the effect of Zn2+ overrides the influence of cytosolic Ca2+, and removal of activating levels of cytosolic Ca2+ no longer influences channel Po. When Zn2+ is elevated to toxic levels (in the millimolar range), it inhibits RyR2 channel function. Further evidence for the modulation of RyR2 function by Zn2+ in isolated permeabilized cardiomyocytes was acquired through measurement of Ca2+ waves. In these recordings, spontaneous Ca2+ waves were markedly increased in both frequency and amplitude in a Zn2+ concentration-dependent manner (between 100 pm and 10 nm Zn2+). Removal of Ca2+ by chelation with 1 mm BAPTA did not abolish Ca2+ waves in the presence of Zn2+, showing that Zn2+ can directly influence Ca2+ dynamics. These findings suggest an important pathophysiological consequence of dysregulated Zn2+ homeostasis, whereby even a small increase in RyR2 open probability during diastole can have major consequences for normal cardiac function and the generation of arrhythmias (37).
It is well established that micromolar concentrations of cytosolic Ca2+ are required to activate RyR2 (38, 39). The potentiation of RyR2 activity by physiological concentrations of Zn2+ that we now show suggests that Zn2+ plays a key role in shaping intracellular Ca2+ responses in cardiac muscle. Tight regulation of intracellular Zn2+ will therefore be required for the controlled release of Ca2+ from SR stores because very small changes in the concentration of free Zn2+ will modify the opening of RyR2 channels in response to very small rises in cytosolic Ca2+, and this may cause channels to become leaky during diastole. Under pathophysiological conditions, where Zn2+ homeostasis is disturbed and levels of Zn2+ may reach >1 nm, we propose that Zn2+ becomes the main activating ligand of RyR2. Under these conditions, RyR2 starts to gate in exceptionally long open states. The ability of 1 nm Zn2+ to activate RyR2 reveals that Zn2+ is a high affinity activator of RyR2 displaying approximately 3 orders of magnitude higher affinity for RyR2 than Ca2+. This alters the current model of cardiac excitation-contraction coupling where it is accepted that a transient changes in the concentration of intracellular Ca2+ is the only trigger for SR Ca2+ release and reveals a new role for Zn2+ in shaping intracellular calcium responses in cardiac muscle.
Determination of the exact levels of intracellular Zn2+ under physiological and pathophysiology conditions is an ongoing area of research, but the development of more sensitive Zn2+ probes that are able to measure low concentrations of free Zn2+ in single living cells is rapidly advancing (40). At reported pathophysiological levels of cytosolic Zn2+ (∼30 nm) (33–36), we show that increases in RyR2 activity are the result of large increases in the lifetime duration of the open channel state rather than an increase in the frequency of openings and this is characteristic of a Ca2+-independent component of channel gating. In the presence of ≥10 nm free Zn2+, we show that chelation of cytosolic Ca2+ is no longer able to close RyR2 channels. This is because there is sufficient Zn2+ present to directly open the channel. The balance of intracellular Zn2+ is therefore crucial in switching RyR2 channels from a “Ca2+-dependent” to a “Ca2+-independent” mode of gating.
Our mechanistic approach has for the first time revealed that Zn2+ is a potent activator of RyR2. Previous studies in which cardiac muscle [3H]ryanodine binding assays were used to assess the effects of Zn2+ on RyR2 function failed to see any effect of Zn2+ between 10 nm and 1 μm in the presence of 50 μm activating Ca2+ (41). This is not surprising because our data now reveal that in the presence of peak levels of Ca2+, the effect of Zn2+ on channel Po is masked. Only by lifetime analysis of our single channel data could we reveal that the mode of channel gating in the presence of Zn2+ (>1 nm) was altered and that this type of gating was consistent with Ca2+-independent openings. Our single channel data also provide the first evidence that when the concentration of Zn2+ is elevated above 1 nm, RyR2 becomes directly activated by Zn2+, and the dependence of Ca2+ on channel activation is abolished.
It is notable that previous studies have shown a slow accumulation of intracellular Zn2+ following prolonged exposure of cardiomyocytes to micromolar levels of extracellular Zn2+ and that this results in a reduced SR Ca2+ load (42). One plausible explanation for these findings, based on our current data, is that an elevation in intracellular Zn2+ removes the Ca2+ dependence of RyR2 causing channels to gate in exceptionally long-lived open states. Further evidence for this is demonstrated in intact cardiomyocytes where, in a 100 pm Zn2+-Tyrode's solution, the caffeine-induced elevation in intracellular Ca2+ was potentiated only after application of 10 μm ZnPy. Potentially this will lead to inappropriate Ca2+ release from the SR and likely result in reduced SR Ca2+ load. Such a mechanism would be particularly problematic under conditions where levels of intracellular Zn2+ are chronically elevated, including diabetes (36) and certain models of dystrophy (43). In line with previous studies (41, 44), we also demonstrated an inhibitory action of Zn2+ on RyR2 gating when cytosolic Zn2+ is raised to nonphysiological and toxic millimolar concentrations. It is possible that the inhibitory action of Zn2+ on RyR2 may be a consequence of Zn2+ binding to the low affinity unspecific divalent inhibitory site of RyR2 (29, 45).
Aberrant Zn2+ homeostasis has been shown to be associated with cardiomyopathy (46–48), but the underlying mechanism of how Zn2+ contributes to cardiac pathology is unknown. Chronic oxidative stress is a major contributor in the pathophysiology of heart failure and under these conditions intracellular levels of Zn2+ increase by as much as 30-fold (36). This increase in Zn2+ is primarily the result of altered metallothionein function, which perturbs its binding of Zn2+, resulting in raised levels (49, 50). Also, recently, a polymorphism in a human gene coding for a member of the metallothionein family has been discovered, and individuals with diabetes who carry this polymorphism have been shown to be more likely to develop cardiovascular complications including chronic heart failure and myocardial damage (51). Diabetic cardiomyopathy is characterized by dysregulation of intracellular Ca2+ release, which consequently reduces cardiac contractility and significantly prolongs the rise of systolic Ca2+ leading to chronic heart failure (16). The controlled release of Ca2+ from the SR during cardiac excitation-contraction coupling is known to govern contractility of the heart. RyR2 plays a fundamental role as the main pathway for the release of Ca2+ and drives cellular contraction. Consequently dysfunction in the release of Ca2+ and modulatory influences that control RyR2 function are identified as contributory to the pathophysiology of heart failure and fatal cardiac arrhythmias (1). The presented study therefore provides a mechanistic explanation as to how raised levels of intracellular Zn2+ may contribute to the progression of contractile dysfunction and heart failure through the alteration of RyR2 gating, which will result in perturbed Ca2+ release.
A simple model to explain how cytosolic Zn2+ modulates RyR2 gating is shown in Fig. 10. We suggest that under physiological conditions, cytosolic Zn2+ is able to fine-tune the release of Ca2+ from the SR while in parallel enabling flexibility and control of SR Ca2+ dynamics mediated through RyR2. Under pathophysiological conditions, where Zn2+ homeostasis is disturbed and levels of Zn2+ may reach >1 nm, RyR2 starts to gate in exceptionally long open states, which leads to very high Po potentially leading to arrhythmia. In this conformation, RyR2 is uncoupled from the usual regulatory effects of cytosolic Ca2+ and RyR2 channels are now activated by solely by Zn2+.
FIGURE 10.
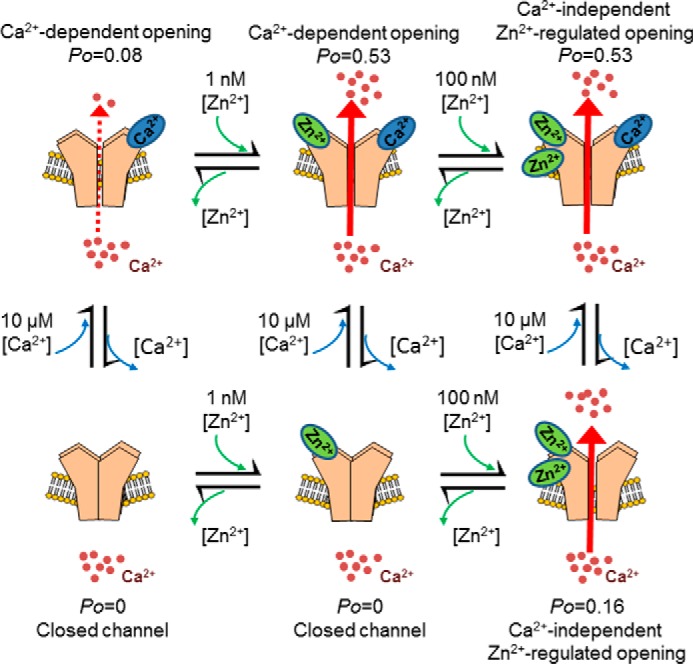
Proposed model of how Zn2+ regulates RyR2-mediated Ca2+ release. When 10 μm cytosolic Ca2+ is the sole ligand, channel openings are brief, and the channel gates with a low Po. Addition of 1 nm Zn2+ to the cytosolic face of the channel causes an increase in the channel Po, and the channel gates with high frequency openings. In both of these conditions, RyR2 gating is regulated by cytosolic Ca2+, and lowering the concentration of free Ca2+ to subactivating levels (<10 nm) reduces RyR2 Po to 0. If the Zn2+ concentration is elevated above 1 nm, RyR2 becomes uncoupled from the usual regulatory effects of cytosolic Ca2+, and Zn2+ becomes the sole activating ligand.
Our study reveals that RyR2-mediated Ca2+ homeostasis is intimately related to intracellular Zn2+ levels and provides a mechanistic explanation as to how altered Zn2+ homeostasis can modulate cardiac RyR2 function. We reveal that Zn2+ is a high affinity activator of RyR2 which challenges our understanding of cardiac excitation-contraction coupling. We suggest that under normal physiological conditions, intracellular Zn2+ is essential in fine-tuning the release of Ca2+ from the SR. Pathological perturbations in Zn2+ homoeostasis will lead to inappropriate release of Ca2+ as observed in certain cardiac abnormalities including heart failure and fatal arrhythmias.
Author Contributions
J. W. and R. D. R. performed experiments and analyzed data; A. J. S. designed the experiments and contributed toward writing the manuscript; and S. J. P. designed the experiments, performed experiments, analyzed data, wrote the manuscript, and supervised the project. All authors discussed the results and implications and commented on the manuscript at all stages.
Acknowledgments
With special thanks to the Dunblane and Shotts abattoirs for donating sheep heart for use in this study. We also thank Benedict Reilly O'Donnell and Gavin Robertson for providing technical assistance with measurements of free Zn2+ and assisting with the single channel TPEN data. We are grateful to Dr. Walter Geibert (University of Edinburgh, Grant Institute) for technical assistance with the ICP-OES analysis. We also thank Prof. Richard Thompson (University of Maryland) for advice on performing accurate measurements of free zinc at very low concentrations.
This work was supported by University of St. Andrews, Tenovus Scotland Grant T14/35 (to S. J. P.), British Heart Foundation Grant FS/14/69/31001 (to S. J. P.), a grant from the John and Lucille van Geest Cardiovascular Diseases Research Fund (to the University of Leicester and R. D. R.), and Biotechnology and Biological Sciences Research Council Grant BB/J006467/1 (to A. J. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- RyR2
- type 2 ryanodine receptor
- SR
- sarcoplasmic reticulum
- BAPTA
- 2,2′-(ethylenedioxy)dianiline-N,N,N′,N′-tetraacetic acid
- TPEN
- N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine
- ZnPy
- zinc pyrithione
- ANOVA
- analysis of variance.
References
- 1. Belevych A. E., Radwański P. B., Carnes C. A., Györke S. (2013) “Ryanopathy”: causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc. Res. 98, 240–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eisner D., Bode E., Venetucci L., Trafford A. (2013) Calcium flux balance in the heart. J. Mol. Cell. Cardiol. 58, 110–117 [DOI] [PubMed] [Google Scholar]
- 3. Kranias E. G., Bers D. M. (2007) Calcium and cardiomyopathies. Subcell. Biochem. 45, 523–537 [DOI] [PubMed] [Google Scholar]
- 4. Maret W. (2001) Crosstalk of the group IIa and IIb metals calcium and zinc in cellular signaling. Proc. Natl. Acad. Sci. U.S.A. 98, 12325–12327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fukada T., Yamasaki S., Nishida K., Murakami M., Hirano T. (2011) Zinc homeostasis and signaling in health and diseases. J. Biol. Inorg. Chem. 16, 1123–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamasaki S., Sakata-Sogawa K., Hasegawa A., Suzuki T., Kabu K., Sato E., Kurosaki T., Yamashita S., Tokunaga M., Nishida K., Hirano T. (2007) Zinc is a novel intracellular second messenger. J. Cell Biol. 177, 637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colvin R. A., Holmes W. R., Fontaine C. P., Maret W. (2010) Cytosolic zinc buffering and muffling: their role in intracellular zinc homeostasis. Metallomics 2, 306–317 [DOI] [PubMed] [Google Scholar]
- 8. Krezel A., Maret W. (2006) Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J. Biol. Inorg. Chem. 11, 1049–1062 [DOI] [PubMed] [Google Scholar]
- 9. Mocchegiani E., Giacconi R., Malavolta M. (2008) Zinc signalling and subcellular distribution: emerging targets in type 2 diabetes. Trends Mol. Med. 14, 419–428 [DOI] [PubMed] [Google Scholar]
- 10. Lichten L. A., Cousins R. J. (2009) Mammalian zinc transporters: nutritional and physiologic regulation. Annu. Rev. Nutr. 29, 153–176 [DOI] [PubMed] [Google Scholar]
- 11. Chabosseau P., Tuncay E., Meur G., Bellomo E. A., Hessels A., Hughes S., Johnson P. R., Bugliani M., Marchetti P., Turan B., Lyon A. R., Merkx M., Rutter G. A. (2014) Mitochondrial and ER-targeted eCALWY probes reveal high levels of free Zn2+. ACS Chem. Biol. 9, 2111–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Turan B., Fliss H., Désilets M. (1997) Oxidants increase intracellular free Zn2+ concentration in rabbit ventricular myocytes. Am. J. Physiol. 272, H2095–H2106 [DOI] [PubMed] [Google Scholar]
- 13. Tuncay E., Bilginoglu A., Sozmen N. N., Zeydanli E. N., Ugur M., Vassort G., Turan B. (2011) Intracellular free zinc during cardiac excitation-contraction cycle: calcium and redox dependencies. Cardiovasc. Res. 89, 634–642 [DOI] [PubMed] [Google Scholar]
- 14. Alvarez-Collazo J., Díaz-García C. M., López-Medina A. I., Vassort G., Alvarez J. L. (2012) Zinc modulation of basal and β-adrenergically stimulated L-type Ca2+ current in rat ventricular cardiomyocytes: consequences in cardiac diseases. Pflugers Arch. 464, 459–470 [DOI] [PubMed] [Google Scholar]
- 15. Atar D., Backx P. H., Appel M. M., Gao W. D., Marban E. (1995) Excitation-transcription coupling mediated by zinc influx through voltage-dependent calcium channels. J. Biol. Chem. 270, 2473–2477 [DOI] [PubMed] [Google Scholar]
- 16. Reuter H., Grönke S., Adam C., Ribati M., Brabender J., Zobel C., Frank K. F., Wippermann J., Schwinger R. H., Brixius K., Müller-Ehmsen J. (2008) Sarcoplasmic Ca2+ release is prolonged in nonfailing myocardium of diabetic patients. Mol. Cell. Biochem. 308, 141–149 [DOI] [PubMed] [Google Scholar]
- 17. Kleinfeld M., Stein E. (1968) Action of divalent cations on membrane potentials and contractility in rat atrium, Am. J. Physiol. 215, 593–599 [DOI] [PubMed] [Google Scholar]
- 18. Ciofalo F. R., Thomas L. J., Jr. (1965) The effects of zinc on contractility, membrane potentials, and cation content of rat atria. J. Gen. Physiol. 48, 825–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kalfakakou V. P., Evangelou A. M., Benveniste J., Arnoux B. (1993) The effects of Zn2+ on guinea pig isolated heart preparations. Biol. Trace Elem. Res. 38, 289–299 [DOI] [PubMed] [Google Scholar]
- 20. Sitsapesan R., Montgomery R. A., MacLeod K. T., Williams A. J. (1991) Sheep cardiac sarcoplasmic reticulum calcium release channels: modification of conductance and gating by temperature. J. Physiol. 434, 469–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carter S., Pitt S. J., Colyer J., Sitsapesan R. (2011) Ca2+-dependent phosphorylation of RyR2 can uncouple channel gating from direct cytosolic Ca2+ regulation. J. Membr. Biol. 240, 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Galfré E., Pitt S. J., Venturi E., Sitsapesan M., Zaccai N. R., Tsaneva-Atanasova K., O'Neill S., Sitsapesan R. (2012) FKBP12 activates the cardiac ryanodine receptor Ca2+-release channel and is antagonised by FKBP12.6. PLoS One 7, e31956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bozym R., Hurst T. K., Westerberg N., Stoddard A., Fierke C. A., Frederickson C. J., Thompson R. B. (2008) Determination of zinc using carbonic anhydrase-based fluorescence biosensors, Methods Enzymol. 450, 287–309 [DOI] [PubMed] [Google Scholar]
- 24. Colquhoun D., Sigworth F. J. (1983) Fitting and statistical analysis of single-channel recording. In Single-channel Recording (Sakmann B., Neher E., eds) pp. 191–263, Plenum, New York [Google Scholar]
- 25. Sigworth F. J., Sine S. M. (1987) Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys. J. 52, 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sims M. W., Winter J., Brennan S., Norman R. I., Ng G. A., Squire I. B., Rainbow R. D. (2014) PKC-mediated toxicity of elevated glucose concentration on cardiomyocyte function. Am. J. Physiol. Heart Circ. Physiol. 307, H587–H597 [DOI] [PubMed] [Google Scholar]
- 27. Somlyo A. V., Gonzalez-Serratos H. G., Shuman H., McClellan G., Somlyo A. P. (1981) Calcium release and ionic changes in the sarcoplasmic reticulum of tetanized muscle: an electron probe study. J. Cell Biol. 90, 577–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415, 198–205 [DOI] [PubMed] [Google Scholar]
- 29. Laver D. R., Baynes T. M., Dulhunty A. F. (1997) Magnesium inhibition of ryanodine-receptor calcium channels: evidence for two independent mechanisms. J. Membr. Biol. 156, 213–229 [DOI] [PubMed] [Google Scholar]
- 30. Ashley R. H., Williams A. J. (1990) Divalent cation activation and inhibition of single calcium release channels from sheep cardiac sarcoplasmic reticulum. J. Gen. Physiol. 95, 981–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stewart R., Song L., Carter S. M., Sigalas C., Zaccai N. R., Kanamarlapudi V., Bhat M. B., Takeshima H., Sitsapesan R. (2008) Single-channel characterization of the rabbit recombinant RyR2 reveals a novel inactivation property of physiological concentrations of ATP. J. Membr. Biol. 222, 65–77 [DOI] [PubMed] [Google Scholar]
- 32. Sitsapesan R., Williams A. J. (1990) Mechanisms of caffeine activation of single calcium-release channels of sheep cardiac sarcoplasmic reticulum. J. Physiol. 423, 425–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sensi S. L., Paoletti P., Bush A. I., Sekler I. (2009) Zinc in the physiology and pathology of the CNS. Nat Rev. Neurosci. 10, 780–791 [DOI] [PubMed] [Google Scholar]
- 34. Aizenman E., Stout A. K., Hartnett K. A., Dineley K. E., McLaughlin B., Reynolds I. J. (2000) Induction of neuronal apoptosis by thiol oxidation. J. Neurochem. 75, 1878–1888 [DOI] [PubMed] [Google Scholar]
- 35. Li Y., Maret W. (2009) Transient fluctuations of intracellular zinc ions in cell proliferation. Exp. Cell Res. 315, 2463–2470 [DOI] [PubMed] [Google Scholar]
- 36. Ayaz M., Turan B. (2006) Selenium prevents diabetes-induced alterations in [Zn2+]i and metallothionein level of rat heart via restoration of cell redox cycle. Am. J. Physiol. Heart Circ. Physiol. 290, H1071–H1080 [DOI] [PubMed] [Google Scholar]
- 37. Bers D. M. (2008) Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49 [DOI] [PubMed] [Google Scholar]
- 38. Rousseau E., Smith J. S., Henderson J. S., Meissner G. (1986) Single channel and 45Ca2+ flux measurements of the cardiac sarcoplasmic reticulum calcium channel. Biophys. J. 50, 1009–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu L., Mann G., Meissner G. (1996) Regulation of cardiac Ca2+ release channel (ryanodine receptor) by Ca2+, H+, Mg2+ and adenine nucleotides under normal and simulated ischemic conditions. Circ. Res. 79, 1100–1109 [DOI] [PubMed] [Google Scholar]
- 40. Vinkenborg J. L., Nicolson T. J., Bellomo E. A., Koay M. S., Rutter G. A., Merkx M. (2009) Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat. Methods 6, 737–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang H., Wei Q. Q., Cheng X. Y., Chen K. Y., Zhu P. H. (2001) Inhibition of ryanodine binding to sarcoplasmic reticulum vesicles of cardiac muscle by Zn2+ ions. Cell Physiol. Biochem. 11, 83–92 [DOI] [PubMed] [Google Scholar]
- 42. Yi T., Vick J. F., Vecchio M. F., Begin K. F., Bell S. F., Delay R. F., Palmer B. M. (2013) Identifying cellular mechanisms of zinc-induced relaxation in isolated cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 305, H706–H715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Crawford A. J., Bhattacharya S. K. (1987) Excessive intracellular zinc accumulation in cardiac and skeletal muscles of dystrophic hamsters. Exp. Neurol. 95, 265–276 [DOI] [PubMed] [Google Scholar]
- 44. Xie H., Chen K. Y., Zhu P. H. (2004) Effect of Zn2+ ions on ryanodine binding to sarcoplasmic reticulum of striated muscles in the presence of pyrithione. Acta Pharmacol. Sin. 25, 1647–1651 [PubMed] [Google Scholar]
- 45. Diaz-Sylvester P. L., Porta M., Copello J. A. (2011) Modulation of cardiac ryanodine receptor channels by alkaline earth cations. PLoS One 6, e26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Little P. J., Bhattacharya R., Moreyra A. E., Korichneva I. L. (2010) Zinc and cardiovascular disease. Nutrition 26, 1050–1057 [DOI] [PubMed] [Google Scholar]
- 47. Sandstead H. H. (1995) Requirements and toxicity of essential trace elements, illustrated by zinc and copper. Am. J. Clin. Nutr. 61, 621S–624S [DOI] [PubMed] [Google Scholar]
- 48. Alexanian I., Parissis J., Farmakis D., Athanaselis S., Pappas L., Gavrielatos G., Mihas C., Paraskevaidis I., Sideris A., Kremastinos D., Spiliopoulou C., Anastasiou-Nana M., Lekakis J., Filippatos G. (2014) Clinical and echocardiographic correlates of serum copper and zinc in acute and chronic heart failure. Clin. Res. Cardiol. 103, 938–949 [DOI] [PubMed] [Google Scholar]
- 49. Maret W. (1994) Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc. Natl. Acad. Sci. U.S.A. 91, 237–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maret W. (1995) Metallothionein/disulfide interactions, oxidative stress, and the mobilization of cellular zinc. Neurochem. Int. 27, 111–117 [DOI] [PubMed] [Google Scholar]
- 51. Giacconi R., Bonfigli A. R., Testa R., Sirolla C., Cipriano C., Marra M., Muti E., Malavolta M., Costarelli L., Piacenza F., Tesei S., Mocchegiani E. (2008) +647 A/C and +1245 MT1A polymorphisms in the susceptibility of diabetes mellitus and cardiovascular complications. Mol. Genet. Metab 94, 98–104 [DOI] [PubMed] [Google Scholar]