

Abstract
The term microbiome refers to the collective genome of the microbes living in and on our bodies, but it has colloquially come to mean the bacteria, viruses, archaea, and fungi that make up the microbiota (previously known as microflora). We can identify the microbes present in the human body (membership) and their relative abundance using genomics, characterize their genetic potential (or gene pool) using metagenomics, and describe their ongoing functions using transcriptomics, proteomics, and metabolomics. Epidemiologists can make a major contribution to this emerging field by performing well-designed, well-conducted, and appropriately powered studies and by including measures of microbiota in current and future cohort studies to characterize natural variation in microbiota composition and function, identify important confounders and effect modifiers, and generate and test hypotheses about the role of microbiota in health and disease. In this review, we provide an overview of the rapidly growing literature on the microbiome, describe which aspects of the microbiome can be measured and how, and discuss the challenges of including the microbiome as either an exposure or an outcome in epidemiologic studies.
Keywords: bioinformatics, diversity, genomics, microbiome, microbiota
The term microbiome refers to the collective genome of the microbes living in and on our bodies, but it has colloquially come to mean the bacteria, viruses, archaea, and fungi that make up the microbiota (previously known as microflora). “-Omic” technologies have transformed our perception of the microbiota by characterizing the microbes present and their relevant abundance, as well as their ongoing functions. We can identify the microbes present in the body (membership) and their relative abundance using genomics, characterize their genetic potential (or gene pool) using metagenomics, and describe their ongoing functions using transcriptomics, proteomics, and metabolomics.
The field of microbiomics is very new, and its application in epidemiology has barely begun, but excitement about its potential is high. Every day, new articles appear in the scientific literature—and often in the newspaper—touting the role of the microbiome in human health. Microbiota have been associated with obesity, the metabolic syndrome, and even autism (1). Disruptions in the microbiota, termed dysbioses, are hypothesized to cause periodontal disease (2), cause inflammatory bowel disease (3), and potentially increase the risk of cancer (4).
Unlike other reviews in this series on -omic technologies, the microbiome is not a technique but a reconceptualization of humans as superorganisms consisting of human cells and microorganisms. We argue that microbiota can be a marker of exposure and a prognostic factor as well as a factor in disease etiology. However, this will require the incorporation of laboratory analyses that generate data characterizing the presence and function of microbes in epidemiologic studies, assessments of the reliability and validity of these analyses and the putative biomarkers, and knowledge about how to best use these data to address questions of clinical and public health importance.
Microbiota are dynamic, and the variation within an individual can be high. As yet, we do not know what magnitude of difference in microbial membership and relative abundance (jointly known as community structure) or function corresponds to a clinically meaningful difference. This lack of knowledge creates challenges for good study design and sample size estimation. Further, because our understanding of the factors that affect the microbiome is limited, so too is our understanding of what factors might confound or modify observed associations between the microbiome and health and disease. This makes it difficult to differentiate between risk markers and causal factors and between microbiomic changes that result from human disease and those that cause human disease. Well-conducted, population-based longitudinal studies are essential to filling these knowledge gaps. In this review, we provide an overview of the rapidly growing literature on the microbiome, describe which aspects of the microbiome can be measured and how, and discuss the challenges of including the microbiome as either an exposure or an outcome in epidemiologic studies.
WHY ALL THE EXCITEMENT ABOUT THE MICROBIOME?
Through the miracle of genetic sequencing, we now have the tools with which to identify the myriad bacteria, viruses, archaea, and fungi that live in and on our bodies—the microbiota. The ability to conduct a census of human microbiota is unprecedented; until the development of -omics technologies, we were able to identify only those microbes that could be grown in the laboratory (as a point of reference, an estimated 95%–99% of all bacteria cannot be grown in pure culture in the laboratory (5)). The results of these -omics microbiota censuses have given us a different perspective on ourselves. All surfaces of our bodies with portals to the outside are either covered in microbes or are subject to a variety of mechanisms designed to limit microbial growth. These surfaces include parts of our anatomy previously believed to be sterile, such as the blood (6), uterus (7), and lung (8). Further, it is now clear that we are outnumbered: Each of us carries 10 times more bacterial cells than human cells, and 100 times more viral particles. Good estimates are not available for eukaryotes, but we all have mites living on the sides of our noses and fungi in our hair. We are infested with microbes. However, this is generally a good thing. We depend upon our microbial communities to help us digest food, resist invasion by pathogens, and synthesize essential vitamins. Microbes are also essential for immune system development and response (9).
This new self-perspective has also led to new explanations for diseases previously resistant to treatment. There are several disease conditions (e.g., bacterial vaginosis, periodontitis, and inflammatory bowel disease) for which the disease course suggests an infectious etiology but no necessary and sufficient microbe has been identified. An alternative hypothesis is that rather than a single causal factor, there is a disruption in the system that causes and maintains the disease. A disrupted microbiotic system is termed dysbiosis. Dysbiosis may manifest as a microbial community that is either much more diverse (e.g., periodontitis) or much less diverse (e.g., antibiotic-associated diarrhea) than a healthy microbial community in the same biological niche, resulting in increased (or chronic) inflammation and reduced pathogen resistance. One area of active study is the identification of factors sufficient to perturb a healthy system and cause disease or sufficient to perturb a dysbiotic system and regenerate health.
Use of broad-spectrum antibiotics, even for short periods of time (3–5 days), is sufficient to perturb the gut microbiota in ways that are identifiable as much as 6 months later (10, 11). Repeated exposure to antibiotics can decrease the ability of microbiota to resist invasion by other microbes, increasing risk of infection. For example, broad-spectrum antibiotic use can result in antibiotic-associated diarrhea, most often caused by an overgrowth of Clostridium difficile (12). The primary treatment for C. difficile infection is (ironically) antibiotics, but C. difficile infection that is unresponsive to antibiotic therapy can be successfully treated by fecal transplantation. A fecal transplant can replace the dysbiotic gut microbiota with healthy ones (13–15).
The vast majority of published studies on the microbiome have been limited to studies of bacteria. However, the field of viromics is rapidly growing (16). Similarly to the bacterial microbiome, the virome varies by body site (Figure 1), is modified by host factors (17, 18), and may be correlated with host disease states (19–21). Notably, we now know that the vast majority of viruses in healthy individuals are bacteriophages, viruses that infect bacterial cells and may play a role in the regulation of microbial communities in humans (22–24). This and other insights derived from sequencing technologies have led to the notion that viruses, like bacteria, may be part of the normal microbiota of healthy individuals (25, 26). Even less developed are studies of the mycobiome (the fungal microbiome) and archaea, but all existing evidence suggests that they are also part of the normal microbiota of healthy individuals (27, 28).
Figure 1.
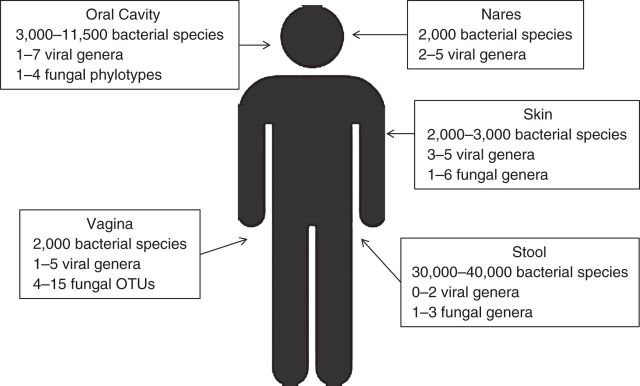
Estimated diversity of bacterial, viral, and fungal microorganisms in the human body as described in the literature (70–76), by body site. OTU, operational taxonomic unit.
There are many excellent reviews of the current state of knowledge on the microbiota of the human gut (29), skin (30), lung (8), and vagina (31). These reviews are primarily descriptive, and many are the result of studies funded by the National Institutes of Health Human Microbiome Project (32) and the European Microbiome Project (33). There also have been studies of previously isolated human populations (34) and a few studies of twins (35, 36), particularly with respect to obesity. Nonetheless, our understanding of the breadth and variation of human microbiota and how it relates to risk of human health and disease is very limited. The most interesting and compelling findings have been results from mouse models (e.g., see Cox et al. (37)) and human fecal transplant studies (e.g., see Kassam et al. (14), Van Nood et al. (15), and Youngster et al. (38)). However, mice are not humans, and human fecal transplant studies have been limited to treatment of antibiotic-associated diarrhea (although there are case reports of fecal transplants for other conditions) (39).
Although we have much descriptive information about the microbes included in the microbiome, what they do and how they do it is less clear. We propose that the major functions of microbiota in the human body can be divided into 3 categories (Figure 2). First, microbiota can act as a shield. The types of species present, their relative abundance, and their ongoing functions can influence the body's ability to resist invasion by other microbes (pathogen resistance) or neutralize the effects of exposure to a drug or toxin. These influences can be either direct or indirect via interactions with the host. Second, the microbiota can act as a filter, keeping out some things but allowing others to enter. Lastly, microbiota can enhance the effects of exposures. Microbiota can facilitate invasion by pathogens and, via microbial processes, amplify the effects of drugs or other exposures.
Figure 2.
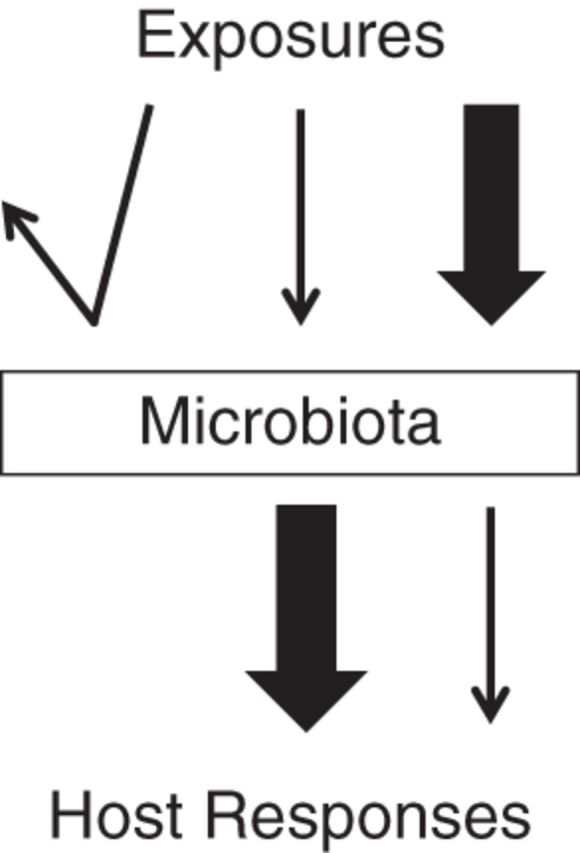
Conceptual model of the potential roles of microbiota in the causal pathway between exposures and host response. The widths of the lines correspond to the magnitudes of the exposures.
The generation time of microbes is rapid; for Escherichia coli in the human gut, it is estimated to average 40 hours (40), but it can be as short as 20 minutes in the laboratory. This means that microbiota can rapidly adapt to short-term changes in habitat that occur when we bathe, eat different foods, or change from tight-fitting clothing to looser clothing. A short-term change in nutrient sources changes the relative abundance of existing microbes but probably not which microbes are present (membership) (41). It might also change their ongoing functions. These changes in microbiota abundance and function may be short-term following a transient exposure, such as eating a hamburger (42), or may be identifiable weeks or months later following exposure to broad-spectrum antibiotics (43). Since microbiota respond quickly to the environment, changes in microbial function or the microbes present or their relative abundance can be a sensitive indicator of exposure or disease pathogenesis. Additionally, because microbiota act as a barrier, filter, or transformer, respond quickly to perturbation, and perform essential functions, they can serve as a diagnostic or prognostic indicator or a component of a disease pathway. Therefore, our understanding of the contribution of the microbiota to an exposure or outcome of interest determines which attribute of the microbiota is relevant for study.
MICROBIOTIC GENETICS ARE MORE DYNAMIC THAN HUMAN GENETICS
The term “microbiota” refers to which organisms are present, and “microbiome” refers to the genomes of those organisms. That collective genome, and even the genome of individual microbes, is not directly analogous to the human genome (Table 1). There is only 1 human genome; although gene deletions may occur, these genetic changes usually have health consequences (44). By contrast, the microbiome has a dynamic gene pool. Each of the microorganisms present has its own genes, so as new species join and others leave, the gene pool changes. Mobile genetic elements (e.g., plasmids) move between microbes of the same species and microbes of different species, making it possible for very different species to have identical genes and functions. Further, within a given microbial species, the gene pool might be quite diverse: The percentage of genes shared among all E. coli is estimated to be less than 30% (45). Therefore, even microbiomes with similar microbial community structures can have different functions, and microbiomes with different community structures can have similar functions. For now, at least, the function of most sequenced microbial genes is unknown.
Table 1.
Characteristics of the Human Microbiome As Compared With the Human Genome
| Human Microbiome | Human Genome |
|---|---|
| Many genomes | A single genome |
| Recombination occurs independently of reproduction | Recombination occurs only during reproduction |
| Functional capacity is dynamic | Basic functions are always present |
| Changes in response to the environment | Stable in response to the environment |
| Highly mutable | Mutations are rare |
| Generation time approximately 30 minutes | Generation time approximately 30 years |
Human reproduction results in gene exchange and the potential for mutation, but once a person is born, his or her functional capacity becomes essentially fixed. By contrast, although mutation occurs in bacteria during reproduction, bacteria exchange genes independently of reproduction. Their functional capacity is dynamic. Ignoring their ability to acquire new functions outside of reproduction, microbes' short generation time allows them to rapidly adapt (on the human time scale) to new environments, even relying solely on mutation. The implication is that if we lose parts of our microbiota, we may or may not lose some functions essential for health. Alternatively, we might be able to gain desirable functions by adding to our microbial communities or their gene pools.
Although many (perhaps most) of us have descendants of microbes we obtained from our mothers during birth (46), from breastfeeding (47), and from daily interactions (48), our microbiome is in a constant state of flux (49). Our microbes are constantly reproducing, mutating, and fighting infection by viruses. They also adapt to changes in their environment and nutrient sources caused by antibiotic therapy, diet, and invasion by pathogens and our body's host response to invasion.
Despite the dynamic nature of microbiota, the environment of the human host—temperature, nutrients, and the immune response—limits which microbes can live in the various ecological niches found in and on us. Further, given that microbiota co-evolved with their human hosts, human genetic variation may select for certain microbial communities. A study of the fecal microbiota of 416 United Kingdom twin pairs suggests that the presence of some taxa is influenced by host genetics (50). Moreover, it seems that each of us has his or her own microbiome, and this—like the human genome—can be uniquely identifying (48, 51).
MICROBIAL COMMUNITY STRUCTURE OR MICROBIAL FUNCTIONS?
Studies colloquially referred to as studies of the “microbiome” can be directed at detecting which microbes are present at various levels of resolution (from phyla to species) and their relative abundance, the genes present (metagenomics), ongoing gene expression (transcriptomics, proteomics), and ongoing processes (metabolomics). These levels of investigation accordingly come with their own terminology and have their own advantages and disadvantages for addressing specific research questions.
The technologies can be dichotomized into those that identify which microbes are present (taxonomic screens, most commonly using the sequence of ribosomal RNA for bacteria, archaea, and eukaryotes) and those that identify what the microbes are doing (shotgun sequencing of the entire genome (metagenomics), transcriptomics, proteomics, and metabolomics) (Table 2). Using a taxonomic screen, every person has a unique microbiome (especially when resolved to the species/strain level), but when they are compared among people, microbiota from specific body sites are similar between people (52). Depending on the body site, the similarities are modified by sociodemographic factors, health behaviors, and medical history.
Table 2.
Key Differences Between Taxonomic (Microbes Present) and Functional (Microbial Functions) Analyses of the Human Microbiome
| Microbes Present | Microbial Functions |
|---|---|
| High variation among individuals | Less variation among individuals |
| Colonization dynamics over time and space | Results of microbial interactions |
| Key microbes associated with health | Key functions associated with health |
| Selection, evolution, succession | Diagnosis, prognosis |
The human body has several distinct biological niches that vary widely in terms of the nutrients available, surveillance by the human host, and exposure to the environment. Although every human has a unique microbiome specific to each site, the characteristics of a specific site require that the microbial communities present perform certain functions. Note that microbial functions are not uniquely defined by species or even within species. Therefore, classification by function should be less variable than classification by species (53). Although this has not been definitely shown—and one study using inferred bacterial function suggested that this may not be the case (54)—measuring functions can give us insight into ongoing microbial interactions, functions associated with health or disease, and potential diagnostics or prognostics.
HOW DO YOU MEASURE THE MICROBIOME?
Taxonomic screens, which are relatively inexpensive, permit characterization of colonization dynamics over time and space, the identification of microbial communities associated with health or disease, and the collection of information for addressing questions regarding selection, evolution, and succession. At the time of this writing, the most common method used for characterizing the bacteria present is a taxonomic screen based on variable portions of the genetic sequence that codes for the ribosome (16S ribosomal RNA (rRNA)).
All cells have ribosomes, and because ribosomes perform the essential function of translating messenger RNA into protein, the genetic sequence is highly conserved. This makes it possible to use primer sets that cross most of the bacterial species present (universal primers) and phylogenetics to classify bacteria into taxonomic groups (55). The 16S rRNA sequence data are compared with sequence databases to determine the genus and species of bacteria that are present. Relative quantities of different 16S rRNA sequences can also be obtained to establish the relative abundance of each species. These data provide a snapshot of who is present from the bacterial world and in what quantities, but they are unable to characterize the relationships between community members. However, the ability to resolve the obtained genetic sequence to the species level varies with the region chosen; depending on the body site and bacterial composition, different regions are preferred. Further, while 16S rRNA analysis can characterize the members of a bacterial community, its use of a single bacterial gene precludes the detection of potential members from other kingdoms, including viruses, fungi, and archaea, and it can also limit resolution to the species level for bacteria. For fungi, ribosomal analysis is also used, but 18S rRNA is sequenced. Other methods are required to capture the viruses and archaea present (56).
Broader strategies for sequencing the genetic material of microbiota allow investigators to describe all organisms present in a community, encompassing bacteria, viruses (57, 58), fungi (59), and archaea (28). This set of all genomes from a diverse set of microbial sources (i.e., the “metagenome”) can be viewed as the gene pool of the functioning of the microbial community at that particular body site (i.e., the “functional potential”). In addition, like taxonomic screens, metagenomics can provide a sense of the relative abundance of different organisms. The usual strategy for whole-microbiome sequencing is to randomly sequence genomic fragments and then compile them to represent whole genomes (shotgun sequencing). Because these methods do not target a single region, they require additional care to ensure that both RNA (which must be reverse-transcribed) and DNA present in small-sized genomes in small quantities are appropriately captured from the sample (60) and that human DNA is not processed and mistaken for organism data. Microbial community structure can also be estimated with metagenomic data, using the ribosomal genes. Metagenomic data sets are very large and the analysis is challenging, but appropriate software is increasingly becoming available (61).
Measuring function is considerably more expensive than conducting taxonomic screens, and each of the different methods for assessing function has strengths and limitations. Measuring the metabolic products present (metabolomics) is the only way to directly assess the ongoing interactions among all of the microbes present and with the human host, but there is considerable technical variation. Transcriptomic studies require targeting transcripts from specific groups (e.g., bacteria) and do not directly correspond to functions. Human messenger RNA is much larger and more stable, and thus (since transcripts are sequenced for detection) can overwhelm the microbial transcriptome. Metagenomics enables characterization of gene potential but not ongoing functions.
Analytical software packages are available for analyzing the results of taxonomic screens (e.g., see Schloss et al. (62) and Hamady et al. (63)), and there is a software package that allows one to infer bacterial functions from taxonomy (64). Software with which to analyze the metagenome is also increasingly available (65). However, beyond the ability to process huge amounts of data from microbiomic studies, the real challenge lies in the best way to achieve the data reduction needed to use these data as an epidemiologic parameter. Epidemiologists can make an important contribution to microbiomic research by working to develop and evaluate methods of producing meaningful parameters from complex microbiomic data.
WHAT ARE THE CHALLENGES FOR EPIDEMIOLOGIC STUDIES?
Recent advances in molecular and computing technology make it possible to incorporate assessments of the microbiome into clinical and epidemiologic studies. However, the learning curve for “-omics” technologies is steep, and the technology for measurement is continually developing. It is essential to have outstanding collaborators who understand the necessity of ensuring the validity and reliability of the methods and the implementation of ongoing quality control and quality assurance procedures. There are considerable technical challenges in this regard (Appendix 1). Collection, storage, and preparation of specimens for testing can affect the results, sometimes to a large degree (66). The polymerase chain reaction method is not without error; sequencing technologies vary in sequence length, error rates, and costs. The high sensitivity of polymerase chain reaction makes contamination across wells or even across runs a continuing worry in a high-throughput setup. For example, a recent study (67) identified DNA in extraction kits and other laboratory reagents. These sequences could be identified in 16S rRNA taxonomic screens and shotgun sequencing, leading to erroneous study results. These findings, among others, have prompted several recommendations for minimizing bias due to differential contamination, including the use of technical controls throughout all processes and the importance of processing samples in a blinded fashion and in random order (67).
The amounts of data involved are large, with correspondingly large computation requirements for initial data cleaning and classification. There is no consensus in the literature on the best methods for binning, for comparing community structures, or for data reduction (Appendix 2). Further, because the measures are highly discriminatory, it is possible to obtain statistically significant results from relatively small samples for which correspondence to meaningful clinical differences may be questionable.
THE WAY FORWARD
Some of the most promising applications of microbiomic research to human health have arisen from studies with substantial rigor in both their technical methods and their epidemiologic design. We highlight 2 exemplar studies here. Antony et al. (68) used metagenomics to compare placental microbial community composition and putative functions between preterm pregnancies and term pregnancies and explored the potentially modifying effects of excess gestational weight gain and obesity. There was significant variation in the placental microbial communities of preterm births by excess gestational weight gain but not by obesity, suggesting that the association between gestational weight gain and preterm birth may be mediated specifically through changes in the placental microbiota. Further, excess gestational weight gain was associated with decreased microbial folate biosynthesis pathways and decreased butanoate metabolism. Because of this study's relatively large sample size (n = 320), the authors were able to make a robust epidemiologic comparison between women with obesity and women with gestational weight gain with respect to an outcome (preterm birth) that clearly followed the exposure.
Longitudinal follow-up of study participants is particularly important when evaluating whether microbial diversity is a risk factor for future disease. A notable example can be found in a study of the association between intestinal diversity and increased patient mortality following allogenic hematopoietic stem-cell transplantation (69). Patients who died following transplantation had significantly lower intestinal diversity, suggesting that the maintenance of intestinal microbiota at the time of transplantation may have an impact on posttransplant survival. While the study was small (n = 80), the longitudinal design allowed for a clear epidemiologic analysis of the association of microbiota changes with the subsequent outcome (mortality).
These carefully designed studies highlight the power of microbiomic research to determine the mechanistic processes by which exposures contribute to disease. By evaluating host microbiota within the context of carefully selected study groups and temporally appropriate outcomes, such studies serve as models for optimal use of the microbiome in future epidemiologic research.
CONCLUSIONS
Our newfound ability to characterize individual microbiota in a relatively quick and inexpensive manner is truly exciting, and the opportunities for productive interdisciplinary collaboration are great. Epidemiologists can make a major contribution to this emerging field by performing well-designed, well-conducted, and appropriately powered studies and by including measures of microbiota in current and future cohort studies to characterize natural variation in microbiota composition and function, identify important confounders and effect modifiers, and generate and test hypotheses about the role of microbiota in health and disease. However, we have a long way to go before the promise of understanding of the microbiome translates into new diagnostic, therapeutic, and preventive measures (70). To achieve that promise requires well-conducted prospective studies that characterize the distribution of microbial communities' structure and function over time and space and identify the determinants of those distributions. We also must temper the enthusiasm that arises from each new characterization by critically appraising each new microbiomic study and placing it within a greater public health context. Since the time of Semmelweiss, millions of lives have been saved—and continue to be saved (Ebola is a case in point) through good public health practices: hand hygiene, sanitation, vaccination, and antibiotics. It is incumbent upon us, as public health workers, to have a firm understanding of the role of microbiota before suggesting any policy changes that might undo sound public health practice.
ACKNOWLEDGMENTS
Author affiliations: Department of Epidemiology and Center for Molecular and Clinical Epidemiology of Infectious Diseases, School of Public Health, University of Michigan, Ann Arbor, Michigan (Betsy Foxman, Emily T. Martin).
This work was supported by the Center for Molecular and Clinical Epidemiology of Infectious Diseases at the University of Michigan (B.F.) and by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, under award K01AI099006 (E.T.M.).
We thank Anna Cronenwett for manuscript preparation.
Conflict of interest: none declared.
APPENDIX 1
Technical Challenges in Microbiomic Studies
Data management
Variation due to differences in sequencing technologies
Quality control and assurance
Computational requirements
Reference databases
Analytical strategies are under development
APPENDIX 2
Epidemiologic Challenges in Microbiomic Studies
High level of variation within and between individuals (dynamic)
Determining which microbiomic differences are clinically significant
Differentiating between microbiomic differences that result from rather than cause a disease process
REFERENCES
- 1.Arrieta M-C, Stiemsma LT, Amenyogbe N, et al. The intestinal microbiome in early life: health and disease. Front Immunol. 2014;5:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curtis MA, Zenobia C, Darveau RP. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe. 2011;104:302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Missaghi B, Barkema HW, Madsen KL, et al. Perturbation of the human microbiome as a contributor to inflammatory bowel disease. Pathogens. 2014;33:510–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zitvogel L, Galluzzi L, Viaud S, et al. Cancer and the gut microbiota: an unexpected link. Sci Transl Med. 2015;7271:271ps1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagier JC, Hugon P, Khelaifia S, et al. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin Microbiol Rev. 2015;281:237–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinakaran V, Rathinavel A, Pushpanathan M, et al. Elevated levels of circulating DNA in cardiovascular disease patients: metagenomic profiling of microbiome in the circulation. PLoS One. 2014;98:e105221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Payne MS, Bayatibojakhi S. Exploring preterm birth as a polymicrobial disease: an overview of the uterine microbiome. Front Immunol. 2014;5:595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickson RP, Erb-Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med. 2013;73:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sommer F, Bäckhed F. The gut microbiota—masters of host development and physiology. Nat Rev Microbiol. 2013;114:227–238. [DOI] [PubMed] [Google Scholar]
- 10.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(suppl 1):4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pallav K, Dowd SE, Villafuerte J, et al. Effects of polysaccharopeptide from Trametes versicolor and amoxicillin on the gut microbiome of healthy volunteers: a randomized clinical trial. Gut Microbes. 2014;54:458–467. [DOI] [PubMed] [Google Scholar]
- 12.Viswanathan VK, Mallozzi MJ, Vedantam G. Clostridium difficile infection: an overview of the disease and its pathogenesis, epidemiology and interventions. Gut Microbes. 2010;14:234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandt LJ, Aroniadis OC, Mellow M, et al. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am J Gastroenterol. 2012;1077:1079–1087. [DOI] [PubMed] [Google Scholar]
- 14.Kassam Z, Lee CH, Yuan Y, et al. Fecal microbiota transplantation for Clostridium difficile infection: systematic review and meta-analysis. Am J Gastroenterol. 2013;1084:500–508. [DOI] [PubMed] [Google Scholar]
- 15.Van Nood E, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;3685:407–415. [DOI] [PubMed] [Google Scholar]
- 16.Virgin HW. The virome in mammalian physiology and disease. Cell. 2014;1571:142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Vlaminck I, Khush KK, Strehl C, et al. Temporal response of the human virome to immunosuppression and antiviral therapy. Cell. 2013;1555:1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robles-Sikisaka R, Ly M, Boehm T, et al. Association between living environment and human oral viral ecology. ISME J. 2013;79:1710–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ly M, Abeles SR, Boehm TK, et al. Altered oral viral ecology in association with periodontal disease. MBio. 2014;53:e01133-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wylie KM, Mihindukulasuriya KA, Sodergren E, et al. Sequence analysis of the human virome in febrile and afebrile children. PLoS One. 2012;76:e27735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willner D, Furlan M, Haynes M, et al. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PLoS One. 2009;410:e7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Paepe M, Leclerc M, Tinsley CR, et al. Bacteriophages: an underestimated role in human and animal health? Front Cell Infect Microbiol. 2014;4:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pride DT, Salzman J, Haynes M, et al. Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J. 2012;65:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minot S, Bryson A, Chehoud C, et al. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A. 2013;11030:12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lecuit M, Eloit M. The human virome: new tools and concepts. Trends Microbiol. 2013;2110:510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;1381:30–50. [DOI] [PubMed] [Google Scholar]
- 27.Cui L, Morris A, Ghedin E. The human mycobiome in health and disease. Genome Med. 2013;57:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaci N, Borrel G, Tottey W, et al. Archaea and the human gut: new beginning of an old story. World J Gastroenterol. 2014;2043:16062–16078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lozupone CA, Stombaugh JI, Gordon JI, et al. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;4897415:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science. 2014;3466212:954–959. [DOI] [PubMed] [Google Scholar]
- 31.Van de Wijgert JHHM, Borgdorff H, Verhelst R, et al. The vaginal microbiota: what have we learned after a decade of molecular characterization? PLoS One. 2014;98:e105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Methé BA, Nelson KE, Pop M, et al. A framework for human microbiome research. Nature. 2012;4867402:215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;4647285:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Contreras M, Costello EK, Hidalgo G, et al. The bacterial microbiota in the oral mucosa of rural Amerindians. Microbiology. 2010;156(Pt 11):3282–3287. [DOI] [PubMed] [Google Scholar]
- 35.Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;3416150:1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;4577228:480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cox LM, Yamanishi S, Sohn J, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;1584:705–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Youngster I, Russell GH, Pindar C, et al. Oral, capsulized, frozen fecal microbiota transplantation for relapsing Clostridium difficile infection. JAMA. 2014;31217:1772–1778. [DOI] [PubMed] [Google Scholar]
- 39.Ianiro G, Bibbò S, Scaldaferri F, et al. Fecal microbiota transplantation in inflammatory bowel disease: beyond the excitement. Medicine (Baltimore). 2014;9319:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savageau MA. Escherichia coli habitats, cell types, and molecular mechanisms of gene control. Am Nat. 1983;1226:732–744. [Google Scholar]
- 41.Schloissnig S, Arumugam M, Sunagawa S, et al. Genomic variation landscape of the human gut microbiome. Nature. 2013;4937430:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;5057484:559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jakobsson HE, Jernberg C, Andersson AF, et al. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One. 2010;53:e9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mills RE, Pittard WS, Mullaney JM, et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 2011;216:830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Touchon M, Hoede C, Tenaillon O, et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009;51:e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dominguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;10726:11971–11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogier EW, Frantz AL, Bruno MEC, et al. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc Natl Acad Sci U S A. 2014;1118:3074–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lax S, Smith DP, Hampton-Marcell J, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;3456200:1048–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flores GE, Caporaso JG, Henley JB, et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014;1512:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodrich JK, Waters JL, Poole AC, et al. Human genetics shape the gut microbiome. Cell. 2014;1594:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fierer N, Lauber CL, Zhou N, et al. Forensic identification using skin bacterial communities. Proc Natl Acad Sci U S A. 2010;10714:6477–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature. 2014;5097500:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaishampayan PA, Kuehl JV, Froula JL, et al. Comparative metagenomics and population dynamics of the gut microbiota in mother and infant. Genome Biol Evol. 2010;2:53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu Z, Malmer D, Langille MGI, et al. Which is more important for classifying microbial communities: who's there or what they can do? ISME J. 2014;812:2357–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jumpstart Consortium Human Microbiome Project Data Generation Working Group. Evaluation of 16S rDNA-based community profiling for human microbiome research. PLoS One. 2012;76:e39315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Norman JM, Handley SA, Virgin HW. Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities. Gastroenterology. 2014;1466:1459–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delwart E. A roadmap to the human virome. PLoS Pathog. 2013;92:e1003146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abeles SR, Pride DT. Molecular bases and role of viruses in the human microbiome. J Mol Biol. 2014;42623:3892–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Willger SD, Grim SL, Dolben EL, et al. Characterization and quantification of the fungal microbiome in serial samples from individuals with cystic fibrosis. Microbiome. 2014;2:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wylie KM, Weinstock GM, Storch GA. Emerging view of the human virome. Transl Res. 2012;1604:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morgan XC, Huttenhower C. Meta'omic analytic techniques for studying the intestinal microbiome. Gastroenterology. 2014;1466:1437–1448.e1. [DOI] [PubMed] [Google Scholar]
- 62.Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;7523:7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010;41:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langille MGI, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;319:814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Segata N, Boernigen D, Tickle TL, et al. Computational meta'omics for microbial community studies. Mol Syst Biol. 2013;9:666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goodrich JK, Di Rienzi SC, Poole AC, et al. Conducting a microbiome study. Cell. 2014;1582:250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salter SJ, Cox MJ, Turek EM, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Antony KM, Ma J, Mitchell KB, et al. The preterm placental microbiome varies in association with excess maternal gestational weight gain. Am J Obstet Gynecol. 2015;2125:653.e1–653.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taur Y, Jenq RR, Perales M-A, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood. 2014;1247:1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huttenhower C, Knight R, Brown CT, et al. Advancing the microbiome research community. Cell. 2014;1592:227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huse SM, Ye Y, Zhou Y, et al. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One. 2012;76:e34242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wylie KM, Mihindukulasuriya KA, Zhou Y, et al. Metagenomic analysis of double-stranded DNA viruses in healthy adults. BMC Biol. 2014;12:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Foulongne V, Sauvage V, Hebert C, et al. Human skin microbiota: high diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS One. 2012;76:e38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scanlan PD, Marchesi JR. Micro-eukaryotic diversity of the human distal gut microbiota: qualitative assessment using culture-dependent and -independent analysis of faeces. ISME J. 2008;212:1183–1193. [DOI] [PubMed] [Google Scholar]
- 75.Findley K, Oh J, Yang J, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;4987454:367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Monteiro-da-Silva F, Sampaio-Maia B, Pereira M de L, et al. Characterization of the oral fungal microbiota in smokers and non-smokers. Eur J Oral Sci. 2013;1212:132–135. [DOI] [PubMed] [Google Scholar]
- 77.Drell T, Lillsaar T, Tummeleht L, et al. Characterization of the vaginal micro- and mycobiome in asymptomatic reproductive-age Estonian women. PLoS One. 2013;81:e54379. [DOI] [PMC free article] [PubMed] [Google Scholar]