

Introduction
Mobile genetic elements have been repeatedly called to duty in life-and-death struggles between hosts and their pathogens (1–4). One of their greatest utilities is the capacity to create DNA sequence diversity in protein-encoding genes, thereby generating protective shields to defend against enemies, or arsenals of weapons to exploit potential hosts. After decades of research considerable evidence now suggests that the V(D)J recombination system, which is essential for generating adaptive immunity in vertebrates, has evolved from an ancestral DNA transposon (2–4). The site-specific recombinases responsible for V(D)J recombination, RAG1 and RAG2, are able to catalyze DNA transposition in a manner analogous to DNA transposons (2), and the RAG1 core and V(D)J recombination signals are likely derived from the transposase and terminal repeats of an ancient DNA transposon similar to Transib (3, 4). Ironically, pathogens also exploit mobile genetic elements to generate protein diversity, altering their antigenic characteristics to evade host immunity (1). This process of antigenic variation is employed by Borrelia species, Neisseria gonorrhoeae and other pathogens. Bacterial antigenic variation often involves a single, highly expressed gene encoding an abundant surface protein and dozens of archived ones that are homologous but different from each other. Replacing all or part of the expressed copy by DNA transposition leads to antigenic variation on the surface of the pathogen.
Diversity-generating retroelements (DGRs) are a recently discovered class of beneficial mobile elements that diversify DNA sequences and the proteins they encode (5, 6). DGRs function through a template-dependent, reverse transcriptase (RT)-mediated mechanism that introduces nucleotide substitutions at defined locations in specific genes (5–7). DGRs were initially discovered during studies of pathogenesis by Bordetella species, which cause respiratory diseases in humans and other animals (5). The cell surfaces of these bacteria are highly dynamic due to changes in gene expression that accompany their infectious cycles (8). In a search for transducing vectors, a group of temperate bacteriophage were discovered that possess a remarkable ability to generate tropic variants that use different cell-surface molecules for infection (5, 9). Subsequent genetic and genomic studies with the prototype phage, BPP-1, showed that tropism switching is mediated by a phage-encoded DGR. This DGR introduces nucleotide substitutions in a gene that specifies a host cell-binding protein, which is positioned at the distal tips of phage tail fibers (Figure 1) (5, 9). As a result, BPP-1 can adapt to dynamic changes on the surfaces of Bordetella species. Guided by the sequences of phage DGR components, homologous elements have been identified in numerous bacterial, plasmid, and phage genomes (6, 10–13). Most DGRs are bacterial chromosomal elements and they are distributed throughout the bacterial domain, with representatives in all phyla that have significant sequence coverage. Although variations in architectures and associated components appear to mediate adaptations to particular needs, all DGRs are predicted to function in a fundamentally similar way. The BPP-1 phage serves as a model for this entire family of retroelements and our discussion begins with a brief description of its features.
Figure 1.
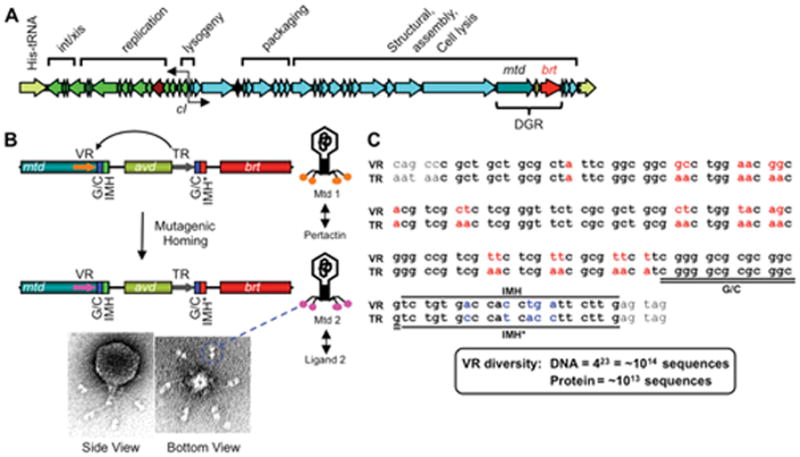
BPP-1 phage and its diversity-generating retroelement (DGR). (A) The BPP-1 genome is represented in the prophage form flanked by a duplication of the His-tRNA gene formed during integration. Functional assignments for most gene clusters are indicated, along with the cI-like repressor and the DGR cassette. (B) Schematic representation of the DGR cassette and its function in phage tropism switching. The cassette contains three genes (mtd, avd and brt) and two 134 bp repeats (template and variable repeats, or TR and VR, respectively). VR is located at the 3′ end of the mtd gene, which encodes the distal tail fiber protein responsible for receptor recognition. Located at the 3′ ends of VR and TR are IMH (Initiation of Mutagenic Homing) and IMH* elements, respectively, in addition to a GC-rich element. Phage tropism switching occurs through DGR-mediated mutagenic homing, in which TR sequence information is transferred to VR with adenine residues in TR appearing as random nucleotides in VR. Shown on the bottom are electron micrographs of the BPP-1 phage; globular structures at the distal ends of tail fibers are Mtd trimers (two per fiber). (C) Comparison of BPP-1 TR and VR. TR and VR sequences are shown in bold. VR variable positions and the corresponding adenine residues in TR are shown in red. IMH, IMH* and GC-rich elements are also indicated. There are 23 adenines in TR which can theoretically generate ~1014 different DNA sequences, or ~1013 different peptides. (Adapted from references 7 & 9)
Tropism-switching Bordetella phage
Bordetella species are aerobic, Gram-negative bacterial pathogens that colonize ciliated respiratory epithelial surfaces. B. pertussis and B. parapertussis are human-restricted and cause whooping cough (pertussis), while B. bronchiseptica infects a broad range of wild and domesticated mammals (8). The infectious cycle of these closely related species is regulated by a conserved, environmentally responsive phosphorelay system composed of the BvgS sensor protein and the BvgA response regulator, which control expression of an extensive array of cell surface and secreted molecules. In the so-called Bvg+ phase, BvgAS is active and induces expression of adhesins, toxins, a type III secretion system and numerous additional factors involved in colonization of respiratory surfaces. In the Bvg− phase the BvgAS phosphorelay is suppressed, virulence genes are quiescent, and a distinct set of loci are induced. In B. bronchiseptica, Bvg− phase genes are responsible for motility, chemotaxis, and survival under conditions of nutrient deprivation, presumably representing an ex vivo phase of the infectious cycle. Dynamic changes in surface molecule expression are critical for the lifestyles of Bordetella species, and they likely pose a challenge to infecting phage.
BPP-1 (Bvg plus-trophic phage 1), isolated from a B. bronchiseptica strain cultured from the upper respiratory tract of a rabbit, preferentially forms plaques on Bvg+ as opposed to Bvg− phase Bordetella (5, 9). Using a combination of deletion, complementation, and cell binding assays, Liu et al. identified the receptor for BPP-1 as pertactin (Prn), an outer membrane autotransporter protein that is included as a protective antigen in acellular pertussis vaccines (5). Expression of prn is activated by BvgAS, thereby explaining BPP-1 tropism. At a frequency of ~10−5, however, BPP-1 formed normal size plaques on Bvg− phase Bordetella (5). Since plaque formation requires repeated rounds of infection, replication, and re-infection, this simple observation suggested that a tropism switch had occurred (5). Indeed, tropic variants fell into two classes. The first, designated BMP (Bvg minus-tropic phage), preferentially infected Bvg− phase cells while the second, BIP (Bvg indiscriminant phage), infected both Bvg+ and Bvg− cells with similar efficiencies. Although these phenotypes are inheritable, tropic variants are continuously generated at characteristic frequencies by BPP, BMP and BIP.
The BPP-1 DGR
To identify genetic changes responsible for tropism switching, whole genome sequences were obtained for a collection of BPP, BIP and BMP variants (5, 9). In each case, tropism switching was accompanied by nucleotide substitutions within a 134 bp sequence, designated the variable repeat (VR), located at the 3′ end of the mtd (major tropism determinant) gene (Figure 1). Mtd is a trimeric tail fiber protein responsible for phage binding to Bordetella surfaces, and changes in its coding sequence confer new ligand specificities (14). Comparison of phage variants showed that variable nucleotides were located at a subset of positions in VR and most often at the first two positions of codons, thereby maximizing amino acid diversity (5, 14). There are 23 variable positions at which any of the 4 nucleotides can be found, giving a theoretical DNA diversity of 1014 sequences and a resulting repertoire of ~10 trillion polypeptides. This rivals the estimated diversity that can be generated by mammalian antibody and T cell receptor genes (15, 16).
An imperfect repeat of VR, designated the template repeat (TR), is found downstream of mtd (Figure 1) (5). Liu et al. made the simple but seminal observation that variable residues in VRs corresponded to adenine residues in TR, which remained unchanged during phage tropism switching (5). This prompted the hypothesis that tropism switching is associated with a mutagenic mechanism that is adenine specific, and that TR somehow provides a template for this process. Indeed, precise deletion of the BPP-1 TR resulted in fully infectious phage particles for Bvg+ Bordetella, but abolished the ability to switch tropism. Furthermore, single nucleotide substitutions introduced into TR, which corresponded to silent mutations in VR, appeared in VR during tropism switching and were accompanied by mutations at adenines (5). These results demonstrated that DNA sequence information was “transferred” from TR to VR during tropism switching and accompanied by adenine-specific mutagenesis.
An additional piece of the puzzle came from the unexpected finding of a reverse transcriptase gene, brt (Bordetella reverse transcriptase), in the dsDNA BPP-1 genome (Figure 1) (5). Its position adjacent to the VR/TR repeats prompted the hypothesis that reverse transcription could be involved in tropism switching. To test this, an in-frame deletion was introduced into brt and the resulting phenotype was identical to the ΔTR deletion: infectious phage particles were produced but tropism switching was abolished. This suggested an intriguing link between reverse transcription and adenine mutagenesis. The brt gene encodes a 38 kDa protein with similarity to RTs found in other retroelements, including a conserved YMDD box found in RT catalytic centers. Substitution of the YMDD box with SMAA resulted in the loss of phage tropism switching in vivo, and RT activity in vitro. It was proposed that tropism switching occurs through an RT-dependent transfer of sequence information from an invariant template (TR) to a region of variability (VR), and is accompanied by adenine mutagenesis in a process called “mutagenic homing.” The RT-dependency predicted the involvement of an RNA intermediate, leading Liu et al. to designate the tropism-switching cassette shown in Figure 1 as a diversity-generating retroelement (DGR) (5). An additional gene in the BPP-1 DGR, designated avd (accessory variability determinant), encodes a small polypeptide (Avd) that plays an essential role in DGR function as described below (6, 9).
DGRs are widespread in bacterial and phage genomes
The ability of the BPP-1 DGR to accelerate the evolution of novel ligand-binding specificities suggested that similar elements would be found elsewhere in nature. Not surprisingly, using Brt as a template Doulatov et al. identified additional DGRs in phage and bacterial genomes (6). These included a Vibrio harveyi phage and the chromosomes of human commensals (Bifidobacterium longum and Bacteroides thetaiotaomicron), a human oral pathogen (Treponema denticola), and cyanobacteria. Some cyanobacterial species contained multiple DGRs, or multiple target ORFs that are diversified in trans by a single DGR. DGR RTs were found to be closely related to RTs of mobile group II introns, and in all cases repeated elements corresponding to VR/TR cognate pairs could be found in nearby loci. Predicted target proteins and VR sequences were diverse, but VRs differed from cognate TRs almost exclusively at sites corresponding to TR adenines, suggesting that DGRs function through a conserved mechanism.
More recently, Minot et al. performed high-throughput DNA sequencing on the gut virome of healthy humans (12). Metagenomic analysis of phage populations from stool samples identified 36 unique TR/VR pairs, 29 of which were adjacent to a DGR-type RT gene. In total, DGRs were identified in 11 out of 12 subjects studied. As with other DGRs, TR/VR sequences differed almost exclusively at sites corresponding to TR adenines. This discovery demonstrated that DGRs are common in the genomes of bacteriophages found in the lower gastrointestinal tract. Using a custom-made script called DiGReF, Schillinger et al. conducted a large scale search of sequence databases and identified 155 DGRs in phage and bacteria, both Gram-positive and Gram-negative (13). DGRs are particularly abundant in certain phyla, with bacteroides (27.7%), firmicutes (31.0%) and proteobacteria (25.2%) containing the greatest numbers of unique elements. DGRs were also found in actinobacteria, cyanobacteria, deinococcus-thermus, nitrospirae, spirochaetes, chlamydiae/verrumicrobia, and other bacterial groups. These observations indicated that DGR host organisms are highly diverse and occupy varied environmental niches. In this large dataset, nearly all VRs were located at the 3′ end of protein-coding genes and they ranged from 50–150 bp. The relatively short length of VRs might reflect constraints on DNA sequence hypervariation in protein-encoding genes, as highly dense nucleotide substitutions over a long stretch would be more likely to result in a loss of function. This study confirmed the earlier observation by Doulatov et al. that many bacterial genomes contain multiple DGRs, or encode single DGRs that diversify multiple target genes (6).
Although DGRs are often considered in the context of Bordetella phage, this is an artifact of their discovery and the choice of BPP-1 as a prototype for mechanistic studies. In reality, DGRs are widely distributed in bacterial chromosomes in addition to phage and plasmid genomes.
DGR target proteins
The cellular localization and physiological functions of the vast majority of DGR-diversified target proteins are uncharacterized, however, common themes are beginning to emerge. Arambula et al. recently analyzed a DGR found on the chromosome of Legionella pneumophila, an opportunistic pathogen (Figure 2) (17). The L. pneumophila DGR is located on a conjugative transposable element and was found to be capable of mutagenic homing with characteristic adenine mutagenesis. Remarkably, its TR contains 43 adenine residues and is theoretically capable of generating 1026 unique DNA sequences, creating a repertoire of 1019 distinct proteins. The DGR-encoded target protein, LdtA, contains both a TAT (twin arginine transport) motif and a lipobox at its N-terminus. The TAT pathway is an alternative secretion system that can translocate folded proteins or protein complexes across bacterial cytoplasmic membranes (18), and lipobox motifs mediate signal peptide cleavage, lipid modification, and anchoring to inner or outer membranes (19). Genetic and biochemical analysis showed that LdtA is indeed a TAT-secreted protein that is lipid modified and localized to the outer leaflet of the Legionella outer membrane, with C-terminal VR sequences exposed to the extracellular milieu (Figure 2) (17). Bioinformatic analysis predicts that target proteins of many bacterial DGRs, including those of T. denticola, Bacteroides species, Vibrio angustum, and S. baltica, also contain N-terminal lipobox motifs preceded by either TAT or Sec secretion signals (17). This suggests that lipid modification and surface display on bacterial outer membranes will be a common feature of proteins diversified by bacterial DGRs.
Figure 2.
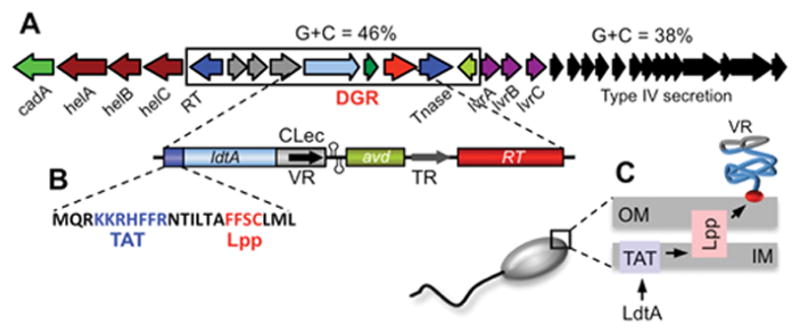
Diversification of a surface-displayed lipoprotein by a Legionella DGR. (A) The L. pneumophila strain Corby DGR is encoded on a genomic island that differs in G+C content from the rest of the genome. VR sequences at the 3′ end of the diversified locus, ldtA, are flanked by tandem hairpin/cruciform structures that are essential for efficient homing. TR contains 43 adenine residues which can create a potential repertoire of 1026 different VR DNA sequences. (B) LdtA contains atypical TAT (twin arginine transport) and Lpp (lipobox, lipid modification) signals at the N-terminus. (C) Cellular localization studies demonstrated that LdtA is exported through the inner membrane via the TAT pathway, lipid modified, and anchored on the outer surface of the outer membrane via an Lpp-like lipoprotein processing pathway. VR-encoded residues are surface displayed by a C-terminal CLec fold. (Adapted from reference 17)
From both structural and functional perspectives, the BPP-1 Mtd protein is by far the most extensively characterized DGR target protein (14, 20, 21). To understand structure-function relationships, McMahon et al. determined the atomic structures of five different Mtd tropic variants (Figure 3) (14). The Mtd variants formed intertwined, pyramid-shaped homotrimers with nearly identical secondary and tertiary structures, indicating that VR diversification did not lead to gross conformational changes. Each monomer was organized into three discrete domains: a β-prism, β-sandwich, and C-type lectin (CLec), arranged from N- to C-terminus. The β-prism domains converge to form a vertex on the top, and the CLec domains interact with each other to form the bottom part of the pyramid-shaped trimer. VR-encoded variable amino acids are presented by CLec folds on the bottom surface of the Mtd trimer, with their side chains solvent exposed and accessible to ligand interactions. The most remarkable feature of the comparative analysis of Mtd structures is that the five distinct tropic variants showed virtually no conformational variation in the VR-encoded regions. As shown in Figure 3, the main chain conformations of these tropic variants are nearly superimposable. This is in striking contrast to antigen-binding regions of immunoglobulin (Ig) molecules, where conformational flexibility is associated with the ability to recognize diverse antigens.
Figure 3.
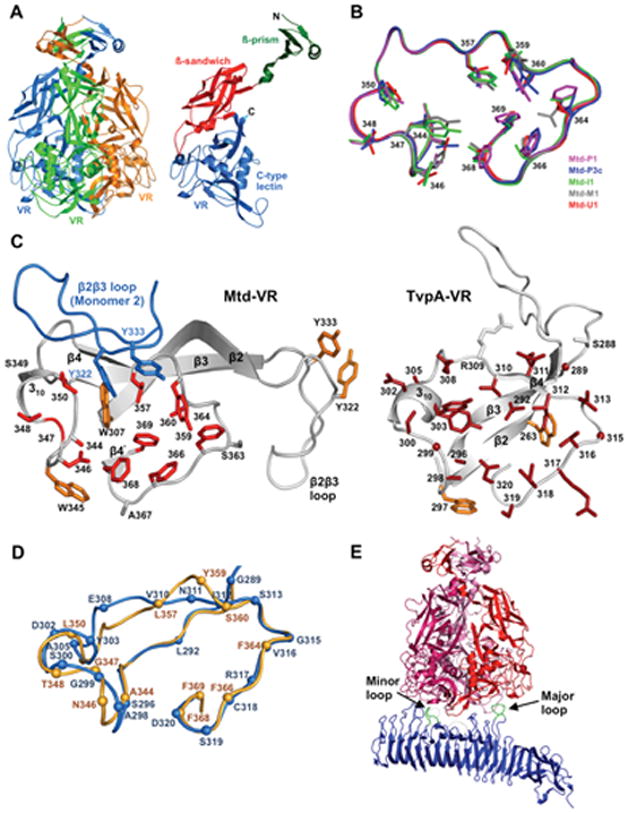
The CLec fold as a scaffold for display of DGR-generated protein diversity. (A) The BPP-1 Mtd protein forms a pyramid-shaped homotrimer (Left) with VR-encoded residues exposed on the bottom surface. (Right) An Mtd monomer containing β-prism, β-sandwich, and VR-encoded CLec domains, from N- to C-terminus. (B) Backbone structures of the VR regions of 5 Mtd variants with different ligand specificities are shown. Despite side chain variations in diversified VR residues, the backbone structures are nearly superimposable. (C) Comparison of the CLec VR regions of BPP-1 Mtd and a Treponema denticola variable protein, TvpA. For Mtd-VR, the β2β3 loop of a second monomer is also shown (blue). (D) Superposition of the VR regions of BPP-1 Mtd (light orange) and T. denticola TvpA (blue). (E) Interaction of an Mtd homotrimer with the receptor protein pertactin. See text for details. (Adapted from references 14, 20 and 22)
The CLec fold appears to be a conserved feature of many DGR diversified proteins. Although DGR target proteins generally share little sequence similarity, structure-based sequence alignments predicted that VR-encoded variable residues are often presented in the context of C-terminal CLec domains (14). This was recently confirmed by X-ray crystallography using the T. denticola TvpA protein (Figure 3C,D), which is predicted to be a DGR-diversified lipoprotein localized to the spirochaetal surface (22). TvpA contains a CLec fold that is highly homologous to Mtd, and VR residues are positioned in a remarkably similar manner. Instead of forming homotrimers like BPP-1 Mtd, however, TvpA exists as a monomer and does not contain β-prism or β-sandwich domains as found in Mtd.
Interestingly, a subset of phage DGRs were predicted to use Ig folds, similar to those found in immunoglobulins, instead of CLec folds to display variable residues (12). These Ig folds were usually located in the middle, as opposed to the 3′ ends of VR-containing ORFs. Although immunoglobulins and these predicted DGR variable proteins both use Ig folds, they appear to have evolved different means for displaying variable residues (12). In the former, diversified residues are located in flexible loop regions positioned between β-sheets. In contrast, structural modeling of Ig-type DGR target proteins indicated that diversified residues are displayed on one face of a β-sheet and the linker region connecting it to the adjacent domain. These observations suggest that DGR target proteins and antigen receptors may have evolved different solutions to accommodate sequence diversity in the context of Ig folds.
To better understand the basis of ligand recognition by DGR variable proteins, Miller et al. co-crystalized BPP-1 Mtd and its outer membrane ligand, pertactin (Figure 3E) (20). Structural analysis showed that each Mtd trimer bound to one molecule of pertactin, whose extracellular domain has an extended β-helix structure. An asymmetric mode of interaction was observed: Two identical VR regions from two of the Mtd monomers in the trimer each bound a different loop from pertactin, with these loops having no sequence similarity to one another.
The fundamental basis of the evolvability of Mtd-ligand interactions was revealed, at least in part, through an analysis of binding constants (20). The Kd for the interaction between purified Mtd trimers and the ectodomain of pertactin was 3.5 μM as determined by surface plasmon resonance. In contrast, the Kd for the intact phage was ~6.9 pM, reflecting a nearly 106-fold increase in binding strength. The BPP-1 phage contains six tail fibers with two Mtd trimers at their tips (21). With 12 Mtd trimers on each phage particle, and the ectodomain of pertactin displayed at high density on the Bordetella surface, ligand-receptor binding is multivalent and driven by avidity, which results in the exponential amplification of individual binding strengths (20). This amplification relaxes the demand for optimal complementarity between partners, greatly expanding the scope of molecules that can be recognized by DGR-diversified proteins. Avidity-driven interactions are inherently evolvable, and are predicted to characterize most, if not all DGR variable protein-ligand interactions.
Directionality of DGR mutagenic homing
Mutagenic homing is an RT-mediated, adenine specific, error-prone process that unidirectionally transfers sequence information from TR to VR (5, 6). In BPP-1, substitution of adenine residues in TR with non-adenine nucleotides eliminated VR mutagenesis at cognate positions, while replacing non-adenine residues with adenines resulted in novel sites of mutagenesis in VR, proving that sequence diversification is intrinsic to TR adenines.
A prominent feature of mutagenic homing is that diversity is specifically targeted to VR while TR sequences remain unchanged (5, 6). What determines this directionality? A comparison of the two repeats in BPP-1 revealed that in addition to differences corresponding to adenine residues in TR, they also differ by five base pairs at their 3′ ends (6). These polymorphisms are located within a 21 bp segment downstream of a 14 bp GC-rich element common to both TR and VR (Figures 1&4). During mutagenic homing, the polymorphisms are never co-converted. Swapping experiments by Doulatov et al. revealed that they are required for the unidirectional transfer of sequence information (6). When the 21-bp element at the 3′ end of VR was swapped with analogous sequences from TR, VR was no longer diversified. Conversely, replacing the 21-bp TR element with the corresponding VR sequences resulted in TR diversification at adenines, albeit at a low level. The 21-bp element in VR was named the Initiation of Mutagenic Homing (or IMH) element, while the corresponding sequence in TR was called IMH*. Further studies showed that similar polymorphisms could be found at the 3′ ends of cognate TRs and VRs in many DGRs (6). As detailed below, DNA structural determinants that follow IMH are also required for efficient homing.
Figure 4.
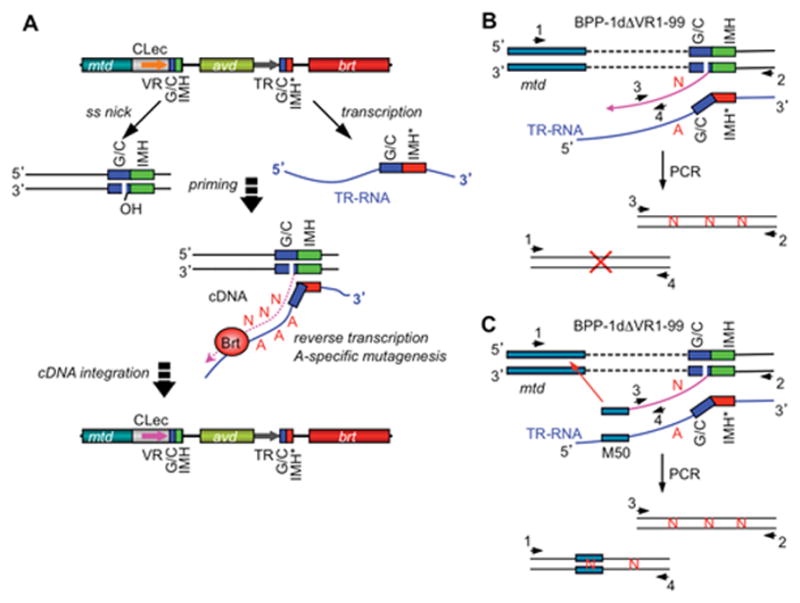
The TPRT model of BPP-1 DGR-mediated mutagenic homing. (A) Mutagenic homing occurs through a TR-RNA intermediate and is RecA-independent, similar to group II intron homing. A marker coconversion assay mapped the cDNA transfer boundary to a narrow region within the GC-rich element at the 3′ end, which may represent 3′ cDNA integration site(s). The marker transfer boundary at the 5′ end was more heterogeneous. A target DNA-primed reverse transcription model, similar to that of group II intron homing, has been proposed to explain these observations. The DNA target site was hypothesized to be nicked within the GC-rich element, with the exposed 3′ hydroxyl group serving as a primer for adenine-specific error-prone reverse transcription of the TR RNA. Integration of cDNA products at the 5′ end requires short stretches of homology between VR and the cDNA and may occur through strand displacement or template switching followed by break repair. Subsequent DNA replication would then create progeny genomes with mutagenized variable regions. (B) Deletion of VR sequence upstream of GC and IMH elements appeared to block 5′ cDNA integration but not 3′ cDNA integration, as analyzed by PCR with primer sets 1&4 and 2&3, respectively. Sequence analysis showed adenine mutagenesis in PCR products generated with primers 2&3. (C) 5′ cDNA integration in ΔVR1-99 was restored by inserting a 50 bp mtd sequence, which is homologous to the region upstream of the deletion junction, in TR. (Adapted from reference 7)
DGR homing occurs through an RNA intermediate
The presence of conserved RTs in DGRs and the required role of Brt in tropism switching suggested that mutagenic homing occurs through an RNA intermediate. To test this hypothesis, the BPP-1 TR was “tagged” with a self-splicing group I intron from bacteriophage T4 (the td intron) (7). Intron tagging was first used by Boeke et al. (23) to probe retrotransposition by the Saccharomyces cerevisiae transposon Ty1, and has subsequently been used to prove that other retroelements, including human long interspersed nuclear elements (LINEs) and mobile group II introns, function through RNA intermediates (24–26). As intron excision takes place only at the RNA level, detection of ligated exons in DNA homing products provides genetic proof that sequence information flows from DNA to RNA to DNA.
PCR-based assays were used to detect homing products (i.e. VR sequences) derived from td intron-tagged TRs supplied in trans on a replicating plasmid (7). As predicted, homing products consisted of VR sequences that contained ligated td exons, and their detection required functional Brt and a splicing-competent td intron. Sequence analysis verified precise intron excision and adenine mutagenesis in progeny VRs. These observations conclusively showed that DGR homing had occurred through a TR RNA intermediate. Regions of TR that are important for function were analyzed by similar PCR-based assays in which donor TRs were sequence-tagged to allow detection and characterization of homing products (7). Mutational analysis demonstrated that sequences internal to TR are largely dispensable, while sequences at either terminus are essential for the function of the RNA intermediate (7). By expressing wild type avd on a compatible plasmid in trans to TR and brt, a 300 bp portion of the avd coding sequence that extends upstream of the limit of TR-VR homology was found to be required for optimal TR RNA function (27). Shorter sequences upstream of TR, of as little as 48 bp, provided partially functional TR RNA species that supported DGR homing at lower efficiencies (27). Sequences downstream of TR that are required for optimal function extend for at least 110 bp (7). Although the TR-containing RNA species that serves as an intermediate in the retrotransposition reaction has yet to be characterized, the observation that sequences that lie beyond the limits of TR/VR homology influence activity suggests that RNA stability and/or structural determinants are important for DGR-mediated homing.
cDNA synthesis and integration
cDNA synthesis could occur through one of several mechanisms. First, it could be initiated through a target DNA-primed reverse transcription (TPRT) reaction similar to those used by non-LTR (long terminal repeat) retroelements and mobile group II introns, which are closely related to DGRs (28–31). Group II introns are site-specific retrotransposons found in bacterial, fungal and plant organelle genomes that insert at cognate intronless alleles in a process called intron homing (32). Intron mobility is catalyzed by a ribonucleoprotein complex consisting of both the spliced intron RNA and the intron-encoded protein with both RT and endonuclease activities. The intron RNA cleaves the DNA sense strand in an RNA-catalyzed reverse splicing reaction, while the intron-encoded protein cleaves the DNA antisense strand shortly downstream of the exon junction and uses it as a primer to reverse transcribe the RNA, leading to intron mobility. Alternative mechanisms for cDNA initiation during DGR homing include priming by an RNA moiety, a non-target DNA, or even a protein capable of donating a free hydroxyl group. The close evolutionary relationship between RTs from DGRs and group II introns, the site-specificity of mutagenic homing and other observations led to the hypothesis that DGRs also function through a TPRT-type mechanism (7).
To determine the site at which TR-derived cDNA integrates at the 3′ end of VR, a marker coconversion assay was developed (7). Single nucleotide substitutions were introduced into sequence-tagged TRs, and DGR-mediated transfer to VR was determined by sequencing homing products amplified by PCR. A clear boundary for marker transfer was detected and localized to a 5 bp sequence within the GC element at the 3′ end of VR (Figure 4). This could represent the site at which cDNA synthesis initiates in a TPRT reaction, or possibly the site of integration of cDNA that was primed by a non-TPRT mechanism. Marker coconversion at the 5′ end of VR was more heterogeneous, suggesting that 5′ cDNA integration can occur at different VR locations, possibly mediated by homology between VR and cDNA sequences.
The TPRT model predicts that homing occurs through sequential steps, with 3′ cDNA integration taking place first (7). Indeed, when the first 99 bp of the BPP-1 VR were deleted (ΔVR1-99) to prevent 5′ cDNA integration, cDNA products linked to VR sequences at their 3′ end with free, unlinked 5′ ends were detected (Figure 4B). These products were IMH- and Brt-dependent and contained adenine-specific mutations, suggesting that adenine mutagenesis occurs during reverse transcription and is an intrinsic property of the DGR RT. In addition, these observations suggested that cDNA integration at the 3′ end of VR can occur independently of 5′ integration, consistent with the hypothesis that DGR homing occurs through a TPRT reaction. It is important to note, however, that neither single-strand nicks nor double-strand breaks have been detected within the GC-rich element where marker coconversion begins, and no endonuclease gene, domain, or activity has been identified in a DGR cassette, raising the possibility that DGR homing could occur through an alternative mechanism.
Is 5′ cDNA integration homology-dependent? To test this, Guo et al. inserted mtd sequences into TR which were homologous to a 50 bp region immediately upstream of the deletion junction in the ΔVR1-99 recipient (Figure 4C) (7). This restored 5′ cDNA integration and DGR homing as confirmed by PCR-based assays and sequence analysis. cDNA integration into cryptic sites was observed at low frequency, and the sites of integration mapped to short stretches of identity (4–12 bp) between TR-mtd and sequences upstream of the ΔVR1-99 deletion. These results showed that 5′ integration can be mediated by short stretches of homology between TR-derived cDNA and target DNA. One model that could account for this is template switching during reverse transcription. It has previously been observed that RTs encoded by the group II intron Ll.LtrB and other non-LTR retroelements, including the Mauriceville retroplasmid and the silkworm Bombyx mori R2 element, are capable of template switching during reverse transcription (33–40). Interestingly, these reactions appear to occur primarily from the 5′ end of the first template to the 3′ end of the second template (DNA or RNA), and involve little or no base-pairing interactions between the latter and the 3′ end of the cDNA. Whatever the mechanism of 5′ cDNA integration might be, it appears to be RecA-independent as DGR homing and phage tropism switching occur at similar efficiencies in wild-type and RecA-deficient Bordetella (7).
DGR target recognition
In addition to the GC-rich sequence and IMH, deletion analysis revealed a third element located downstream of the BPP-1 VR that is required for efficient homing (Figure 5) (41). This region contains two 8 bp GC-rich inverted repeats capable of forming a DNA hairpin or cruciform structure with a 4 nt loop. Mutations that disrupt stem formation greatly decreased DGR homing and phage tropism switching, while restoration of base pairing with heterologous complementary sequences restored DGR activity. This suggested that the DNA stem-loop structure, rather than its primary sequence, is important for target recognition. Further characterization of the BPP-1 structure showed that the length and GC content of the stem modulate the efficiency of target recognition. The sequence of the 4 nt loop, however, appeared to be critical as changes in either sequence or size dramatically reduced homing. Structure-specific nuclease digestion assays confirmed DNA structure formation in vitro in dsDNA, which required negative supercoiling. The position of the hairpin/cruciform, which is normally located 4 bp downstream of VR, is also important. Moving it 4 bp in the 5′ direction or 15 bp in the 3′ direction significantly reduced DGR homing, although shorter insertions were tolerated. Marker transfer studies showed that extending the length between VR and the structured element did not affect the 3′ marker coconversion boundary, demonstrating that the cruciform influences the efficiency of homing, but not the site of cDNA integration. Although the exact function of the stem-loop structure is unknown, it is predicted to represent a binding site for host and/or DGR-encoded factors involved in cDNA priming and integration. Comparisons with other phage DGRs, as well as DGRs in bacterial chromosomes, suggested that similar structures are a conserved feature of target sequences (Figure 5C). This was proven correct by Arambula et al. who demonstrated the required roles of analogous stem-loop sequences for homing by the L. pneumophila DGR (Figure 2&5C) (17).
Figure 5.
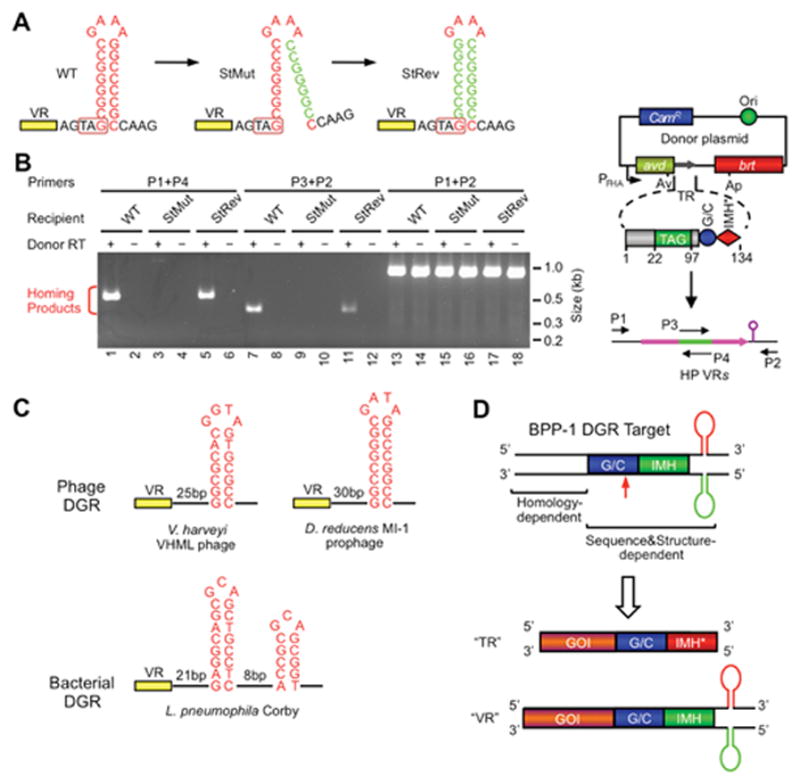
Role of a DNA secondary structure in DGR target recognition. (A) A DNA hairpin/cruciform structure downstream of VR is required for BPP-1 DGR target recognition. The wild type (WT) structure contains an 8 bp GC-rich stem and a 4 nt GAAA loop and is located 4 bp downstream of VR. Mutating the 3′ half of the stem (StMut) dramatically reduced DGR mutagenic homing (B) and phage tropism switching (not shown), while complementary changes to the 5′ half of the stem (StRev) restored DGR activity in both assays. (B) PCR-based DGR homing assays with sequence-tagged TRs and VRs flanked by WT or mutant stem sequences. Shown on the right is a diagram of the PCR assay. Green represents the tag sequence transferred from TR to VR. P1-4 are primers annealing to the tag or flanking regions. (C) Similar DNA structures are found at analogous positions in a number of other phage (two shown) and bacterial (one shown) DGRs. The phage stems are GC-rich and range from 7 to 10 bp, and loops have a conserved 4 nt sequence, G(A/G)NA. The L. pneumophila Corby DGR has a more complex tandem structure that is required for homing. (D) BPP-1 DGR target recognition at the 3′ end is both sequence and structure-dependent, requiring GC, IMH and a hairpin/cruciform structure. Target recognition at the 5′ end is homology-mediated. By inserting a gene of interest (GOI) upstream of GC, IMH and the DNA structure, the heterologous gene can be diversified by the BPP-1 DGR through appropriate engineering of TR. (Adapted from reference 41)
As shown in Figure 4, mutagenic homing is a “copy-and-replace” process which replaces parental VRs with TR-derived cDNAs that are mutagenized at specific sites (7). Target recognition at the 3′ end is both sequence- and structure-dependent, while target recognition at the 5′ end is dependent on short stretches of homology (Figure 5D) (7, 41). A feature of the mechanism that seems key to its beneficial nature is that all cis acting sequences and trans acting factors required for additional rounds of diversification are precisely reconstituted during mutagenic homing. This allows repeated rounds of diversification and, presumably, the optimization of beneficial traits.
Guided by the TPRT model and the VR recognition rules described above (Figure 5D), the BPP-1 DGR was engineered to target a kanamycin resistance gene (aph3′ Ia) (41). A defective allele with a 3′ truncation was placed upstream of GC, IMH and the cruciform structure to form a recipient VR. A donor TR was then engineered to contain the missing aph3′ Ia sequences along with a short stretch of upstream homology. As predicted, the donor TR was able to repair the defective gene, conferring kanamycin resistance in an RT-dependent reaction. Thus, DGRs can be designed to target genes of interest, with potentially broad applications for protein engineering.
Structure and function of the Avd accessory protein
The BPP-1 DGR includes the avd locus, which encodes a positively charged 15 kDa protein that is essential for homing and is conserved in numerous bacterial and phage DGRs (6, 9, 10, 27). An X-ray crystal structure of BPP-1 Avd was determined by Alayyoubi et al. (Figure 6A) to provide a guide for functional analysis (27). Avd forms a barrel-shaped homopentamer which is positively charged on all surfaces, including an hourglass-shaped pore that runs through the center of the barrel and constricts to an ~8 Å diameter. Each Avd monomer forms a four-helix bundle with the α helices running up and down in anti-parallel fashion.
Figure 6.
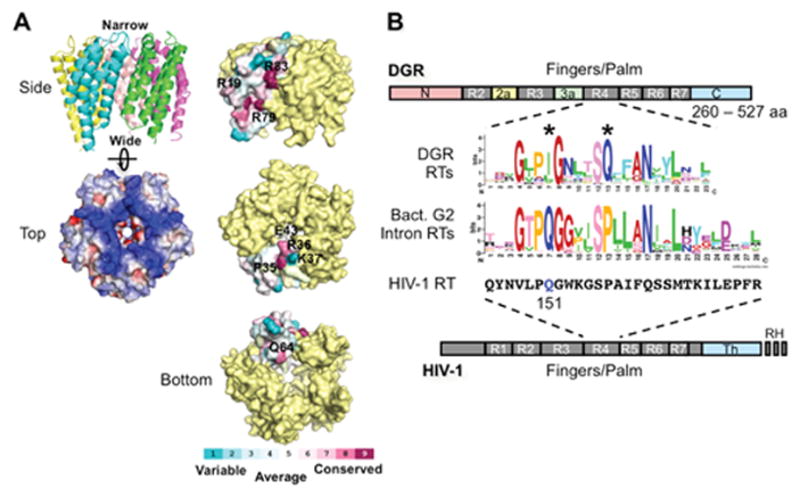
Avd and Brt. (A) (Left) The BPP-1 Avd protein forms a homopentameric structure, with each monomer containing 4 helices running up and down (side view). The pentamer is highly positively charged (top view; blue, positively charged; red, negatively charged). (Right) Amino acid residues on the side, top, and bottom of the Avd pentamer that were tested for Avd-Brt binding and/or DGR homing (27). (B) DGR RT domains and the sequence logo of its highly conserved domain R4. R1-R7 are conserved sequence blocks found in the finger and palm domains of retroviral RTs, such as HIV-1 RT (bottom). DGR RTs contain sequence insertions between R2 and R3 (R2a), and between R3 and R4 (R3a), as well as divergent N- and C-termini. They do not contain the thumb (Th) and RNase H (RH) domains that are found in HIV-1 RT. The domain R4 sequence logo of 155 DGR RTs was generated by Schillinger et al. using WebLogo (13, 49, 50). Comparison with the domain R4 sequence logo that we generated from 93 bacterial group II intron RTs [group II intron database: http://webapps2.ucalgary.ca/~groupii/orf/orfalignment.html; (51–53)], which are most closely related to DGR RTs, showed several characteristic differences, including the two highly conserved positions labeled with *. Also included for comparison is the corresponding amino acid sequence block of HIV-1 RT (Strain BRU; accession # K02013). The glutamine residue at position 151, which plays a role in nucleotide and template preference during reverse transcription, is highlighted in blue. (Adapted from reference 13)
A number of amino acid residues located on the surface or near the pore of the BPP-1 Avd pentamer were subjected to mutagenesis (27). Mutations at two conserved residues on the side of the pentamer, R79A and R83A, eliminated detectable homing, and mutations of two residues on the top of the pentamer, R36A and K37A, resulted in a partial loss of DGR activity. Defects in homing were not due to a loss of protein structure or stability, as identical CD spectra were obtained with wild type and homing-defective Avd mutant proteins. Other mutations, including R19A (side), P35A (top), E43A (near the pore), and Q64A (bottom), had little effect on activity. These results suggest that positively charged residues on the side and top of the Avd pentamer have important functional roles.
As predicted by its positive charge, BPP-1 Avd was found to associate in vitro with ssRNA, ssDNA, DNA:RNA complexes and dsDNA, although the interactions were nonspecific (27). In contrast, Avd pentamers associated with purified Brt, which is also predicted to be positively charged, in a manner that was abolished by the R79A and R83A mutations (i.e., the ones that eliminated DGR homing), but not by other point mutations. These results suggested a correlation between Avd-Brt binding and DGR function. From these and other results, Alayyoubi et al. proposed that the pentameric nature of Avd may be involved in organizing a multivalent assembly consisting of Brt and nucleic acid components to somehow coordinate the multiple events required for homing (27).
DGR RTs and adenine mutagenesis
Adenine mutagenesis is a unique hallmark of DGRs, but its mechanism remains enigmatic. In the td intron-tagging experiment described above, no adenine-specific changes were observed in spliced RNA, suggesting that adenine mutagenesis does not involve RNA editing (7). However, adenine mutagenesis was observed in cDNA products when VR1-99 was deleted to “trap” cDNA intermediates between the 3′ and 5′ integration steps, suggesting that adenine mutagenesis occurs during cDNA synthesis. These observations, coupled with the fact that DGR RTs are both highly conserved and unique, support the hypothesis that adenine mutagenesis is a property inherent to DGR RTs.
DGR RTs range from 260 to 527 amino acids, with an average length of ~380 residues (Figure 6B) (13). They have divergent sequences at their N- and C-termini, with a conserved central core that includes common structural motifs found in most other RTs. They do not contain domains encoding RNase H activity, as found in retroviral RTs, or a DNA endonuclease activity as found in RTs of group II introns and LINEs. Within their shared sequences, DGR RTs have some intriguing features. The most prominent is located within the Finger 4 region, which differs from those of group II intron RTs and HIV-1 RT at several highly conserved positions (13). The Finger 4 region of HIV-1 RT forms part of the nucleotide-binding pocket that positions incoming dNTPs and influences specificity and error-prone polymerization (13, 42). Mutations at Q151 (highlighted in blue in Figure 6B) in HIV-1 RT, which corresponds to conserved isoleucine/leucine/valine residues in DGR RTs, change nucleotide and template preferences and confer resistance to inhibitory nucleoside analogs like AZT (43–45). It seems likely that unique features of this motif found in DGR RTs play a role in adenine mutagenesis.
At least two potential mechanisms could account for adenine mutagenesis. In the first, DGR RTs are hypothesized to carry out a variation of error-prone reverse transcription that inserts random deoxyribonucleotides opposite adenines, but rarely other residues, while synthesizing cDNA from TR-containing RNA templates. An alternative mechanism could be imagined in which DGR RTs have an increased propensity to incorporate dUTP, as opposed to dTTP when copying adenines. dUTP residues in cDNA products would then be recognized by host-encoded uracil DNA glycosylases (UDGs) and excised, leaving abasic sites. If these cDNA products are subsequently used as templates for second-strand cDNA sysnthesis, which could be catalyzed by a DGR RT or a host-encoded DNA polymerase, random nucleotides would be incorporated opposite to these abasic sites.
Understanding the mechanism of adenine mutagenesis may have broad implications. During mutagenic homing by the BPP-1 DGR, about 30% of TR adenines are converted to other nucleotides in progeny VRs (7). This error rate is far greater (~10,000 times higher) than that of HIV-1 RT, which is ~1.4–3.0 × 10−5 in vivo (46, 47). The low fidelity of HIV-1 RT enables the virus to generate vast numbers of sequence variants, enabling escape from immune surveillance and drug resistance. If DGR RTs are truly responsible for adenine mutagenesis, they are likely to be the most error-prone DNA-polymerizing enzymes yet discovered.
Summary
DGRs are beneficial retroelements present in diverse bacteria and phage (6, 10–13). Their apparent function is to accelerate the evolution of ligand-binding interactions. Structural, bioinformatic, and biochemical studies indicate that CLec folds are a conserved solution to display variable residues of DGR-diversified proteins (14, 20, 22). Unexpectedly, this structural fold is highly static in DGR diversified proteins studied to date. Despite major differences in amino acid side chains, the backbones of BPP-1 Mtd variants are nearly identical to each other and their CLec folds superimpose with the diversified domain in T. denticola TvpA. This limited structural variability suggests that most DGR variable proteins will have relatively weak interactions with their respective ligands. For the BPP-1 phage, successful host recognition relies on multivalent interactions (i.e. avidity) between Mtd trimers on phage particles and arrays of receptors on the bacterial surface, which results in immense amplification of an otherwise weak monovalent ligand-receptor interaction. Avidity-driven binding is likely a conserved feature that contributes to the evolvability of DGR variable proteins (20).
A significant amount has been learned regarding the mechanism of DGR-mediated mutagenic homing (5–7, 17, 27, 41). Sequence diversification is mediated by a unique class of RTs associated with the conversion of adenine residues in an RNA intermediate into random nucleotides in a cDNA that ultimately replaces the variable region of a target gene with a diversified derivative. This process is independent of the host RecA-mediated homologous recombination machinery, similar to the mobility mechanism of group II introns (7). Current evidence is consistent with the hypothesis that mutagenic homing initiates through a TPRT mechanism, although other possibilities exist. BPP-1 DGR target recognition is both sequence- and structure-dependent at the 3′ end, requiring GC and IMH sequence elements and a DNA cruciform structure, while target recognition at the 5′ end is mediated by short stretches of homology (41). Based on these and other observations, the BPP-1 DGR was successfully engineered to target a heterologous gene.
Despite these advances, many questions remain unanswered. Hundreds of DGRs have been identified in diverse bacteria and phage, but their biological functions remain largely unknown (6, 10, 13, 17, 48). Understanding the functions of diverse DGRs is a major challenge. On the mechanistic side, the TPRT model is valuable for guiding experiments, but it is only one of several models that could explain DGR homing and the evidence supporting it is incomplete. In addition, it is unclear what specific role Avd plays in the homing reaction. The mechanism of adenine mutagenesis is of great interest as it represents a hallmark of DGR function. It has been hypothesized to be an intrinsic property of DGR-encoded RTs and if true, understanding how it occurs may help us understand RT fidelity in general. Mutagenic homing is clearly a complex process that requires the participation of host factors, although none have been identified to date. And finally, the utility of DGRs for protein engineering has yet to be exploited. In addition to providing prodigious levels of diversity, mutagenic homing is a regenerative process that can operate through unlimited rounds to optimize protein functions. This may be particularly advantageous for directed protein evolution since desired traits can be selected and continuously evolved in iterative cycles, without the need for library construction or other interventions, and through a process that takes place entirely within bacterial cells.
Acknowledgments
This work was supported by NIH grant (R01 AI069838) to J.F.M. and P.G. and University of Missouri startup funds to H.G.. J.F.M. is a co-founder of AvidBiotics Corp. and a member of its Board of Directors and Scientific Advisory Board.
References
- 1.Vink C, Rudenko G, Seifert HS. Microbial antigenic variation mediated by homologous DNA recombination. FEMS Microbiol Rev. 2012;36:917–948. doi: 10.1111/j.1574-6976.2011.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agrawal A, Eastman QM, Schatz DG. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 1998;394:744–751. doi: 10.1038/29457. [DOI] [PubMed] [Google Scholar]
- 3.Kapitonov VV, Jurka J. RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol. 2005;3:e181. doi: 10.1371/journal.pbio.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hencken CG, Li X, Craig NL. Functional characterization of an active Rag-like transposase. Nat Struct Mol Biol. 2012;19:834–836. doi: 10.1038/nsmb.2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu M, Deora R, Doulatov SR, Gingery M, Eiserling FA, Preston A, Maskell DJ, Simons RW, Cotter PA, Parkhill J, Miller JF. Reverse transcriptase-mediated tropism switching in Bordetella bacteriophage. Science. 2002;295:2091–2094. doi: 10.1126/science.1067467. [DOI] [PubMed] [Google Scholar]
- 6.Doulatov S, Hodes A, Dai L, Mandhana N, Liu M, Deora R, Simons RW, Zimmerly S, Miller JF. Tropism switching in Bordetella bacteriophage defines a family of diversity-generating retroelements. Nature. 2004;431:476–481. doi: 10.1038/nature02833. [DOI] [PubMed] [Google Scholar]
- 7.Guo H, Tse LV, Barbalat R, Sivaamnuaiphorn S, Xu M, Doulatov S, Miller JF. Diversity-generating retroelement homing regenerates target sequences for repeated rounds of codon rewriting and protein diversification. Mol Cell. 2008;31:813–823. doi: 10.1016/j.molcel.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melvin JA, Scheller EV, Miller JF, Cotter PA. Bordetella pertussis pathogenesis: current and future challenges. Nat Rev Microbiol. 2014;12:274–288. doi: 10.1038/nrmicro3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu M, Gingery M, Doulatov SR, Liu Y, Hodes A, Baker S, Davis P, Simmonds M, Churcher C, Mungall K, Quail MA, Preston A, Harvill ET, Maskell DJ, Eiserling FA, Parkhill J, Miller JF. Genomic and genetic analysis of Bordetella bacteriophages encoding reverse transcriptase-mediated tropism-switching cassettes. J Bacteriol. 2004;186:1503–1517. doi: 10.1128/JB.186.5.1503-1517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medhekar B, Miller JF. Diversity-generating retroelements. Curr Opin Microbiol. 2007;10:388–395. doi: 10.1016/j.mib.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon DM, Zimmerly S. A diversity of uncharacterized reverse transcriptases in bacteria. Nucleic Acids Res. 2008;36:7219–7229. doi: 10.1093/nar/gkn867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minot S, Grunberg S, Wu GD, Lewis JD, Bushman FD. Hypervariable loci in the human gut virome. Proc Natl Acad Sci U S A. 2012;109:3962–3966. doi: 10.1073/pnas.1119061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schillinger T, Lisfi M, Chi J, Cullum J, Zingler N. Analysis of a comprehensive dataset of diversity generating retroelements generated by the program DiGReF. BMC Genomics. 2012;13:430. doi: 10.1186/1471-2164-13-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMahon SA, Miller JL, Lawton JA, Kerkow DE, Hodes A, Marti-Renom MA, Doulatov S, Narayanan E, Sali A, Miller JF, Ghosh P. The C-type lectin fold as an evolutionary solution for massive sequence variation. Nat Struct Mol Biol. 2005;12:886–892. doi: 10.1038/nsmb992. [DOI] [PubMed] [Google Scholar]
- 15.Murphy K. Janeway’s Immunobiology. 8. Garland Science, Taylor & Francis Group, LLC; London and New York: 2012. The Generation of Lymphocyte Antigen Receptors; p. 171. [Google Scholar]
- 16.Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. 7. Elsevier Saunders; Philadelphia, PA: 2012. Lymphocyte Development and Antigen Receptor Gene Rearrangement; p. 186. [Google Scholar]
- 17.Arambula D, Wong W, Medhekar BA, Guo H, Gingery M, Czornyj E, Liu M, Dey S, Ghosh P, Miller JF. Surface display of a massively variable lipoprotein by a Legionella diversity-generating retroelement. Proc Natl Acad Sci U S A. 2013;110:8212–8217. doi: 10.1073/pnas.1301366110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stanley NR, Palmer T, Berks BC. The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J Biol Chem. 2000;275:11591–11596. doi: 10.1074/jbc.275.16.11591. [DOI] [PubMed] [Google Scholar]
- 19.Narita S, Tokuda H. Sorting of bacterial lipoproteins to the outer membrane by the Lol system. Methods Mol Biol. 2010;619:117–129. doi: 10.1007/978-1-60327-412-8_7. [DOI] [PubMed] [Google Scholar]
- 20.Miller JL, Le Coq J, Hodes A, Barbalat R, Miller JF, Ghosh P. Selective ligand recognition by a diversity-generating retroelement variable protein. PLoS Biol. 2008;6:e131. doi: 10.1371/journal.pbio.0060131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai W, Hodes A, Hui WH, Gingery M, Miller JF, Zhou ZH. Three-dimensional structure of tropism-switching Bordetella bacteriophage. Proc Natl Acad Sci U S A. 2010;107:4347–4352. doi: 10.1073/pnas.0915008107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Coq J, Ghosh P. Conservation of the C-type lectin fold for massive sequence variation in a Treponema diversity-generating retroelement. Proc Natl Acad Sci U S A. 2011;108:14649–14653. doi: 10.1073/pnas.1105613108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boeke JD, Garfinkel DJ, Styles CA, Fink GR. Ty elements transpose through an RNA intermediate. Cell. 1985;40:491–500. doi: 10.1016/0092-8674(85)90197-7. [DOI] [PubMed] [Google Scholar]
- 24.Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH., Jr High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 25.Cousineau B, Smith D, Lawrence-Cavanagh S, Mueller JE, Yang J, Mills D, Manias D, Dunny G, Lambowitz AM, Belfort M. Retrohoming of a bacterial group II intron: mobility via complete reverse splicing, independent of homologous DNA recombination. Cell. 1998;94:451–462. doi: 10.1016/s0092-8674(00)81586-x. [DOI] [PubMed] [Google Scholar]
- 26.Guo H, Karberg M, Long M, Jones JP, 3rd, Sullenger B, Lambowitz AM. Group II introns designed to insert into therapeutically relevant DNA target sites in human cells. Science. 2000;289:452–457. doi: 10.1126/science.289.5478.452. [DOI] [PubMed] [Google Scholar]
- 27.Alayyoubi M, Guo H, Dey S, Golnazarian T, Brooks GA, Rong A, Miller JF, Ghosh P. Structure of the essential diversity-generating retroelement protein bAvd and its functionally important interaction with reverse transcriptase. Structure. 2013;21:266–276. doi: 10.1016/j.str.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- 29.Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002;21:5899–5910. doi: 10.1093/emboj/cdf592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimmerly S, Guo H, Perlman PS, Lambowitz AM. Group II intron mobility occurs by target DNA-primed reverse transcription. Cell. 1995;82:545–554. doi: 10.1016/0092-8674(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 31.Zimmerly S, Guo H, Eskes R, Yang J, Perlman PS, Lambowitz AM. A group II intron RNA is a catalytic component of a DNA endonuclease involved in intron mobility. Cell. 1995;83:529–538. doi: 10.1016/0092-8674(95)90092-6. [DOI] [PubMed] [Google Scholar]
- 32.Lambowitz AM, Zimmerly S. Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol. 2011;3:a003616. doi: 10.1101/cshperspect.a003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennell JC, Wang H, Lambowitz AM. The Mauriceville plasmid of Neurospora spp. uses novel mechanisms for initiating reverse transcription in vivo. Mol Cell Biol. 1994;14:3094–3107. doi: 10.1128/mcb.14.5.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen B, Lambowitz AM. De novo and DNA primer-mediated initiation of cDNA synthesis by the mauriceville retroplasmid reverse transcriptase involve recognition of a 3′ CCA sequence. J Mol Biol. 1997;271:311–332. doi: 10.1006/jmbi.1997.1185. [DOI] [PubMed] [Google Scholar]
- 35.George JA, Burke WD, Eickbush TH. Analysis of the 5′ junctions of R2 insertions with the 28S gene: implications for non-LTR retrotransposition. Genetics. 1996;142:853–863. doi: 10.1093/genetics/142.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bibillo A, Eickbush TH. The reverse transcriptase of the R2 non-LTR retrotransposon: continuous synthesis of cDNA on non-continuous RNA templates. J Mol Biol. 2002;316:459–473. doi: 10.1006/jmbi.2001.5369. [DOI] [PubMed] [Google Scholar]
- 37.Bibillo A, Eickbush TH. End-to-end template jumping by the reverse transcriptase encoded by the R2 retrotransposon. J Biol Chem. 2004;279:14945–14953. doi: 10.1074/jbc.M310450200. [DOI] [PubMed] [Google Scholar]
- 38.Stage DE, Eickbush TH. Origin of nascent lineages and the mechanisms used to prime second-strand DNA synthesis in the R1 and R2 retrotransposons of Drosophila. Genome Biol. 2009;10:R49. doi: 10.1186/gb-2009-10-5-r49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhuang F, Mastroianni M, White TB, Lambowitz AM. Linear group II intron RNAs can retrohome in eukaryotes and may use nonhomologous end-joining for cDNA ligation. Proc Natl Acad Sci U S A. 2009;106:18189–18194. doi: 10.1073/pnas.0910277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White TB, Lambowitz AM. The retrohoming of linear group II intron RNAs in Drosophila melanogaster occurs by both DNA ligase 4-dependent and -independent mechanisms. PLoS Genet. 2012;8:e1002534. doi: 10.1371/journal.pgen.1002534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo H, Tse LV, Nieh AW, Czornyj E, Williams S, Oukil S, Liu VB, Miller JF. Target site recognition by a diversity-generating retroelement. PLoS Genet. 2011;7:e1002414. doi: 10.1371/journal.pgen.1002414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 43.Kaushik N, Talele TT, Pandey PK, Harris D, Yadav PN, Pandey VN. Role of glutamine 151 of human immunodeficiency virus type-1 reverse transcriptase in substrate selection as assessed by site-directed mutagenesis. Biochemistry. 2000;39:2912–2920. doi: 10.1021/bi991376w. [DOI] [PubMed] [Google Scholar]
- 44.Sarafianos SG, Hughes SH, Arnold E. Designing anti-AIDS drugs targeting the major mechanism of HIV-1 RT resistance to nucleoside analog drugs. Int J Biochem Cell Biol. 2004;36:1706–1715. doi: 10.1016/j.biocel.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 45.Boyer PL, Sarafianos SG, Clark PK, Arnold E, Hughes SH. Why do HIV-1 and HIV-2 use different pathways to develop AZT resistance? PLoS Pathog. 2006;2:e10. doi: 10.1371/journal.ppat.0020010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J Virol. 2010;84:9864–9878. doi: 10.1128/JVI.00915-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schillinger T, Zingler N. The low incidence of diversity-generating retroelements in sequenced genomes. Mob Genet Elements. 2012;2:287–291. doi: 10.4161/mge.23244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dai L, Toor N, Olson R, Keeping A, Zimmerly S. Database for mobile group II introns. Nucleic Acids Res. 2003;31:424–426. doi: 10.1093/nar/gkg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simon DM, Clarke NA, McNeil BA, Johnson I, Pantuso D, Dai L, Chai D, Zimmerly S. Group II introns in eubacteria and archaea: ORF-less introns and new varieties. RNA. 2008;14:1704–1713. doi: 10.1261/rna.1056108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Candales MA, Duong A, Hood KS, Li T, Neufeld RA, Sun R, McNeil BA, Wu L, Jarding AM, Zimmerly S. Database for bacterial group II introns. Nucleic Acids Res. 2012;40:D187–190. doi: 10.1093/nar/gkr1043. [DOI] [PMC free article] [PubMed] [Google Scholar]