

Summary
Group A streptococci (GAS) express soluble and surface-bound virulence factors. Secreted streptokinase (SK) allelic variants exhibit varying abilities to activate host plasminogen (Pg), and GAS pathogenicity is associated with Pg activation and localisation of the resulting plasmin (Pm) on the bacterial surface to promote dissemination. The various mechanisms by which GAS usurp the host proteolytic system are discussed, including the molecular sexuality mechanism of conformational activation of the Pg zymogen (Pg*), and subsequent proteolytic activation of substrate Pg by the S•KPg* and SK•Pm catalytic complexes. Substantial progress has been made to delineate both processes in a unified mechanism. Pm coats the bacteria by direct and indirect binding pathways involving plasminogen-binding group A streptococcal M-like protein (PAM), and host fibrin(ogen). Transgenic mouse models using human Pg are being optimised to mimic infections by SK variants in humans, and to define in vivo combined mechanisms of these variants and PAM.
Keywords: fibrinolysis, plasminogen, streptococcus, streptokinase, enzymology
Streptokinase from Group A Streptococcus Species
Group A, beta-hemolytic Streptococcus pathogens (GAS) are responsible for common throat and skin infections, but they are also the major cause of pneumonia, meningitis, sepsis, endocarditis, toxic shock syndrome, and necrotising fasciitis. These invasive infectious pathologies can trigger severe complications such as stroke and renal failure, and affect 18 million people globally, resulting in 517,000 deaths per year [1]. The human host-specific S. pyogenes enhances its pathogenicity by a multitude of cell wall-linked and secreted virulence factors [2–5], one of which is streptokinase (SK). SK usurps the host’s fibrinolytic system and uses unique molecular mechanisms to bind and ultimately activate the host zymogen, plasminogen (Pg) into plasmin (Pm). Phylogenetic studies of SK from GAS isolates classify sequence variants into clusters SK1, SK2a and SK2b [6]. The strains expressing these allelic variants exhibit varying pathogenicity, and different interpretations of their individual molecular mechanisms of Pg activation, fibrin(ogen) binding, and susceptibility to α2-antiplasmin (α2-AP) have been proposed [3, 4, 7]. Our mechanism studies reported here, have mainly focused on SK from the S. equisimilis strain H46A used to treat myocardial infarction [8]. SKH46A is 86% identical in sequence and ~90% homologous to SK1 variants. The SK allelic sequence variants most likely evolved under selective pressure for survival in the hostile host environment to encode critical differences in their mechanisms of Pg activation pathways to generate Pm. Long-term goals will include defining critical mechanism differences for these allelic variants.
Roles of Plasminogen Activation
Plasminogen is the zymogen precursor of plasmin, the central proteinase in the fibrinolytic system. Intravascular fibrinolysis, or fibrin clot dissolution by plasmin, occurs after wound repair, or can be pharmacologically induced by intravenous infusion of plasminogen activators after thrombosis and blood vessel occlusion. [Glu]Pg circulates as a compact α-form, with its spiral array of 5 kringle (K) domains stabilised by lysine-binding site (LBS) interactions of K2, K4 and K5 with basic residues and chloride ions [9]. The N-terminal PAN (Pg/Apple/Nematode) module binds to LBS of K4 and K5, and this interaction keeps [Glu]Pg in the spiral conformation. K1, the only kringle with an accessible LBS, extends from the structure and interacts with fibrin(ogen). Solving the crystal structure of compact [Glu]Pg provided a significant advance in understanding the conformational properties of circulating Pg [9, 10]. Small molecules and lysine analogues such as benzamidine and 6-aminohexanoic acid (6-AHA) can bind the kringles, and cause a conformational change to the partially extended β-conformation (benzamidine) or the fully extended γ-conformation (6-AHA) [11, 12]. The Pg activation cleavage site Arg561-Val562 in compact [Glu]Pg is shielded by the 350s loop, a K3-K4 linking segment. The Lys77-Lys78 bond, not readily accessible in compact [Glu]Pg, can be cleaved by plasmin, generating a more extended N-truncated Pg form, [Lys]Pg, which adopts a β-conformation and is more reactive with physiological and bacterial plasminogen activators [11]. In the absence of chloride ions [Glu]Pg adopts the γ-conformation.
In addition to degrading fibrin clots, plasmin also activates metalloproteinases and plays a role in tissue remodelling, angiogenesis, and processes of pathogen and tumor cell dissemination. Bacterial pathogens and cancer cells are capable of recruiting host plasmin to their cell surfaces, which facilitates spreading through tissues [13, 14]. Whereas tumor invasion and physiological fibrinolysis use proteolytic Pg activation, pathogens have developed unique mechanisms of host Pg binding to secreted and surface-bound virulence factors, resulting in conformational Pg activation without the need for proteolytic cleavage of the zymogen.
Proteolytic and Cofactor-Induced Activation Mechanisms
Physiological tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA) use the classical cleavage mechanism to activate Pg, by proteolysis of the Arg561-Val562 bond (chymotrypsinogen numbering Arg15-Val16, or the P1–P1′ site) in the Pg activation loop that is sterically restrained by a disulphide bridge. This sterical restraint only allows the residues between P3 and P2′ of the activation loop to make contact with the specificity pocket of tPA and uPA [15]. The S2′ specificity pocket of these proteases is limited in size by the presence of a bulky tyrosine residue, which poses a restriction on the residues that can occupy this subsite. Upon cleavage of the Pg activation loop, the new N-terminal Val16 residue inserts into the Val16 binding pocket and engages the side chain carboxylate of Asp194 (Pg numbering 740) to form the substrate recognition site and the oxyanion hole. This is the established mechanism for chymotrypsin(ogen)-like serine proteinase zymogens [16]. A model for the classic cleavage mechanism of such a zymogen is shown in Figures 1A and B.
FIGURE 1.
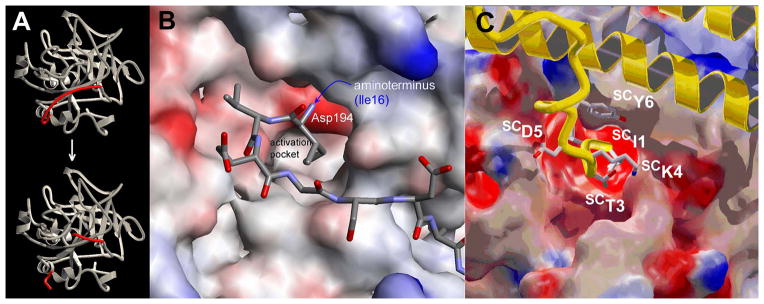
A,B. Proteolytic zymogen activation mechanism. Proteolytic cleavage of the Arg15-Ile16 of Pre2 generates a new N-terminal Ile16, which insets into the N-terminal binding pocket in the zymogen, forming a salt bridge with Asp194. This induces a conformational change involving the folding of 3 segments of the catalytic domain. The conformational change forms the substrate binding site and oxyanion hole required for catalysis. C. Binding of the N-terminal SCIle1-SCTyr6 sequence to Pre2. The SC backbone is in yellow, side chains have coloured atoms, and the bound Pre2 is coloured according to its electrostatic surface potential. This figure was first published in Nature. Friedrich R, Panizzi P, Fuentes-Prior P, Richter K, Verhamme I, Anderson PJ, Kawabata S, Huber R, Bode W, Bock PE. Staphylocoagulase is a prototype for the mechanism of cofactor-induced zymogen activation. Nature. 2003; 425: 535–9.
By contrast, the secreted streptococcal virulence factor streptokinase (SK) forms a stoichiometric complex with host Pg, and activates it by a mechanism that does not involve proteolysis. The N-terminal sequence Ile1-Ala-Gly of SK mimics the Ile1-Val-Gly/Asn sequences of trypsin and chymotrypsin, and the plasmin Val1-Val-Gly sequence, which insert into the activation pocket of the nascent proteases during proteolytic activation. Insertion of the SK N-terminal sequence is proposed to trigger zymogen activation, according to the ‘molecular sexuality’ hypothesis of zymogen activation by intrusion of N-terminal sequences of bacterial staphylocoagulase (SC) and SK into the activation pockets of prothrombin and Pg, respectively [17]. Upon insertion of the SK N-terminus in the Ile16 N-terminal binding cleft (chymotrypsinogen numbering) of Pg, Ile1 of SK forms a salt bridge with Asp194, resulting in a conformational change, and formation of the substrate-binding site and the oxyanion hole, required for catalytic activity. Loss-of-function studies with SK N-terminal deletion mutants, by us and others [18, 19] have supported this hypothesis, favouring it over an alternative mechanism in which SK binding was proposed to move the ε-amino group of the conserved Lys156 (Lys698 in Pg) into the binding pocket, and trigger conformational activation [20]. The molecular sexuality mechanism of SK-induced conformational activation of Pg was postulated in 1976, when SK and SC were the only known conformational zymogen activators [17]. We showed clear structural evidence for such a molecular sexuality mechanism, by determining for the first time the high resolution crystal structure of the complex between a large N-terminal SC fragment, SC(1-325), and prethrombin 2 (Pre2), the prothrombin catalytic domain and immediate zymogen precursor of thrombin [21]. The N-terminus of SC(1-325) inserts into the Ile16 pocket and triggers folding of the zymogen activation domain (residues 142-152, 186-194 and 216-226) by forming a salt bridge with Asp194, which generates substrate recognition, subsites and forms the oxyanion hole. The Ile1-Val2 amino-terminal residues of SC(1-325) inserting into the Pre2 binding pocket, and the critical salt bridge with Asp194 are clearly resolved (Figure 1C). We also solved the structure of SC(1-325) bound to human thrombin, in which the Ile1-Val2 residues of the thrombin heavy chain occupy the specificity pocket. This study reported similar loss-of-function of SC N-terminal deletion and substitution mutants, further lending credence to the SK-triggered Pg conformational activation for which no crystal structure is yet available. A model of this activation is shown in Figure 2A. Based on this structure, we proposed a family of staphylococcal and streptococcal homologs named zymogen activator and adhesion proteins (ZAAPs) [22], that also includes the conformational prothrombin activator, von Willebrand factor-binding protein (VWbp), secreted by Staphylococcus aureus.
FIGURE 2.

A. Crystal structure of the SK•μPm complex, and model of Pg activation by SK. Top: The α (red), β (yellow) and γ (purple) domains of SK surround the catalytic domain of plasmin (green). The catalytic triad is shown in pink. The top and side views show the crater-like arrangement of the SK domains surrounding the catalytic site of μPm. The N-terminal 11 residues are disordered because the Pm N-terminus is inserted. This is the only structure available of a SK•Pm complex [29]. Bottom: A model was constructed of the SK complex with the Pg catalytic domain, based on the SK•μPm structure, showing the N-terminal insertion that activates Pg. Evidence for the molecular sexuality mechanism is that deletion of the N-terminal Ile residue of SK inactivates its ability to activate Pg conformationally. There is currently no structure of the SK•Pg* zymogen complex to support this. B. The trigger-and-bullet mechanism of Pg activation by SK. Rapid SK binding to the catalytic domain of Pg, and conformational Pg activation in the SK•Pg* catalytic complex form the first phase of the catalytic cycle (trigger). SK•Pg* binds a second Pg molecule in the substrate mode and proteolytically activates it to Pm. Pm displaces Pg from SK•Pg* because of its higher affinity compared to Pg. This causes transition of the catalytic complexes from SK•Pg* to SK•Pm as the sole catalyst, which initiates the bullet cycle. SK•Pm proteolytically converts the remaining free Pg to Pm. This figure was originally published in The Journal of Biological Chemistry. Boxrud PD, Bock PE. Coupling of conformational and proteolytic activation in the kinetic mechanism of plasminogen activation by streptokinase. J Biol Chem. 2004; 279: 36642–36649. © the American Society for Biochemistry and Molecular Biology. C. Specificity of SK(2-414) reactivation by Ala-substituted SK(1-10) peptides. The activities of SK(1-10) peptides with Ala substituted at the residues indicated were determined relative to the wild-type peptide (Ala at residue 2, 100% rescue). Peptide-initiated rescue of Pg activation by SK(2-414) is shown without (solid) and with (open) 6-AHA. This figure was originally published in The Journal of Biological Chemistry. Boxrud PD, Verhamme IM, Fay WP, Bock PE. Streptokinase triggers conformational activation of plasminogen through specific interactions of the amino-terminal sequence and stabilises the active zymogen conformation. J Biol Chem. 2001; 276: 26084–26089. © the American Society for Biochemistry and Molecular Biology. D. Rescue of Pg activation by Y4P substitution in the ALAB49 variant. ALAB49 shows weak conformational Pg activation (open circles) that is significantly enhanced by substitution of Tyr4 to Pro, the residue in wild-type SK (closed circles).
A Unified Mechanism Coupling Conformational and Proteolytic Activation
Our studies [23, 24] that couple conformational Pg activation to subsequent proteolytic Pm formation in a unified mechanism of SK action have clarified molecular events that were previously subject to different interpretations, such as Pm formation by intramolecular proteolytic activation within the complex with SK [25]. We demonstrated that SK binds rapidly to Pg and induces the activated Pg* conformation in the SK•Pg* catalytic complex by the molecular sexuality mechanism. This process is considered the trigger of the mechanism (Figure 2B). The SK•Pg* complex then binds another Pg molecule in the substrate mode, and converts this substrate Pg to Pm by the classical proteolytic mechanism. Pm binds SK with 500–900-fold higher affinity, thereby displacing Pg* from the complex. The resulting catalytic SK•Pm complex becomes the sole catalyst, which terminates the trigger cycle and initiates the bullet cycle (Figure 2B). The SK•Pm catalytic complex then proteolytically converts remaining free Pg to Pm. The very slow off-rate for the dissociation of Pm bound in the SK•Pm catalytic complex (t1/2 ~800 s) suggests that the SK•Pm bullet cycle may turn without significant dissociation of SK•Pm [26]. Activation of Pg can be analysed by kinetic analysis of continuous hydrolysis of a plasmin-specific chromogenic substrate, D-VLK-pNA, that is also cleaved by the SK•Pg* and SK•Pm complexes. Progress curves of Pg activation are fit by the parabolic equation: ΔA405 nm=v2t2/2 + v1t to obtain the initial rate of substrate hydrolysis (v1) due to the conformationally activated SK•Pg* complex, and the rate of the increase in activity with time due to Pm formation (v2) [23, 24, 27]. The hyperbolic SK dependence of v1 has a KD that reflects SK•Pg* formation and a limiting rate representing the bimolecular rate (kcat/Km) of D-VLK-pNA hydrolysis by SK•Pg*. The SK dependence of v2, the rate of Pm formation, increases to a maximum and decreases to zero as SK is increased. Pg acts both as the catalyst in the SK•Pg* complex and as the substrate of this complex, and Pm generation decreases as free Pg is depleted by higher SK. The bimolecular rate constant for Pm generation is calculated from v2. An intramolecular cleavage mechanism cannot account for this behaviour of v2. This approach allows determining the binding and kinetic parameters of both SK•Pg* and SK•Pm formation. This analysis was validated independently by full time-courses of Pg activation, and three discontinuous chromogenic substrate initial rate assays, orthogonal to the assays described above [28]. The assays use different conditions of quenching with active site-blocked Pm (FFR-Pm), 6-AHA, and α2-AP to allow quantitation of time-courses of Pg depletion, transient formation of conformationally activated SK•Pg*, and formation of Pm and SK•Pm. Results of both kinetic approaches are in excellent agreement, supporting the unified mechanism that couples conformational and proteolytic Pg activation.
Different Interactions of SK with Pg/Pm in the Catalytic and Substrate Mode
SK consists of three tightly folded domains, α, β, and γ, linked by segments that allow flexible movement of the domains in solution. When bound to Pg or Pm, the SK domains exhibit a vastly increased level of organisation into a highly ordered structure resembling a three-sided crater that surrounds the catalytic site, and provides a binding site for substrate Pg (Figure 2A) [29, 30]. In the crystal structure of the catalytic complex of SK with μ-plasmin (the catalytic domain only), 15 N-terminal and 32 C-terminal residues are among the disordered regions, and the β-domain shows significantly less contacts with μ-plasmin than the α- and γ-domains, suggesting a possible role in binding of substrate Pg to the complex.
Studies with the deletion mutant SKΔK414 showed that the C-terminal Lys414 residue of SK interacts with an LBS on a Pg/Pm kringle domain, which causes enhancement of the affinity of SK•Pg* and SK•Pm catalytic complex formation [31]. Binding of native SK and SKΔK414 to [Glu]Pg was LBS-independent, due to shielding of critical LBS in the compact α-conformation of [Glu]Pg [9, 10]. However, SKΔK414 binding to [Lys]Pg and [Lys]Pm in the catalytic mode was significantly weaker than native SK binding. SKΔK414 binding to [Lys]Pg and [Lys]Pm and conformational [Lys]Pg activation were LBS-independent, whereas [Lys]Pg substrate binding and proteolytic [Lys]Pm generation were LBS-dependent. This indicates that SK Lys414 binding to [Lys]Pg and [Lys]Pm kringles enhances SK• [Lys]Pg* and SK• [Lys]Pm catalytic complex formation, and other residues are involved in substrate Pg binding. Kinetic analysis of mini-Pg activation by SKΔK414 indicated no difference in affinity between native SK and the mutant binding to this Pg form, lacking kringles 1-4, which suggests that Lys414 may interact with K4 [27]. Stopped-flow fluorescence binding of SK to active-site-labelled Pg and Pm demonstrated that Lys414 binding to a Pg/Pm kringle, likely K4, enhances formation of an initial rapid equilibrium encounter complex, succeeded by two sequential conformational changes [12, 26]. The weaker encounter complex affinity, and the faster off-rates for the second conformational step of SK•Pg* formation compared to SK•Pm, result in a lower affinity of the SK•Pg* complex, and expression of a weaker “pro”-exosite for binding of Pg in the substrate mode. Numerical analysis of forward and reverse rates and affinities for initial, intermediate and final complexes suggested that the Pg encounter also involves significant non-LBS interactions with the protease domain, whereas Pm binding may include additional contributions of other lysines, consistent with tight SK•Pm binding.
Previous studies reported LBS dependence of Pg substrate recognition [32], but were not in agreement on the putative SK domains involved in this interaction [33, 34]. The SK β-domain contains a protruding hairpin loop called the 250-loop (Ala251–Ile264), which is ordered in the structure of the isolated β-domain [35], but disordered in the SK•μPm complex [29]. Early studies implicating Lys256 and Lys257 of the 250-loop in [Glu]Pg conformational activation and substrate recognition used constructs of recombinant SK with a methionine residue or a maltose-binding protein added onto the N-terminus, which led to diverse conclusions about Pg conformational activation and substrate binding [36, 37]. Binding of the isolated β-domain to Pg and kringle 5 (K5) was LBS-dependent [33], implicating K5 in Pg substrate recognition. We used site-directed mutagenesis of Ala substitutions on the SKΔK414 backbone to identify Arg253, Lys256, and Lys257 in the SK β-domain 250-loop, critical for recognition of Pg, and mini-Pg (lacking K1-K4), as substrates of the catalytic SK•Pg* complex. These data confirmed K5 of Pg as the site of interaction of these residues [27]. Molecular modelling suggested that the residues bound K5 in a non-canonical fashion, rather than through a pseudo-lysine motif formed by residues with cationic (Arg and His) and anionic side chains (Glu) arranged spatially on a helix. This motif is seen in binding of VEK-30, a 30-residue peptide from PAM, to K2 of Pg [38, 39], and also in α-enolase from Streptococcus pneumoniae, which has an internal Pg binding site containing two Lys residues, and acidic Asp and Glu residues located on a surface loop [40]. Lys256 and Lys257 were also critical for substrate recognition of bovine Pg (BPg), which is not activated conformationally by SK, but binds as a substrate to human SK•Pm. The complex of the double Ala mutant with fluorescently labelled Pm did not bind BPg, and the complex with active Pm did not support BPg activation. It is noteworthy that SK2a and 2b variants were found to be weak Pg activators, consistent with the non-conservation of Arg253 and Lys256 in their 250-loop [3, 27, 41].
Another bacterial activator, staphylokinase (SAK), with no sequence similarity to SK, but sharing a similar domain fold, forms a tight stoichiometric SAK•Pm complex that cleaves Pg bound in the substrate mode [15]. However, there is no clear functional evidence for Pg binding and conformational activation by SAK. We identified a protein, secreted by Streptococcus agalactiae, with moderate sequence identity to SK and SAK, and named it skizzle (SkzL) [42]. SkzL binds Pg, and deletion of its C-terminal Lys415 reduces its binding to [Lys]Pg ~30-fold, indicative of a kringle interaction. SkzL enhances the activation of [Glu]Pg by uPA, activation of [Glu]Pg and [Lys]Pg by single chain tPA, and plasma clot lysis by these Pg activators. These results suggest a role for SkzL in S. agalactiae pathogenesis through fibrinolytic enhancement.
Specific Interactions of the SK Amino-Terminal Sequence with Pg
Equilibrium binding and kinetic studies of Pg activation with SK(2-414), an SK mutant lacking Ile1, demonstrated that SK(2-414) bound to [Lys]Pg and [Lys]Pm with similar affinity as native SK, but induced no detectable conformational Pg activation [18]. This activity was partially rescued by the peptides SK(1-2), SK(1-5), SK(1-10), and SK(1-15). A mixture of SK(1-10) and SK(2-414) triggered formation of the active catalytic site in the Pg zymogen, allowing active site-specific fluorescence labelling of Pg, whereas sequence-scrambled SK(1-10) was inactive. Ala scanning of the SK1-10 residues (Figure 2C) showed that the rescue effect of SK(1-10) is completely lost by substituting Ile1 and Gly3, partially by substituting Trp6, Leu7 and Arg10, and 2-fold enhanced by Asp9 to Ala substitution. For some residues the effect was 6-AHA-dependent: a peptide with Pro4 changed to Ala behaved like the wild-type peptide in the absence of 6-AHA, but lost its rescue capability in 6-AHA, which supports obligatory interactions with LBS on Pg. These data indicate the requirement of sequence-specific interactions of at least the first 10 SK residues to guide insertion of Ile1 of SK into the Pg N-terminal binding cleft during conformational Pg activation by SKH46A. SK(2-414) binding to Pm is ~1,500-fold tighter than to Pg, and together with the N-terminal sequence specificity, may be part of a cooperative mechanism to trigger the conformational change and stabilise the active zymogen conformation.
The N-terminal Ile-Ala-Gly sequence is conserved in SKH46A and the allelic variants. SK2a and SK2b have Pro4→Tyr, Val22→Met, and Thr25→Ile substitutions, and a non-conserved 30 – 36 sequence cluster [3] that may affect binding to Pg and Pm. The ALAB49 SK2b variant, isolated from impetigo lesions, does not activate [Glu]Pg [3], but we observed that it causes weak conformational [Lys]Pg activation, with KD for the catalytic SK•Pg* complex of 3 μM. Upon substitution of Tyr4 with Pro, Pg activation was partially restored (Figure 2D, unpublished results), with KD ~0.6 μM, a 5-fold gain of function. However, making the corresponding Pro4→Tyr substitution in SKH46A had no effect on the affinity of the SK•Pg* complex, suggesting differential binding effects of the N-terminal sequences on Pg activation by the allelic SK variants. Interactions extending further into the α-domain may play a role in high affinity binding of SKH46A to Pm, as shown by a differential, <360-fold loss of affinity of the SK55–414 truncation mutant for Pm compared to the <2-fold weaker binding to [Lys]Pg [43]. The SK(1–10) peptide failed to reactivate conformational Pg activation by this mutant, suggesting additional binding interactions of the α-domain. Binding of SK N-terminal deletion mutants that lack the complete α-domain was significantly more LBS-dependent [43], and thermodynamic analysis indicated that α-domain binding interactions with Pg/Pm were mainly LBS-independent.
Surface Localisation of Pg/Pm by Bacterial Virulence Factors, and Role of Fibrin
The cell wall-linked Plasminogen-binding group A streptococcal M-like protein (PAM) of many skin-tropic GAS strains binds host Pg/Pm and promotes tissue dissemination through Pm generation mediated by SK [13, 44]. Soluble PAM exhibits a temperature-dependent equilibrium between the dimeric and monomeric state, a general property of Fbg-binding M proteins, whereas surface-bound PAM is a homodimer [45]. PAM uses a pseudo-lysine motif to capture Pg and Pm at two sites, allowing interaction with SK for inducing Pg activation to Pm that remains bound on the bacterial surface. This suggests that the Pg binding sites for PAM and SK do not overlap. Inactivation of genes encoding PAM or SK results in loss of infectivity, illustrating a mechanism of cooperation between surface-bound and secreted virulence factors [6]. PAM was also reported to bind Fbg/Fbn that may initiate an alternative pathway to Pm formation. However, the status of Fbg/Fbn binding to PAM is unclear, with some studies reporting no binding [44], and others documenting qualitative Fbg interaction [6]. One possible conformation of Fbg/Fbn binding to PAM is suggested by the crystal structure of an M1 protein fragment binding four Fbg/Fbn Fragment D (FgD) molecules, perpendicular to its two B-repeats. The triple α-helical coiled-coil of FgD interacts with the coiled-coil of the dimeric M1-fragment [45]. M1 does not bind Pm, and Fbg/Fbn binding may be an indirect mechanism to recruit SK•Pg* and SK•Pm complexes to the surface, by binding these in ternary complexes. It is likely that varying affinities for Fbg/Fbn binding to PAM variants and M1 proteins on GAS may help define their degree of infectivity. A recent study showed that Fbg binding enhances the SK•Pg* affinity by 200-fold, signalled by linkage to the SK•Pg* active site in the ternary SK•Pg*•Fbg complex [28]. SK variants may differ in how they subvert host proteins Fbg/Fbn in a dissemination mechanism by assembly of high affinity SK•Pg*•Fbg/Fbn catalytic complexes that induce a different Pg conformational/proteolytic pathway to Pm, and are resistant to inactivation by α2-AP [4, 7].
In Vivo Assessment of SK in Streptococcal Infections
Recent advances in understanding the molecular mechanisms that govern human Pg activation by SK and the resultant Pm burst triggered at the host-pathogen surface has led many to question how exactly S. pyogenes uses SK to promote dissemination. It is thought that once host Pg is activated to Pm by SK•Pg*/Pm, the SK•Pm complex coats S. pyogenes, thereby providing directed proteolysis, allowing the pathogen to cross host fibrin-barriers [46]. It is now known that SK does not activate mouse Pg [46–50], and, as a result, mice have some level of resistance to S. pyogenes infections [51]. Mouse Pg is also a substrate for SK complexes with human Pg/Pm [52], although its turnover is slow [48, 50, 51]. There is ample evidence documenting the enhanced lethality of S. pyogenes in the presence of human Pg, either provided genetically through transgenic mice [51] or via intraperitoneal human Pg bolus injections [46]. Both strategies may be aided by the prolonged blood half-life of the human zymogen [53]. The identity between human Pg and mouse Pg is high at 79% [54], indicating that modest amino acid substitution drives the preferential ability of SK to activate human Pg. Human Pg or Pm bind PAM with higher affinity than mouse Pg [48, 55], alleviating the concern that mouse Pg might competitively inhibit human Pg complex formation in vivo through saturating the PAM receptors. It is, however, unclear whether human Pg activation is a strict requirement for pathogen spreading in mice, and whether lack of this activation potential precludes disseminative infections. Plasma concentrations of human Pg in the transgenic models do not exceed ~17% of the normal level, and may not be fully representative of what occurs during infections in humans. Furthermore, it is interesting to speculate that co-inheritance of SK2b and PAM may foster GAS virulence potential [7], or that SpeB regulation may trigger pathogen dissemination [56]. GAS expressing SK1 and SK2b showed different levels of virulence in transgenic mouse models expressing human Pg [3, 51], with pathogens expressing SK2b having little effect, but SK1 producers exhibiting significant virulence. Depending on the human Pg levels in these mice, all SK variants may be virulent to some degree, and a model with physiological circulating human [Glu]Pg concentrations may be required to compare in vivo mechanisms of allelic SK infection, growth, dissemination, and virulence.
Future Prospects
SK from GAS isolates proves to be a secreted virulence factor with no known physiological inhibitors. The SK•Pg* and SK•Pm complexes by themselves, and bound to fibrin or PAM on the bacterial surface, are resistant to the physiological serpin, α2-AP, making plasmin generation essentially unstoppable [57]. Usurping host Pg activation by multiple pathways allows GAS such as S. pyogenes to disseminate through tissues and the circulation for colonisation and growth. With the advent of new small animal imaging techniques, it is becoming possible to track GAS infections over time in a single animal. Bioluminescence-expressing bacteria can be used to track GAS populations in vivo through light production driven by over-expression of either the genomically incorporated Photorhabdus luminescens lux operon [58] or by a stabilised plasmid encoding the firefly luciferase gene [59]. Tracking of GAS is also possible through administration of imaging probes that target the bacteria themselves, or reflect heightened neutrophil and monocyte function. Tracking an infection in a single animal over time (e.g. days to weeks) will ultimately allow for better assessment of GAS dissemination from initial infection sites (i.e. skin, muscle, and lung), and will facilitate evaluating adjunctive therapies to disrupt assembly of the (Pg/Pm) •SK2b•PAM or SK1•Pg*•Fbg•M1-protein complexes on the pathogen surface. Due to the ever-increasing occurrence of antibiotic resistance, there is an urgent need for mechanism-based therapeutics to combat GAS infections. Targeting biosynthesis of SK with small molecules while sparing growth is a novel way to address antibiotic resistance [60]. Mouse monoclonal antibodies that target virulence factors, e.g. critical regions of SK and PAM, may be humanised, and engineered to exhibit greater affinity. This approach may represent a first step in innovative translational research toward development of effective inhibitors of host Pg activation by bacterial pathogens.
Acknowledgments
The authors thank Dr. A. Maddur and Ms. B. Gibson for preparing the ALAB49 Y4P mutant and performing the Pg activation experiments. This work was supported by NIH grants R01HL056181 and R01HL071544 (P.E. Bock) from the National Heart, Lung and Blood Institute.
Footnotes
Disclosure of Conflict of Interests
The authors state that they have no conflict of interests.
References
- 1.Ralph AP, Carapetis JR. Group a streptococcal diseases and their global burden. Curr Top Microbiol Immunol. 2013;368:1–27. doi: 10.1007/82_2012_280. [DOI] [PubMed] [Google Scholar]
- 2.Bisno AL, Brito MO, Collins CM. Molecular basis of group A streptococcal virulence. Lancet Infect Dis. 2003;3:191–200. doi: 10.1016/s1473-3099(03)00576-0. [DOI] [PubMed] [Google Scholar]
- 3.Cook SM, Skora A, Gillen CM, Walker MJ, McArthur JD. Streptokinase variants from Streptococcus pyogenes isolates display altered plasminogen activation characteristics – implications for pathogenesis. Mol Microbiol. 2012;86:1052–62. doi: 10.1111/mmi.12037. [DOI] [PubMed] [Google Scholar]
- 4.Cook SM, Skora A, Walker MJ, Sanderson-Smith ML, McArthur JD. Site-restricted plasminogen activation mediated by group A streptococcal streptokinase variants. Biochem J. 2014;458:23–31. doi: 10.1042/BJ20131305. [DOI] [PubMed] [Google Scholar]
- 5.McArthur JD, Cook SM, Venturini C, Walker MJ. The role of streptokinase as a virulence determinant of Streptococcus pyogenes--potential for therapeutic targeting. Curr Drug Targets. 2012;13:297–307. doi: 10.2174/138945012799424589. [DOI] [PubMed] [Google Scholar]
- 6.Kalia A, Bessen DE. Natural selection and evolution of streptococcal virulence genes involved in tissue-specific adaptations. J Bacteriol. 2004;186:110–21. doi: 10.1128/JB.186.1.110-121.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Liang Z, Hsueh H-T, Ploplis VA, Castellino FJ. Characterization of Streptokinases from Group A Streptococci Reveals a Strong Functional Relationship That Supports the Coinheritance of Plasminogen-binding M Protein and Cluster 2b Streptokinase. J Biol Chem. 2012;287:42093–103. doi: 10.1074/jbc.M112.417808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sikri N, Bardia A. A history of streptokinase use in acute myocardial infarction. Tex Heart Inst J. 2007;34:318–27. [PMC free article] [PubMed] [Google Scholar]
- 9.Law Ruby HP, Caradoc-Davies T, Cowieson N, Horvath Anita J, Quek Adam J, Encarnacao Joanna A, Steer D, Cowan A, Zhang Q, Lu Bernadine GC, Pike Robert N, Smith AI, Coughlin Paul B, Whisstock James C. The X-ray Crystal Structure of Full-Length Human Plasminogen. Cell Reports. 2012;1:185–90. doi: 10.1016/j.celrep.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 10.Xue Y, Bodin C, Olsson K. Crystal structure of the native plasminogen reveals an activation-resistant compact conformation. J Thromb Haemost. 2012;10:1385–96. doi: 10.1111/j.1538-7836.2012.04765.x. [DOI] [PubMed] [Google Scholar]
- 11.Boxrud PD, Bock PE. Streptokinase binds preferentially to the extended conformation of plasminogen through lysine binding site and catalytic domain interactions. Biochemistry. 2000;39:13974–81. doi: 10.1021/bi000594i. [DOI] [PubMed] [Google Scholar]
- 12.Verhamme IM, Bock PE. Rapid binding of plasminogen to streptokinase in a catalytic complex reveals a three-step mechanism. J Biol Chem. 2014;289:28006–18. doi: 10.1074/jbc.M114.589077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker MJ, McArthur JD, McKay F, Ranson M. Is plasminogen deployed as a Streptococcus pyogenes virulence factor? Trends Microbiol. 2005;13:308–313. doi: 10.1016/j.tim.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 14.Stillfried GE, Saunders DN, Ranson M. Plasminogen binding and activation at the breast cancer cell surface: the integral role of urokinase activity. Breast Cancer Res. 2007;9:R14. doi: 10.1186/bcr1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parry MA, Zhang XC, Bode I. Molecular mechanisms of plasminogen activation: bacterial cofactors provide clues. Trends Biochem Sci. 2000;25:53–9. doi: 10.1016/s0968-0004(99)01521-2. [DOI] [PubMed] [Google Scholar]
- 16.Huber R, Bode W. Structural Basis of Activation and Action of Trypsin. Acc Chem Res. 1978;11:114–22. [Google Scholar]
- 17.Bode W, Huber R. Induction of the bovine trypsinogen-trypsin transition by peptides sequentially similar to the N-terminus of trypsin. FEBS Lett. 1976;68:231–6. doi: 10.1016/0014-5793(76)80443-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boxrud PD, Verhamme IM, Fay WP, Bock PE. Streptokinase triggers conformational activation of plasminogen through specific interactions of the amino-terminal sequence and stabilizes the active zymogen conformation. J Biol Chem. 2001;276:26084–9. doi: 10.1074/jbc.M101966200. [DOI] [PubMed] [Google Scholar]
- 19.Wang S, Reed GL, Hedstrom L. Deletion of Ile1 changes the mechanism of streptokinase: evidence for the molecular sexuality hypothesis. Biochemistry. 1999;38:5232–40. doi: 10.1021/bi981915h. [DOI] [PubMed] [Google Scholar]
- 20.Renatus M, Engh RA, Stubbs MT, Huber R, Fischer S, Kohnert U, Bode W. Lysine 156 promotes the anomalous proenzyme activity of tPA: X-ray crystal structure of single-chain human tPA. EMBO J. 1997;16:4797–805. doi: 10.1093/emboj/16.16.4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedrich R, Panizzi P, Fuentes-Prior P, Richter K, Verhamme I, Anderson PJ, Kawabata S, Huber R, Bode W, Bock PE. Staphylocoagulase is a prototype for the mechanism of cofactor-induced zymogen activation. Nature. 2003;425:535–9. doi: 10.1038/nature01962. [DOI] [PubMed] [Google Scholar]
- 22.Panizzi P, Friedrich R, Fuentes-Prior P, Bode W, Bock PE. The staphylocoagulase family of zymogen activator and adhesion proteins. Cell Mol Life Sci. 2004;61:2793–8. doi: 10.1007/s00018-004-4285-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boxrud PD, Bock PE. Coupling of conformational and proteolytic activation in the kinetic mechanism of plasminogen activation by streptokinase. J Biol Chem. 2004;279:36642–9. doi: 10.1074/jbc.M405265200. [DOI] [PubMed] [Google Scholar]
- 24.Boxrud PD, Verhamme IM, Bock PE. Resolution of conformational activation in the kinetic mechanism of plasminogen activation by streptokinase. J Biol Chem. 2004;279:36633–41. doi: 10.1074/jbc.M405264200. [DOI] [PubMed] [Google Scholar]
- 25.Kosow DP. Kinetic mechanism of the activation of human plasminogen by streptokinase. Biochemistry. 1975;14:4459–65. doi: 10.1021/bi00691a018. [DOI] [PubMed] [Google Scholar]
- 26.Verhamme IM, Bock PE. Rapid-reaction kinetic characterization of the pathway of streptokinase-plasmin catalytic complex formation. J Biol Chem. 2008;283:26137–47. doi: 10.1074/jbc.M804038200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tharp AC, Laha M, Panizzi P, Thompson MW, Fuentes-Prior P, Bock PE. Plasminogen substrate recognition by the streptokinase-plasminogen catalytic complex is facilitated by Arg253, Lys256, and Lys257 in the streptokinase beta-domain and kringle 5 of the substrate. J Biol Chem. 2009;284:19511–21. doi: 10.1074/jbc.M109.005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nolan M, Bouldin SD, Bock PE. Full time course kinetics of the streptokinase-plasminogen activation pathway. J Biol Chem. 2013;288:29482–93. doi: 10.1074/jbc.M113.477935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Lin X, Loy JA, Tang J, Zhang XC. Crystal structure of the catalytic domain of human plasmin complexed with streptokinase. Science. 1998;281:1662–5. doi: 10.1126/science.281.5383.1662. [DOI] [PubMed] [Google Scholar]
- 30.Boxrud PD, Fay WP, Bock PE. Streptokinase binds to human plasmin with high affinity, perturbs the plasmin active site, and induces expression of a substrate recognition exosite for plasminogen. J Biol Chem. 2000;275:14579–89. doi: 10.1074/jbc.275.19.14579. [DOI] [PubMed] [Google Scholar]
- 31.Panizzi P, Boxrud PD, Verhamme IM, Bock PE. Binding of the COOH-terminal lysine residue of streptokinase to plasmin(ogen) kringles enhances formation of the streptokinase. plasmin(ogen) catalytic complexes. J Biol Chem. 2006;281:26774–8. doi: 10.1074/jbc.C600171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin LF, Houng A, Reed GL. Epsilon amino caproic acid inhibits streptokinase-plasminogen activator complex formation and substrate binding through kringle-dependent mechanisms. Biochemistry. 2000;39:4740–5. doi: 10.1021/bi992028x. [DOI] [PubMed] [Google Scholar]
- 33.Loy JA, Lin X, Schenone M, Castellino FJ, Zhang XC, Tang J. Domain interactions between streptokinase and human plasminogen. Biochemistry. 2001;40:14686–95. doi: 10.1021/bi011309d. [DOI] [PubMed] [Google Scholar]
- 34.Liu L, Sazonova IY, Turner RB, Chowdhry SA, Tsai J, Houng AK, Reed GL. Leucine 42 in the fibronectin motif of streptokinase plays a critical role in fibrin-independent plasminogen activation. J Biol Chem. 2000;275:37686–91. doi: 10.1074/jbc.M003963200. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Tang J, Hunter B, Zhang XC. Crystal structure of streptokinase beta-domain. FEBS Lett. 1999;459:85–9. doi: 10.1016/s0014-5793(99)01214-4. [DOI] [PubMed] [Google Scholar]
- 36.Dhar J, Pande AH, Sundram V, Nanda JS, Mande SC, Sahni G. Involvement of a nine-residue loop of streptokinase in the generation of macromolecular substrate specificity by the activator complex through interaction with substrate kringle domains. J Biol Chem. 2002;277:13257–67. doi: 10.1074/jbc.M108422200. [DOI] [PubMed] [Google Scholar]
- 37.Chaudhary A, Vasudha S, Rajagopal K, Komath SS, Garg N, Yadav M, Mande SC, Sahni G. Function of the central domain of streptokinase in substrate plasminogen docking and processing revealed by site-directed mutagenesis. Protein Sci. 1999;8:2791–805. doi: 10.1110/ps.8.12.2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rios-Steiner JL, Schenone M, Mochalkin I, Tulinsky A, Castellino FJ. Structure and binding determinants of the recombinant kringle-2 domain of human plasminogen to an internal peptide from a group A Streptococcal surface protein. J Mol Biol. 2001;308:705–19. doi: 10.1006/jmbi.2001.4646. [DOI] [PubMed] [Google Scholar]
- 39.Cnudde SE, Prorok M, Castellino FJ, Geiger JH. X-ray crystallographic structure of the angiogenesis inhibitor, angiostatin, bound to a peptide from the group A streptococcal surface protein PAM. Biochemistry. 2006;45:11052–60. doi: 10.1021/bi060914j. [DOI] [PubMed] [Google Scholar]
- 40.Bergmann S, Wild D, Diekmann O, Frank R, Bracht D, Chhatwal GS, Hammerschmidt S. Identification of a novel plasmin(ogen)-binding motif in surface displayed alpha-enolase of Streptococcus pneumoniae. Mol Microbiol. 2003;49:411–23. doi: 10.1046/j.1365-2958.2003.03557.x. [DOI] [PubMed] [Google Scholar]
- 41.McArthur JD, McKay FC, Ramachandran V, Shyam P, Cork AJ, Sanderson-Smith ML, Cole JN, Ringdahl U, Sjöbring U, Ranson M, Walker MJ. Allelic variants of streptokinase from Streptococcus pyogenes display functional differences in plasminogen activation. Faseb J. 2008;22:3146–53. doi: 10.1096/fj.08-109348. [DOI] [PubMed] [Google Scholar]
- 42.Wiles KG, Panizzi P, Kroh HK, Bock PE. Skizzle is a novel plasminogen- and plasmin-binding protein from Streptococcus agalactiae that targets proteins of human fibrinolysis to promote plasmin generation. J Biol Chem. 2010;285:21153–64. doi: 10.1074/jbc.M110.107730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bean RR, Verhamme IM, Bock PE. Role of the streptokinase alpha-domain in the interactions of streptokinase with plasminogen and plasmin. J Biol Chem. 2005;280:7504–10. doi: 10.1074/jbc.M411637200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berge A, Sjöbring U. PAM, a novel plasminogen-binding protein from Streptococcus pyogenes. J Biol Chem. 1993;268:25417–24. [PubMed] [Google Scholar]
- 45.Macheboeuf P, Buffalo C, Fu CY, Zinkernagel AS, Cole JN, Johnson JE, Nizet V, Ghosh P. Streptococcal M1 protein constructs a pathological host fibrinogen network. Nature. 2011;472:64–8. doi: 10.1038/nature09967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khil J, Im M, Heath A, Ringdahl U, Mundada L, Cary Engleberg N, Fay WP. Plasminogen enhances virulence of group A streptococci by streptokinase-dependent and streptokinase-independent mechanisms. J Infect Dis. 2003;188:497–505. doi: 10.1086/377100. [DOI] [PubMed] [Google Scholar]
- 47.Sun H. Exploration of the host haemostatic system by group A streptococcus: implications in searching for novel antimicrobial therapies. J Thromb Haemost. 2011;9 (Suppl 1):189–94. doi: 10.1111/j.1538-7836.2011.04316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu Q, Figuera-Losada M, Ploplis VA, Cnudde S, Geiger JH, Prorok M, Castellino FJ. The lack of binding of VEK-30, an internal peptide from the group A streptococcal M-like protein, PAM, to murine plasminogen is due to two amino acid replacements in the plasminogen kringle-2 domain. J Biol Chem. 2008;283:1580–87. doi: 10.1074/jbc.M705063200. [DOI] [PubMed] [Google Scholar]
- 49.Marcum JA, Kline DL. Species specificity of streptokinase. Comp Biochem Physiol B. 1983;75:389–94. doi: 10.1016/0305-0491(83)90345-0. [DOI] [PubMed] [Google Scholar]
- 50.Yakovlev SA, Rublenko MV, Izdepsky VI, Makogonenko EM. Activating effect of the plasminogen activators on plasminogens of different mammalia species. Thromb Res. 1995;79:423–8. doi: 10.1016/0049-3848(95)00131-a. [DOI] [PubMed] [Google Scholar]
- 51.Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjöbring U, Ginsburg D. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science. 2004;305:1283–6. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 52.Li Z, Ploplis VA, French EL, Boyle MD. Interaction between group A streptococci and the plasmin(ogen) system promotes virulence in a mouse skin infection model. J Infect Dis. 1999;179:907–14. doi: 10.1086/314654. [DOI] [PubMed] [Google Scholar]
- 53.Gonias SL, Einarsson M, Pizzo SV. Catabolic pathways for streptokinase, plasmin, and streptokinase activator complex in miceIn vivo reaction of plasminogen activator with alpha 2-macroglobulin. J Clin Invest. 1982;70:412–23. doi: 10.1172/JCI110631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Degen SJ, Bell SM, Schaefer LA, Elliott RW. Characterization of the cDNA coding for mouse plasminogen and localization of the gene to mouse chromosome 17. Genomics. 1990;8:49–61. doi: 10.1016/0888-7543(90)90225-j. [DOI] [PubMed] [Google Scholar]
- 55.Ringdahl U, Svensson M, Wistedt AC, Renne T, Kellner R, Muller-Esterl W, Sjöbring U. Molecular co-operation between protein PAM and streptokinase for plasmin acquisition by Streptococcus pyogenes. J Biol Chem. 1998;273:6424–30. doi: 10.1074/jbc.273.11.6424. [DOI] [PubMed] [Google Scholar]
- 56.Cole JN, McArthur JD, McKay FC, Sanderson-Smith ML, Cork AJ, Ranson M, Rohde M, Itzek A, Sun H, Ginsburg D, Kotb M, Nizet V, Chhatwal GS, Walker MJ. Trigger for group A streptococcal M1T1 invasive disease. FASEB J. 2006;20:1745–7. doi: 10.1096/fj.06-5804fje. [DOI] [PubMed] [Google Scholar]
- 57.Cederholm-Williams SA, De Cock F, Lijnen HR, Collen D. Kinetics of the reactions between streptokinase, plasmin and alpha 2-antiplasmin. Eur J Biochem. 1979;100:125–32. doi: 10.1111/j.1432-1033.1979.tb02040.x. [DOI] [PubMed] [Google Scholar]
- 58.Francis KP, Yu J, Bellinger-Kawahara C, Joh D, Hawkinson MJ, Xiao G, Purchio TF, Caparon MG, Lipsitch M, Contag PR. Visualizing pneumococcal infections in the lungs of live mice using bioluminescent Streptococcus pneumoniae transformed with a novel gram-positive lux transposon. Infect Immun. 2001;69:3350–8. doi: 10.1128/IAI.69.5.3350-3358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loh JM, Proft T. Toxin-antitoxin-stabilized reporter plasmids for biophotonic imaging of Group A streptococcus. Appl Microbiol Biotechnol. 2013;97:9737–45. doi: 10.1007/s00253-013-5200-7. [DOI] [PubMed] [Google Scholar]
- 60.Sun H, Xu Y, Sitkiewicz I, Ma Y, Wang X, Yestrepsky BD, Huang Y, Lapadatescu MC, Larsen MJ, Larsen SD, Musser JM, Ginsburg D. Inhibitor of streptokinase gene expression improves survival after group A streptococcus infection in mice. Proc Natl Acad Sci U S A. 2012;109:3469–74. doi: 10.1073/pnas.1201031109. [DOI] [PMC free article] [PubMed] [Google Scholar]