

Abstract
The mechanism underlying acrylamide-induced neurotoxicity remains controversial. Previous studies have focused on acrylamide-induced toxicity in adult rodents, but neurotoxicity in weaning rats has not been investigated. To explore the neurotoxic effect of acrylamide on the developing brain, weaning rats were gavaged with 0, 5, 15, and 30 mg/kg acrylamide for 4 consecutive weeks. No obvious neurotoxicity was observed in weaning rats in the low-dose acrylamide group (5 mg/kg). However, rats from the moderate- and high-dose acrylamide groups (15 and 30 mg/kg) had an abnormal gait. Furthermore, biochemical tests in these rats demonstrated that glutamate concentration was significantly reduced, and γ-aminobutyric acid content was significantly increased and was dependent on acrylamide dose. Immunohistochemical staining showed that in the cerebral cortex, γ-aminobutyric acid, glutamic acid decarboxylase and glial fibrillary acidic protein expression increased remarkably in the moderate- and high-dose acrylamide groups. These results indicate that in weaning rats, acrylamide is positively associated with neurotoxicity in a dose-dependent manner, which may correlate with upregulation of γ-aminobutyric acid and subsequent neuronal degeneration after the initial acrylamide exposure.
Keywords: nerve regeneration, γ-aminobutyric acid, glial fibrillary acidic protein, glutamic acid decarboxylase, neurotoxicity, weaning, organ index, cerebrum, cortex, glutamate, neural regeneration
Introduction
Acrylamide, a water-soluble ethenyl monomer, is extracted from hydrated acrylonitrile and has been widely used in industrial production. A large number of animal experiments show that acrylamide is carcinogenic and has neuronal and reproductive toxic properties (Tyl et al., 2000; Pourentezari et al., 2014; Sen et al., 2015). In 2002, researchers at the Swedish National Food Administration and Stockholm University found that the concentration of acrylamide in fried and grilled food exceeded the concentration in drinking water stipulated by WHO by 500 times (Tareke et al., 2000; Rosen and Hellenas, 2002). The World Food and Agriculture Organization estimates that ordinarily, the daily intake of acrylamide from food is a maximum of 0.3–0.8 mg/kg, but in fact most people take in more than that average value. More importantly, children's daily intake of acrylamide may be 2–3 times more than that of adults, which means 4–5 times more than the average values (WHO, 2002). One possible reason is that children frequently eat more potato chips and cookies which contain high levels of acrylamide (Dybing and Sanner, 2003). Therefore, we should pay more attention to the effect of acrylamide on children.
Previous studies have mainly focused on the effect of acrylamide on adult rodents. There are few papers reporting neurotoxicity of acrylamide on weaning rats. Therefore, in the present study, we used a weaning rat model to study neurotoxic effects of acrylamide on developmental brain.
Neurochemical synaptic transmission is an essential function of the central nervous system and neurotransmitters play a critical role in this process. γ-Aminobutyric acid (GABA) is the most important inhibitory neurotransmitter in the mammalian central nervous system. It has been shown that acrylamide affects the receptor binding levels of dopamine, adrenocorticotropic hormone, 5-hydroxytryptamine, glycine, GABA and benzodiazepine (Agrawal et al., 1981; Bondy et al., 1981). 17–30% of axons use GABA as a transmitter and cerebral GABA is generated by decarboxylation of glutamate by a rate-limiting enzyme glutamic acid decarboxylase (GAD). Activity of GAD has a direct impact on the level of GABA in the brain. GAD exists in two isoforms based on its molecular weight: GAD65 and GAD67. GAD65 is located in synaptic vesicle membranes, triggering fast and point-to-point GABA neural transmission (Tian et al., 1999). GAD67 is spread evenly throughout cell bodies, catalyzing GABA as a nutritional factor for neurodevelopment (Kaufman et al., 1991; Owens and Kriegstein, 2002). Because GAD is essential for the synthesis of GABA, we used GABA and GAD concentrations as indicators to reflect the impact of acrylamide on GABA.
Acrylamide was shown to block transportation of GABA receptor in cells and consequently led to intracellular accumulation of the receptor (Ho et al., 2002). Previous studies also showed that some neurotoxins that inhibited the central nervous system function may affect GABA, indicating that GABA may be a target of acrylamide (Li et al., 2007; Zhou et al., 2009). The effect of acrylamide on the function and impact of GABA requires further study.
In addition, astrocytes play an important role in the response to a variety of central nervous system injuries. Glial fibrillary acidic protein (GFAP) is an astrocytic marker and upregulation of GFAP is reported in all types of central nervous system injury. Reactive astrocytosis is characterized by increased GFAP-immunoreactive cells and branches, hyperplasy, and strengthened GFAP expression. Therefore, this study also examined the neurotoxic mechanisms of acrylamide on the cerebrum from the perspective of GFAP and reactive astrocytes.
Materials and Methods
Animal model preparation
Forty male Sprague-Dawley rats aged 3–4 weeks and weighing 59–80 g were purchased from the Laboratory Animal Center of Guangzhou University of Chinese Medicine in China (certification No. SCXK (Yue) 2008-C020). After 7 days of acclimation, rats were equally and randomly divided into a control group, a low-dose acrylamide group (5 mg/kg), a moderate-dose acrylamide group (15 mg/kg), and a high-dose acrylamide group (30 mg/kg). Acrylamide (purity 99.9 %) was purchased from Tianjin Yongda Chemical Reagent Development Center, Tianjin, China. Acrylamide was dissolved in saline and rats were gavaged at 5 mL/kg, 5 times per week for 4 weeks. The control rats were treated with the same volume of saline. Rats were individually housed at 23 ± 2°C and 50 ± 10% humidity with 12-hour light/dark cycles, and allowed free access to drinking water and rodent laboratory food. Animal weight was recorded once a week and the changes in daily behavior were observed and recorded.
Organ index
Six rats from each group were weighed and decapitated under anesthesia. The cerebral cortex was separated on ice and weighed quickly. The organ index of cerebral cortex was calculated (i.e., tissue weight/body weight). After weighing, the cortex was immediately placed in liquid nitrogen and stored at −80°C for glutamate and GABA biochemical assay.
Enzyme-linked immunosorbent assay (ELISA) for glutamate and GABA concentrations
Samples from cerebral cortex tissues were placed in a precooled glass homogenizer and homogenate buffer (0.1 M PBS, pH 7.0) was added at a ratio of 1:9 (w/v). Homogenates were transferred to centrifuge tubes and preserved at −20°C until use. The optical densities of glutamate and GABA were detected and then their concentrations were calculated by ultraviolet spectroscopy and ELISA assay. In accordance with the ELISA kit for glutamate and GABA (USCNLIFE, Wuhan, China), all experimental procedures were conducted following the manufacturer's instructions. The microplate reader (TECAN TYPE: Sunrise Remote/Touchscreen) was from Nanjing Huadong Electronics Group Co., Ltd. (Nanjing, China).
Paraffin embedding and section preparation
Four rats from each group were anesthetized with sodium pentobarbital (50 mg/kg) intraperitoneally and transcardially perfused with 4% paraformaldehyde. Brains were isolated, post-fixed in 4% paraformaldehyde for 24 hours at 4°C, and sliced coronally into 6-μm-thick sections as directed by the rat cerebrum stereotaxic atlas (Bregma +1.8 to −0.8) (Bao and Chu, 1991). Sample sections were divided into two groups. One group was used to perform hematoxylin-eosin staining for comparison of morphological changes in neurons, and the other was used for immunohistochemical staining.
Immunohistochemistry
Sections were deparaffinized, incubated in 3% H2O2 solution for 15 minutes to deactivate endogenous peroxidase and antigen retrieval was performed in 0.01 M citrate butter solution (pH 6.0) using a microwave. Sections were incubated with the following primary polyclonal antibodies: mouse anti-GFAP 1:400; rabbit anti-GABA 1:100; mouse anti-GAD65 1:100 at 4°C overnight. Polyclonal antibodies anti-GABA, anti-GAD65 and anti-GFAP, horseradish peroxidase-streptavidin-biotin-peroxidase complex kit and 3,3′-diaminobenzidine kit were all from Wuhan Boster Biological Technology, Ltd. (Wuhan, China). After washing, sections were then incubated with their respective homologous secondary antibodies (1:100; Wuhan Boster Biological Technology, Ltd.) for 2 hours at room temperature followed by streptavidin-biotin-peroxidase complex solution for 1 hour and then visualized with 3,3′-diaminobenzidine solution.
Imaging analysis
The sections were observed and photographed under Olympus BX51 microscope (Olympus, Tokyo, Japan). Four rats were chosen from each group, and four slices were taken from each brain. Four high-power fields were taken randomly on each section. The image pro-plus 6.0 software (Media Cybernetics, Rockville, MD, USA) was used to count cell and measure optical density.
Statistical analysis
All data were presented as the mean ± SD and analyzed by SPSS 16.0 software (SPSS, Chicago, IL, USA). The differences between groups were compared with one-way analysis of variance, followed by Dunnett's post hoc test. A value of P < 0.05 was considered statistically significant.
Results
Acrylamide effects on behavior of weaning rats
The weight of rats in the control and low-dose acrylamide groups increased gradually during the exposure period without any abnormal behavior. Rats from the moderate-dose acrylamide group presented slight uncoordinated motor movement. Splayed hind limb feet were observed at 4 weeks. Rats from the high-dose acrylamide group showed splay hind limb feet at 2 weeks, which developed to complete splay, inability to support the body and inversed foot. No paralysis was observed among all groups.
The organ index of cortex of rats from four groups (0, 5, 15, 30 mg/kg acrylamide) was 0.187 ± 0.015%, 0.189 ± 0.026%, 0.191 ± 0.021%, and 0.211 ± 0.023%, respectively. The statistical data showed no significant difference in organ index during acrylamide treatment (P > 0.05).
Acrylamide effects on concentrations of glutamate and GABA in the cerebrum of weaning rats
After acrylamide treatment, there was no difference in glutamate and GABA concentrations in the cerebrum between the low-dose acrylamide group and the control group (P > 0.05). A significant decrease in glutamate concentration was observed in the cerebrum of rats from the moderate- and high-dose acrylamide groups (P < 0.05). GABA concentrations were significantly increased (P < 0.05), presenting an obvious dose-effect relationship (Figure 1).
Figure 1.
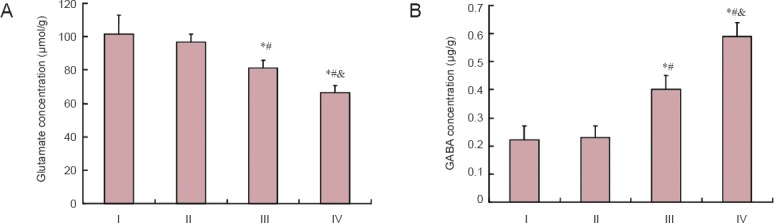
Effect of acrylamide on glutamate (A) and GABA (B) concentrations in the cerebral cortex at 4 weeks.
Data are expressed as the mean ± SD (n = 6). The differences between groups were compared with one-way analysis of variance, followed by Dunnett's post hoc test. *P < 0.05, vs. I (0 mg/kg); #P < 0.05, vs. II (5 mg/kg); &P < 0.05, vs. III (15 mg/kg). GABA: Gamma aminobutyric acid; I: control group; II: low-dose acrylamide group (5 mg/kg); III: moderate-dose acrylamide group (15 mg/kg); IV: high-dose acrylamide group (30 mg/kg).
Acrylamide effects on neuronal morphology in cerebral cortex of weaning rats
As shown in Figure 2, neurons were in a neat arrangement and the neuronal count did not decrease with administration of acrylamide. However, abnormal neuronal morphology was observed with increased acrylamide dose. Neurons in the moderate-dose acrylamide group showed concentrated nuclei, increased basophilic chromatin and absent nucleoli. Neurons in the high-dose acrylamide group showed increased concentrated nuclei in neurons, darkly-stained basophilic chromatin and small cell bodies.
Figure 2.
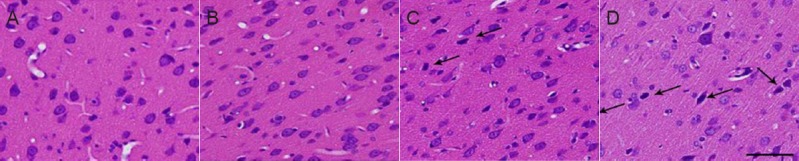
Neuronal morphology in the cerebral cortex of weaning rats at 4 weeks after acrylamide treatment (hematoxylin-eosin staining).
The images show the changes of neurons in the cerebral cortex in the control group (A), low- (B), moderate- (C) and high-dose acrylamide groups (D). Arrows represent abnormal neurons. Scale bar: 25 μm.
Acrylamide effects on GFAP, GABA and GAD65 immunoreactivities in cerebral cortex of weaning rats
GFAP-immunoreactive cells were observed under a light microscope. Astrocytic processes were widely present throughout the cerebral cortex, particularly in the granular layer. GFAP-immunoreactive cells in rats from the control and low-dose acrylamide groups were stained brown with small sized thin axons (Figure 3). Those from the moderate- and high-dose acrylamide groups were stained dark brown with long processes and there were more in number. Analysis of Figure 3 showed that compared with the control group, the number of GFAP-immunoreactive cells and mean optical density were higher in rats from moderate-dose acrylamide group (P < 0.05). GFAP-immunoreactive cell counts in cerebral cortex of the high-dose acrylamide group presented the same change as seen with the moderate-dose group (P < 0.05).
Figure 3.
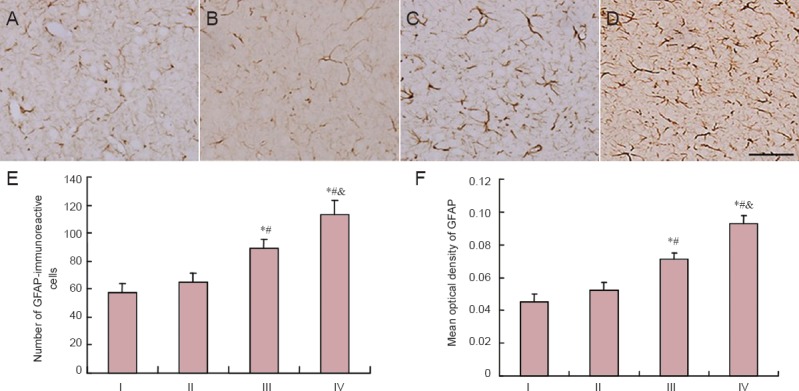
GFAP-immunoreactive cells in cerebral cortex at 4 weeks after acrylamide treatment.
The images show the expression of GFAP in the cerebral cortex in the control (A), low- (B), moderate- (C), and high-dose acrylamide groups (D). Scale bar: 25 μm. (E) Number of GFAP-immunoreactive cells. (F) Densitometry of GFAP immunoreactivity. Data are expressed as the mean ± SD (n = 6). The differences between groups were compared with one-way analysis of variance, followed by Dunnett's post hoc test. *P < 0.05, vs. I (0 mg/kg); #P < 0.05, vs. II (5 mg/kg); &P < 0.05, vs. III (15 mg/kg). GFAP: Glial fibrillary acidic protein; I: control group; II: low-dose acrylamide group (5 mg/kg); III: moderate-dose acrylamide group (15 mg/kg); IV: high-dose acrylamide group (30 mg/kg).
GABA-immunoreactive substances were brown and round and distributed in the cytoplasm of neurons. GABA-immunoreactive neurons were spread widely in the cerebral cortex. Rats from the low-dose acrylamide group showed light GABA staining in neurons. Rats from the moderate- and high-dose acrylamide groups displayed intense GABA staining. GABA-immunoreactive neurons from all groups did not exhibit obvious morphological changes (Figure 4). Quantitative analysis showed that the mean optical density of GABA-immunoreactive neurons in moderate- and high-dose acrylamide groups increased significantly (P < 0.05) when compared with the control group (Figure 4).
Figure 4.
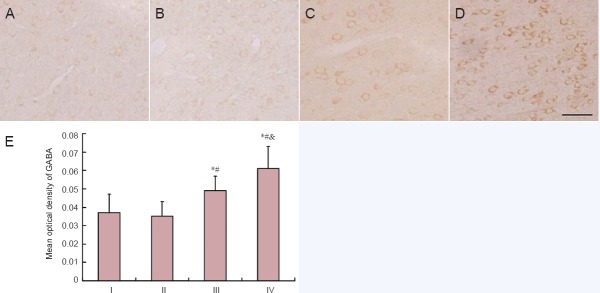
GABA-immunoreactive neurons in cerebral cortex at 4 weeks after acrylamide treatment.
The images show the immunoreactivity of GABA in the cerebral cortex in the control (A), low- (B), moderate- (C), and high-dose acrylamide groups (D). Scale bar: 25 μm. (E) Densitometry of GABA immunoreactivity. Data are expressed as the mean ± SD (n = 6). The differences between groups were compared with one-way analysis of variance, followed by Dunnett's post hoc test. *P < 0.05, vs. I (0 mg/kg); #P < 0.05, vs. II (5 mg/kg); &P < 0.05, vs. III (15 mg/kg). GABA: Gamma aminobutyric acid; I: control group; II: low-dose acrylamide group (5 mg/kg); III: moderate-dose acrylamide group (15 mg/kg); IV: high-dose acrylamide group (30 mg/kg).
GAD65 immunoreactive staining was located mainly in the plasma of neurons, which was similar to the distribution of GABA expression in neurons. Rats from the control and low-dose acrylamide groups showed light GAD65 immunoreactivity in the cortex (Figure 5). Quantitative analysis showed that the mean optical density of GAD65 immunoreactivity in the moderate- and high-dose acrylamide groups was significantly greater compared with the low-dose acrylamide group (P < 0.05; Figure 5).
Figure 5.
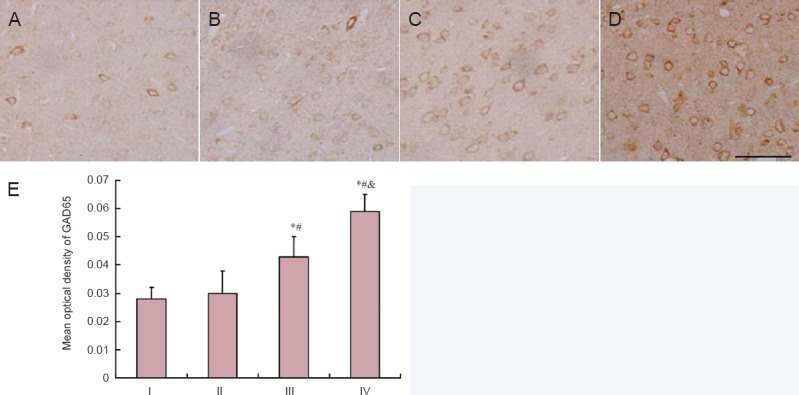
GAD65-immunoreactive neurons in cerebral cortex at 4 weeks after acrylamide treatment.
The images show the immunoreactivity of GAD65 in the cerebral cortex in the control (A), low- (B), moderate- (C), and high-dose acrylamide groups (D). (E) Densitometry of GAD65 immunoreactivity. Data are expressed as the mean ± SD (n = 6). The differences between groups were compared with one-way analysis of variance, followed by Dunnett's post hoc test. *P < 0.05, vs. I (0 mg/kg), #P < 0.05, vs. II (5 mg/kg); &P < 0.05, vs. III (15 mg/kg). GAD65: Glutamic acid decarboxylase 65; I: control group; II: low-dose acrylamide group (5 mg/kg); III: moderate-dose acrylamide group (15 mg/kg); IV: high-dose acrylamide group (30 mg/kg).
Discussion
As a commonly used chemical material since the 1980s, the toxicity of acrylamide has attracted more and more attention in recent years (Agrawal et al., 1981). Acrylamide can be absorbed into the body by many ways, such as via the respiratory system, digestive system, skin, and muscle. Additionally, acrylamide can easily spread throughout the whole body very quickly. Long exposure to acrylamide may lead to toxic accumulation and result in toxic symptoms (Bondy et al., 1981). Recently, high doses of acrylamide have been detected in fried food, which is frequently eaten by children. Our study aimed to investigate the potential impact of acrylamide in a weaning rat model.
A previous study has demonstrated that mild acrylamide intoxication is exhibited by slowly increased sensation and motor nerve abnormality, and moderate intoxication portrays deep sensory disturbance and ataxia (Costa et al., 1992). Severe intoxication shows sub-acute development of the syndrome; first cerebral dysfunction, which disappears upon stopping exposure, then multiple motor dysfunctions appear, including numbness, decreased myodynamia and paralysis. In this study, by the end of acrylamide exposure, rats from the low-dose acrylamide group did not have obvious changes in behavior. Rats from the high-dose acrylamide group experienced nervous function defects, such as typical anxiety and weak hind limbs.
We did not observe a decrease in neuronal number in the cerebral cortex using hematoxylin-eosin staining; however, we observed an increased number of abnormal neurons that were distributed in varied layers of the cerebral cortex and which is consistent with a previous study (He et al., 2001). Astrocytes are widely distributed in the brain and GFAP is an intermediate filament protein located mainly in astrocytes, often used as a marker of astrocytes. As GFAP expression increases in response to almost all types of brain injury, it has been regarded as a biomarker of injury in the central nervous system (Deng and Yuan, 2005). In our experiment, GFAP immunoreactivity increased significantly in response to acrylamide in a dose-effective manner. This is in accordance with the aforementioned pathomorphological changes. As to why GFAP increases following central nervous system injury is still unclear, we assume that cerebral cortex injury induced by acrylamide may stimulate reactive hyperplasia of GFAP.
The mechanism underlying acrylamide-induced neuronal injury remains unclear. An earlier study has suggested that it might be related to acrylamide-induced apoptosis (Li et al., 2008). Li et al. (2008) found that degenerated neurons exposed to low-dose acrylamide tended to be in the early stages of, or on the verge of apoptosis, while neurons exposed to high-dose acrylamide did not show obvious changes in apoptosis. Acrylamide can suppress metabolism and axonal transport in neurons, causing deficiency of nutritional factors (Honig and Rosenberg, 2000; LoPachin et al., 2004). In addition, acrylamide can inhibit the function of 3-phosphoglyceraldehyde dehydrogenase, phosphofructokinase and neuron specific enolase, interfere with metabolic processes and inhibit rapid axonal transport. This further leads to deficient nutrition supply, axon degeneration in the central and peripheral nervous systems, and shedding of the myelin sheath (Gold et al., 1992; He et al., 2002).
There are three types of GABA in the cerebrum: free, half-bound and bound GABA. Bound GABA is considered the major form, and free GABA is considered the transmitter of the receptor. The release of GABA is dependent on Ca2+ release when newly generated GABA is first released. The absorption is the principle cause of GABA inactivation, owing to the highly efficient re-absorptive mechanism that leads to the termination of synaptic transmission. An in vitro experiment reported that acrylamide decreased glutamate in the synapse and increased GABA in the synaptic cleft (Costa et al., 1992). Acrylamide also caused a decrease in adenosine triphosphate level of cerebrum, which in turn weakened the ability of neurons to reuptake GABA. The abnormally high GABA concentration in the synaptic cleft may over-stimulate the corresponding receptor in the postsynaptic membrane, causing depression of normal neuron activity. In this study, biochemical and immunohistochemical results showed no significant changes in the levels of glutamate, GABA and GAD65 in cerebral cortex of rats from the low-dose acrylamide and control groups. In contrast, the moderate- and high-dose acrylamide groups exhibited a dose-dependent decrease in glutamate and an increase in GABA and GAD65, suggesting that acrylamide may upregulate GABA synthesis by increasing GAD65 expression and consequently decrease the concentration of glutamate, the precursor of GABA synthesis. Acrylamide-induced increase of GABA concentration may cause synaptic depression and be involved in slow, long-term neuronal damage. In conclusion, this study provides experimental evidence to indicate the mechanism of neurotoxicity induced by acrylamide.
Footnotes
Funding: This study was supported by the Medical Scientific Research Foundation of Guangdong Province in China, No. B2014202; and the Natural Science Foundation of Guangdong Province in China, No. 2014A030310455.
Conflicts of Interest: None declared.
Copyedited by Paul P, Pack M, Wang J, Qiu Y, Li CH, Song LP, Zhao M
References
- Agrawal AK, Seth PK, Squibb RE, Tilson HA, Uphouse LL, Bondy SC. Neurotransmitter receptors in brain regions of acrylamide-treated rats. I: Effects of a single exposure to acrylamide. Pharmacol Biochem Behav. 1981;14:527–531. doi: 10.1016/0091-3057(81)90312-9. [DOI] [PubMed] [Google Scholar]
- Bao X, Chu S. Beijing: People's Medical Publishing House; 1991. The Rat Barin Atlas. [Google Scholar]
- Bondy SC, Tilson HA, Agrawal AK. Neurotransmitter receptors in brain regions of acrylamide-treated rats. II: effects of extended exposure to acrylamide. Pharmacol Biochem Behav. 1981;14:533–537. doi: 10.1016/0091-3057(81)90313-0. [DOI] [PubMed] [Google Scholar]
- Costa LG, Deng H, Gregotti C, Manzo L, Faustman EM, Bergmark E, Calleman CJ. Comparative studies on the neuro and reproductive toxicity of acrylamide and its epoxide metabolite glycidamide in the rat. Neurotoxicology. 1992;13:219–224. [PubMed] [Google Scholar]
- Deng L, Yuan Q. The study progress of GFAP in the neuronal system. Luzhou Yixueyuan Xuebao. 2005;2:189–192. [Google Scholar]
- Dybing E, Sanner T. Risk assessment of acrylamide in foods. Toxicol Sci. 2003;75:7–15. doi: 10.1093/toxsci/kfg165. [DOI] [PubMed] [Google Scholar]
- Gold BG, Griffin JW, Price DL. Somatofugal axonal atrophy precedes development of axonal degeneration in acrylamide neuropathy. Arch Toxicol. 1992;66:57–66. doi: 10.1007/BF02307271. [DOI] [PubMed] [Google Scholar]
- He Q, Han M, Rao M. Pathological changes in Purkinje cells of the cerebellum in acrylamide-intoxicated Ola mice and 6J mice. Zhongguo Laodong Weisheng Zhiyebing Zazhi. 2001;2:102–104. [Google Scholar]
- He Q, Han M, Rao M. Effect of acrylamide on creatine kinase and adenosine triphosphate in brain of mice and its significance. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2002;20:195–196. [PubMed] [Google Scholar]
- Ho WH, Wang SM, Yin HS. Acrylamide disturbs the subcellular distribution of GABAA receptor in brain neurons. J Cell Biochem. 2002;85:561–571. doi: 10.1002/jcb.10159. [DOI] [PubMed] [Google Scholar]
- Honig LS, Rosenberg RN. Apoptosis and neurologic disease. Am J Med. 2000;108:317–330. doi: 10.1016/s0002-9343(00)00291-6. [DOI] [PubMed] [Google Scholar]
- Kaufman DL, Houser CR, Tobin AJ. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J Neurochem. 1991;56:720–723. doi: 10.1111/j.1471-4159.1991.tb08211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, He X, Deng S. Effect of methyl tertiary butyl ether on amino butyric acid in cerebral of rat. Zhongguo Gonggong Weisheng. 2007;8:968–969. [Google Scholar]
- Li S, Jiang H, Cui N. Effects of bcl-2 and bax expression and neuronal denegreation in the cerebral cortex in rats with acrylamide treatment. J Toxicol. 2008;1:31–32. [Google Scholar]
- LoPachin RJ, Lehning EJ. Acrylamide-induced distal axon degeneration: a proposed mechanism of action. Neurotoxicology. 1994;15:247–259. [PubMed] [Google Scholar]
- LoPachin RM, Schwarcz AI, Gaughan CL, Mansukhani S, Das S. In vivo and in vitro effects of acrylamide on synaptosomal neurotransmitter uptake and release. Neurotoxicology. 2004;25:349–363. doi: 10.1016/S0161-813X(03)00149-9. [DOI] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Pourentezari M, Talebi A, Abbasi A, Khalili MA, Mangoli E, Anvari M. Effects of acrylamide on sperm parameters, chromatin quality, and the level of blood testosterone in mice. Iran J Reprod Med. 2014;12:335–342. [PMC free article] [PubMed] [Google Scholar]
- Rosen J, Hellenas KE. Analysis of acrylamide in cooked foods by liquid chromatography tandem mass spectrometry. Analyst. 2002;127:880–882. doi: 10.1039/b204938d. [DOI] [PubMed] [Google Scholar]
- Sen E, Tunali Y, Erkan M. Testicular development of male mice offsprings exposed to acrylamide and alcohol during the gestation and lactation period. Hum Exp Toxicol. 2015;34:401–414. doi: 10.1177/0960327114542883. [DOI] [PubMed] [Google Scholar]
- Tareke E, Rydberg P, Karlsson P, Eriksson S, Tornqvist M. Acrylamide: a cooking carcinogen? Chem Res Toxicol. 2000;13:517–522. doi: 10.1021/tx9901938. [DOI] [PubMed] [Google Scholar]
- Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci U S A. 1999;96:12911–12916. doi: 10.1073/pnas.96.22.12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilson HA. The neurotoxicity of acrylamide: an overview. Neurobehav Toxicol Teratol. 1981;3:445–461. [PubMed] [Google Scholar]
- Tyl RW, Friedman MA, Losco PE, Fisher LC, Johnson KA, Strother DE, Wolf CH. Rat two-generation reproduction and dominant lethal study of acrylamide in drinking water. Reprod Toxicol. 2000;14:385–401. doi: 10.1016/s0890-6238(00)00097-6. [DOI] [PubMed] [Google Scholar]
- Tyl RW, Marr MC, Myers CB, Ross WP, Friedman MA. Relationship between acrylamide reproductive and neurotoxicity in male rats. Reprod Toxicol. 2000;14:147–157. doi: 10.1016/s0890-6238(00)00066-6. [DOI] [PubMed] [Google Scholar]
- WHO. Geneva, Switzerland: FAO/WHO; 2002. Health implications of acrylamide in food. [Google Scholar]
- Zhou Y, Kang X, Wang M. Influence of propofol on amino neurotransmitters in different brain regions of rats in vivo. J Clin Anesthesiol. 2009;4:329–331. [Google Scholar]