

Abstract
Objectives
SMT19969 is a novel antimicrobial under clinical development for the treatment of Clostridium difficile infection (CDI). The objective was to determine the comparative susceptibility of 82 C. difficile clinical isolates (which included ribotype 027 isolates and isolates with reduced metronidazole susceptibility) to SMT19969, fidaxomicin, vancomycin and metronidazole and to determine the killing kinetics and post-antibiotic effects of SMT19969, fidaxomicin and vancomycin against C. difficile.
Methods
MICs were determined by agar incorporation. Killing kinetics and post-antibiotic effects were determined against C. difficile BI1, 630 and 5325 (ribotypes 027, 012 and 078, respectively).
Results
SMT19969 showed potent inhibition of C. difficile (MIC90=0.125 mg/L) and was markedly more active than either metronidazole (MIC90 = 8 mg/L) or vancomycin (MIC90 = 2 mg/L). There were no differences in susceptibility to SMT19969 between different ribotypes. Fidaxomicin was typically one doubling dilution more active than SMT19969 and both agents maintained activity against isolates with reduced susceptibility to metronidazole. In addition, SMT19969 was bactericidal against the C. difficile strains tested, with reductions in viable counts to below the limit of detection by 24 h post-inoculation. Vancomycin was bacteriostatic against all three strains. Fidaxomicin was bactericidal although reduced killing was observed at concentrations <20 × MIC against C. difficile BI1 (ribotype 027) compared with other strains tested.
Conclusions
These data demonstrate that SMT19969 is associated with potent and bactericidal activity against the strains tested and support further investigation of SMT19969 as potential therapy for CDI.
Keywords: C. difficile, PAE, antimicrobial
Introduction
Clostridium difficile infection (CDI) is a significant cause of morbidity and mortality in both the acute care setting and the wider healthcare system.1,2 The global increase in the incidence of CDI is driven, in part, by the emergence of fluoroquinolone-resistant ribotype 027 C. difficile strains,3 which continue to account for ∼30% of CDI cases in North America.4 These strains are associated with poor outcomes, including reduced cure rates and increased rates of recurrent disease.5,6 Although the prevalence of ribotype 027 was thought to have declined in Europe,7 the recent EUCLID study demonstrates the dominance of ribotype 027 in Central and Eastern Europe.8 Also, new hyper-virulent strains such as ribotype 244 continue to emerge.9
CDI pathogenesis is associated with antimicrobial use that causes reduced diversity of the gut microbiota, thus reducing the host's ability to resist colonization by, and expansion of, C. difficile. These conditions allow C. difficile spores to germinate, with resultant toxin production leading to disease symptoms. Vancomycin and metronidazole, the mainstay antibiotics used in CDI, have been shown to cause further collateral damage to the gut microbiota,10–12 and analysis of the gut microbiome of CDI patients has shown that recurrent disease is associated with a markedly decreased diversity in the bacterial populations of the gut.13 As such, therapeutic approaches that minimize further deleterious effects to the gut microbiota may reduce rates of recurrent CDI.
SMT19969 is a novel antimicrobial under specific development for CDI that shows potent inhibition of C. difficile, but is associated with minimal growth inhibition of both Gram-positive and Gram-negative components of the indigenous gut microflora;14,15 such focused activity may result in reduced rates of recurrent disease. The following studies further assessed and compared growth inhibition by, and the in vitro pharmacodynamics of, SMT19969, fidaxomicin, vancomycin and metronidazole against C. difficile.
Materials and methods
Test agents
SMT19969 (supplied by Summit plc, Abingdon, UK), fidaxomicin [obtained from Cubist Pharmaceuticals, Lexington, MA, USA (for MIC determination) and from Santa Cruz Biotechnology, Santa Cruz, CA, USA (for kill curve and post-antibiotic effect, PAE, assays)] and metronidazole (supplied by Sigma) were reconstituted in DMSO prior to further dilution. Vancomycin (supplied by Sigma) stock solutions were prepared in water.
Bacterial strains
Susceptibility testing was carried out using C. difficile clinical isolates collected in the UK from subjects with CDI. All isolates had been submitted to the C. difficile ribotyping network. A total of 82 C. difficile isolates were assessed across five panels comprising 30 distinct isolates of different PCR ribotypes (referred to as the genotypically distinct group), 10 PCR ribotype 001 isolates, 11 PCR ribotype 027 isolates, 10 PCR ribotype 106 isolates and 21 PCR ribotype 001 isolates showing reduced metronidazole susceptibility (MIC 4–8 mg/L).16 Staphylococcus aureus ATCC 29213, Bacteroides fragilis ATCC 25285 and C. difficile PCR ribotype 010 E4 were used as control organisms. C. difficile E4 is a non-toxigenic PCR ribotype 010 internal control strain.17
Kill curve and PAE studies were performed using C. difficile 630 (ATCC BAA 1382; ribotype 012), BI1 (NCTC 13366; ribotype 027) and 5325 (ATCC BAA-1875; ribotype 078).
MICs
Comparative susceptibility testing was carried out on Wilkins Chalgren agar due to the superior growth compared with Brucella agar and also the ability to detect reduced susceptibility to metronidazole.16 MICs were determined by agar incorporation according to a previously validated method.16 All isolates were tested in duplicate for susceptibility to SMT19969, fidaxomicin, vancomycin and metronidazole.
For the kill curve and PAE studies, MIC values (Table 1) were established for SMT19969, fidaxomicin and vancomycin by broth microdilution using BHIS medium (brain heart infusion broth supplemented with 5 g/L yeast extract and 0.025% l-cysteine); this medium was used in order to be consistent with later experiments.
Table 1.
Reference MICs established by broth microdilution for killing kinetics and PAE studies
| C. difficile strain (ribotype) | MIC (mg/L) |
||
|---|---|---|---|
| SMT19969 | vancomycin | fidaxomicin | |
| BI1 (027) | 0.125 | 4 | 0.25 |
| 630 (012) | 0.125 | 4 | 0.25 |
| 5325 (078) | 0.125 | 1 | 0.25 |
Killing kinetics
Cultures of C. difficile were prepared by inoculating fresh, pre-reduced BHIS medium with a single colony of the required strain. Following overnight (18–20 h) incubation, cultures were back-diluted 1: 100 into fresh BHIS broth (∼106 cfu/mL) containing either DMSO (1% v/v; vehicle) or 1, 2, 5, 10 or 20 × MIC of SMT19969, vancomycin or fidaxomicin. Viable counts were determined at 0, 2, 4, 6, 8 and 24 h post-inoculation on BHIS agar. Data presented are the means of triplicate experiments. The limit of detection (LOD) for these assays was 500 cfu/mL. A ≥3 log10 reduction in viability relative to the starting inoculum was considered bactericidal.
PAE
Cultures of C. difficile were prepared in 15 mL polypropylene centrifuge tubes in the presence of DMSO or antibiotic, as described above. Cultures were incubated for 1 h before being collected by centrifugation (4696 g, 5 min, 25°C), washed once with BHIS to remove test articles and finally re-suspended in fresh BHIS. For assays involving exposure to fidaxomicin, cultures were transferred to sterile glass bijou tubes following removal of the test article in order to minimize drug carryover due to non-specific binding as previously reported.18 Centrifugation was the only step performed outside the anaerobic workstation. At this point, samples were removed for determination of viable counts (0 h) and further samples were removed for counting after 2, 4, 6, 8 and 24 h. The LOD for these experiments was 50 cfu/mL. The PAE of each test article was defined as PAE = T − C, where T is the time required for determination of the viable count to increase 10-fold over the post-washing viable count in the presence of antibiotic, and C is the time required for the viable count to increase 10-fold over the post-washing viable count in the absence of antibiotic. The mean values obtained from three independent experiments were used for T and C. PAE (in hours) was estimated to the nearest timepoint. Where the PAE endpoint was reached overnight, the endpoint was defined as the limits of the PAE (i.e. 8–20 h). No interpolation between timepoints was attempted.
Results
Susceptibility testing
The comparative susceptibilities of 82 C. difficile clinical isolates to SMT19969, fidaxomicin, metronidazole and vancomycin are shown in Table 2. Vancomycin generally maintained a consistent level of activity against the majority of the panels assessed with an MIC90 of 2 mg/L (typical range = 0.5–4 mg/L) for each panel except for the metronidazole-susceptible ribotype 001 panel where an MIC90 = 4 mg/L was recorded. Although the vast majority of the 82 isolates had vancomycin MIC values of 1 or 2 mg/L, seven isolates (ribotypes 056, 001, 027 and 106) showed an elevated MIC value of 4 mg/L. Susceptibility to metronidazole was more varied across the panels. For ribotypes 027 and 106, MIC50 values were one dilution higher than for the metronidazole-susceptible ribotype 001 panel, and two dilutions higher than MIC50 values recorded for the genotypically distinct group.
Table 2.
MICs of SMT19969, fidaxomicin, metronidazole and vancomycin for 82 C. difficile clinical isolates
| Panel (n) | Agent | MIC range (mg/L) | MIC50 (mg/L) | MIC90 (mg/L) |
|---|---|---|---|---|
| Genotypically distinct (30)a | SMT19969 | 0.06–0.125 | 0.125 | 0.125 |
| vancomycin | 1–4 | 2 | 2 | |
| metronidazole | 0.25–2 | 0.5 | 2 | |
| fidaxomicin | 0.008–0.125 | 0.03 | 0.06 | |
| Ribotype 001 (10) | SMT19969 | 0.06–0.125 | 0.125 | 0.125 |
| vancomycin | 0.5–4 | 1 | 4 | |
| metronidazole | 0.125–1 | 1 | 1 | |
| fidaxomicin | 0.008–0.06 | 0.03 | 0.06 | |
| Ribotype 027 (11) | SMT19969 | 0.125–0.25 | 0.125 | 0.125 |
| vancomycin | 0.5–4 | 1 | 2 | |
| metronidazole | 1–2 | 2 | 2 | |
| fidaxomicin | 0.03–0.06 | 0.06 | 0.06 | |
| Ribotype 106 (10) | SMT19969 | 0.125–0.25 | 0.125 | 0.125 |
| vancomycin | 0.5–4 | 1 | 2 | |
| metronidazole | 1–2 | 2 | 2 | |
| fidaxomicin | 0.03–0.125 | 0.06 | 0.125 | |
| Reduced metronidazole susceptibility (21)b | SMT19969 | 0.06–0.125 | 0.125 | 0.125 |
| vancomycin | 0.5–4 | 1 | 2 | |
| metronidazole | 4–8 | 4 | 8 | |
| fidaxomicin | 0.015–0.03 | 0.03 | 0.03 | |
| Overall total (82) | SMT19969 | 0.06–0.25 | 0.125 | 0.125 |
| vancomycin | 0.5–4 | 1 | 2 | |
| metronidazole | 0.125–8 | 2 | 8 | |
| fidaxomicin | 0.008–0.125 | 0.03 | 0.06 |
aThirty distinct isolates of different PCR ribotype.
bRibotype 001.
Both fidaxomicin and SMT19969 showed potent activity against the 82 isolates tested, with MIC90 values of 0.06 mg/L (range = 0.008–0.125 mg/L) and 0.125 mg/L (range = 0.06–0.25 mg/L), respectively, which was typically 16–32-fold lower than the MIC90 values recorded for either vancomycin against the 82 isolates or metronidazole against the metronidazole-susceptible isolates. Fidaxomicin showed increased potency compared with SMT19969 with MIC90 values one dilution and MIC50 values two dilutions lower than those recorded for SMT19969. SMT19969 activity was consistent across different ribotypes, including ribotype 027 isolates; the MIC90 for each panel of clinical isolates was 0.125 mg/L. Similarly, fidaxomicin showed highly consistent activity against the strains tested, although it was slightly less active against ribotype 106 compared with the other isolates. For the PCR ribotype 001 isolates showing reduced susceptibility to metronidazole, both vancomycin and SMT19969 maintained activity levels comparable to those observed against the metronidazole-susceptible ribotype 001 strains, whilst fidaxomicin MIC90 values were one dilution lower (0.03 versus 0.06 mg/L).
Overall SMT19969 showed potent growth inhibition of the 82 isolates tested, with an MIC90 of 0.125 mg/L, which was 16-fold lower than the MIC90 values recorded for vancomycin against the 82 isolates and the 61 metronidazole-susceptible isolates.
Killing kinetics
The comparative killing kinetics of SMT19969, vancomycin and fidaxomicin were assessed against C. difficile strains BI1 (ribotype 027), 630 (ribotype 012) and 5325 (ribotype 078). Kill curves for vancomycin, fidaxomicin and SMT19969 against C. difficile BI1 are shown in Figures 1–3, with reductions in cfu/mL for C. difficile 630 and 5325 following 24 h of exposure to antibiotic shown in Table 3. It should be noted that the starting inoculum for experiments with fidaxomicin and C. difficile 630 and 5325 was somewhat lower (3.72 × 105 and 2.17 × 105 cfu/mL, respectively) than for vancomycin (1.94 × 106 and 9.00×105 cfu/mL) or SMT19969 (3.28 × 106 and 1.42 × 106 cfu/mL). Starting inocula for experiments with C. difficile BI1 for SMT19969, fidaxomicin and vancomycin were 7.50 × 105, 1.30 × 106 and 3.17 × 106 cfu/mL, respectively.
Figure 1.
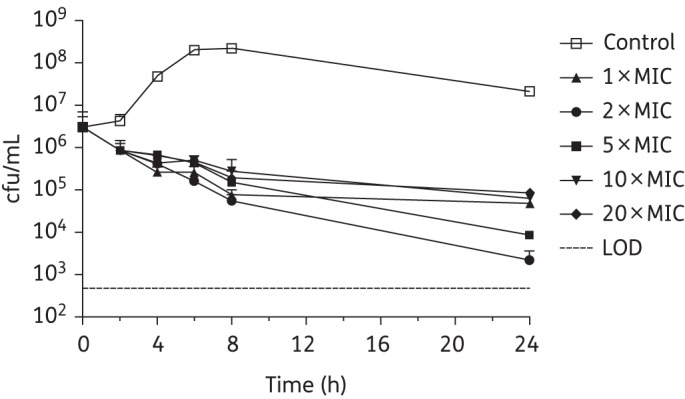
Twenty-four hour time–kill curves for vancomycin against C. difficile BI1. Data are the means (+SD) of triplicate experiments.
Figure 3.
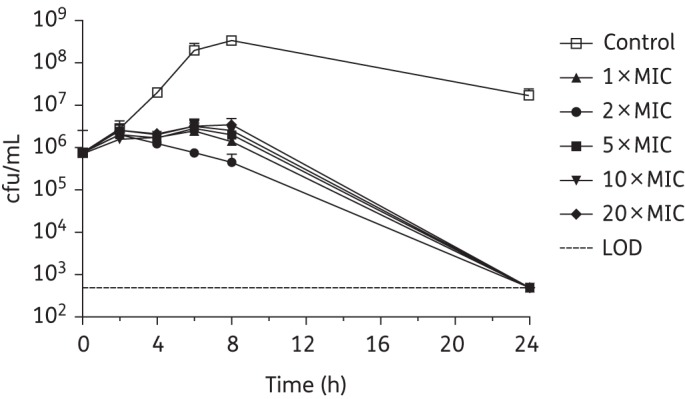
Twenty-four hour time–kill curves for SMT19969 against C. difficile BI1. Data are the means (+SD) of triplicate experiments.
Table 3.
Mean log10 cfu/mL reductions for C. difficile 630 and 5325 following 24 h of exposure to SMT19969, vancomycin or fidaxomicin
| Strain | Agent | Mean log10 reductions in cfu/mL |
||||
|---|---|---|---|---|---|---|
| 1 × MIC | 2 × MIC | 5 × MIC | 10 × MIC | 20 × MIC | ||
| 630 | SMT19969 | 3.61 | 2.17 | >3.82a | >3.82a | >3.82a |
| vancomycin | 3.25 | 3.22 | 2.99 | 3.46 | 3.59 | |
| fidaxomicin | 2.45 | >2.87a | >2.87a | >2.87a | >2.87a | |
| 5325 | SMT19969 | 2.60 | 1.62 | 2.73 | 3.16 | 3.33 |
| vancomycin | 1.14 | 2.65 | 1.82 | 1.81 | 1.46 | |
| fidaxomicin | 2.41 | 2.16 | >2.64a | >2.64a | >2.64a | |
aReduced to below LOD.
Vancomycin generally achieved a slow reduction in bacterial counts of C. difficile BI1, resulting in a 1.5–2.5 log10 reduction in viability after 24 h (Figure 1); a 3.1 log10 reduction in cfu/mL was observed at 24 h at 2 × MIC. Similarly, fidaxomicin at 1–10 × MIC was bacteriostatic against C. difficile BI1 with a 1.5–2.0 log10 reduction in viable counts. At 20 × MIC, fidaxomicin was bactericidal, resulting in a 3.3 log10 reduction in viable counts after 24 h of exposure (Figure 2). All concentrations of SMT19969 resulted in a reduction in viable counts to below the LOD (>3.2 log10 reduction in cfu/mL) at 24 h (Figure 3). Killing by SMT19969 was independent of drug concentration.
Figure 2.
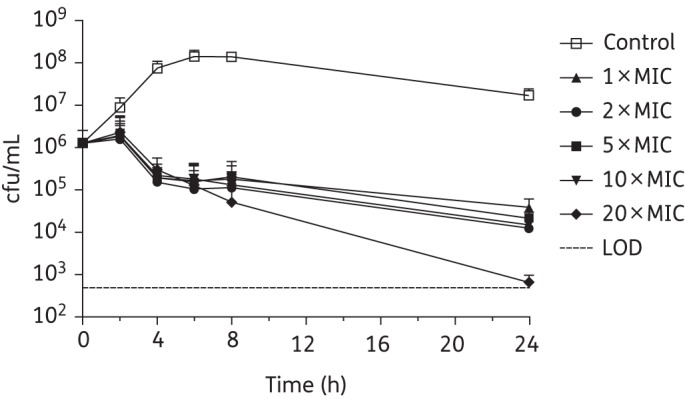
Twenty-four hour time–kill curves for fidaxomicin against C. difficile BI1. Data are the means (+SD) of triplicate experiments.
The viability of C. difficile strain 630 when exposed to vancomycin and SMT19969 over 24 h was similar to that observed for C. difficile BI1. Vancomycin gradually reduced the viability of C. difficile 630 over time in a concentration-independent manner, although a greater reduction in viable counts, compared with C. difficile BI1, of 3.0–3.6 log10 was observed at 24 h (Table 3). As observed with BI1, SMT19969 was bacteriostatic to 8 h with reductions in viable counts to below the LOD (>3.8 log10) at concentrations ≥5 × MIC. Fidaxomicin was more effective at killing C. difficile 630 than C. difficile BI1, with counts reduced below the LOD by 6 h (20 × MIC) or 24 h (2–10 × MIC).
Against C. difficile 5325, vancomycin was bacteriostatic at all concentrations tested (Table 3). Consistent with observations for strains 630 and BI1, all tested concentrations of SMT19969 were bacteriostatic for ∼8 h. Between 8 and 24 h, counts of C. difficile 5325 fell by ≥3 log10 cfu/mL at concentrations >5 × MIC (Table 3). Fidaxomicin resulted in a marked decrease in viable counts, with concentrations ≥5 × MIC resulting in loss of viability of C. difficile to below the LOD by 24 h (Table 3). At 20 × MIC, fidaxomicin rapidly reduced cell viability to below the LOD by 6 h.
PAE
The PAE of each antibiotic was established against C. difficile BI1, 630 and 5325 by monitoring the growth of each strain following a 1 h exposure to 1, 2, 5, 10 or 20 × MIC of the drug. Recovery of C. difficile BI1 following pre-exposure to SMT19969 is shown in Figure 4, with calculated PAEs for all drug–strain combinations shown in Table 4.
Figure 4.
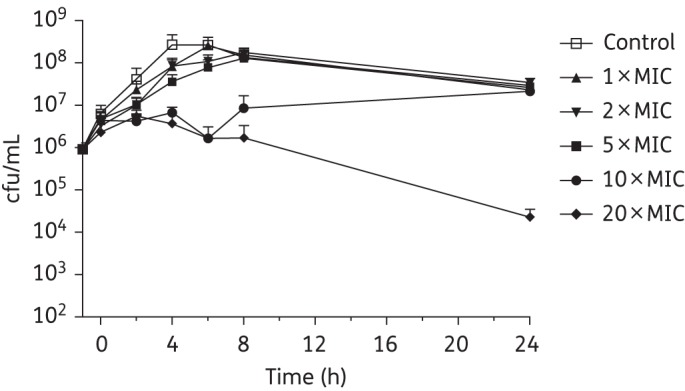
PAE of SMT19969 against C. difficile BI1. Data are the means (+SD) of triplicate experiments. Drug removed at 0 h.
Table 4.
PAE for SMT19969, vancomycin and fidaxomicin against C. difficile BI1, 630 and 5325
| Strain | Agent | PAE (h) |
||||
|---|---|---|---|---|---|---|
| 1 × MIC | 2 × MIC | 5 × MIC | 10 × MIC | 20 × MIC | ||
| BI1 | SMT19969 | 0 | 0 | 2 | 8–20 | >20 |
| vancomycin | 0 | 0 | 0 | 0 | 0 | |
| fidaxomicin | 0 | 8–20 | 8–20 | 8–20 | NE | |
| 5325 | SMT19969 | 0 | 0 | 0 | 4 | NE |
| vancomycin | 0 | 2 | 2 | 0 | 0 | |
| fidaxomicin | 8–20 | 8–20 | 8–20 | 8–20 | >20 | |
| 630 | SMT19969 | 0 | 2 | 2 | 4 | >20 |
| vancomycin | 0 | 0 | 0 | 0 | 2 | |
| fidaxomicin | 8–20 | 8–20 | 8–20 | >20 | >20 | |
NE, not established due to inconsistent growth recovery (see the text).
SMT19969 had a PAE of 2 h for C. difficile BI1 at 5 × MIC with concentrations of 10 or 20 × MIC suppressing growth for ≥8 h. At 20 × MIC, C. difficile BI1 did not recover with a 2 log10 reduction in viable counts at 24 h post-removal of SMT19969 (Figure 4). Fidaxomicin had a pronounced PAE (8–20 h) at concentrations ≥2 × MIC. At 20 × MIC of fidaxomicin, inconsistent recovery of bacteria was observed with a 1–2 log10 reduction in viability noted in two out of three replicates (complete recovery was seen in the third replicate; Table 4). Vancomycin showed no measurable PAE against C. difficile BI1 (Table 4).
At all tested concentrations, a 1 h exposure to vancomycin resulted in a PAE of 0–2 h for C. difficile 5325 (Table 4). SMT19969 concentrations of 10 × MIC were required to produce a quantifiable PAE of 4 h. At 20 × MIC of SMT19969, inconsistent recovery of C. difficile 5325 was observed (viability in two out of three replicates reduced below the LOD). Fidaxomicin concentrations ≥MIC had a prolonged PAE of 8–20 h. At 20 × MIC, the growth of the culture remained suppressed at 24 h post-removal of antibiotic, with a concomitant 2 log10 reduction in viable counts.
For C. difficile 630, the PAE of vancomycin was <2 h for concentrations from 1 to 10 × MIC, whilst at 20 × MIC a PAE of 2 h was observed. SMT19969 exhibited a PAE of 2–4 h up to 10 × MIC (Table 4). At 20 × MIC of SMT19969, counts of C. difficile 630 fell below the LOD by 24 h post-removal of SMT19969. Fidaxomicin showed a prolonged (8–20 h) PAE against strain 630 at concentrations between 1 and 5 × MIC whilst at higher concentrations no growth was observed with the viability of the culture reduced 2.4 log10 in the 24 h following removal of antibiotic.
Discussion
Current antimicrobial therapy options for CDI are very limited.19 Clinical response to metronidazole is inferior at the end of therapy compared with vancomycin in patients with severe CDI.20,21 Isolates showing reduced susceptibility to metronidazole have been reported,16 and whilst no link between clinical outcome and the MIC of metronidazole has been established, elevated MICs in conjunction with low intraluminal antibiotic concentrations following oral administration may affect efficacy. Both vancomycin and metronidazole are associated with high rates of recurrent disease with 20%–30% of subjects experiencing a recurrent infection following the primary episode; recurrence rates may be >65% following a third episode of CDI.22 Recurrent CDI remains frustratingly difficult to treat, with a deleterious impact on patient welfare and healthcare system resources. The recently approved fidaxomicin has been shown in Phase 3 clinical studies to be non-inferior in clinical response to vancomycin at the end of treatment and to be superior in sustained clinical response to 25 days post end of therapy with an overall reduction in rates of recurrence compared with vancomycin. However, recurrence rates were comparable for vancomycin and fidaxomicin for subjects infected with C. difficile BI (ribotype 027) strains.23,24 New agents, particularly those that reduce rates of recurrent disease, are required to effectively manage CDI.
The studies described here show that SMT19969 resulted in potent inhibition of a range of C. difficile clinical isolates, consistent with previously reported in vitro activity against C. difficile.14,15 Compared with vancomycin and metronidazole, SMT19969 was considerably more potent; MIC90 values were typically 16-fold lower, with consistent activity against a range of clinically relevant ribotypes. Fidaxomicin showed increased potency compared with SMT19969 with MIC90 values one dilution and MIC50 values two dilutions lower than those recorded for SMT19969.
In addition to potent growth inhibition, SMT19969 was bactericidal against three C. difficile clinical isolates representing ribotypes 027, 012 and 078. Against C. difficile BI1, SMT19969 resulted in a >3 log10 reduction in viable counts following 24 h of exposure, with viable counts typically reduced to below the LOD of the assay; fidaxomicin showed reduced killing at 24 h with typical log10 reductions in cfu/mL of 1.5–2.0 (although at 20 × MIC viable counts were reduced by 3.3 log10 cfu/mL). Slightly reduced killing by SMT19969 of strain 5325 was observed at concentrations ≤5 × MIC. Against C. difficile 630 and 5325, fidaxomicin was bactericidal at all concentrations above the MIC and, consistent with previous reports, killing by fidaxomicin was time dependent.25
Killing by vancomycin was largely independent of drug concentration, and in accordance with previous results,26 vancomycin was bacteriostatic. In addition, vancomycin was shown to be associated with a minimal PAE (typically 0–2 h), as reported previously.18,26 SMT19969, at concentrations ≥5 × MIC, was associated with a pronounced PAE with no growth recovery observed following a 1 h exposure to 20 × MIC. Although higher concentrations of SMT19969 were required to effect a prolonged PAE, mean faecal concentrations of 1363 μg/g (SD ±446), more than three orders of magnitude higher than the MIC90 of SMT19969 for C. difficile, were observed in Phase 1 healthy volunteers receiving 200 mg twice daily for 10 days.27 As previously reported, fidaxomicin showed a very prolonged PAE, typically 8–20 h, at all concentrations.18
The data reported here show that SMT19969 has potent and consistent activity against the examined C. difficile strains. In addition, SMT19969 was bactericidal against all strains tested, and was associated with a pronounced PAE at higher concentrations, as would be expected in the gastrointestinal tract of CDI subjects. In conjunction with its narrow spectrum of activity,14,15 these data support continued investigation of SMT19969 as a potential therapy for CDI.
Funding
This study was initiated and financially supported by Summit plc through a Seeding Drug Discovery Award (grant number 091055) and a Translation Award from the Wellcome Trust (grant number 099444).
Transparency declarations
S. D. B. has received funding for consultancy research from Procarta Biosystems. M. H. W. has received consulting fees from Actelion, Astellas, Cubist, The Medicines Company, Merck, Novartis, Optimer, Pfizer, Sanofi-Pasteur, Summit, Synthetic Biologics and VH Squared, lecture fees from Alere, Astellas and Pfizer, and grant support from Actelion, Astellas, bioMérieux, Cubist, Da Volterra, Merck and Summit. D. C., A. W., S. B. and P. W. are employees of Evotec and R. J. V. is an employee of Summit plc and holds share options. All other authors: none to declare.
Acknowledgements
These data were presented, in part, at the Fifty-fourth Interscience Conference on Antimicrobial Agents and Chemotherapy, Washington, DC, 2014 (Abstract F-240) and at the Fifty-first Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, 2011 (Abstract B-1194).
References
- 1.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 2009; 7: 526–36. [DOI] [PubMed] [Google Scholar]
- 2.Kelly CP, Lamont JT. Clostridium difficile—more difficult than ever. New Engl J Med 2008; 359: 1932–40. [DOI] [PubMed] [Google Scholar]
- 3.He M, Miyajima F, Roberts P, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet 2013; 45: 109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tickler IA, Goering RV, Whitmore JD, et al. Strain types and antimicrobial resistance patterns of Clostridium difficile isolates from the United States, 2011 to 2013. Antimicrob Agents Chemother 2014; 58: 4214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrella LA, Sambol SP, Cheknis A, et al. Decreased cure and increased recurrence rates for Clostridium difficile infection caused by the epidemic C. difficile BI strain. Clin Infect Dis 2012; 55: 351–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker AS, Eyre DW, Wyllie DH, et al. Relationship between bacterial strain type, host biomarkers, and mortality in Clostridium difficile infection. Clin Infect Dis 2013; 56: 1589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer MP, Notermans DW, van Benthem BHB, et al. Clostridium difficile infection in Europe: a hospital-based survey. The Lancet 2011; 377: 63–73. [DOI] [PubMed] [Google Scholar]
- 8.Davies KA, Longshaw CM, Davis GL, et al. Underdiagnosis of Clostridium difficile across Europe: the European, multicentre, prospective, biannual, point-prevalence study of Clostridium difficile infection in hospitalised patients with diarrhoea (EUCLID). Lancet Infect Dis 2014; 14: 1208–19. [DOI] [PubMed] [Google Scholar]
- 9.Lim SK, Stuart RL, Mackin KE, et al. Emergence of a ribotype 244 strain of Clostridium difficile associated with severe disease and related to the epidemic ribotype 027 strain. Clin Infect Dis 2014; 58: 1723–30. [DOI] [PubMed] [Google Scholar]
- 10.Edlund C, Barkholt L, Olsson-Liljequist B, et al. Effect of vancomycin on intestinal flora of patients who previously received antimicrobial therapy. Clin Infect Dis 1997; 25: 729–32. [DOI] [PubMed] [Google Scholar]
- 11.Louie TJ, Emery J, Krulicki W, et al. OPT-80 eliminates Clostridium difficile and is sparing of bacteroides species during treatment of C. difficile infection. Antimicrob Agents Chemother 2009; 53: 261–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rea MC, Dobson A, O'Sullivan O, et al. Effect of broad- and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc Natl Acad Sci USA 2011; 108 Suppl 1: 4639–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang JY, Antonopoulos DA, Kalra A, et al. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis 2008; 197: 435–8. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein EJ, Citron DM, Tyrrell KL, et al. Comparative in vitro activities of SMT19969, a new antimicrobial agent, against Clostridium difficile and 350 Gram-positive and Gram-negative aerobic and anaerobic intestinal flora isolates. Antimicrob Agents Chemother 2013; 57: 4872–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baines SD, Crowther GS, Freeman J, et al. SMT19969 as a treatment for Clostridium difficile infection: an assessment of antimicrobial activity using conventional susceptibility testing and an in vitro gut model. J Antimicrob Chemother 2015; 70: 182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baines SD, O'Connor R, Freeman J, et al. Emergence of reduced susceptibility to metronidazole in Clostridium difficile. J Antimicrob Chemother 2008; 62: 1046–52. [DOI] [PubMed] [Google Scholar]
- 17.Brazier J, Fawley WN, Freeman J, et al. Reduced susceptibility of Clostridium difficile to metronidazole. Journal of Antimicrobial Chemotherapy 2001; 48: 741–2. [DOI] [PubMed] [Google Scholar]
- 18.Babakhani F, Gomez A, Robert N, et al. Postantibiotic effect of fidaxomicin and its major metabolite, OP-1118, against Clostridium difficile. Antimicrob Agents Chemother 2011; 55: 4427–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debast SB, Bauer MP, Kuijper EJ. European Society of Clinical Microbiology and Infectious Diseases (ESCMID): update of the treatment guidance document for Clostridium difficile infection (CDI). Clin Microbiol Infect 2013; 20: 1–26. [DOI] [PubMed] [Google Scholar]
- 20.Zar FA, Bakkanagari SR, Moorthi KM, et al. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis 2007; 45: 302–7. [DOI] [PubMed] [Google Scholar]
- 21.Johnson S, Louie TJ, Gerding DN, et al. Vancomycin, metronidazole, or tolevamer for Clostridium difficile infection: results from two multinational, randomized, controlled trials. Clin Infect Dis 2014; 59: 345–54. [DOI] [PubMed] [Google Scholar]
- 22.Kelly CP. Can we identify patients at high risk of recurrent Clostridium difficile infection? Clin Microbiol Infect 2012; 18 Suppl 6: 21–7. [DOI] [PubMed] [Google Scholar]
- 23.Louie TJ, Miller MA, Mullane KM, et al. Fidaxomicin versus vancomycin for Clostridium difficile Infection. N Engl J Med 2011; 364: 422–31. [DOI] [PubMed] [Google Scholar]
- 24.Cornely OA, Crook DW, Esposito R, et al. Fidaxomicin versus vancomycin for infection with Clostridium difficile in Europe, Canada, and the USA: a double-blind, non-inferiority, randomised controlled trial. Lancet Infect Dis 2012; 12: 281–9. [DOI] [PubMed] [Google Scholar]
- 25.Babakhani F, Gomez A, Robert N, et al. Killing kinetics of fidaxomicin and its major metabolite, OP-1118, against Clostridium difficile. J Med Microbiol 2011; 60: 1213–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Odenholt I, Walder M, Wullt M. Pharmacodynamic studies of vancomycin, metronidazole and fusidic acid against Clostridium difficile. Chemotherapy 2007; 53: 267–74. [DOI] [PubMed] [Google Scholar]
- 27.Vickers RJ, Robinson N, Tinsley J, et al. SMT19969 for Clostridium difficile infection: Phase 1 study to investigate safety and pharmacokinetics of single and multiple oral escalating doses in healthy male subjects. In: Abstracts of the Fifty-third Interscience Conference on Antimicrobial Agents and Chemotherapy, Denver, CO, 2013 Abstract F626 American Society for Microbiology, Washington, DC, USA. [Google Scholar]