

Abstract
Aims
Notch and activin receptor-like kinase 1 (ALK1) have been implicated in arterial specification, angiogenic tip/stalk cell differentiation, and development of arteriovenous malformations (AVMs), and ALK1 can cooperate with Notch to up-regulate expression of Notch target genes in cultured endothelial cells. These findings suggest that Notch and ALK1 might collaboratively program arterial identity and prevent AVMs. We therefore sought to investigate the interaction between Notch and Alk1 signalling in the developing vertebrate vasculature.
Methods and results
We modulated Notch and Alk1 activities in zebrafish embryos and examined effects on Notch target gene expression and vascular morphology. Although Alk1 is not necessary for expression of Notch target genes in arterial endothelium, loss of Notch signalling unmasks a role for Alk1 in supporting hey2 and ephrinb2a expression in the dorsal aorta. In contrast, Notch and Alk1 play opposing roles in hey2 expression in cranial arteries and dll4 expression in all arterial endothelium, with Notch inducing and Alk1 repressing these genes. Although alk1 loss increases expression of dll4, AVMs in alk1 mutants could neither be phenocopied by Notch activation nor rescued by Dll4/Notch inhibition.
Conclusion
Control of Notch targets in arterial endothelium is context-dependent, with gene-specific and region-specific requirements for Notch and Alk1. Alk1 is not required for arterial identity, and perturbations in Notch signalling cannot account for alk1 mutant-associated AVMs. These data suggest that AVMs associated with ALK1 mutation are not caused by defective arterial specification or altered Notch signalling.
Keywords: Activin receptor-like kinase 1, Notch, Arteriovenous malformation, Zebrafish, Angiogenesis
1. Introduction
When activated by transmembrane ligands of the Delta and Jagged families, the Notch intracellular domain (NICD) is cleaved, translocates to the nucleus, binds to the DNA binding protein, recombination signal binding protein for immunoglobulin kappa J (RBPJ), and induces target gene expression.1 In the vasculature, delta-like ligand 4 (Dll4)/Notch signalling controls arterial specification and angiogenic tip/stalk cell selection,2–5 and in mouse and zebrafish models, decreased Dll4/Notch function leads to direct connections between arteries and veins, or arteriovenous malformations (AVMs).2,6–9 Because Dll4/Notch signalling transcriptionally up-regulates the arterial endothelial marker, ephrinb2 (Efnb2),10 and decreased Dll4/Notch signalling results in loss of Efnb2 and ectopic arterial expression of the venous marker, Ephb4,2,6–8,10–13 AVMs resulting from decreased Dll4/Notch signalling are generally attributed to disruption of arterial-venous identity. Notch loss-of-function (Notchlof) generates small calibre AVMs that are associated with thin, nearly atretic arteries.6,7,14 Notch gain-of-function (Notchgof), which enhances Efnb2 and represses Ephb4, also results in AVMs in mice,2,6–8,10–13 and human brain AVMs exhibit increased Notch signalling.15,16 AVMs associated with Notchgof involve enlarged arteries containing supernumerary endothelial cells,8,9,11,12,17 suggesting that failed repulsion mediated by EfnB2/EphB4, which is required for segregation of venous and arterial cells in developing vessels,18,19 may be responsible for these AVMs. Thus, both Notchlof and Notchgof result in AVMs associated with disrupted arterial-venous identity, but the morphological characteristics of these AVMs are distinct, with low flow, small calibre shunts associated with Notchlof and high flow, large calibre shunts associated with Notchgof.
Similar to Notch signalling, bone morphogenetic protein (BMP) signalling has also been implicated in AVM prevention. BMP ligands bind to a heterotetrameric complex of type I and type II serine/threonine kinase receptors; non-signalling type III receptors facilitate ligand binding. Upon complex formation, type II receptors phosphorylate type I receptors, which in turn phosphorylate Smad1, Smad5, and Smad9. Phosphorylated Smads bind to Smad4, translocate to the nucleus, and bind to DNA to regulate gene expression.20 In humans, heterozygous loss of endoglin (ENG, encoding a type III receptor), activin receptor-like kinase 1 (ACVRL1 or ALK1, encoding a type I receptor), or SMAD4 results in hereditary haemorrhagic telangiectasia (HHT), a disease characterized by a predisposition to development of telangiectasias and AVMs.21–24 Alk1 mutant mice exhibit decreased Efnb2 expression in the dorsal aorta (DA),25 and BMP9/ALK1 transcriptionally induces EFNB2 in cultured human umbilical artery endothelial cells,13 suggesting that ALK1, similar to Notch, is required for arterial identity. Also like Notch, ALK1 has been implicated in maintenance of stalk cell identity.26 Thus, ALK1 and Notch might function in a common pathway to control arterial and/or stalk cell identity and prevent AVMs.
Several lines of evidence suggest that Notch and ALK1 interact in endothelial cells. Activation of either DLL4/Notch or BMP9/ALK1 in cultured endothelial cells enhances expression of canonical Notch targets HEY1, HEY2, and HES1,13,26,27 and simultaneous activation of these pathways synergistically increases expression of HEY1 and HEY2.26 A less dramatic effect on HEY2 expression is observed in response to combined stimulation by constitutively active Notch (NICD) and ALK1 (ALK1CA).10 Induction of HEY1 and HEY2 by BMP9/ALK1 requires SMAD4 but not RBPJ,26 and BMP9 induces SMAD1/5 binding to HEY1, HEY2, and HES1 promoters in cultured human umbilical vein endothelial cells (HUVECs).28 These findings suggest that BMP9/ALK1 directly stimulates canonical Notch targets by a SMAD-dependent and NICD/RBPJ-independent pathway. Notch and ALK1 additively induce VEGFR1, a stalk cell marker, and inhibit APELIN, a tip cell marker, and endothelial cells lacking ALK1, SMAD4, or HEY2 are more likely to be in the tip position.26 Together, these results suggest that BMP9/ALK1/SMAD signalling induces canonical Notch targets independently of NICD/RBPJ and reinforces Notch-mediated acquisition of arterial identity and maintenance of stalk cell fate.
Although Notch and ALK1 exhibit synergistic interactions with respect to Notch target gene expression in cultured endothelial cells, evidence for synergy is less compelling for phenotypic endpoints. Both NICD and ALK1CA dampen endothelial cell sprouting, but simultaneous pathway activation shows no further effects, and inhibition of one pathway does not inhibit effects of activation of the other pathway.13 Similarly, both ALK1 and Notch inhibition enhance VEGF-stimulated tube formation in HUVECs and increase vascular area in the postnatal retina; however, combined inhibition of these pathways shows less than additive effects.26 Thus, the synergy between Notch and ALK1 in controlling gene expression may not directly translate to synergistic effects on endothelial cell behaviour.
To better understand the interaction between Notch and Alk1, we assayed Notch target gene expression and vascular phenotype in zebrafish embryos with altered Notch and/or Alk1 activity. Results demonstrate that Alk1 is not necessary for maintenance of Notch target genes or arterial identity in vivo. However, concomitant inhibition of Notch and Alk1 revealed context-dependent interactions, with cooperative maintenance of hey2 and efnb2a in the DA but opposing effects on hey2 and no effect on efnb2a in cranial arteries. In addition, we observed increased dll4 expression in both trunk and cranial arterial endothelium in the absence of Alk1 signalling, in contrast to the loss of expression observed with Notch inhibition. These molecular data suggested that AVMs in alk1 mutants might arise due to Notch gain-of-function; however, Notch activation failed to phenocopy and Notch inhibition failed to rescue AVMs associated with loss of alk1. Taken together, these data demonstrate that Notch and Alk1 exhibit context-specific and target-specific interactions in controlling Notch target gene expression in vivo, and that AVMs associated with Alk1 deficiency do not result from perturbations in Notch activity.
2. Methods
Additional details can be found in Supplementary material online.
2.1. Zebrafish lines and maintenance
All zebrafish (Danio rerio) experiments conformed to NIH/NRC guidelines (Guide for the Care and Use of Laboratory Animals) and were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (protocol 12070690690). Animals were maintained according to standard protocols.29 Mutant lines alk1ft09e (p.Y88X), alk1y6 (p.L240F), and alk1s407 (g.IVS8-2A>T) are functional nulls and have indistinguishable phentoyptes.30–32 Transgenic lines Tg(fli1a:GAL4FF)ubs3, Tg(UAS:Kaede)rk8, Tg(5xUAS-E1b:6xMYC-notch1a)kca3, Tg(EPV.TP1-Mmu.Hbb:egfp)um14 [abbreviated Tg(tp1:egfp)um14], Tg(kdrl:gfp)la116, Tg(gata1a:dsred)sd2 and Tg(fli1a.ebs:alk1CA-mCherry) have been described.33–38 Tg(fli1a.ebs:alk1CA-mCherry) is embryonic lethal; therefore, F1 embryos were analysed from mosaic P0 founders. Tg(fli1a.ep:mRFP-CAAX)pt504, with mRFP-labelled endothelial cell membranes, was generated using a previously described construct.31
2.2. Morpholinos and drug treatments
Morpholinos targeting alk131 and dll44 have been described and validated; sequences are provided in the Supplementary material online. These morpholinos phenocopy their respective genotypic mutants,5,31,39,40 supporting the validity of these tools.41 For Notch inhibition experiments, embryos were exposed to 10 µmol/L LY411575 (Stemgent, Cambridge, MA, USA), 50 µmol/L DAPT (Sigma, St Louis, MO, USA), or 1% dimethyl sulfoxide (DMSO) from 23 to 36 h post-fertilization (hpf) or 23–48 hpf.
2.3. In situ hybridization
Whole mount in situ hybridization and vibratome sectioning were carried out as described30,39 using an InSituPro VSi liquid handler (Intavis Inc., Chicago, IL, USA).
2.4. Confocal and two-photon imaging and image analysis
Fluorescence imaging was performed using a Leica TCS SP5 multiphoton/confocal microscope (Leica Microsystems, Wetzlar, Germany) as previously described.39 LAS AF software, version 3.0.0 build 8134, was used to measure fluorescence intensity in Tg(tp1:egfp)um14 embryos. Effects of Notch inhibition and alk1 mutation on forebrain and midbrain central artery sprouting at 36 hpf were analysed from confocal micrographs using Adobe Photoshop CS6.
2.5. Statistical analysis
GraphPad Prism v6.0 was used to analyse data using Student's t-test (fluorescence intensities) or one-way ANOVA followed by Tukey's post-hoc test (BCA areas, central artery sprouting). Significance was set at P < 0.05.
3. Results
3.1. Notch is active concomitant with Alk1 in cranial arterial endothelium
Because our goal was to determine whether Alk1 and Notch signalling interact during vascular development, and because alk1 plays a critical role in zebrafish cranial arterial development,30,31,39 we first assessed Notch activity in cranial endothelium using a double transgenic line, Tg(tp1:egfp)um14;Tg(fli1a.ep:mRFP-CAAX)pt504. These fish report Notch activity, as visualized by EGFP expression,33 on a background of mRFP-labelled endothelial cells. Embryos were imaged at 36 hpf, a time point when Alk1 is active in cranial arterial endothelium.39 Notch activity was weak to moderate in cranial arteries, including the first aortic arch (AA1), internal carotid artery (ICA), caudal division of the internal carotid artery (CaDI), optic artery (OA), and basal communicating artery (BCA) (Figure 1A). All of these arteries are alk1-positive at 36 hpf.31 Notch activity was also detected in the alk1-negative posterior communicating segments (PCS) and metencephalic arteries (MtA), but was absent in cranial veins (Figure 1A). These data demonstrate that cranial vascular Notch activity is arterial-specific, and that all alk1-positive arteries have active Notch signalling.
Figure 1.
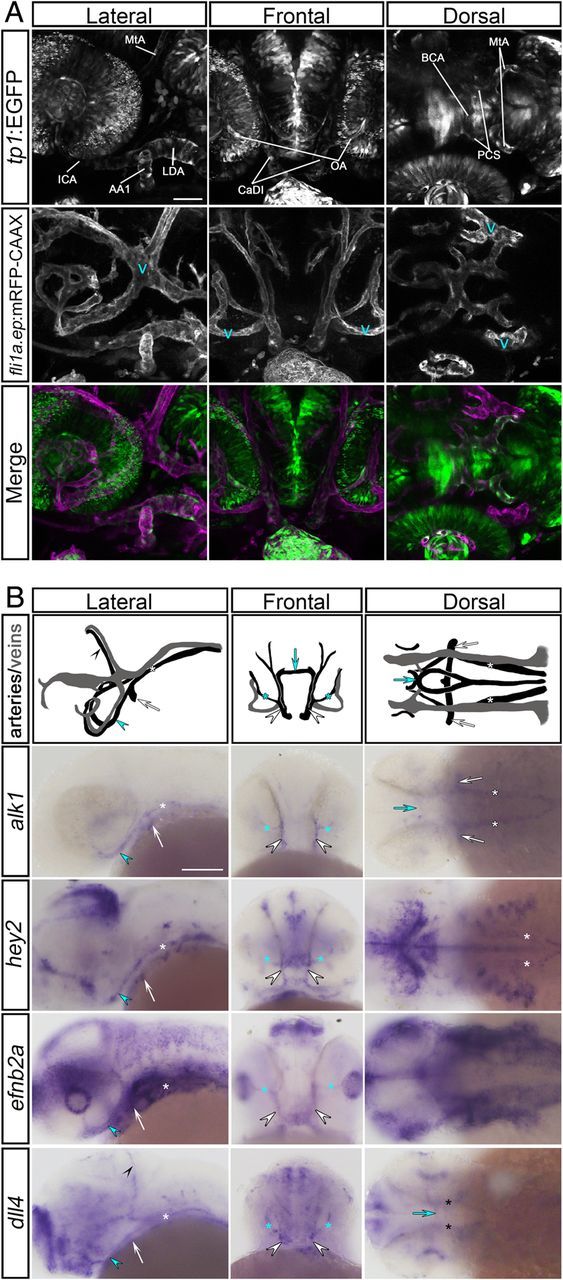
Notch is active concomitant with Alk1 in cranial arterial endothelium. (A) Notch activity is detectable in cranial arterial but not venous endothelium (‘v’) at 36 hpf. Two-dimensional confocal projections of Tg(tp1:egfp)um14 (green in merge) and Tg(fli1a.ep:mRFP-CAAX) pt504 (magenta in merge). alk1-positive arteries: AA1, aortic arch 1; ICA, internal carotid artery; LDA, lateral dorsal aorta; CaDI, caudal division of internal carotid artery; OA, optic artery; BCA, basal communicating artery. alk1-negative arteries: PCS, posterior communicating segment; MtA, metencephalic artery. Images represent N = 20 embryos. Lateral and dorsal views, anterior leftward; frontal view, anterior up. Scale bar, 50 μm. (B) Wiring diagrams (arteries black, veins grey) and representative whole mount in situ hybridization for alk1, hey2, efnb2a, and dll4 at 36 hpf. Expression of Notch targets is detected in the alk1-positive AA1 (white arrow), ICA (blue arrowhead), LDA (white asterisk), CaDI (white arrowhead), OA (blue asterisk), and BCA (blue arrow). dll4 is also expressed in the alk1-negative PCS (black asterisk) and MtA (black arrowhead). Plane of focus of dll4, frontal view, is deeper than other frontal images because of interfering dll4 brain expression. Images represent N > 63 embryos. Lateral and dorsal views, anterior leftward; frontal view, anterior up. Scale bar, 100 μm.
Next, we assayed cranial vessel expression of endogenous Notch targets. Hairy and enhancer of split (HES)-related proteins are transcriptional repressors that are induced by NICD/RBPJ,42 and the HES-related genes HES1, HEY1, and HEY2 are up-regulated by BMP9/ALK1 in cultured endothelial cells.10,26,28 The zebrafish genome contains two hes1 paralogs, her6 and her9.43 However, neither these genes nor hey1 were detectable in endothelium at 24–36 hpf (data not shown). In contrast, hey2 was expressed in all alk1-positive cranial arteries at 36 hpf (Figure 1B). efnb2a, another Notch target,10 as well as dll4, which encodes a Notch ligand that is positively regulated by Notch signalling,44–46 were also expressed in all alk1-positive cranial arteries at 36 hpf (Figure 1B). dll4 was additionally expressed in the alk1-negative PCS and MtA. These data demonstrate that Notch targets hey2, efnb2a, and dll4 are expressed in cranial arterial endothelium concomitant with active Alk1 signalling and are good candidates for cooperative regulation by Notch and Alk1.
3.2. Notch and Alk1 cooperatively regulate hey2 and efnb2a but oppositely regulate dll4 in the dorsal aorta
To investigate the interaction between Notch and Alk1 in the regulation of Notch targets in vivo, we assayed tp1:egfp, hey2, efnb2a, and dll4 in embryos with impaired Notch and/or Alk1 signalling. To inhibit Notch signalling, we treated embryos with 10 µmol/L LY411575, a gamma-secretase inhibitor, between 23 and 36 hpf. This time period brackets the critical time period of Alk1 function: alk1 is first detectable around 26 hpf, and alk1 mutation results in enlargement of cranial arteries by 32 hpf.31 LY411575 treatment had no effect on heartbeat or blood flow but resulted in severe trunk curvature (data not shown) as expected in Notch-inhibited embryos.2,4,47
Notch signalling is active in the zebrafish DA throughout embryonic development,2,5,48 and despite no obvious requirement for Alk1, alk1 is indeed expressed in the DA as early as 26 hpf.31 LY411575 treatment resulted in complete loss of DA tp1:egfp expression (Figure 2A), demonstrating effective abrogation of NICD/RBPJ-mediated transcription. However, the effect of Notch inhibition on expression of endogenous Notch targets was variable. LY411575 treatment had no effect on hey2, moderately decreased efnb2a, and completely abolished dll4 (Figure 2A). These observations agree with published data2,40 and suggest that among these DA genes, dll4 is most sensitive to perturbation of Notch signalling, whereas other pathways maintain hey2 and efnb2a in the absence of Notch.
Figure 2.
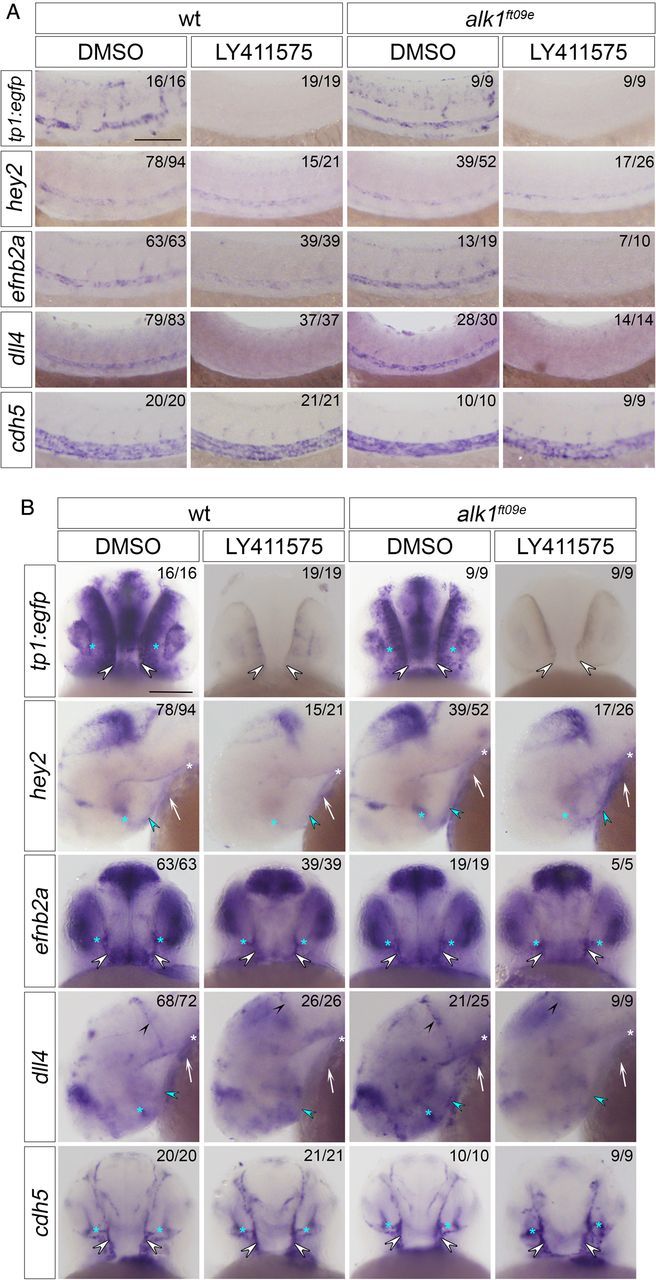
Notch- and Alk1-mediated control of Notch target gene expression is gene-specific and context-dependent. Whole mount in situ hybridization for tp1:egfp, hey2, efnb2a, dll4, and cdh5 in 36 hpf wt and alk1ft09e embryos treated with 1% DMSO or 10 μmol/L LY411575, 23–36 hpf. (A) Trunk. Lateral view, anterior leftwards. (B) Head. AA1, white arrow; ICA, blue arrowhead; LDA, white asterisk; CaDI, white arrowheads; OA, blue asterisks; MtA, black arrowheads. tp1:egfp, efnb2a, cdh5: frontal views, anterior up. hey2, dll4: lateral views, anterior left. Numbers in upper right corners indicate number of embryos with similar phenotype/total number of embryos assayed. Scale bars, 100 μm.
Next, we examined Notch target gene expression in the DA in alk1ft09e mutants (Figure 2A). alk1 mutation had no effect on expression of tp1:egfp, hey2, or efnb2a in the DA (Figure 2A), demonstrating that Alk1 is not necessary for expression of these arterial-specific Notch targets or for acquisition of arterial identity. In contrast, dll4 expression was up-regulated in the DA in alk1ft09e mutants (Figure 2A). This observation was confirmed in alk1y6 and alk1s407 mutants and in alk1 morphants (Supplementary material online, Figure S1A). Furthermore, endothelial-specific expression of alk1CA (fli1a.ebs:alk1CA-mCherry) dramatically repressed dll4 (Supplementary material online, Figure S1B). These data suggest that Alk1 opposes Notch in dll4 regulation in the DA.
Compared with Notch inhibition alone, combined alk1ft09e mutation and Notch inhibition had no effect on tp1:egfp or dll4 expression (Figure 2A), supporting the idea that Notch is required for expression of these genes. In contrast, concomitant abrogation of Alk1 and Notch signalling decreased hey2 and nearly eliminated efnb2a (Figure 2A). hey2 results, originally obtained in LY411575-treated alk1ft09e embryos, were recapitulated in DAPT-treated alk1y6 mutants and LY411575-treated alk1 morphants (data not shown). These findings suggest cooperative support of hey2 and efnb2a expression by Notch and Alk1 in the DA and agree with published data suggesting that both genes are regulated independently via NICD/RBPJ and ALK1/Smad1,5.2,13,26,28
3.3. Notch and Alk1 exhibit gene-specific antagonistic interactions in regulation of cranial arterial endothelial gene expression
We next examined regulation of Notch target genes in cranial arteries, which enlarge upstream of AVMs in alk1 mutants. LY411575 treatment abrogated tp1:egfp expression in cranial arterial endothelium and neural domains (Figure 2B, Supplementary material online, Figure S2), as expected. However, whereas Notch inhibition dramatically decreased hey2 in cranial arterial endothelium, efnb2a was refractory to this treatment (Figure 2B, Supplementary material online, Figure S2). These effects were different from those observed in the DA (Figure 2A), suggesting context-specific gene regulation. LY411575 treatment eliminated arterial dll4 expression in cranial arteries, similar to the DA, but increased dll4 in neural tissues (Figure 2B, Supplementary material online, Figure S2). These observations support the idea of a positive feedback loop specific to arterial endothelial cells in which Notch signalling directly regulates DLL4 expression.40,44–46
As in the DA, in cranial arteries tp1:egfp, hey2, and efnb2a were unaffected by alk1 mutation or knockdown, whereas dll4 was markedly up-regulated, and a fli1a.ebs:alk1CA-mCherry transgene dramatically repressed dll4 (Figure 2B, Supplementary material online, Figures S1C, D and S2). Also similar to the DA, loss of alk1 failed to rescue abrogation of tp1:egfp or dll4 expression induced by LY411575 treatment (Figure 2B, Supplementary material online, Figure S2). In contrast, hey2 and efnb2a behaved differently in the absence of both Notch and Alk1 signalling in trunk vs. cranial arterial endothelial domains. In cranial arteries, efnb2a expression proved refractory to combined loss of Notch and Alk1 signalling, whereas this same treatment increased hey2 expression compared with Notch inhibition alone (Figure 2B, Supplementary material online, Figure S2). hey2 results, originally obtained in LY411575-treated alk1ft09e embryos, were recapitulated in DAPT-treated alk1y6 mutants and LY411575-treated alk1 morphants (Supplementary material online, Figure S3). These results suggest that neither Notch nor Alk1 is necessary for cranial arterial efnb2a expression, whereas Notch activates and Alk1 dampens hey2 expression in cranial arteries, with Alk1 acting either downstream of NICD or independently of Notch. These data support the notion that region-specific regulatory networks control arterial expression of Notch target genes. Effects of Notch and Alk1 manipulation on Notch target gene expression are summarized in Table 1.
Table 1.
Qualitative changes in arterial gene expression in response to altered Notch and/or Alk1 signallinga
| Dorsal aorta |
Cranial arteries |
|||||
|---|---|---|---|---|---|---|
| Wild-type |
alk1 mutant/morphant |
Wild-type |
alk1 mutant/morphant |
|||
| LY411575 | DMSO | LY411575 | LY411575 | DMSO | LY411575 | |
| tp1:egfp | ↓↓↓ | NC | ↓↓↓ | ↓↓↓ | NC | ↓↓↓ |
| hey2 | NC | NC | ↓ | ↓↓ | NC | NC |
| efnb2a | ↓↓ | NC | ↓↓↓ | NC | NC | NC |
| dll4 | ↓↓↓ | ↑ | ↓↓↓ | ↓↓↓ | ↑ | ↓↓↓ |
aEmbryos were treated with 1% DMSO or 10 µmol/L LY411575, 23–36 hpf, and analysed by in situ hybridization at 36 hpf. Table indicates increased (up arrows), decreased (down arrows; number of arrows indicates qualitative strength of response), or no change (NC) in expression compared with DMSO-treated wild-type embryos.
3.4. Notchgof and alk1lof generate vascular morphologies with some phenotypic overlap but with independent aetiologies
Our in vivo gene expression studies demonstrated that loss of alk1 is associated with increased endothelial dll4 expression. Therefore, we reasoned that enhanced Dll4/Notch signalling might phenocopy cranial AVMs in alk1 mutants. To investigate this possibility, we compared cranial vascular development in wild-type embryos, Tg(fli1a:GAL4FF)ubs3;Tg(5xUAS-E1b:6xMYC-notch1a)kca3 embryos [which ectopically express Notch1a ICD in all endothelial cells; hereafter referred to as Tg(endo:N1ICD)], and alk1 mutant or morphant embryos.
In wild-type embryos, single sprouts emerged from the most posterior aspect of each bilateral venous primordial midbrain channel (PMBC) ∼22 hpf and migrated medially to connect to a sprout emanating from the apex of the paired CaDIs by ∼ 26 hpf, forming the BCA (Supplementary material online, Movie S1). These transient BCA/PMBC connections serve as the primary drainage for the CaDI/BCA between 26 and 36 hpf but regress thereafter as downstream arteries develop (Supplementary material online, Movie S2, and Corti et al.31).
In Tg(endo:N1ICD) embryos, the CaDIs developed and lumenized normally, but sprouting from the PMBC was impaired, with BCA/PMBC connections delayed up to 8 h compared with wild-type [Figure 3, compare A–E, wt to F–J, Tg(endo:N1ICD); Supplementary material online, Movies S3 and S4]. Vascular morphology was variable in 36 hpf Tg(endo:N1ICD) embryos, with establishment of early alternative drainage connections (e.g. BCA to PCS/MtA, Figure 3G and H) associated with relatively normal calibre vessels, and establishment of late connections associated with CaDI/BCA engorgement and enlargement (Figure 3P and Q). Regardless of 36 hpf phenotype, BCA area was not significantly increased compared with control at 48 hpf (Figure 3S), but all Tg(endo:N1ICD) embryos maintained at least one BCA/PMBC connection, resulting in a small calibre AVM (Figure 3P′, Q′, T).
Figure 3.
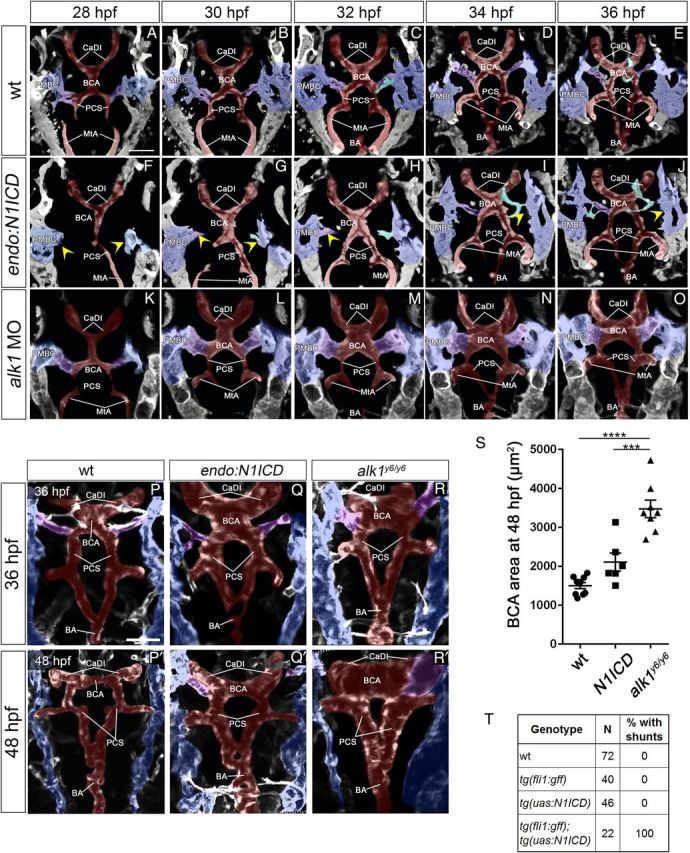
notchgof and alk1lof cranial AVMs have independent aetiologies. (A–J) Cranial arterial development in wt (A–E), Tg(endo:N1ICD) (F–J), and alk1 morphant (K–O) embryos, 28–36 hpf. See also Supplementary material online, Movies S4–S5. In Tg(endo:N1ICD) embryos (F–J), PMBC-derived sprouts (yellow arrowheads) are delayed, compromising CaDI/BCA drainage. In alk1 morphant embryos (K–O), connections form normally, but BCA enlargement is evident by 30 hpf. Two-dimensional projections of Z-stacks from two photon/confocal time-lapse imaging, frontal views, anterior up. Images represent N = 8 wt, 6 Tg(endo:N1ICD), 10 alk1 MO. Endothelial transgenes imaged: wt and Tg(endo:N1ICD), Tg(fli1a:GAL4FF;UAS:kaede); alk1 MO, Tg(fli1a:mrfp-caax)pt504. (P–R′) Two-photon imaging of wt (P, P′), Tg(endo:N1ICD) (Q, Q′), and alk1y6 mutant (R, R′) embryos at 36 and 48 hpf. Tg(endo:N1ICD) embryos show phenotypic overlap with alk1 mutants, with variable enlargement of the CaDI (36 hpf) and consistent retention of BCA/PMBC connections (48 hpf). Two-dimensional projections of Z-stacks, dorsal views, anterior up. Images represent N = 10 wt, 8 Tg(endo:N1ICD), and 8 alk1y6. Imaged transgene is Tg(kdrl:gfp)la116. (A–R) Pseudocolouring: PMBC (blue), CtA (cyan), CaDI/BCA/PCS/BA (red), BCA/PMBC connection (purple). Scale bars, 50 μm. (S) BCA area at 48 hpf. N = 6–9 for each condition; lines represent mean ± SEM. One-way ANOVA followed by Tukey's post-hoc test, ***P < 0.001, ****P < 0.0001. (T) Presence of shunts at 48 hpf.
Although the Tg(endo:N1ICD) phenotype bears some resemblance to the alk1 mutant phenotype, these phenotypes originate and progress differently. In Tg(endo:N1ICD) embryos, delayed venous sprouting compromises early cranial arterial drainage, resulting in variable changes in CaDI/BCA calibres and small AVMs stemming from persistent BCA/PMBC connections (Figure 3F–J, Q, Q′). In alk1 morphants or mutants, venous-derived angiogenic sprouting is not delayed, and all vessel connections develop normally (Figure 3K–O; Supplementary material online, Movie S5). However, increased endothelial cell number in the CaDI leads to increased calibre and altered haemodynamics, causing downstream vessels to adapt by maintaining normally transient arterial-venous connections (Figure 3R, R′, S), most often between the BCA and PMBC.30,31,39 Although the Tg(endo:N1ICD) phenotype decreases in severity over time (Figure 3Q and Q′), the alk1 mutant phenotype exacerbates over time (Figure 3R and R′), with progressive increases in vessel calibre both upstream and downstream of the AVM.
3.5. Notch activity is not required for AVM development in alk1 mutants
To further explore the role of enhanced Dll4/Notch in AVM development in the absence of Alk1 function, we inhibited Notch activity in alk1 mutants and assessed cranial vascular phenotype. First, we injected a splice-blocking dll4 morpholino4 into alk1y6 mutants. The morpholino generated an aberrant splice product that eliminated exon 3, decreased wild-type transcript to ∼37% of control, and resulted in intersegmental vessel hypersprouting4,40 in 60% of embryos (Supplementary material online, Figure S4). Wild-type dll4 morphants exhibited a small but significant increase in BCA area at 36 hpf (Figure 4A, B and I) but normal cranial vessel morphology, with no AVMs, at 48 hpf (Figure 4E, F and J). dll4 knockdown failed to rescue increased BCA area (36 hpf) or cranial AVMs (48 hpf) in alk1y6 mutants (Figure 4C, D, and G–J).
Figure 4.

dll4 expression is not required for AVM development in alk1 mutants. Embryos from an alk1y6/+;Tg(kdrl:gfp)la116 incross were injected at the 1–4-cell stage with 15 ng dll4 MO or 5-bp mismatch control MO, imaged at 36 (A–D) or 48 (E–H) hpf, and genotyped. Two-dimensional projections of two-photon Z-stacks. Pseudo-colouring: PMBC (blue), CtA (cyan), CaDI/BCA/PCS/BA (red), and BCA/PMBC connection (purple). Dorsal views, anterior up. Images represent N = 7–41 embryos per condition. Scale bars, 50 μm. (I) dll4 knockdown failed to rescue increased BCA area in alk1y6 mutants at 36 hpf. Lines represent mean ± SEM, N = 6–21 embryos per condition. One-way ANOVA followed by Tukey's post-hoc test, * P < 0.05; ****P < 0.0001. (J) dll4 knockdown failed to rescue cranial shunt formation in alk1y6 mutants at 48 hpf.
Because we could not achieve complete knockdown of dll4, we treated alk1 mutants with LY411575 (beginning at 23 hpf) to eliminate Notch activity and assessed cranial vascular phenotype at 36 and 48 hpf (Figure 5). In DMSO-treated wild-type embryos, the anterior central arteries (CtAs)49 sprouted as the BCA/PMBC connection regressed: one or two sprouts emerged from more anterior aspects of each PMBC and migrated medially, with sprouts interacting ipsilaterally but not contralaterally and ultimately connecting to the BCA by 36 hpf (Figure 5A). Notch inhibition in wild-type embryos had no effect on the CaDI/BCA but caused hypersprouting in the PMBC-derived CtAs at 36 hpf, with significant increases in number of PMBC-derived sprouts, connections to the BCA, branch points, and contralateral sprout connections (Figure 5B, Supplementary material online, Figure S5A, A′, B, B′ and E–H).
Figure 5.

Notch activity is not required for AVM development in alk1 mutants. Embryos from an alk1y6/+;Tg(kdrl:gfp)la116 incross were treated with 1% DMSO or 10 µmol/L LY411575 at 23 hpf and cranial vasculature imaged at 36 (A–D) and 48 (E–H) hpf. Two-dimensional projections of two-photon Z-stacks; in (E–H), dorsal planes were removed to highlight BCA/PCS. Pseudo-colouring: PMBC (blue), CtA (cyan), CaDI/BCA/PCS/BA (red), and BCA/PMBC shunt (purple). Dorsal views, anterior up. Scale bars, 50 μm. (I) Notch inhibition failed to rescue increased BCA area in alk1y6 mutants at 36 hpf. Lines represent mean ± SEM, N = 4–7 embryos per condition. One-way ANOVA followed by Tukey's post-hoc test, *P < 0.05; ***P < 0.001. (J) Notch inhibition failed to rescue cranial shunt formation in alk1y6 mutants at 48 hpf.
In contrast to Notch-inhibited embryos, DMSO-treated alk1 mutants exhibited enlarged CaDIs/BCAs at 36 hpf, as previously reported,30,31,39 but PMBC-derived CtAs were unaffected (Figure 5C, Supplementary material online, Figure S5C, C′, D, D′ and E–H). Notch-inhibited alk1 mutants were indistinguishable from DMSO-treated alk1 mutants in terms of increased BCA calibre at 36 hpf (Figure 5C, D, and I). At 48 hpf, LY411575 treatment decreased PCS calibre in both wild-type and alk1 mutant embryos but failed to rescue alk1 mutant AVMs (Figure 5E–H and J). Furthermore, despite enhanced dll4 expression, Notch signalling, as quantified by mean BCA EGFP fluorescence intensity in Tg(tp1:egfp)um14 embryos, was unchanged in alk1 morphants compared with control (Supplementary material online, Figure S6). These data support dll4 morphant data and suggest that enhanced Dll4/Notch signalling does not underlie AVM development in the absence of alk1.
Although Notch inhibition failed to rescue the alk1 mutant phenotype, alk1 mutation dampened Notch inhibitor-induced effects on CtAs, decreasing number of PMBC-derived sprouts, number of connections to the BCA, and CtA branch points to levels between wild-type DMSO-treated and wild-type LY411575-treated embryos (Figure 5A–D, Supplementary material online, Figure S5). However, effects did not achieve statistical significance. Further studies are required to determine whether this result reflects a genetic interaction between Alk1 and Notch pathways or an effect of altered vascular haemodynamics.
4. Discussion
Our results in zebrafish embryos demonstrate context-specific effects of Notch signalling on arterial endothelial gene expression. Although Notch inhibition abrogated transcription of a synthetic Notch reporter and endogenous dll4 in both trunk and cranial arteries, other arterial Notch targets were less sensitive to Notch inhibition, suggesting that additional control elements sustain expression of these genes in the absence of Notch. Furthermore, Notch sensitivity of particular genes showed regional variation, suggesting unique regulatory mechanisms in different vascular beds. These data suggest that context-specific regulation, which may be lost or dampened in cultured endothelial cells, plays an important role in the control of Notch target gene expression in vivo.
Based on published work demonstrating cooperative interactions between DLL4/Notch and BMP9/ALK1 in enhancing arterial Notch target gene expression in cultured endothelial cells,26 we had anticipated that combined Notch and Alk1 inhibition might additively if not synergistically decrease Notch target gene expression and impair arterial specification. However, abrogation of Alk1 signalling failed to decrease expression of Notch targets or disrupt arterial (hey2, efnb2a, dll4) or venous (data not shown) identity, and we uncovered only minor cooperative interactions between Alk1 and Notch, with both contributing to maintenance of efnb2a and hey2 expression in trunk but not cranial arteries. Furthermore, we uncovered opposing roles of Notch and Alk1 in expression of the Notch target and arterial marker, dll4. Although dll4 is up-regulated in the zebrafish DA in the absence of blood flow,50 lack of blood flow cannot account for increased dll4 in alk1 mutants: blood flow remains strong in alk1 mutants at 36 hpf, with only a subtle redistribution of flow towards cranial vessels.31 Together with previous data demonstrating increased cxcr4a and decreased edn1 in the absence of blood flow or alk1 expression,31 these data support the idea that Alk1 mediates a flow-based signal that controls expression of a subset of arterial genes. It is also possible that dll4 up-regulation in the absence of alk1 might be attributed to the loss of the Smad1/5 target, id1. In the DA, id1 is down-regulated in the absence of alk1 or blood flow (data not shown), and ID1 stabilizes HES1 (Her6 in zebrafish), which in turn represses DLL4.51,52 Therefore, decreased Id1 might decrease Her6, thereby resulting in increased dll4. However, we were unable to detect her6 in zebrafish arterial endothelial cells by in situ hybridization.
In addition to the unanticipated opposing effects of Notch and Alk1 on dll4 expression, we demonstrated a paradoxical interaction with respect to cranial arterial hey2 regulation, with loss of alk1 restoring hey2 to near control levels in Notch-inhibited embryos. This finding suggests that Alk1 may repress hey2 in cranial arterial endothelial cells either downstream of NICD cleavage or via an independent mechanism. Although it is possible that enhanced blood flow through enlarged, alk1-dependent vessels might increase hey2 expression, this seems unlikely given that hey2 expression is unchanged in alk1 mutants with intact Notch signalling (Table 1) or in the absence of blood flow (data not shown).
Dll4 is a critical arterial endothelial Notch ligand5,10 and Notchgof and in particular Dll4 overexpression results in large calibre arteries and AVMs similar in morphology to AVMs in Alk1 knockout mice.8,9,11,12,17 Therefore, our finding that Alk1 inhibited dll4 expression initially suggested to us that increased Notch signalling might contribute to AVMs in alk1 mutants. However, multiple lines of evidence fail to support this hypothesis. First, although ectopic endothelial expression of N1ICD results in small cranial AVMs involving the same vessels as in alk1 mutants, the origin and progression of these AVMs differs dramatically. The primary defect leading to AVMs in N1ICD-expressing embryos is delayed venous-derived sprouting, whereas the primary defect in alk1 mutants is increased endothelial cell number in and calibre of upstream arteries.31 Differences in the spatiotemporal expression of fli1a-driven N1ICD vs. alk1 limit the utility of our approach; however, we would expect that earlier N1ICD expression would cause an even more pronounced arterial phenotype if enhanced arterial Notch signalling could serve as a proxy for arterial alk1 loss. Secondly, neither dll4 knockdown nor Notch inhibition rescues alk1 mutant AVMs. Thirdly, despite increased dll4 expression, we failed to detect increased canonical Notch activity in alk1 mutants. Together, these results suggest that AVMs arise independently in Notchgof and Alk1lof embryos (Figure 6).
Figure 6.
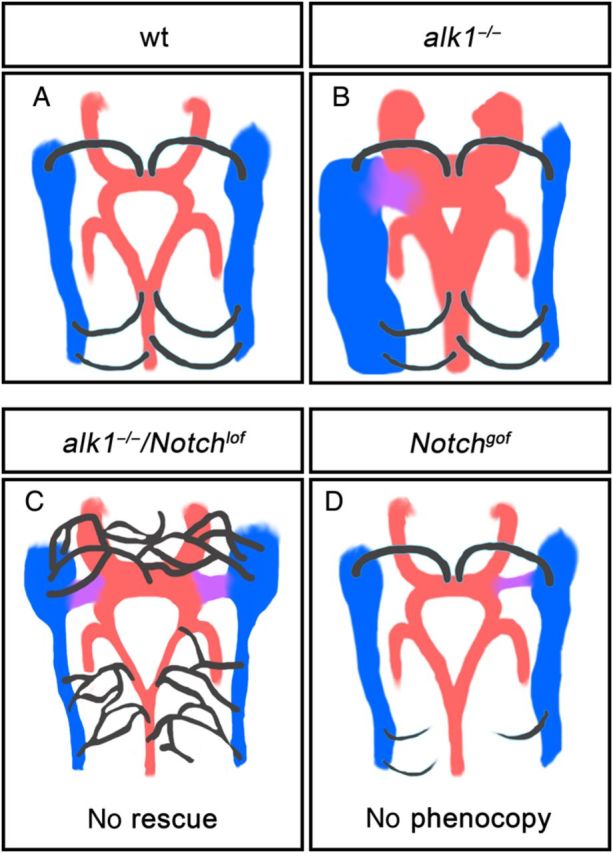
Cranial vessel architecture resulting from Alk1 and/or Notch manipulation. alk1 mutants develop enlarged cranial arteries (red) that drain through an aberrantly retained connection (purple) to major primitive drainage veins (blue). Although this phenotype is associated with increased arterial dll4, notchgof fails to phenocopy and notchlof fails to rescue this defect. notchgofcauses impaired venous-derived sprouting (black), whereas Notchlof causes enhanced venous-derived sprouting. 48 hpf, dorsal views, anterior up.
Although AVMs initiate via independent mechanisms in Notchgof and Alk1lof embryos, both represent retention of normally transient BCA/PMBC connections. In alk1 mutants, maintenance of BCA/PMBC connections occurs as an adaptive response to increased shear stress caused by enlargement of upstream arteries.31 In Notchgof embryos, impaired venous-derived sprouting delays CaDI/BCA drainage, which likely alters cranial vascular haemodynamics and affects remodelling. Thus, an adaptive response to altered haemodynamics, downstream of independent primary molecular and cellular defects, may be a unifying factor in the development of Notchgof and Alk1lof AVMs.
Although we failed to rescue vascular defects in alk1 mutants via Notch inhibition, we partially rescued CtA hypersprouting defects in Notch-inhibited embryos via alk1 mutation, suggesting some interaction between Notch and Alk1 in the cranial vasculature. It is possible that restored hey2 expression in the absence of both Notch and Alk1 signalling might contribute to this phenomenon, as knockdown of HEY2 enhances sprouting in cultured HUVECs.26 However, whether these observations represent a true genetic interaction or a response to changes in vascular haemodynamics remains to be determined.
In summary, our in vivo analysis of Notch and Alk1 signalling demonstrates gene-specific and context-specific interactions, with examples of both cooperative and antagonistic control of gene expression. However, results fail to support the idea that these pathways interact synergistically to control Notch target genes, program arterial identity, and prevent AVMs.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the National Institutes of Health (R01 HL079108 to B.L.R.).
Conflict of interest: none declared.
Acknowledgements
We thank Zachary Kupchinsky for fish care; Sarah Young, Teresa Capasso, Donna Unke, and Harinee Suthakar for technical contributions; and Drs Nathan Lawson (University of Massachusetts Medical School, Worcester, MA, USA), Brant Weinstein (NIH/NICHD, Bethesda, MD, USA), Heinz-Georg Belting, and Markus Affolter (University of Basel, Switzerland) for reagents.
References
- 1.Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet 2012;13:654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA, Weinstein BM. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 2001;128:3675–3683. [DOI] [PubMed] [Google Scholar]
- 3.Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalen M, Gerhardt H, Betsholtz C. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007;445:776–780. [DOI] [PubMed] [Google Scholar]
- 4.Siekmann AF, Lawson ND. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 2007;445:781–784. [DOI] [PubMed] [Google Scholar]
- 5.Quillien A, Moore JC, Shin M, Siekmann AF, Smith T, Pan L, Moens CB, Parsons MJ, Lawson ND. Distinct Notch signaling outputs pattern the developing arterial system. Development 2014;141:1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krebs LT, Shutter JR, Tanigaki K, Honjo T, Stark KL, Gridley T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev 2004;18:2469–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duarte A, Hirashima M, Benedito R, Trindade A, Diniz P, Bekman E, Costa L, Henrique D, Rossant J. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev 2004;18:2474–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlson TR, Yan Y, Wu X, Lam MT, Tang GL, Beverly LJ, Messina LM, Capobianco AJ, Werb Z, Wang R. Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. Proc Natl Acad Sci USA 2005;102:9884–9889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krebs LT, Starling C, Chervonsky AV, Gridley T. Notch1 activation in mice causes arteriovenous malformations phenocopied by ephrinB2 and EphB4 mutants. Genesis 2010;48:146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iso T, Maeno T, Oike Y, Yamazaki M, Doi H, Arai M, Kurabayashi M. Dll4-selective Notch signaling induces ephrinB2 gene expression in endothelial cells. Biochem Biophys Res Commun 2006;341:708–714. [DOI] [PubMed] [Google Scholar]
- 11.Kim YH, Hu H, Guevara-Gallardo S, Lam MT, Fong SY, Wang RA. Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development 2008;135:3755–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trindade A, Kumar SR, Scehnet JS, Lopes-da-Costa L, Becker J, Jiang W, Liu R, Gill PS, Duarte A. Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood 2008;112:1720–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JH, Peacock MR, George SC, Hughes CC. BMP9 induces EphrinB2 expression in endothelial cells through an Alk1-BMPRII/ActRII-ID1/ID3-dependent pathway: implications for hereditary hemorrhagic telangiectasia type II. Angiogenesis 2012;15:497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benedito R, Trindade A, Hirashima M, Henrique D, da Costa LL, Rossant J, Gill PS, Duarte A. Loss of Notch signalling induced by Dll4 causes arterial calibre reduction by increasing endothelial cell response to angiogenic stimuli. BMC Dev Biol 2008;8:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy PA, Lu G, Shiah S, Bollen AW, Wang RA. Endothelial Notch signaling is upregulated in human brain arteriovenous malformations and a mouse model of the disease. Lab Invest 2009;89:971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ZhuGe Q, Zhong M, Zheng W, Yang GY, Mao X, Xie L, Chen G, Chen Y, Lawton MT, Young WL, Greenberg DA, Jin K. Notch-1 signalling is activated in brain arteriovenous malformations in humans. Brain 2009;132:3231–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy PA, Lam MT, Wu X, Kim TN, Vartanian SM, Bollen AW, Carlson TR, Wang RA. Endothelial Notch4 signaling induces hallmarks of brain arteriovenous malformations in mice. Proc Natl Acad Sci USA 2008;105:10901–10906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herbert SP, Huisken J, Kim TN, Feldman ME, Houseman BT, Wang RA, Shokat KM, Stainier DY. Arterial-venous segregation by selective cell sprouting: an alternative mode of blood vessel formation. Science 2009;326:294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindskog H, Kim YH, Jelin EB, Kong Y, Guevara-Gallardo S, Kim TN, Wang RA. Molecular identification of venous progenitors in the dorsal aorta reveals an aortic origin for the cardinal vein in mammals. Development 2014;141:1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu P, Liu J, Derynck R. Post-translational regulation of TGF-beta receptor and Smad signaling. FEBS Lett 2012;586:1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996;13:189–195. [DOI] [PubMed] [Google Scholar]
- 22.McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management, and pathogenesis. Genet Med 2011;13:607–616. [DOI] [PubMed] [Google Scholar]
- 23.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, McCormick MK, Pericak-Vance MA, Heutnik P, Oostra BA, Haitjema T, Westerman CJJ, Porteous ME, Guttmacher AE, Letarte M, Marchuk DA. Endoglin, a TGF-b binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994;8:345–351. [DOI] [PubMed] [Google Scholar]
- 24.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004;363:852–859. [DOI] [PubMed] [Google Scholar]
- 25.Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet 2000;26:328–331. [DOI] [PubMed] [Google Scholar]
- 26.Larrivee B, Prahst C, Gordon E, del Toro R, Mathivet T, Duarte A, Simons M, Eichmann A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell 2012;22:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor KL, Henderson AM, Hughes CC. Notch activation during endothelial cell network formation in vitro targets the basic HLH transcription factor HESR-1 and downregulates VEGFR-2/KDR expression. Microvasc Res 2002;64:372–383. [DOI] [PubMed] [Google Scholar]
- 28.Morikawa M, Koinuma D, Tsutsumi S, Vasilaki E, Kanki Y, Heldin CH, Aburatani H, Miyazono K. ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res 2011;39:8712–8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westerfield M. The Zebrafish Book. Eugene: University of Oregon Press, 1995. [Google Scholar]
- 30.Roman BL, Pham VN, Lawson ND, Kulik M, Childs S, Lekven AC, Garrity DM, Moon RT, Fishman MC, Lechleider RJ, Weinstein BM. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development 2002;129:3009–3019. [DOI] [PubMed] [Google Scholar]
- 31.Corti P, Young S, Chen CY, Patrick MJ, Rochon ER, Pekkan K, Roman BL. Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development 2011;138:1573–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin SW, Beis D, Mitchell T, Chen JN, Stainier DY. Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development 2005;132:5199–5209. [DOI] [PubMed] [Google Scholar]
- 33.Parsons MJ, Pisharath H, Yusuff S, Moore JC, Siekmann AF, Lawson N, Leach SD. Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech Dev 2009;126:898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi J, Dong L, Ahn J, Dao D, Hammerschmidt M, Chen JN. FoxH1 negatively modulates flk1 gene expression and vascular formation in zebrafish. Dev Biol 2007;304:735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheer N, Campos-Ortega JA. Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mech Dev 1999;80:153–158. [DOI] [PubMed] [Google Scholar]
- 36.Herwig L, Blum Y, Krudewig A, Ellertsdottir E, Lenard A, Belting HG, Affolter M. Distinct cellular mechanisms of blood vessel fusion in the zebrafish embryo. Curr Biol 2011;21:1942–1948. [DOI] [PubMed] [Google Scholar]
- 37.Traver D, Paw BH, Poss KD, Penberthy WT, Lin S, Zon LI. Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat Immunol 2003;4:1238–1246. [DOI] [PubMed] [Google Scholar]
- 38.Hatta K, Tsujii H, Omura T. Cell tracking using a photoconvertible fluorescent protein. Nat Protoc 2006;1:960–967. [DOI] [PubMed] [Google Scholar]
- 39.Laux DW, Young S, Donovan JP, Mansfield CJ, Upton PD, Roman BL. Circulating Bmp10 acts through endothelial Alk1 to mediate flow-dependent arterial quiescence. Development 2013;140:3403–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leslie JD, Ariza-McNaughton L, Bermange AL, McAdow R, Johnson SL, Lewis J. Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development 2007;134:839–844. [DOI] [PubMed] [Google Scholar]
- 41.Schulte-Merker S, Stainier DY. Out with the old, in with the new: reassessing morpholino knockdowns in light of genome editing technology. Development 2014;141:3103–3104. [DOI] [PubMed] [Google Scholar]
- 42.Borggrefe T, Oswald F. The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci 2009;66:1631–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou M, Yan J, Ma Z, Zhou Y, Abbood NN, Liu J, Su L, Jia H, Guo AY. Comparative and evolutionary analysis of the HES/HEY gene family reveal exon/intron loss and teleost specific duplication events. PLoS One 2012;7:e40649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caolo V, van den Akker NM, Verbruggen S, Donners MM, Swennen G, Schulten H, Waltenberger J, Post MJ, Molin DG. Feed-forward signaling by membrane-bound ligand receptor circuit: the case of NOTCH DELTA-like 4 ligand in endothelial cells. J Biol Chem 2010;285:40681–40689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sacilotto N, Monteiro R, Fritzsche M, Becker PW, Sanchez-Del-Campo L, Liu K, Pinheiro P, Ratnayaka I, Davies B, Goding CR, Patient R, Bou-Gharios G, De Val S. Analysis of Dll4 regulation reveals a combinatorial role for Sox and Notch in arterial development. Proc Natl Acad Sci USA 2013;110:11893–11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wythe JD, Dang LT, Devine WP, Boudreau E, Artap ST, He D, Schachterle W, Stainier DY, Oettgen P, Black BL, Bruneau BG, Fish JE. ETS factors regulate Vegf-dependent arterial specification. Dev Cell 2013;26:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang YJ, Brand M, Heisenberg CP, Beuchle D, Furutani-Seiki M, Kelsh RN, Warga RM, Granato M, Haffter P, Hammerschmidt M, Kane DA, Mullins MC, Odenthal J, van Eeden FJ, Nusslein-Volhard C. Mutations affecting neurogenesis and brain morphology in the zebrafish, Danio rerio. Development 1996;123:205–216. [DOI] [PubMed] [Google Scholar]
- 48.Zhong TP, Rosenberg M, Mohideen MA, Weinstein B, Fishman MC. Gridlock, an HLH gene required for assembly of the aorta in zebrafish. Science 2000;287:1820–1824. [DOI] [PubMed] [Google Scholar]
- 49.Isogai S, Horiguchi M, Weinstein BM. The vascular anatomy of the developing zebrafish: an atlas of embryonic and early larval development. Dev Biol 2001;230:278–301. [DOI] [PubMed] [Google Scholar]
- 50.Watson O, Novodvorsky P, Gray C, Rothman AM, Lawrie A, Crossman DC, Haase A, McMahon K, Gering M, Van Eeden FJ, Chico TJ. Blood flow suppresses vascular Notch signalling via dll4 and is required for angiogenesis in response to hypoxic signalling. Cardiovasc Res 2013;100:252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moya IM, Umans L, Maas E, Pereira PN, Beets K, Francis A, Sents W, Robertson EJ, Mummery CL, Huylebroeck D, Zwijsen A. Stalk cell phenotype depends on integration of Notch and Smad1/5 signaling cascades. Dev Cell 2012;22:501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bai G, Sheng N, Xie Z, Bian W, Yokota Y, Benezra R, Kageyama R, Guillemot F, Jing N. Id sustains Hes1 expression to inhibit precocious neurogenesis by releasing negative autoregulation of Hes1. Dev Cell 2007;13:283–297. [DOI] [PubMed] [Google Scholar]