

Abstract
Bone remodeling, the fundamental process for bone renewal, is targeted by treatments of osteoporosis to correct the imbalance between bone resorption and bone formation and reduce the risk of fractures and associated clinical consequences. Currently available therapeutics affect bone resorption and bone formation in the same direction and either decrease (inhibitors of bone resorption) or increase (parathyroid hormone [PTH] peptides) bone remodeling. Studies of patients with rare bone diseases and genetically modified animal models demonstrated that bone resorption and bone formation may not necessarily be coupled, leading to identification of molecular targets in bone cells for the development of novel agents for the treatment of osteoporosis. Application of such agents to the treatment of women with low bone mass confirmed that bone resorption and bone formation can be modulated in different directions and so far two new classes of therapeutics for osteoporosis have been defined with distinct mechanisms of action. Such treatments, if combined with a favorable safety profile, will offer new therapeutic options and will improve the management of patients with osteoporosis.
Key Points
| In osteoporosis, there is an imbalance between bone resorption and bone formation leading to bone loss and structural decay of the skeleton. |
| Currently available therapeutics affect bone resorption and bone formation in the same direction and either decrease (inhibitors of bone resorption) or increase (PTH peptides) bone remodeling. |
| New classes of therapeutics for osteoporosis with different mechanisms of action are in clinical development. |
| Inhibitors of Cathepsin K reduce bone resorption while preserving bone formation and increase bone mineral density at the spine and the hip continuously for at least 5 years of treatment. |
| Inhibitors of sclerostin increase bone formation while reducing bone resorption and impressively increase bone mineral density at the spine and the hip. |
Introduction
Osteoporosis is characterized by reduced bone mass and strength leading to increased risk of fractures. Pharmacological interventions aim to decrease this risk and the associated clinical consequences by correcting the imbalance between bone resorption and bone formation that constitutes the pathophysiological basis of the disease. Most currently available agents inhibit bone resorption and formation to varying degrees and decrease the risk of fractures but cannot replace already lost bone, and they only modestly decrease the risk of non-vertebral fractures, the most frequent osteoporotic fractures. Parathyroid hormone (PTH) peptides, the only approved bone-forming agents, stimulate bone formation but also bone resorption and have not been shown to reduce the risk of hip fractures, the most devastating clinical consequence of osteoporosis. These unmet needs have led to efforts for the development of new therapeutics for osteoporosis based on improved knowledge of the local regulation of bone remodeling arising mainly from the study of rare bone diseases and genetically modified animal models [1]. We review here the information that led to the rational design and clinical application of new agents for the pharmacological management of osteoporosis.
General Considerations
Bone remodeling occurs in an orderly fashion by the basic multicellular units (BMUs), temporary anatomical structures comprising a team of osteoclasts in the front and a team of osteoblasts in the back, supported by blood vessels, nerves, and connective tissue. Osteoclasts resorb bone by removing bone mineral and degrading the organic matrix, while osteoblasts move to the resorbed area and lay down new bone matrix that subsequently mineralizes, a process known as coupling. The mechanisms regulating this coupling are not entirely clear but it is thought that growth factors mobilized from the bone matrix during resorption might contribute to intercellular signaling and subsequent stimulation of bone formation. Alternatively or in addition, the osteoclasts produce factors that might contribute to generation and differentiation of osteoblast precursors [2, 3]. It is now generally accepted that osteocytes are the main regulators of bone remodeling due to their location in bone allowing them to sense mechanical signals and to respond to chemical signals regulating bone and mineral metabolism by secreting factors that can modulate the number and function of osteoblasts and osteoclasts [4–6].
An increased number and life span of osteoclasts and a decrease in the formation and life span of osteoblasts induce an imbalance between bone resorption and bone formation, the cellular basis of osteoporosis. This imbalance, in favor of resorption, results in bone loss and deterioration of bone architecture. The decline in the ability of osteoblasts to refill the resorption cavity leads to reduction of the thickness of the bone packets and thinning of the trabeculae. In addition, the enhanced osteoclastic resorption per unit time that occurs at the menopause results in perforation and removal of trabeculae and loss of their connectivity [7]. Cortical bone becomes wider in diameter and thinner, due to the move of the endosteal surface outwards at a greater pace than bone placed in the periosteum, but also more porotic due to enhanced intracortical remodeling [8]. The net outcome of these changes is increased bone fragility and this provides the rationale for the development of agents for the pharmacological management of osteoporosis. It is clear from the described changes that reduction of bone resorption must be an essential component of any therapeutic approach for the maintenance or improvement of bone strength. However, this approach cannot replace already lost bone, which is required for better fracture protection in women with severe disease. For this, specific stimulation of bone formation is essential. Thus, in theory, optimal pharmacological management of osteoporosis should aim at decreasing bone resorption (endosteal and intracortical) and stimulating bone formation at all skeletal envelopes, including the periosteum. Such approach will not only prevent the structural decay of bone tissue but will also increase bone mass and may lead to improved reduction of the risk of non-vertebral fractures, which occur predominantly at cortical bone sites.
Inhibitors of Bone Resorption
Inhibitors of osteoclastic bone resorption, such as bisphosphonates, denosumab and selective estrogen receptor modulators (SERMs), reduce the rate of bone resorption to varying degrees by different mechanisms of action. The reduction of the rate of bone resorption is invariably followed by reduction of the rate of bone formation due to the coupling of the two processes. The final result is an overall decrease of the rate of bone turnover to a level that depends on the potency of the individual agent used and is maintained during the whole period of treatment. The introduction of the most potent inhibitor of bone resorption, denosumab, into clinical practice made any further development of this class of agents obsolete. However, studies of humans and animals with osteopetrosis indicated that reduction of bone resorption may not necessarily be coupled with reduced bone formation if the osteoclasts remain intact [9]. Loss of function of a number of molecules regulating removal of bone mineral or degradation of bone matrix were shown to be associated with a decrease of bone resorption without, however, affecting or even stimulating bone formation [10, 11]. Cathepsin K (CatK), a protease abundantly expressed in osteoclasts responsible for the degradation of the organic matrix of bone, is the most extensively studied molecule in preclinical and clinical studies.
Cathepsin K Inhibitors
CatK is a member of a family of cysteine proteases that is synthesized as a pro-enzyme before being transported to lysosomes where it is cleaved to produce the active enzyme that degrades collagen type I and other bone matrix proteins within the acidic environment of resorption lacunae [12]. Congenital absence of CatK in patients with pycnodysostosis, a rare, autosomal, recessive osteochondrodysplasia, is characterized by increased bone density, bone deformities, and increased bone fragility, complications that are not present in heterozygotes [13]. CatK-deficient mice develop a high bone mass phenotype in the presence of fully differentiated osteoclasts, while mice over-expressing CatK had increased bone turnover and decreased trabecular bone volume [14, 15]. The discovery that loss of function of CatK decreases bone resorption with increased number of viable osteoclasts and the surprising finding of preservation or even increase in bone formation provided the rationale for the development of a new class of antiresorptive agents that target this enzyme (Fig. 1) [16–18]. The mechanism responsible for the maintenance or increase in bone formation in the presence of reduced bone resorption by CatK inhibition may be due to stimulation of osteoblasts by osteoclast-derived factors (clastokines, such as sphingosine-1-phosphate) or matrix-derived growth factors (such as IGF-1) that are not degraded [19, 20]. Initial studies of CatK inhibitors showed off-target inhibition of other cathepsins due either to their lack of specificity for CatK or to their accumulation in lysosomes of cells other than osteoclasts and led to the design of new agents potentially devoid of such effects. Two CatK inhibitors are currently in clinical development for the treatment of osteoporosis, namely odanacatib (Merck & Co) and ONO-5334 (Ono Pharmaceutical Company), which in phase II clinical trials had similar effects on bone turnover and bone mineral density (BMD) without any off-target effects. We will further discuss only odanacatib, as an example of pharmacological inhibition of CatK, because it is the only one tested so far in a phase III clinical study.
Fig. 1.
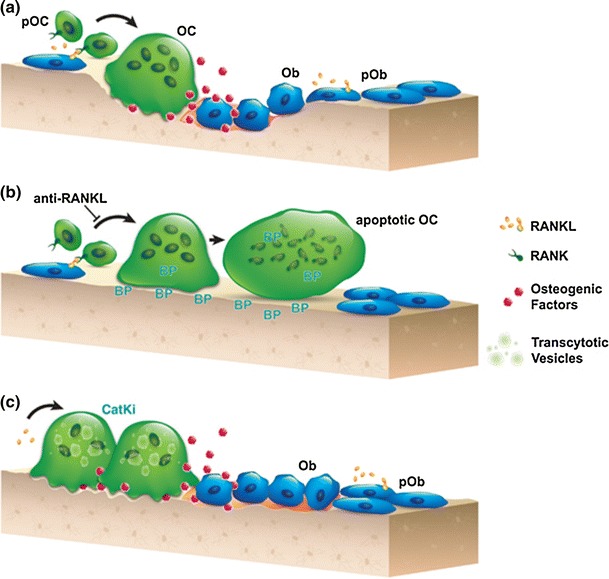
Schematic representations of a the normal coupling process of bone resorption and formation during the remodeling process. RANKL promotes differentiation and activation of osteoclasts at remodeling sites. Coupling factors derived from the resorbed bone matrix or directly from the activated osteoclasts stimulate the recruitment and maturation of osteoblasts to initiate bone formation on the existing resorption surface. b Denosumab blocks osteoclastogenesis, and bisphosphonate induces the loss of ruffled border and eventual osteoclast apoptosis. These therapies lead to little-to-no resorption surface and fewer numbers of osteoclasts on bone. c Treatment with a CatK inhibitor reduces osteoclastic resorption efficiency and retards transcytotic trafficking of matrix removal. This does not prevent other osteoclast functions, such as the generation of a shallow resorption surface and the release of osteogenic factors; together, these functions initiate osteoblast bone formation. BP bisphosphonate, CatKi cathepsin K inhibitor, Ob osteoblast, OC osteoclast, pOb osteoblast progenitor, pOC osteoclast progenitor, RANK receptor activator of nuclear factor kappa-B, RANKL RANK ligand. Reproduced with permission from [17]
Odanacatib is a selective, orally administered CatK inhibitor [21]. Unlike basic CatK inhibitors, odanacatib is neutral and does not accumulate in the acidic environment of lysosomes which could lead to off-target inhibition of other cathepsins [16, 22]. Odanacatib is metabolized by CYP3A4 and its absorption is not impaired by food intake [17, 23]. In animal models, odanacatib reduced bone resorption while preserving bone formation in trabecular and endocortical surfaces. In addition, odanacatib reduced cortical remodeling and increased modeling-based bone formation, and improved the cortical area of the femur and its strength [24, 25]. Odanacatib was further superior to alendronate in increasing cortical thickness, possibly through increased periosteal bone formation, an action that was also observed during treatment with another CatK inhibitor [26, 27].
A 2-year (with a 3-year extension) phase IIb dose-finding clinical trial of postmenopausal women with low bone mass identified 50 mg once weekly as the optimal oral dose of odanacatib [28–30]. In this study, treatment with odanacatib 50 mg once weekly reduced biochemical markers of bone resorption by about 55 %, while markers of bone formation were mildly and transiently reduced, returning to baseline after about 2 years. Serum levels of the osteoclast marker TRAP5b increased with treatment, confirming osteoclast viability. These changes of bone markers were associated with continuous increases in BMD by 11.9 % at the spine and more importantly at the hip by 8.5 and 9.8 % at the total hip (TH) and femoral neck (FN), respectively, after 5 years. Different from bisphosphonates but similar to other antiresorptives, the effects of odanacatib on bone turnover and BMD were rapidly reversible upon discontinuation of treatment. Odanacatib treatment exhibited a generally favorable safety and tolerability profile, with incidence of adverse events similar to placebo.
In another 2-year, placebo-controlled study, the effects of odanacatib on the cortical and trabecular compartments and strength of bone were assessed by quantitative computed tomography (QCT) and high-resolution QCT (HR-QCT) in 214 postmenopausal women with low areal BMD [31, 32]. As in the phase II study, odanacatib decreased bone resorption, maintained bone formation, and increased areal BMD. In addition, it increased volumetric BMD and estimated bone strength of both the hip and the spine as well as cortical and trabecular density, cortical thickness, and estimated strength of the distal radius and distal tibia. In this study, adverse effects were also similar between placebo- and odanacatib-treated women.
The antifracture efficacy of odanacatib was examined in the Long-Term Odanacatib Fracture Trial (LOFT), the largest clinical trial in osteoporosis. LOFT is a phase III, event-driven clinical trial, with a preplanned extension, of women aged >65 years with osteoporosis [33]. Participants had either a prior radiographic vertebral fracture and BMD T-score ≤−1.5 at the FN or TH or no prior vertebral fracture and BMD T-score ≤−2.5 at the FN or TH. The trial was designed to test the hypothesis that treatment with odanacatib 50 mg once weekly reduces the risk of new morphometric vertebral, hip and non-vertebral fractures (all primary endpoints) in postmenopausal women with osteoporosis. Women were randomized in a 1:1 ratio to receive placebo or odanacatib 50 mg once weekly. All received weekly vitamin D3 (5600 IU) and daily calcium supplements to ensure a daily intake of approximately 1200 mg. In July 2012, an independent Data Monitoring Committee (DMC) recommended termination of the study since odanacatib displayed a favorable benefit/risk profile as well as efficacy relative to placebo. At the same time, the DMC suggested that additional safety data should be obtained in the preplanned blinded extension study.
The 16,071 women included in the study had a mean age of 72.8 years, 57 % were Caucasian and 46.5 % had prior vertebral fracture. Mean BMD T-scores were: LS −2.7, TH −2.4, and FN −2.7 and mean duration of follow-up was 34 months [34]. Compared with placebo, treatment with odanacatib decreased the incidence of new and worsening morphometric vertebral fractures by 54 %, of hip fractures by 47 %, of non-vertebral fractures by 23 % and of clinical fractures by 72 % (all p < 0.0001). Odanacatib treatment led to progressive increases over 5 years in BMD at LS and TH: 11.2 and 9.5 %, respectively, compared with placebo. Adverse events were generally well balanced between groups. Adjudicated morphea-like skin lesions occurred more frequently in odanacatib-treated patients (12) compared with placebo (3) and resolved/improved after study drug discontinuation. Adjudicated femoral shaft fractures with atypical features occurred only in odanacatib-treated patients (5), while no cases of ONJ were reported. No meaningful differences between groups were observed in adjudicated systemic sclerosis, respiratory infections, or delayed fracture union. Major cardiovascular events overall were generally balanced; however, there were numerically more adjudicated strokes with odanacatib than with placebo; final blinded adjudication of major cardiovascular events is ongoing [35].
Stimulators of Bone Formation
The only currently available bone forming agent, PTH, stimulates bone formation but also bone resorption. PTH binds to the PTH/PTHrP type 1 receptor and activates several signaling pathways, including the canonical Wnt-signaling pathway, having both anabolic and catabolic effects on bone that are probably exerted via signaling in osteocytes [36]. Teriparatide, given by daily subcutaneous injections, increases cancellous and endocortical bone formation, mainly at sites undergoing active bone remodeling, but has limited effect on periosteal bone formation and increases cortical porosity [37]. PTHrP 1–36 and its analog abaloparatide, which bind to the PTH/PTHrP 1 receptor, also increase bone formation and bone resorption markers, but to a lesser extent than teriparatide, and improve hip BMD significantly more than teriparatide [38, 39].
Concurrent treatment of women with osteoporosis with teriparatide and the inhibitor of bone resorption, denosumab, increased BMD at all skeletal sites considerably more than either monotherapy alone after 2 years [40, 41]. The difference in response between teriparatide and teriparatide/denosumab treatment is probably due to inhibition of teriparatide-stimulated RANKL (receptor activator of nuclear factor kappa-B ligand) production by denosumab that reduced bone resorption and allowed teriparatide to exert a stimulatory effect only on bone formation. These results reinforce the hypothesis that for optimal therapeutic outcome, bone formation and bone resorption should be modulated in different directions. The results of the studies of CatK inhibitors illustrated that this may be feasible. CatK inhibitors, however, may preserve bone formation but are not anabolic agents, an important unmet need in the management of osteoporosis.
The design of a genuine anabolic treatment for osteoporosis must address the possibility of stimulating bone formation without concomitant stimulation of bone resorption and ensuring that formation is stimulated at quiescent bone surfaces. Human and animal genetics indicated that this may be feasible. In particular, the recognition of the pivotal role of the Wnt signaling pathway in bone formation provided a number of potential targets for the development of new pharmaceuticals. For clinical use, however, treatments should not only modify the expression of target molecules but need also to have bone specificity to avoid potential off-target effects [1, 42, 43]. One such target is sclerostin, a negative regulator of bone formation produced exclusively in the skeleton by osteocytes [44].
Sclerostin Inhibitors
The role of sclerostin in bone metabolism was identified in studies of patients with sclerosteosis and van Buchem disease, two rare sclerosing bone dysplasias with very similar phenotypes and high bone mass [45]. These diseases are characterized by progressive generalized overgrowth and thickening of bone that is resistant to fracture [46]. The two conditions are due to different defects of the SOST gene which is located on chromosome 17q12–21 and encodes the protein sclerostin [47–50]. Both defects result in impaired production of sclerostin leading to a dramatic increase in bone mass in humans and mice, whereas mice overexpressing sclerostin are osteopenic [51, 52].
Osteocyte-produced sclerostin is transported to the bone surface through the osteocyte dendritic network where it inhibits the proliferation, differentiation, and survival of osteoblasts. Sclerostin also has an autocrine function and upregulates RANKL synthesis in osteocytes, thereby stimulating osteoclastogenesis [53] (Fig. 2). SOST has been identified in the kidney, liver, and heart, but sclerostin expression has not been identified in any of these tissues in humans [45, 47, 48]. Sclerostin decreases bone formation by antagonizing the canonical Wnt signaling pathway in osteoblasts. It binds to the first propeller domain of the LRP5/6 receptor, thereby disabling the formation of the co-receptor complex between LRP5/6 and Frizzled receptor, and inhibiting the Wnt pathway high up in the signaling cascade [54]. The exact mechanism by which sclerostin interacts with the LRP5/6 receptor remains to be established but it is thought that it requires a co-factor to inhibit the Wnt pathway similar to another Wnt antagonist, Dkk1, which needs Kremen to inhibit the pathway. LRP4, produced by osteoblasts and early osteocytes, was recently proposed as a facilitator of the inhibitory function of sclerostin on bone formation, and mutations in LRP4 were identified in patients with a phenotype closely resembling that of sclerosteosis [53]. In addition, inhibition of LRP4 by a specific antibody increased the rate of bone formation and bone mass in mice [55].
Fig. 2.
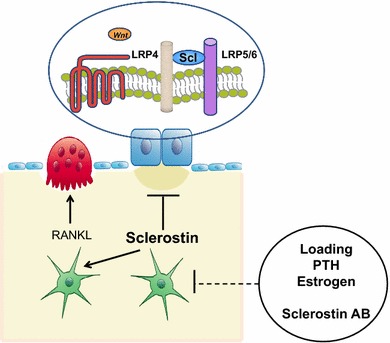
Schematic representation of sclerostin actions. Osteocyte-produced sclerostin inhibits the proliferation, differentiation, and survival of osteoblasts and reduces bone formation; it also stimulates the production of RANKL by neighboring osteocytes and bone resorption. In osteoblasts, sclerostin binds to LRP5/6 and inhibits the Wnt signaling pathway, an action facilitated by LRP4. Production of sclerostin is decreased by mechanical loading, PTH, estrogens and other factors, and antisclerostin antibodies. LRP low-density lipoprotein receptor-related protein, PTH parathyroid hormone, RANKL receptor activator of nuclear factor kappa-B ligand
The restricted expression of sclerostin in the skeleton and the lack of abnormalities in organs other than the skeleton in patients and animals with sclerostin deficiency made this protein an attractive target for the development of a new bone-forming therapy for the management of osteoporosis. This approach was further supported by studies of heterozygous carriers of sclerosteosis who have increased serum levels of P1NP and high, normal, or increased BMD but no clinical symptoms, signs, or complications of sclerosteosis [46, 56]. Two inhibitors of sclerostin are currently being tested for the treatment of osteoporosis; romosozumab or AMG 785 (humanized monoclonal antibody, Amgen and UCB) and blosozumab (humanized monoclonal antibody, Eli Lilly). Romosozumab was used in most reported studies. In aged ovariectomized rats and nonhuman primates, romosozumab stimulated trabecular and cortical bone formation and increased bone mass and strength [57, 58]. Importantly, the majority of new bone formation induced by romosozumab was modeling-based, occurring at quiescent surfaces, demonstrating a clear anabolic response [59, 60]. The increased bone formation induced by romosozumab treatment was not associated with an increase in bone resorption. Instead, a decrease of osteoclast surface was observed, suggesting a functional uncoupling between bone formation and bone resorption, as also shown in the studies of SOST knock-out mice. The effect of sclerostin inhibition on bone formation markers decreased with prolongation of treatment and was reversible upon its discontinuation.
In phase I human studies, administration of single or multiple doses of romosozumab and blosozumab increased bone formation and decreased bone resorption markers associated with significant increases in BMD [61– 63]. In a placebo-controlled, dose-escalating study of 72 healthy men and postmenopausal women, it was shown that a single injection of romosozumab markedly increased bone formation markers and BMD and was well tolerated [62]. Serum levels of the bone formation marker P1NP reached a peak 14–25 days after the antibody administration and returned progressively to baseline after about 2 months. In contrast, serum levels of the bone resorption marker CTX decreased to a minimum about 14 days after the antibody injection and returned to baseline after about 2 months, in agreement with animal data.
Recker et al. recently reported the results of a dose-finding study of blosozumab given subcutaneously for 1 year to 120 postmenopausal women aged 45–85 years with BMD T-scores between −2.0 and −3.5 [64]. All women received calcium and vitamin D supplements and were randomized to receive placebo or blosozumab (180 mg every 4 weeks, 180 mg every 2 weeks, or 270 mg every 2 weeks). Blosozumab treatment induced dose-dependent increases in spine BMD by 8.4, 14.9, and 17.0 %, respectively; TH BMD by 2.1, 4.5, and 6.3 %, respectively; FN BMD by 2.7, 3.9, and 6.3 %, respectively; and total body bone mineral content by 1.7, 4.2, and 7.3 %, respectively, after 1 year; blosozumab treatment had no effect on BMD of the distal radius. Mild injection site reactions were more frequently observed with blosozumab than with placebo and neutralizing antibodies developed in one patient, which affected her response to treatment. Although the frequency of adverse events was similar among all groups, four women (all Japanese) were diagnosed with breast cancer between 3 months of initiating treatment to 1 year after the last dose of blosozumab; none of the investigators considered this adverse event to be related to blosozumab treatment.
Results of a phase II clinical trial of the efficacy and tolerability of romosozumab in postmenopausal women with low bone mass were also reported [65]. In this study, different doses and dosing intervals of subcutaneous injections of romosozumab were compared with placebo, oral alendronate 70 mg weekly, and subcutaneous teriparatide 20 μg daily. The primary efficacy point of the study was the change of spine BMD after 12 months. All doses of romosozumab induced significant increases in BMD. The highest dose of romosuzamab used, 210 mg once monthly, increased BMD at the spine (11.3 %), TH (4.1 %) and FN (3.7 %). These increases were significantly higher than those observed in women treated with either alendronate or teriparatide. For example, the corresponding increases at the spine were 4.1 % for alendronate and 7.1 % with teriparatide after 12 months. No significant differences in BMD of the distal third of the radius were observed at 12 months between any of the romosozumab groups and placebo, alendronate, or teriparatide groups. Adverse events were similar among all groups of studied women except for mild reactions at the injection sites of romosozumab. One patient treated with romosozumab was diagnosed during the trial with breast cancer that was not considered to be treatment-related.
Continuation of treatment for a second year was associated with further increases in LS and TH BMD to total gains of 15.7 and 6.0 %, respectively. Women who transitioned to denosumab after 2 years with romosozumab continued to accrue BMD at a rate similar to that of romosozumab during the second year, while in those who transitioned to placebo, BMD returned towards pretreatment levels; similar results were reported after discontinuation of blosozumab [66]. Serum P1NP and CTX levels remained below baseline values during the second year of romosozumab treatment [67].
Kinetics of biochemical markers of bone turnover in humans and histological data in animals during treatment with sclerostin inhibitors were different from those observed during treatment of patients with other antiosteoporotic agents (Fig. 3). There was an early rapid increase in bone formation markers followed by a progressive decline with time which was not due to the development of neutralizing antibodies. The effect of sclerostin inhibition on bone formation was further associated with a decrease of bone resorption, possibly through an inhibitory effect of the antibody on the production of RANKL by the osteocytes [68]. Treatment prolongation, however, appears to modestly reduce bone resorption but also bone turnover. It may, thus, be that while romosozumab acts as a pure anabolic agent in the beginning of treatment, its continued administration results in mild inhibition of bone resorption and reduction of the remodeling space. Phase III clinical studies are currently investigating the antifracture efficacy and tolerability of romosozumab and blosozumab in patients with osteoporosis (www.clinicaltrials.gov).
Fig. 3.
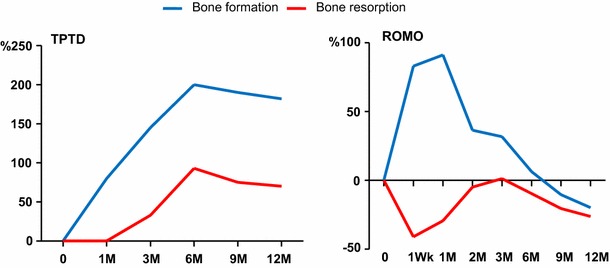
Schematic representation of changes in the levels of biochemical markers of bone turnover during treatment with subcutaneous injections of either teriparatide (TPTD, 20 ug daily) or romosozumab (ROMO, 210 mg once monthly) for 1 year. Bone formation and bone resorption were assessed by measuring serum levels of procollagen type 1 aminoterminal propeptide (P1NP) and carboxy-terminal collagen crosslinking (CTX), respectively. Reproduced from [41]
Positioning inhibitors of sclerostin in the management of patients with osteoporosis will depend not only on their efficacy in increasing bone mass and reducing the risk of fractures at all skeletal sites, but also on their safety profile. The observations that patients with sclerostin deficiency do not have complications from organs other than the skeleton, and that heterozygous carriers do not have any specific clinical phenotype, are reassuring. However, the number of these individuals is small and the Wnt-signaling pathway is involved in growth and differentiation of many cell types and its activation has been linked to tumorinogenesis, osteoarthritis, and cardiovascular calcification.
Conclusions
The two components of bone remodeling, resorption and formation, constitute the primary target of pharmacological interventions for the management of the disease. It is now clear that bone resorption and formation can be differently modulated by new classes of antiosteoporotic medications that provide a novel, personalized perspective for the management of patients in clinical practice.
Disclosures
Dr Appelman-Dijkstra declares no conflicts of interest. Dr Papapoulos has received consulting/speaking fees from Amgen, Merck & Co, and UCB. No funding has been received for this manuscript.
References
- 1.Appelman-Dijkstra NM, Papapoulos SE. Novel approaches to the treatment of osteoporosis. Best Pract Res Clin Endocrinol Metab. 2014;28(6):843–857. doi: 10.1016/j.beem.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 2.Karsdal MA, Martin TJ, Bollerslev J, Christiansen C, Henriksen K. Are nonresorbing osteoclasts sources of bone anabolic activity? J Bone Miner Res. 2007;22(4):487–494. doi: 10.1359/jbmr.070109. [DOI] [PubMed] [Google Scholar]
- 3.Martin TJ, Sims NA. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med. 2005;11(2):76–81. doi: 10.1016/j.molmed.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int. 2014;94(1):25–34. doi: 10.1007/s00223-013-9774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–238. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaffler MB, Cheung WY, Majeska R, Kennedy O. Osteocytes: master orchestrators of bone. Calcif Tissue Int. 2014;94(1):5–24. doi: 10.1007/s00223-013-9790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seeman E, Delmas PD. Bone quality—the material and structural basis of bone strength and fragility. N Engl J Med. 2006;354(21):2250–2261. doi: 10.1056/NEJMra053077. [DOI] [PubMed] [Google Scholar]
- 8.Zebaze RM, Ghasem-Zadeh A, Bohte A, Iuliano-Burns S, Mirams M, Price RI, Mackie EJ, Seeman E. Intracortical remodelling and porosity in the distal radius and post-mortem femurs of women: a cross-sectional study. Lancet. 2010;375(9727):1729–1736. doi: 10.1016/S0140-6736(10)60320-0. [DOI] [PubMed] [Google Scholar]
- 9.Segovia-Silvestre T, Neutzsky-Wulff AV, Sorensen MG, Christiansen C, Bollerslev J, Karsdal MA, Henriksen K. Advances in osteoclast biology resulting from the study of osteopetrotic mutations. Hum Genet. 2009;124(6):561–577. doi: 10.1007/s00439-008-0583-8. [DOI] [PubMed] [Google Scholar]
- 10.Henriksen K, Karsdal MA, Martin TJ. Osteoclast-derived coupling factors in bone remodeling. Calcif Tissue Int. 2014;94(1):88–97. doi: 10.1007/s00223-013-9741-7. [DOI] [PubMed] [Google Scholar]
- 11.Thudium CS, Moscatelli I, Flores C, Thomsen JS, Brüel A, Gudmann NS, Hauge EM, Karsdal MA, Richter J, Henriksen K. A comparison of osteoclast-rich and osteoclast-poor osteopetrosis in adult mice sheds light on the role of the osteoclast in coupling bone resorption and bone formation. Calcif Tissue Int. 2014;95(1):83–93. doi: 10.1007/s00223-014-9865-4. [DOI] [PubMed] [Google Scholar]
- 12.Garnero P, Borel O, Byrjalsen I, Ferreras M, Drake FH, McQueney MS, Foged NT, Delmas PD, Delaissé JM. The collagenolytic activity of cathepsin K is unique among mammalian proteinases. J Biol Chem. 1998;273(48):32347–32352. doi: 10.1074/jbc.273.48.32347. [DOI] [PubMed] [Google Scholar]
- 13.Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273(5279):1236–1238. doi: 10.1126/science.273.5279.1236. [DOI] [PubMed] [Google Scholar]
- 14.Gowen M, Lazner F, Dodds R, Kapadia R, Feild J, Tavaria M, Bertoncello I, Drake F, Zavarselk S, Tellis I, Hertzog P, Debouck C, Kola I. Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J Bone Miner Res. 1999;14(10):1654–1663. doi: 10.1359/jbmr.1999.14.10.1654. [DOI] [PubMed] [Google Scholar]
- 15.Kiviranta R, Morko J, Uusitalo H, Aro HT, Vuorio E, Rantakokko J. Accelerated turnover of metaphyseal trabecular bone in mice overexpressing cathepsin K. J Bone Miner Res. 2001;16(8):1444–1452. doi: 10.1359/jbmr.2001.16.8.1444. [DOI] [PubMed] [Google Scholar]
- 16.Boonen S, Rosenberg E, Claessens F, Vanderschueren D, Papapoulos S. Inhibition of cathepsin K for treatment of osteoporosis. Curr Osteoporos Rep. 2012;10(1):73–79. doi: 10.1007/s11914-011-0085-9. [DOI] [PubMed] [Google Scholar]
- 17.le Duong T. Therapeutic inhibition of cathepsin K-reducing bone resorption while maintaining bone formation. Bonekey Rep. 2012;1:67. doi: 10.1038/bonekey.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yasuda Y, Kaleta J, Brömme D. The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev. 2005;57(7):973–993. doi: 10.1016/j.addr.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 19.Fuller K, Lawrence KM, Ross JL, Grabowska UB, Shiroo M, Samuelsson B, Chambers TJ. Cathepsin K inhibitors prevent matrix-derived growth factor degradation by human osteoclasts. Bone. 2008;42(1):200–211. doi: 10.1016/j.bone.2007.09.044. [DOI] [PubMed] [Google Scholar]
- 20.Lotinun S, Kiviranta R, Matsubara T, Alzate JA, Neff L, Luth A, Koskivirta I, Kleuser B, Vacher J, Vuorio E, Horne WC, Baron R. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest. 2013;123(2):666–681. doi: 10.1172/JCI64840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gauthier JY, Chauret N, Cromlish W, Desmarais S, le Duong T, Falgueyret JP, Kimmel DB, Lamontagne S, Léger S, LeRiche T, Li CS, Massé F, McKay DJ, Nicoll-Griffith DA, Oballa RM, Palmer JT, Percival MD, Riendeau D, Robichaud J, Rodan GA, Rodan SB, Seto C, Thérien M, Truong VL, Venuti MC, Wesolowski G, Young RN, Zamboni R, Black WC. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg Med Chem Lett. 2008;18(3):923–928. doi: 10.1016/j.bmcl.2007.12.047. [DOI] [PubMed] [Google Scholar]
- 22.Pennypacker BL, le Duong T, Cusick TE, Masarachia PJ, Gentile MA, Gauthier JY, Black WC, Scott BB, Samadfam R, Smith SY, Kimmel DB. Cathepsin K inhibitors prevent bone loss in estrogen-deficient rabbits. J Bone Miner Res. 2011;26(2):252–262. doi: 10.1002/jbmr.223. [DOI] [PubMed] [Google Scholar]
- 23.Pennypacker BL, Oballa RM, Levesque S, Kimmel DB, le Duong T. Cathepsin K inhibitors increase distal femoral bone mineral density in rapidly growing rabbits. BMC Musculoskelet Disord. 2013;14:344. doi: 10.1186/1471-2474-14-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cusick T, Chen CM, Pennypacker BL, Pickarski M, Kimmel DB, Scott BB, le Duong T. Odanacatib treatment increases hip bone mass and cortical thickness by preserving endocortical bone formation and stimulating periosteal bone formation in the ovariectomized adult rhesus monkey. J Bone Miner Res. 2012;27(3):524–537. doi: 10.1002/jbmr.1477. [DOI] [PubMed] [Google Scholar]
- 25.Pennypacker BL, Chen CM, Zheng H, Shih MS, Belfast M, Samadfam R, le Duong T. Inhibition of cathepsin K increases modeling-based bone formation, and improves cortical dimension and strength in adult ovariectomized monkeys. J Bone Miner Res. 2014;29(8):1847–1858. doi: 10.1002/jbmr.2211. [DOI] [PubMed] [Google Scholar]
- 26.Cabal A, Jayakar RY, Sardesai S, Phillips EA, Szumiloski J, Posavec DJ, Mathers PD, Savitz AT, Scott BB, Winkelmann CT, Motzel S, Cook L, Hargreaves R, Evelhoch JL, Dardzinski BJ, Hangartner TN, McCracken PJ, le Duong T, Williams DS. High-resolution peripheral quantitative computed tomography and finite element analysis of bone strength at the distal radius in ovariectomized adult rhesus monkey demonstrate efficacy of odanacatib and differentiation from alendronate. Bone. 2013;56(2):497–505. doi: 10.1016/j.bone.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 27.Jerome C, Missbach M, Gamse R. Balicatib, a cathepsin K inhibitor, stimulates periosteal bone formation in monkeys. Osteoporos Int. 2012;23(1):339–349. doi: 10.1007/s00198-011-1593-2. [DOI] [PubMed] [Google Scholar]
- 28.Langdahl B, Binkley N, Bone H, Gilchrist N, Resch H, Rodriguez Portales J, Denker A, Lombardi A, Le Bailly De Tilleghem C, Dasilva C, Rosenberg E, Leung A. Odanacatib in the treatment of postmenopausal women with low bone mineral density: 5 years of continued therapy in a phase 2 study. J Bone Miner Res. 2012;27(11):2251–2258. doi: 10.1002/jbmr.1695. [DOI] [PubMed] [Google Scholar]
- 29.Eisman JA, Bone HG, Hosking DJ, McClung MR, Reid IR, Rizzoli R, Resch H, Verbruggen N, Hustad CM, DaSilva C, Petrovic R, Santora AC, Ince BA, Lombardi A. Odanacatib in the treatment of postmenopausal women with low bone mineral density: 3-year continued therapy and resolution of effect. J Bone Miner Res. 2011;26(2):242–251. doi: 10.1002/jbmr.212. [DOI] [PubMed] [Google Scholar]
- 30.Bone HG, McClung MR, Roux C, Recker RR, Eisman JA, Verbruggen N, Hustad CM, DaSilva C, Santora AC, Ince BA. Odanacatib, a cathepsin-K inhibitor for osteoporosis: a 2-year study in postmenopausal women with low bone density. J Bone Miner Res. 2010;25(5):937–947. doi: 10.1359/jbmr.091035. [DOI] [PubMed] [Google Scholar]
- 31.Brixen K, Chapurlat R, Cheung AM, Keaveny TM, Fuerst T, Engelke K, Recker R, Dardzinski B, Verbruggen N, Ather S, Rosenberg E, de Papp AE. Bone density, turnover, and estimated strength in postmenopausal women treated with odanacatib: a randomized trial. J Clin Endocrinol Metab. 2013;98(2):571–580. doi: 10.1210/jc.2012-2972. [DOI] [PubMed] [Google Scholar]
- 32.Cheung AM, Majumdar S, Brixen K, Chapurlat R, Fuerst T, Engelke K, Dardzinski B, Cabal A, Verbruggen N, Ather S, Rosenberg E, de Papp AE. Effects of odanacatib on the radius and tibia of postmenopausal women: improvements in bone geometry, microarchitecture, and estimated bone strength. J Bone Miner Res. 2014;29(8):1786–1794. doi: 10.1002/jbmr.2194. [DOI] [PubMed] [Google Scholar]
- 33.Bone HG, Dempster DW, Eisman JA, Greenspan SL, McClung MR, Nakamura T, Papapoulos S, Shih WJ, Rybak-Feiglin A, Santora AC, Verbruggen N, Leung AT, Lombardi A. Odanacatib for the treatment of postmenopausal osteoporosis: development history and design and participant characteristics of LOFT, the Long-Term Odanacatib Fracture Trial. Osteoporos Int. 2015;26:699–712. [DOI] [PMC free article] [PubMed]
- 34.McClung M, Langdahl B, Papapoulos S, Saag K, Adami S, Bone H, de Villiers T, Kiel D, Kumar P, Lim S-K, Ling X, Lippuner K, Mautalen C, Nakamura T, Reginster J-Y, Reid I, Portales JR, Roux C, Walliser J, Waats N, Zanchetta JR, Zerbine C, Rybak-Feiglin A, Cohn D, DaSilva C, Massaad R, Santora A, Scott B, Verbruggen N, Leung A, Lomardi A. Odanacatib anti-fracture efficacy and safety in postmenopausal women with osteoporosis. Results from the phase III long-term odanacatib fracture trial (LOFT). JBMR 2014;p. S51 (abstract 1147), Epub.
- 35.McClung MR, Langdahl B, Papapoulos S, Saag K, Bone H, Rybak-Feiglin A, Cohn D, DaSilva CA, Massaad R, Santora AC, Scott BB, Kaufman KD, Verbruggen N, Leung A, Lombardi A. Odanacatib anti-fracture efficacy and safety in postmenopausal women with osteoporosis: results from the phase III long-term odanacatib fracture trial (LOFT) ECTS-IBMS Abstracts; (2015) OC4.4.
- 36.Saini V, Marengi DA, Barry KJ, Fulzele KS, Heiden E, Liu X, Dedic C, Maeda A, Lotinun S, Baron R, Pajevic PD. Parathyroid hormone (PTH)/PTH-related peptide type 1 receptor (PPR) signaling in osteocytes regulates anabolic and catabolic skeletal responses to PTH. J Biol Chem. 2013;288(28):20122–20134. doi: 10.1074/jbc.M112.441360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Compston JE. Skeletal actions of intermittent parathyroid hormone: effects on bone remodelling and structure. Bone. 2007;40(6):1447–1452. doi: 10.1016/j.bone.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Horwitz MJ, Augustine M, Kahn L, Martin E, Oakley CC, et al. A comparison of parathyroid hormone-related protein (1-36) and parathyroid hormone (1-34) on markers of bone turnover and bone density in postmenopausal women: The PrOP study. J Bone Miner Res. 2013;28:2266–2276. doi: 10.1002/jbmr.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leder BZ, O’Dea LSL, Zanchetta JR, Kumar P, Banks K, McKay K, Lyttle CR, Hattersley G. Effects of abaloparatide, a human parathyroid hormone-related peptide analog, on bone mineral density in postmenopausal women with osteoporosis. J Clin Endocrinol Metab. 2015;100(2):697–706. doi: 10.1210/jc.2014-3718. [DOI] [PubMed] [Google Scholar]
- 40.Tsai JN, Uihlein AV, Lee H, Kumbhani R, Siwila-Sackman E, McKay EA, Burnett-Bowie SA, Neer RM, Leder BZ. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet. 2013;382(9886):50–56. doi: 10.1016/S0140-6736(13)60856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leder BZ, Tsai JN, Uihlein AV, Burnett-Bowie SA, Zhu Y, Foley K, Lee H, Neer RM. Two years of Denosumab and teriparatide administration in postmenopausal women with osteoporosis (The DATA Extension Study): a randomized controlled trial. J Clin Endocrinol Metab. 2014;99(5):1694–1700. doi: 10.1210/jc.2013-4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papapoulos SE. Targeting sclerostin as potential treatment of osteoporosis. Ann Rheum Dis. 2011;70(Suppl 1):i119–i122. doi: 10.1136/ard.2010.141150. [DOI] [PubMed] [Google Scholar]
- 43.Papapoulos SE. Anabolic bone therapies in 2014: New bone-forming treatments for osteoporosis. Nat Rev Endocrinol. 2015;11:69–70. doi: 10.1038/nrendo.2014.214. [DOI] [PubMed] [Google Scholar]
- 44.van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Löwik CW. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199(6):805–814. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moester MJ, Papapoulos SE, Lowik CW, van Bezooijen RL. Sclerostin: current knowledge and future perspectives. Calcif Tissue Int. 2010;87(2):99–107. doi: 10.1007/s00223-010-9372-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Lierop AH, Hamdy NA, Hamersma H, van Bezooijen RL, Power J, Loveridge N, Papapoulos SE. Patients with sclerosteosis and disease carriers: human models of the effect of sclerostin on bone turnover. J Bone Miner Res. 2011;26(12):2804–2811. doi: 10.1002/jbmr.474. [DOI] [PubMed] [Google Scholar]
- 47.Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Den Van, Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10(5):537–543. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 48.Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, Mulligan J. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68(3):577–589. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39(2):91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, Gardner JC, Galas D, Schatzman RC, Beighton P, Papapoulos S, Hamersma H, Brunkow ME. A, 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110(2):144–152. doi: 10.1002/ajmg.10401. [DOI] [PubMed] [Google Scholar]
- 51.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 52.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22(23):6267–6276. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, Bouwmeester T, Schirle M, Bueno-Lozano M, Fuentes FJ, Itin PH, Boudin E, Freitas F, Jennes K, Brannetti B, Charara N, Ebersbach H, Geisse S, Lu CX, Bauer A, Van Hul W, Kneissel M. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286(22):19489–19500. doi: 10.1074/jbc.M110.190330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280(20):19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 55.Chang MK, Kramer I, Huber T, Kinzel B, Guth-Gundel S, Leupin O, Kneissel M. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc Natl Acad Sci USA. 2014;111(48):E5187–E5195. doi: 10.1073/pnas.1413828111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gardner JC, van Bezooijen RL, Mervis B, Hamdy NA, Lowik CW, Hamersma H, Beighton P, Papapoulos SE. Bone mineral density in sclerosteosis; affected individuals and gene carriers. J Clin Endocrinol Metab. 2005;90(12):6392–6395. doi: 10.1210/jc.2005-1235. [DOI] [PubMed] [Google Scholar]
- 57.Li X, Warmington KS, Niu QT, Asuncion FJ, Barrero M, Grisanti M, Dwyer D, Stouch B, Thway TM, Stolina M, Ominsky MS, Kostenuik PJ, Simonet WS, Paszty C, Ke HZ. Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J Bone Miner Res. 2010;25(12):2647–2656. doi: 10.1002/jbmr.182. [DOI] [PubMed] [Google Scholar]
- 58.Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, Gong J, Gao Y, Cao J, Graham K, Tipton B, Cai J, Deshpande R, Zhou L, Hale MD, Lightwood DJ, Henry AJ, Popplewell AG, Moore AR, Robinson MK, Lacey DL, Simonet WS, Paszty C. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25(5):948–959. doi: 10.1002/jbmr.14. [DOI] [PubMed] [Google Scholar]
- 59.Li X, Niu QT, Warmington KS, Asuncion FJ, Dwyer D, Grisanti M, Han CY, Stolina M, Eschenberg MJ, Kostenuik PJ, Simonet WS, Ominsky MS, Ke HZ. Progressive increases in bone mass and bone strength in an ovariectomized rat model of osteoporosis after 26 weeks of treatment with a sclerostin antibody. Endocrinology. 2014;155(12):4785–4797. doi: 10.1210/en.2013-1905. [DOI] [PubMed] [Google Scholar]
- 60.Ominsky MS, Niu QT, Li C, Li X, Ke HZ. Tissue-level mechanisms responsible for the increase in bone formation and bone volume by sclerostin antibody. J Bone Miner Res. 2014;29(6):1424–1430. doi: 10.1002/jbmr.2152. [DOI] [PubMed] [Google Scholar]
- 61.McColm J, Hu L, Womack T, Tang CC, Chiang AY. Single- and multiple-dose randomized studies of blosozumab, a monoclonal antibody against sclerostin, in healthy postmenopausal women. J Bone Miner Res. 2014;29(4):935–943. doi: 10.1002/jbmr.2092. [DOI] [PubMed] [Google Scholar]
- 62.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26(1):19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 63.Padhi D, Allison M, Kivitz AJ, Gutierrez MJ, Stouch B, Wang C, Jang G. Multiple doses of sclerostin antibody romosozumab in healthy men and postmenopausal women with low bone mass: a randomized, double-blind, placebo-controlled study. J Clin Pharmacol. 2014;54(2):168–178. doi: 10.1002/jcph.239. [DOI] [PubMed] [Google Scholar]
- 64.Recker R, Benson C, Matsumoto T, Bolognese M, Robins D, Alam J, Chiang AY, Hu L, Krege JH, Sowa H, Mitlak B, Myers S. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res. 2015;30:216–24. [DOI] [PubMed]
- 65.McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A, Langdahl BL, Reginster JY, Zanchetta JR, Wasserman SM, Katz L, Maddox J, Yang YC, Libanati C, Bone HG. Romosozumab in Postmenopausal Women with Low Bone Mineral Density. N Engl J Med. 2014;370(5):412–420. doi: 10.1056/NEJMoa1305224. [DOI] [PubMed] [Google Scholar]
- 66.Recknor CP, Recker RR, Benson CT, Robins DA, Chiang AY, Alam J, Hu L, Matsumoto T, Sowa H, Sloan JH, Konrad RJ, Mitlak BH, Sipos AA. The effect of discontinuing treatment with blosozumab: follow-up results of a phase 2 randomized clinical trial in postmenopausal women with low bone mineral density. J Bone Miner Res. 2015 [Epub ahead of print]. [DOI] [PubMed]
- 67.McClung MR, Chines A, Brown JP, Diez-Perez A, Resch H, Caminis J, Bolognese M, Goemaeres S, Bone HG, Zanchetta JR, Maddox J, Rosen O, Bray S, Gauer A. Effects of 2 years of treatment with romosozumab followed by 1 year of denosumab or placebo in postmenopsusal women with low bone mineral density. JBMR. 2014;29(Suppl. 1):S53 (abstract 1152). doi: 10.1002/jbmr.3452. [DOI] [PubMed] [Google Scholar]
- 68.Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One. 2011;6(10):e25900. doi: 10.1371/journal.pone.0025900. [DOI] [PMC free article] [PubMed] [Google Scholar]