

Abstract
Ovarian sex cord-stromal tumors are infrequent and represent approximately 7% of all primary ovarian tumors. This histopathologic ovarian tumor group differs considerably from the more prevalent epithelial ovarian tumors. Although sex cord-stromal tumors present in a broad age group, the majority tend to present as a low-grade disease that usually follows a nonaggressive clinical course in younger patients. Furthermore, because the constituent cells of these tumors are engaged in ovarian steroid hormone production (e.g., androgens, estrogens, and corticoids), sex cord-stromal tumors are commonly associated with various hormone-mediated syndromes and exhibit a wide spectrum of clinical features ranging from hyperandrogenic virilizing states to hyperestrogenic manifestations. The World Health Organization sex cord-stromal tumor classification has recently been revised, and currently these tumors have been regrouped into the following clinicopathologic entities: pure stromal tumors, pure sex cord tumors, and mixed sex cord-stromal tumors. Moreover, some entities considered in the former classification (e.g., stromal luteoma, stromal tumor with minor sex cord elements, and gynandroblastoma) are no longer considered separate tumors in the current classification. Herein, we discuss and revise the ultrasonography, computed tomography, and magnetic resonance imaging characteristics of the different histopathologic types and clinicopathologic features of sex cord-stromal tumors to allow radiologists to narrow the differential diagnosis when facing ovarian tumors.
Ovarian sex cord-stromal tumors are uncommon neoplasms that represent approximately 7% of all ovarian tumors (1). These tumors comprise a heterogeneous group and are formed by diverse cell types that arise from the primitive sex cords or stromal cells (1, 2). The stromal cells include theca cells, fibroblasts, and Leydig cells whereas the gonadal primitive sex cords include granulosa cells and Sertoli cells (3). These cell types may be present separately or admixed and display different degrees of differentiation (4).
As some of the constituent cells of these types of tumors are engaged in ovarian steroid hormone production (e.g., androgens, estrogens, and corticoids), sex cord-stromal tumors are commonly associated with various hormone-mediated syndromes and exhibit a wide spectrum of clinical features. Tumors formed from ovarian cells (e.g., granulosa cells and theca cells) are often hyperestrogenic, whereas those comprising testicular cell types (e.g., Sertoli and Leydig cells) are usually hyperandrogenic. However, many tumors are nonfunctioning, and those comprising female cells may produce androgens and vice-versa (4).
Tumors that induce hyperandrogenicity may present with virilization signs (e.g., hirsutism, acne, irregular menstrual periods, male-pattern baldness, loss of female fat distribution, and hoarse voice), whereas tumor subtypes associated with abnormal estrogen production may present with hyperestrogenicity (e.g., isosexual precocity in children, abnormal uterine bleeding, endometrial hyperplasia, and carcinoma).
The association of ovarian sex cord-stromal tumors with typical clinical syndromes is not the only characteristic distinguishing these tumors from the more common ovarian epithelial neoplasms. Although sex cord-stromal tumors affect patients throughout a wide age range, the majority tend to present in younger patients and as a low-grade disease (stage I) that usually follows a nonaggressive clinical course. Therefore, the primary treatment is surgical and the prognosis is generally favorable. Furthermore, ovarian sex cord-stromal tumors may exhibit characteristic radiologic features with which radiologists should become familiar. Conversely, recognition of the spectrum of the ultrasonography (US), computed tomography (CT), and magnetic resonance imaging (MRI) appearances as well as clinicopathologic features of ovarian sex cord-stromal tumors may assist radiologists to narrow the differential diagnosis when facing ovarian tumors.
World Health Organization classification of sex-cord stromal tumors
The World Health Organization (WHO) classification of sex cord-stromal tumors has recently been revised in 2014 (5) (Table 1). In the current revision, these tumors were regrouped into the following clinicopathologic entities: pure stromal tumors, pure sex cord tumors, and mixed sex cord-stromal tumors.
Table 1.
WHO classification scheme for ovarian sex cord-stromal tumors (2014)
Pure stromal tumors
|
Pure sex cord tumors
|
Mixed sex cord-stromal tumors
|
WHO, World Health Organization; NOS, not otherwise specified.
The “pure stromal tumors” category comprises entities from the previous thecoma-fibroma group, which had been classified under the granulosa-stromal cell tumor group division, as well as entities from the previous steroid cell group division (6) (Table 2).
Table 2.
Former WHO classification scheme for ovarian sex cord-stromal tumors (2003)
Granulosa-stromal cell tumors
|
Sertoli-stromal cell tumors
|
Sex cord-stromal tumors of mixed or unclassified cell types
|
Steroid cell tumors
|
WHO, World Health Organization; NOS, not otherwise specified.
In the recent classification, luteinized thecoma is no longer considered a separate entity from the thecoma group, and therefore this term should only be used when associated with sclerosing peritonitis. Instead, a separate histopathologic entity titled “luteinized thecoma associated with sclerosing peritonitis” now exists.
A distinct rare ovarian neoplasm, which was recently named “microcystic stromal tumor,” was added to this category whereas the denomination “stromal tumor with minor sex cord elements” is not encompassed by this classification.
Moreover, in terms of steroid cell tumors, the term “stromal luteoma,” which was used to designate small steroid tumors confined to the ovarian cortex, has also been discarded.
The subdivision of pure sex cord tumors now comprises tumors that were previously categorized separately, including adult granulosa cell tumors (GCTs), juvenile GCTs, Sertoli cell tumors, and sex cord tumors with annular tubules.
The different types with respect to Sertoli-Leydig cell tumor (SLCT) differentiation as well as sex cord-stromal tumors, not otherwise specified are categorized as “mixed sex cord-stromal tumors.” Stromal-Leydig cell tumor and gynandroblastomas are no longer considered in the current classification.
Pure stromal tumors
Fibroma, cellular fibroma, and fibrosarcoma
Fibromas are almost always endocrine-inert tumors composed of spindle stromal cells that produce a collagenous stroma (5, 7, 8). These are undeniably the most common sex cord-stromal tumors, representing 4% of all ovarian neoplasms (4, 5, 9) (Fig. 1). Fibromas can present at any age, although the mean age of occurrence is in the late forties, and are associated with nevoid basal carcinoma syndrome (5). Fibromas range in size from small to large lesions. Small lesions are frequently asymptomatic, but women can present with pelvic discomfort or acute abdominal pain due to ovarian torsion as the size increases. Fibromas can mimic malignancy when present in the classic Meigs’ syndrome (hydrothorax, ascites, and benign ovarian tumor), which typically disappears after tumor removal (3, 4, 10, 11). The tumor size is known to correlate with the presence of ascites.
Figure 1.
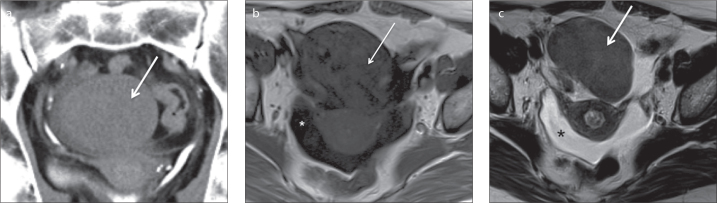
a–c. A 77-year-old female patient with a right ovarian fibroma. Coronal delayed contrast-enhanced computed tomography image (a) shows a solid, well-defined, homogeneous right ovarian mass isodense to the uterus with very sparse contrast uptake (arrow). This fibroma is hypointense on both axial T1-weighted (b) and T2-weighted images (c). A small amount of ascites is observed in the pelvic recesses (asterisks).
Hypercellular fibromas are classified as either cellular fibromas or fibrosarcomas. Cellular fibromas constitute 10% of all ovarian fibromas and have low malignant potential, exhibit mild nuclear atypia, and may present more than four mitotic figures per 10 high-powered fields (5). These generally exhibit the same clinical manifestations but tend to be larger than fibromas, thus potentially leading to necrosis and hemorrhage, particularly due to torsion (4, 5) (Fig. 2).
Figure 2.
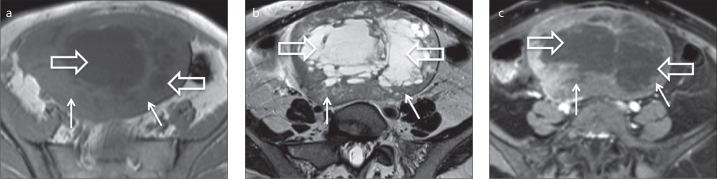
a–c. A 50-year-old female patient with a right ovarian cellular fibroma. Axial T1-weighted (a), T2-weighted (b) and gadolinium-enhanced fat-suppressed T1-weighted (c) images show a large, heterogeneous and well-defined tumor of the right ovary. The tumor features a large-centred cystic/necrotic area with T2 hyperintensity and T1 hypointensity (open arrows). The solid components exhibit an intermediate T2 signal and T1 isointensity to the muscle as well as avid contrast uptake (arrows).
Ovarian fibrosarcomas are rare entities that follow a malignant clinical course and tend to exhibit moderate-to-severe nuclear atypia and mitotic figures (5, 12). These tumors usually present in postmenopausal women as large unilateral masses, often with necrosis and hemorrhage (4, 5, 12).
Although different ovarian fibroma US patterns may be encountered, the US appearance is usually a solid hypoechoic attenuating ovarian mass (13). On CT, fibromas usually appear as homogeneous solid ovarian masses with delayed contrast enhancement (3, 13) (Fig. 1a). Calcification may be present and widespread throughout the tumor (13). Given their predominant collagenous and fibrous components, fibromas exhibit characteristic MRI features such as hypointensity on T1-weighted images and marked hypointensity on T2-weighted images as well as weak and delayed enhancement after gadolinium administration (3) (Fig. 1b, 1c). Notwithstanding, edema and cystic degeneration may be encountered in fibromas; these appear as dispersed hyperintense areas on T2-weighted images (8, 10, 13). These characteristics are most frequently observed in large lesions and fibrosarcomas (8, 10, 13). Both predominantly cystic fibromas and fibromas with T1 and T2 hyperintensity/isointensity have also been described (9, 14).
Regarding the relationship between ovarian fibromas and the ovary, Oh et al. (9) reported that exophytic fibroma growth from the ovarian periphery without altering the normal form of the ipsilateral ovary is not uncommon (46%) and that depicting the rest of the ipsilateral ovary in premenopausal women is normal (83%). In addition, 67% of the 24 studied fibromas exhibited a border of T2 hypointensity or pseudocapsule around the outer ovarian margin that reflected compression of the ovarian tissue (9).
The MRI-based differential diagnosis includes essentially fibrous ovarian tumors such as Brenner tumors and adenofibromas as well as pedunculated nondegenerative subserosal and broad ligament leiomyomas (15). Depiction of the tumor-feeding ovarian arteries or vascular signal voids between the uterus and leiomyoma might help to differentiate these entities (16). Thomassin-Naggara et al. (17), in a retrospective study that accessed the accuracy of dynamic MRI for differentiating ovarian fibromas from uterine subserosal leiomyomas, reported that the dynamic-contrast MRI enhancement of uterine leiomyomas was higher in terms of maximal enhancement and enhancement at 30, 60, and 90 seconds. However, no significant statistical difference existed in a delayed T1 post-contrast sequence (17).
Fibroma may induce adnexal torsion. In such cases, hemorrhagic infarction or necrosis may occur (3, 10). Hemorrhagic infarction may be difficult to define because of the solid nature of the tumor. Nonetheless, an eccentric hyperintense area on T1-weighted images may allow this diagnosis (3).
Excepting a few cellular fibromas and fibrosarcomas, these types of tumors have typically benign courses and are curable via surgical excision (7).
Thecoma and luteinized thecoma associated with sclerosing peritonitis
Thecomas are composed of lipid-laden stromal cells that resemble theca cells, which usually encircle the ovarian follicles, and exhibit estrogenic activity in most cases (4, 7, 18) (Fig. 3). Thecomas account for 0.5%–1% of all primary ovarian tumors (19). These tumors are more likely to occur in postmenopausal women and, with rare exceptions, are considered benign neoplasms (5). Affected women experience estrogen-related symptoms such as uterine bleeding, endometrial hyperplasia, and endometrial carcinoma; the latter has been reported to occur in 21% of cases (4, 8) (Fig. 3).
Figure 3.
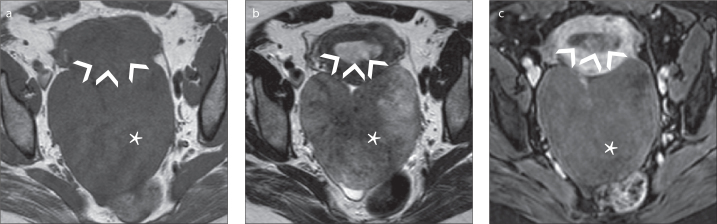
a–c. A 61-year-old postmenopausal female patient who presented with metrorrhagia and was diagnosed with a right ovarian thecoma and endometrial polyps. Axial T1-weighted image (a) shows a large homogeneous right ovarian mass isointense to the myometrium. Axial T2-weighted image of the tumor (b) reveals areas of isointense and hyperintense signal relative to the myometrium, whereas axial gadolinium-enhanced fat-suppressed T1-weighted image (c) shows weak contrast-enhancement of the tumor (*). Endometrial polyps display hypointensity on T2-weighted image (b), isointensity to the endometrium on T1-weighted image (a), and homogeneous gadolinium uptake (arrowheads).
Thecomas associated with androgen production may contain steroid-type cells (lutein cells) and were previously formally classified as “luteinized thecomas” (5). This histopathologic classification is no longer recommended and should only be used if there is an association with sclerosing peritonitis. However, luteinized thecoma associated with sclerosing peritonitis is usually a bilateral hormonally inert tumor that occurs in younger women at an average age of 28 years (5). Thecomas that manifest in combination with fibrous tissue may be classified as fibrothecomas (5).
Generally, pure thecomas or thecomas with scanty fibrotic components do not have distinct US and CT appearances and mimic other solid ovarian tumors (13). A recent study by Zhang et al. (19) prospectively evaluated the MRI characteristics of 18 thecomas/fibrothecomas, namely the diffusion-weighted imaging (DWI) features and apparent diffusion coefficients (ADC) at 3.0 T, and correlated these with the features of other solid ovarian tumors and adnexal leiomyomas. The authors concluded that most thecomas/fibrothecomas (61.1%) were homogenous masses and isointense to the myometrium on DWI-MRI and that the ADC values of thecomas/fibrothecomas did not significantly differ from those of other ovarian solid tumors and leiomyomas (19). When compared with predominant fibrous tumors, pure thecomas tend to exhibit greater hyperintensity on T2-weighted images; more avid contrast-enhancement may be observed because theca cells are greatly vascularized (13, 18) (Fig. 3).
Sclerosing stromal tumor
Sclerosing stromal tumor (SST) is a benign neoplasm that accounts for less than 5% of ovarian sex cord-stromal tumors (20) (Fig. 4). Unlike fibroma, thecoma, and adult GCTs, SSTs are more likely to occur in young women; approximately 80% of reported cases are under 30 years of age (13, 21). Although SSTs most commonly occur after menarche, a few cases have been reported in premenarchal girls (22). Typically, SSTs manifest as unilateral masses. To our knowledge, only two cases of bilateral SSTs have been described: one in an 11-year-old premenarchal girl and the other in a pregnant woman with Gorlin syndrome after clomiphene therapy for infertility (21, 23).
Figure 4.
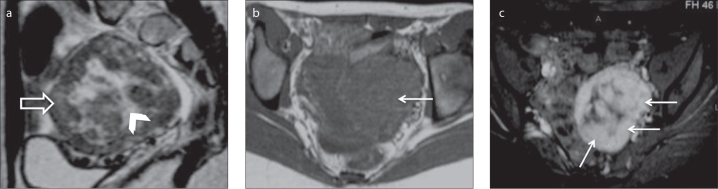
a–c. A 14-year-old female patient with a left ovarian sclerosing stromal tumor. Sagittal T2-weighted image (a) reveals a left ovarian mass with a pseudolobular spoke-wheel pattern characterized by intermediate-signal intensity of the outer nodules (open arrow) surrounding a central hyperintense cystic area (arrowhead). Axial T1-weighted image (b) shows a well-defined, slightly lobulated, smooth-bordered hypointense mass (arrow). Axial gadolinium-enhanced fat-suppressed T1-weighted image (c) shows early and avid contrast uptake by the solid portions of the tumor (arrows).
Pelvic pain and menstrual irregularities are frequent symptoms (20, 24). A few hormonally active tumors that produced androgens and/or estrogens have been documented in the literature (18). SSTs of the ovary have also been associated with pregnancy (25). Although rare, ascites may be present (3).
Upon gross examination, SSTs are mostly solid masses with yellowish foci, edema, and cystic areas (4). The histopathology is characterized by the presence of an ill-defined pseudolobular pattern in the cellular areas, which are separated by edematous fibrous areas. These nodular portions contain collagen-producing spindle-shaped cells and vacuolated lipid-containing lutein cells. Prominent vascularization is seen within these areas (4, 24). Both these typical features and the edema present in SSTs are thought to result from the expression of vascular permeability factor/vascular endothelial growth factor in lutein cells and its receptor fms-like tyrosine kinase 1 in small to middle-sized blood vessels (4, 24, 26).
The imaging findings of SSTs reflect their characteristic histologic features. US commonly reveals unilateral tumors with star-shaped hypoechoic areas enclosed by solid areas or solid tumors with medially located multiple small round or cleft-like hypoechoic areas (15, 24). Multilocular heterogeneous cystic masses and irregular septae have also been described (15). Color Doppler imaging reveals intratumoral blood vessels in the periphery and between the central cystic spaces. Low-resistance waveforms are visible on pulsed Doppler US (24). The described US features may mimic those of malignant ovarian tumors; therefore, further MRI-based radiologic evaluation is usually necessary.
On MRI, a heterogeneous solid tumor with iso- to hyperintensity is observed on T2-weighted imaging (Fig. 4a) (3). The pseudolobular areas in the outer part of the lesion commonly exhibit a spoke-wheel pattern and iso- to hypointensity on T2-weighted imaging in contrast with the hyperintense septa (15, 18) (Fig. 4a). Cystic areas are hyperintense on T2-weighted imaging and hypointense on T1-weighted imaging (15) (Fig. 4a, 4b). A thick, hypointense rim that outlines the tumor on T2-weighted imaging reflects compression of the ovarian cortex by the slow-growing tumor (3). Dynamic contrast-enhanced MRI and CT images reveal early and avid peripheral contrast uptake, reflecting prominent vasculature in the cellular areas, with centripetal progression on late images (16, 18, 20) (Fig. 4c).
- To our knowledge, all ovarian SSTs described in literature were benign, did not recur, and were treated successfully via surgical excision.
Steroid cell tumor and Leydig tumor
Steroid cell tumors are very uncommon neoplasms that account for 0.1% of all ovarian tumors (5). These are defined as ovarian neoplasms composed exclusively of cells resembling steroid-secreting cells without Reinke crystals (5). In contrast, intracytoplasmatic Reinke crystals may characteristically be present in Leydig cell tumors, which are formed from Leydig cells and represent approximately 20% of all steroid cell tumors (5) (Fig. 5). Approximately 80% of steroid tumors are steroid cell tumors, not otherwise specified (5) (Fig. 6). Formally, the term “stromal luteoma” was used to designate a small steroid cell tumor confined to the ovarian cortex; however, this designation was discarded in the most recent WHO classification of ovarian tumors (5).
Figure 5.

a–d. A 53-year-old female patient, who was being monitored for hirsutism, diagnosed with a right ovarian Leydig cell tumor. Axial T1-weighted image (a) shows an increased in size right ovary (arrow). Axial T2-weighted image (b) demonstrates a hypointense, small solid lesion in the right ovary (arrow). Axial gadolinium-enhanced T1-weighted image (c) demonstrates enhancement of the lesion. Note that the hyperintense mass is well delineated against the ovarian stroma (arrow, c). The section surface of the right adnexal specimen (d) contains an ill-defined brown–yellow hilar tumor with a long axis of 10 mm (arrowheads).
Figure 6.
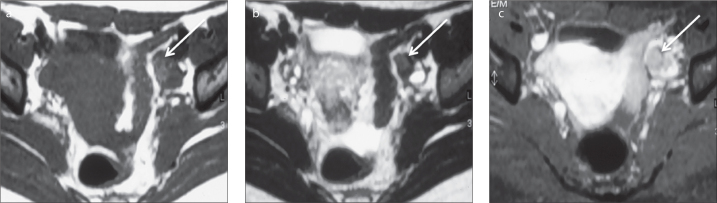
a–c. A 19-year-old female patient who presented with amenorrhea and hirsutism and was diagnosed with a left ovarian steroid cell tumor, not otherwise specified (NOS). Axial T1-weighted image (a) shows a small solid tumor in the left ovary hyperintense to the ovarian stroma (arrow); this lesion displays intermediate/low signal intensity on axial T2-weighted image (b) (arrow); axial gadolinium-enhanced T1-weighted image (c) demonstrates enhancement of the lesion (arrow). Material reproduced from Martins I, et al. (50), with permission.
Steroid cell tumors occur at an average age of 43 years (5). The majority is androgenic, and patients exhibit virilizing symptoms (50% of cases). Occasionally, these tumors are associated with estrogenic manifestations, and a few cases have been associated with hypercortisolism and progestational changes (3, 5, 7). Leydig tumors usually occur in older women, most of whom exhibit hyperandrogenicity (7). Estrogenic Leydig tumors are less frequent but may also occur. Whereas Leydig tumors are benign, approximately one-third of steroid cell tumors, not otherwise specified, are clinically malignant (5).
During imaging evaluations, Leydig and steroid cell tumors appear as unilateral solid masses. Leydig tumors tend to be small (mean, 2.4 cm) and are reportedly isoechoic to the uterus on US and hypoattenuating on CT (5, 27, 28). The signal intensity on T2-weighted imaging differs depending on the amount of fibrous stroma (10, 28). Hyperintense areas may be observed on T1 and T2-weighted images; these reflect the presence of lipid components (10) (Fig. 5). Small virilizing Leydig tumors are sometimes difficult to identify on CT and transabdominal US. Transvaginal US with color Doppler and MRI are important tools for diagnosing these tumors, which often can only be identified by depicting morphologic changes within the ovary (13).
Few cases of steroid cell tumors, not otherwise specified, have been reported in the literature. These have been described as larger tumors (average size, 8.4 cm) that vary from solid masses to multilocular cystic masses with nodular walls (29, 30). On MRI, isointensity on T2-weighted imaging and avid contrast uptake may be observed (18, 28) (Fig. 6).
Pure sex cord tumors
Adult and juvenile granulosa cell tumors
GCTs are low-grade malignant ovarian sex cord-stromal tumors that represent less than 5% of all malignant ovarian tumors (3). Clinicopathologically, these tumors are divided into two histologic subtypes, adult and juvenile, of which the former accounts for 95% of the neoplasms (13, 16). The incidence of adult GCT peaks strikingly in early postmenopausal women, whereas the juvenile form occurs predominantly in children and young women (<30 years) (13, 31).
Although GCTs are the most common estrogen-producing tumors, a small subset is androgenic (13, 18, 31, 32). Women typically present with hyperestrogenicity, including vaginal bleeding and breast tenderness during the postmenopausal stage. During the reproductive years, women frequently present with altered menstrual patterns ranging from amenorrhea to excessive uterine bleeding (4, 13, 31). In the pediatric population, isosexual pseudoprecocity is common (13).
Estrogen overproduction is also responsible for endometrial hyperplasia and concomitant endometrial cancer, which according to the literature occur in 32%–85% and 3%–22% of cases, respectively (33, 34). Uterine cancer is nearly always a low-grade, low-stage endometrioid adenocarcinoma (35). Moreover, women with GCT have a higher risk of breast cancer development (36, 37). Unregulated inhibin production can cause infertility, and androgen secretion may induce virilizing symptoms in a small group of patients (31, 32).
Most patients have palpable pelvic masses upon clinical examination. Mass enlargement and the compression of adjacent structures can occasionally cause abdominal symptoms (4). Mass rupture and consequent hemoperitoneum may be seen at presentation (3, 4, 13).
Despite the different ages of onset, clinical findings, and histologic characteristics, the adult and juvenile subtypes of GCT have similar imaging features (13). GCTs differ from epithelial ovarian neoplasms via predominant unilaterality and confinement to the ovary with no peritoneal seeding at the time of the diagnosis in most cases (3, 31). Moreover, GCTs usually do not feature intracystic papillary projections and rarely contain intratumoral calcifications (13, 31, 38).
GCTs are typically unilateral masses (average size, 12 cm) and can macroscopically range from solid masses to multilocular cystic lesions with solid components lesions and exclusively cystic masses, although homogeneous solid lesions and unilocular cysts are less common (3, 4, 13, 18, 31) (Figs. 7–9). The imaging appearances of these tumors vary according to the gross pathologic tumor characteristics. US and CT imaging usually reveal multicystic masses with solid components and either irregular thickened or thin septations (38) (Fig. 9).
Figure 7.
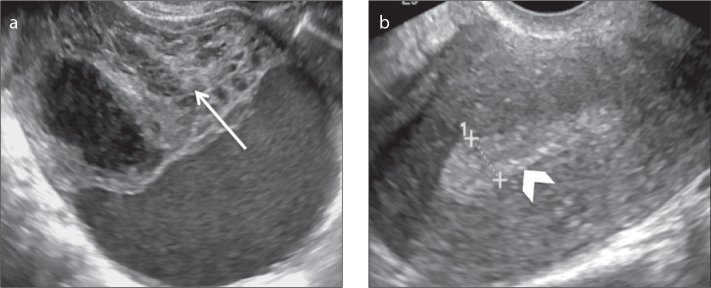
a, b. A 50-year-old postmenopausal female patient with a right ovarian adult granulosa cell tumor and proliferative endometrium who presented with metrorrhagia. Transvaginal US images (a, b) show a heterogeneous and apparently complex mass that is predominantly solid with focal cystic areas (arrow, a). Note the thickness of the endometrium, which exceeded 8 mm (arrowhead, b).
Figure 8.
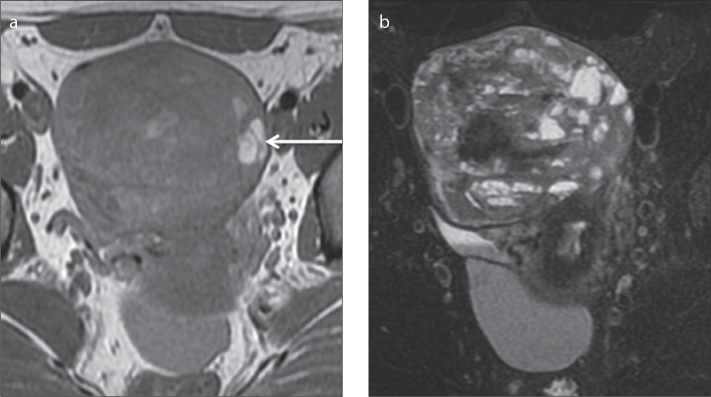
a, b. A 40-year-old female patient with a right ovarian adult granulosa cell tumor. Axial T1-weighted (a) and fat-suppressed T2-weighted (b) images show a large, well-defined, multiloculated cystic tumor with septations of varying thicknesses. Axial T1-weighted image (a) depicts areas of hyperintensity within the cystic locules that reflect hemorrhagic areas (arrow).
Figure 9.
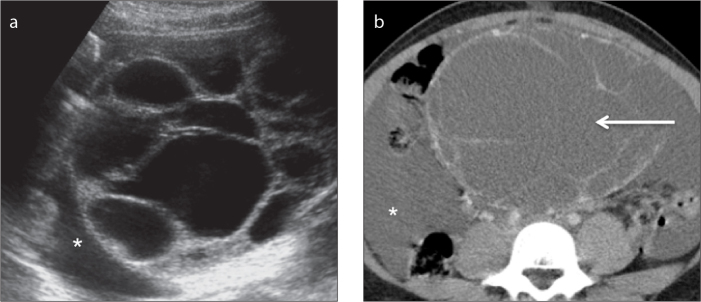
a, b. A 17-year-old female patient with a right ovarian juvenile granulosa cell tumor. A transabdominal US image (a) shows a multilocular cystic ovarian mass. An axial contrast-enhanced CT image (b), shows a multiloculated low attenuating mass with thin septations (arrow). Note the presence of abundant ascites (asterisks).
On MRI, the tumor usually presents a sponge-like appearance, indicating a multilocular cystic mass (18) (Fig. 8). On T2-weighted images, the solid tumor component is usually isointense but thickened septations may be hypointense (15, 38). The cystic portions may exhibit fluid-fluid levels and areas of intracystic hemorrhage are typically hyperintense on T1-weighted images (15, 38). Solid components tend to exhibit contrast uptake on gadolinium-enhanced images, whereas cystic areas are nonenhancing (15).
Continuous estrogenic stimulation is responsible for endometrial thickening, endometrial hemorrhage, and uterine enlargement (31). Because of the associated hormonal activity, the majority of GCTs are detected early and present as stage I disease (>80%), leading to a reported five-year survival rate exceeding 90% (32, 39).
Characteristically, recurrence exhibits a late pattern and has been reported in less than 40% of patients with stage I disease, of whom 56% had fatal outcomes (40). Given the long natural history and rarity of these tumors, information regarding factors that might be predictive of recurrence is limited. Still, the disease stage at presentation has been shown to be the most important factor (40). Most juvenile GCT cases present with stage I disease and are less likely to recur after simple resection; however, the occasional clinically malignant tumors usually exhibit fast growth and early intra-abdominal spread (13, 38).
Mixed sex cord-stromal tumors
Sertoli–Leydig cell tumor
SLCT is a rare sex cord-stromal tumor that accounts for approximately 0.5% of all ovarian neoplasms (3, 18) (Fig. 10). SLCT and SST occur predominantly in the same age group; approximately 75% of SLCTs are encountered in women younger than 30 years (13). However, few cases have been described in postmenopausal women (41, 42). For SLCT, nearly all cases are unilateral (98%) and 80% are restricted to the ovary at diagnosis (4, 18).
Figure 10.
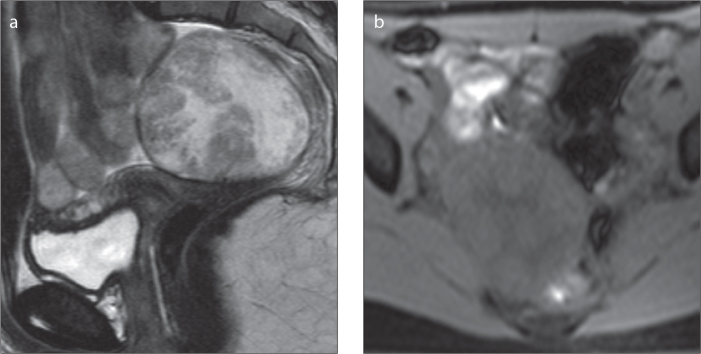
a, b. A 13-year-old female patient with a right ovarian Sertoli-Leydig cell tumor. Sagittal T2-weighted (a) and axial fat-suppressed T1-weighted (b) images show a well-defined, heterogeneous, apparently complex tumor. The solid portions are iso- to hyperintense on T2-weighted image (a), and the cystic portions are hyperintense on T2-weighted image (a) and hypointense on fat-suppressed T1-weighted image (b).
SLCT is the most common virilizing ovarian tumor, as approximately 30%–50% of these tumors produce androgens (testosterone and androgen precursors) and more than one-third of cases develop symptoms of virilization (4, 15, 18). Notwithstanding, SLCT is a rare cause of virilization in premenopausal women. In this age group, other differential diagnosis such as polycystic ovary syndrome and adrenal androgen-secreting tumors should be considered. In cases of androgen-secreting SLCTs, the laboratory data normally indicate elevated serum testosterone levels but, in contrast to masculinizing adrenal tumors, normal or slightly elevated urinary 17-ketosteroid levels (4).
Many SLCTs are hormonally inactive and a small subgroup is estrogenic. Sudden abdominal pain and swelling are frequent symptoms of nonfunctioning tumors.
Very rarely, SLCT have been reported to associate with elevated alpha-fetoprotein serum levels; this is a typical feature of germ cell tumors such as yolk sac neoplasm. Although only approximately 30 cases have been described to date, SLCT is the most commonly reported ovarian nongerm cell tumor associated with elevated serum alpha-fetoprotein levels (43, 44).
SLCT has a nonspecific appearance. On US, these tumors usually present either as a distinct hypoechoic mass or a heterogeneous mass that is primarily solid with multiple cystic spaces. Small virilizing SLCTs may be easily detected using color Doppler US rather than transvaginal US alone (13, 45, 46). On CT images, a soft-tissue attenuating adnexal mass is usually seen (15). The solid tumor portions characteristically exhibit avid contrast uptake.
On MRI, the T2 signals of the solid components differ according to the extent of fibrous components. Nonetheless, strong hypointensity on T2-weighted images is not characteristic (13, 18) (Fig. 10). Hypointense areas on T1-weighted images and hyperintense areas on T2-weighted images reflect cystic areas. Cysts may also exhibit mild hyperintensity on T1-weighted images (47). Striking homogeneous or heterogeneous contrast enhancement of the solid areas is observed on gadolinium-enhanced images.
The prognosis of SLCT is good, and 92% of tumors manifest as stage I (13). The most important prognostic factors are the stage and degree of histologic differentiation (3, 13, 48). In a previous report by Sigismondi et al. (49), five-year survival rates of 92.3% and 33.3% were reported for patients with stage I and stage ≥2 disease, respectively. In the same report, patients with well-differentiated tumors had a five-year survival rate of 100% versus 77.8% for patients with moderately and poorly differentiated tumors.
In contrast to GCTs, which tend to recur late, malignant SLCTs usually recur early in the pelvic and abdominal cavity, with a reported relapse rate of 71.4% within two years after diagnosis (49).
Conclusion
Ovarian sex cord-stromal tumors are infrequent tumors that differ from the more frequent epithelial neoplasms via strong associations with hormone-mediated syndromes, presentation in a broad age range, and the near-ubiquitous diagnosis of low-stage disease with a good outcome. These tumors, which develop from cells arising from the primitive sex cords or stromal cells, comprise a diverse group. As a result, these tumors are currently subdivided as pure stromal tumors, pure sex cord tumors, or mixed sex cord-stromal tumors.
The radiologic appearances of these tumors vary along with their morphologies. Notwithstanding, some radiologic features prevail in certain types of tumors. Fibromas typically present as solid hypoechoic and homogenously isodense masses with characteristic hypointensity on T1-weighed images, strong hypointensity on T2-weighted images, and delayed enhancement after contrast administration. Thecomas and GCTs typically associate with hyperestrogenic states that may induce endometrial hyperplasia and endometrial carcinoma. Steroid cell tumors, Leydig tumors, and SLCTs are characteristic virilizing neoplasms. Leydig cell tumors are usually small tumors that may be difficult to depict via different imaging methods; therefore, radiologists should note morphologic changes within the ovary, especially on MRI and transvaginal US with color Doppler. SSTs frequently manifest on US as unilateral tumors comprising star-shaped hypoechoic areas enclosed by solid areas. On MRI, the pseudolobular solid areas exhibit a spoke-wheel pattern and display iso- to hypointensity on T2-weighted images and early and avid contrast uptake.
The rarity of sex cord-stromal tumors contributes to a low index of suspicion; therefore, a thorough knowledge of the clinicopathologic and radiologic findings of these tumors is important and allows radiologists to narrow the differential diagnoses for ovarian tumors, thus facilitating surgical planning and the avoidance of inappropriate treatments.
Main points.
Ovarian sex cord-stromal tumors are infrequent tumors that differ from the more frequent epithelial neoplasms via strong associations with hormone-mediated syndromes, presentation in a broad age range, and the near-ubiquitous diagnosis of low-stage disease with a good outcome.
The World Health Organization sex cord-stromal tumor classification has recently been revised, and currently these tumors have been regrouped into the following clinicopathologic entities: pure stromal tumors, pure sex cord tumors, and mixed sex cord-stromal tumors.
These types of tumors may exhibit characteristic radiologic and clinical features with which radiologists should become familiar in order to narrow the differential diagnoses for ovarian tumors.
Footnotes
Conflict of interest disclosure
The authors declared no conflicts of interest.
References
- 1.Haroon S, Zia A, Idrees R, Memon A, Fatima S, Kayani N. Clinicopathological spectrum of ovarian sex cord-stromal tumors; 20 years’ retrospective study in a developing country. J Ovarian Res. 2013;6:87. doi: 10.1186/1757-2215-6-87. http://dx.doi.org/10.1186/1757-2215-6-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shim SH, Kim DY, Lee SW, et al. Laparoscopic management of early-stage malignant non-epithelial ovarian tumors: surgical and survival outcomes. Int J Gynecol Cancer. 2013;23:249–255. doi: 10.1097/IGC.0b013e318272e754. http://dx.doi.org/10.1097/IGC.0b013e318272e754. [DOI] [PubMed] [Google Scholar]
- 3.Jung SE, Rha SE, Lee JM, et al. CT and MRI findings of sex cord-stromal tumor of the ovary. Am J Roentgenol. 2005;185:207–215. doi: 10.2214/ajr.185.1.01850207. http://dx.doi.org/10.2214/ajr.185.1.01850207. [DOI] [PubMed] [Google Scholar]
- 4.Pratt J. Pathology of the ovary. 1st ed. Philadelphia: Sounders; 2004. pp. 197–226. [Google Scholar]
- 5.Kurman RJ, Carcangiu ML, Herrington CS, Young RH. WHO Classification of Tumours. 4th ed. Vol. 6. Lyon: IARC; 2014. Classification of tumours of the ovary; pp. 44–56. [Google Scholar]
- 6.Tavassoli FA, Devilee P. WHO Classification of Tumours; Tumours of the Breast and Female Genital Organs. 3th ed. Lyon: IARC; 2003. Tumours of the ovary and the peritoneum; pp. 146–162. [Google Scholar]
- 7.Chen VW, Ruiz B, Killeen JL, Coté TR, Wu XC, Correa CN. Pathology and classification of ovarian tumors. Cancer. 2003;97:2631–2642. doi: 10.1002/cncr.11345. http://dx.doi.org/10.1002/cncr.11345. [DOI] [PubMed] [Google Scholar]
- 8.Shinagare AB, Meylaerts LJ, Laury AR, Mortele KJ. MRI features of ovarian fibroma and fibrothecoma with histopathologic correlation. AJR Am J Roentgenol. 2012;198:W296–303. doi: 10.2214/AJR.11.7221. http://dx.doi.org/10.2214/AJR.11.7221. [DOI] [PubMed] [Google Scholar]
- 9.Oh SN, Rha SE, Byun JY, et al. MRI features of ovarian fibromas: emphasis on their relationship to the ovary. Clin Radiol. 2008;63:529–535. doi: 10.1016/j.crad.2007.10.006. http://dx.doi.org/10.1016/j.crad.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Montoriol PF, Mons A, Da Ines D, Bourdel N, Tixier L, Garcier JM. Fibrous tumours of the ovary: aetiologies and MRI features. Clin Radiol. 2013;68:1276–1283. doi: 10.1016/j.crad.2013.07.005. http://dx.doi.org/10.1016/j.crad.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Yazdani S, Alijanpoor A, Sharbatdaran M, et al. Meigs’ syndrome with elevated serum CA125 in a case of ovarian fibroma/thecoma. Caspian J Intern Med. 2014;5:43–45. [PMC free article] [PubMed] [Google Scholar]
- 12.Ray S, Biswas BK, Mukhopadhyay S. Giant primary ovarian fibrosarcoma: Case report and review of pitfalls. J Cytol. 2012;29:255–257. doi: 10.4103/0970-9371.103946. http://dx.doi.org/10.4103/0970-9371.103946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Outwater EK, Wagner BJ, Mannion C, McLarney JK, Kim B. Sex cord-stromal and steroid cell tumors of the ovary. Radiographics. 1998;18:1523–1546. doi: 10.1148/radiographics.18.6.9821198. http://dx.doi.org/10.1148/radiographics.18.6.9821198. [DOI] [PubMed] [Google Scholar]
- 14.Yen P, Khong K, Lamba R, Corwin MT, Gerscovich EO. Ovarian fibromas and fibrothecomas: sonographic correlation with computed tomography and magnetic resonance imaging: a 5-year single-institution experience. J Ultrasound Med. 2013;32:13–18. doi: 10.7863/jum.2013.32.1.13. [DOI] [PubMed] [Google Scholar]
- 15.Hricak H. Fibrothecoma and Sclerosing Stromal Tumor. In: Hricak H, Akin O, Sala E, Ascher SM, Levine D, Reinhold C, editors. Diagnostic Imaging: Gynecology. 1st ed. Salt Lake City: Amirsys; 2007. pp. 728–731. [Google Scholar]
- 16.Jung SE, Lee JM, Rha SE. CT and MR imaging of ovarian tumors with emphasis on differential diagnosis. Radiographics. 2002;22:1305–1325. doi: 10.1148/rg.226025033. http://dx.doi.org/10.1148/rg.226025033. [DOI] [PubMed] [Google Scholar]
- 17.Thomassin-Naggara I, Daraï E, Nassar-Slaba J, Cortez A, Marsault C, Bazot M. Value of dynamic enhanced magnetic resonance imaging for distinguishing between ovarian fibroma and subserous uterine leiomyoma. J Comput Assist Tomogr. 2007;31:236–242. doi: 10.1097/01.rct.0000237810.88251.9e. http://dx.doi.org/10.1097/01.rct.0000237810.88251.9e. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka YO, Tsunoda H, Kitagawa Y, Ueno T, Yoshikawa H, Saida Y. Functioning ovarian tumors: direct and indirect findings at MR imaging. Radiographics. 2004;24:S147–166. doi: 10.1148/rg.24si045501. http://dx.doi.org/10.1148/rg.24si045501. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, Zhang GF, Wang TP, Zhang H. Value of 3.0 T diffusion-weighted imaging in discriminating thecoma and fibrothecoma from other adnexal solid masses. J Ovarian Res. 2013;6:58. doi: 10.1186/1757-2215-6-58. http://dx.doi.org/10.1186/1757-2215-6-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khanna M, Khanna A, Manjari M. Sclerosing stromal tumor of ovary: a case report. Case Rep Pathol. 2012;2012:592836. doi: 10.1155/2012/592836. http://dx.doi.org/10.1155/2012/592836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang YW, Hong SS, Jeen YM, Kim MK, Suh ES. Bilateral sclerosing stromal tumor of the ovary in a premenarchal girl. Pediatr Radiol. 2009;39:731–734. doi: 10.1007/s00247-009-1190-0. http://dx.doi.org/10.1007/s00247-009-1190-0. [DOI] [PubMed] [Google Scholar]
- 22.Yen E, Deen M, Marshall I. Youngest reported patient presenting with an androgen producing sclerosing stromal ovarian tumor. J Pediatr Adolesc Gynecol. 2014;27:e121–124. doi: 10.1016/j.jpag.2013.09.007. http://dx.doi.org/10.1016/j.jpag.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Ismail SM, Walker SM. Bilateral virilizing sclerosing stromal tumours of the ovary in a pregnant woman with Gorlin’s syndrome: implications for pathogenesis of ovarian stromal neoplasms. Histopathology. 1990;17:159–163. doi: 10.1111/j.1365-2559.1990.tb00688.x. http://dx.doi.org/10.1111/j.1365-2559.1990.tb00688.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee MS, Cho HC, Lee YH, Hong SR. Ovarian sclerosing stromal tumors: gray scale and color Doppler sonographic findings. J Ultrasound Med. 2001;20:413–417. doi: 10.7863/jum.2001.20.4.413. [DOI] [PubMed] [Google Scholar]
- 25.Calabrese M, Zandrino F, Giasotto V, Rissone R, Fulcheri E. Sclerosing stromal tumor of the ovary in pregnancy: clinical, ultrasonography, and magnetic resonance imaging findings. Acta Radiol. 2004;45:189–192. doi: 10.1080/02841850410004148. http://dx.doi.org/10.1080/02841850410004148. [DOI] [PubMed] [Google Scholar]
- 26.Kawauchi S, Tsuji T, Kaku T, Kamura T, Nakano H, Tsuneyoshi M. Sclerosing stromal tumor of the ovary: a clinicopathologic, immunohistochemical, ultrastructural, and cytogenetic analysis with special reference to its vasculature. Am J Surg Pathol. 1998;22:83–92. doi: 10.1097/00000478-199801000-00011. http://dx.doi.org/10.1097/00000478-199801000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Souto SB, Baptista PV, Braga DC, Carvalho D. Ovarian Leydig cell tumor in a post-menopausal patient with severe hyperandrogenism. Arq Bras Endocrinol Metabol. 2014;58:68–75. doi: 10.1590/0004-2730000002461. http://dx.doi.org/10.1590/0004-2730000002461. [DOI] [PubMed] [Google Scholar]
- 28.Sakamoto K, Fujimitsu R, Ida M, Horiuchi S, Hamada Y, Yoshimitsu K. MR diagnosis of steroid cell tumor of the ovary: value of chemical shift imaging. Magn Reson Med Sci. 2009;8:193–195. doi: 10.2463/mrms.8.193. http://dx.doi.org/10.2463/mrms.8.193. [DOI] [PubMed] [Google Scholar]
- 29.Saida T, Tanaka YO, Minami M. Steroid cell tumor of the ovary, not otherwise specified: CT and MR findings. AJR Am J Roentgenol. 2007;188:W393–4. doi: 10.2214/AJR.06.0867. http://dx.doi.org/10.2214/AJR.06.0867. [DOI] [PubMed] [Google Scholar]
- 30.Jiang W, Tao X, Fang F, Zhang S, Xu C. Benign and malignant ovarian steroid cell tumors, not otherwise specified: case studies, comparison, and review of the literature. J Ovarian Res. 2013;6:53. doi: 10.1186/1757-2215-6-53. http://dx.doi.org/10.1186/1757-2215-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kottarathil VD, Antony MA, Nair IR, Pavithran K. Recent advances in granulosa cell tumor ovary: a review. Indian J Surg Oncol. 2013;4:37–47. doi: 10.1007/s13193-012-0201-z. http://dx.doi.org/10.1007/s13193-012-0201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crew KD, Cohen MH, Smith DH, Tiersten AD, Feirt NM, Hershman DL. Long natural history of recurrent granulosa cell tumor of the ovary 23 years after initial diagnosis: a case report and review of the literature. Gynecol Oncol. 2005;96:235–240. doi: 10.1016/j.ygyno.2004.09.023. http://dx.doi.org/10.1016/j.ygyno.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 33.Lee IH, Choi CH, Hong DG, et al. Clinicopathologic characteristics of granulosa cell tumors of the ovary: a multicenter retrospective study. J Gynecol Oncol. 2011;22:188–195. doi: 10.3802/jgo.2011.22.3.188. http://dx.doi.org/10.3802/jgo.2011.22.3.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pautier P, Lhommé C, Culine S, et al. Adult granulosa-cell tumor of the ovary: a retrospective study of 45 cases. Int J Gynecol Cancer. 1997;7:58–65. doi: 10.1046/j.1525-1438.1997.00417.x. http://dx.doi.org/10.1046/j.1525-1438.1997.00417.x. [DOI] [PubMed] [Google Scholar]
- 35.Rabban J, Gupta D, Zaloudek CJ, Chen L. Synchronous ovarian granulosa cell tumor and uterine serous carcinoma: A rare association of a high-risk endometrial cancer with an estrogenic ovarian tumor. Gynecol Oncol. 2006;103:1164–1168. doi: 10.1016/j.ygyno.2006.09.005. http://dx.doi.org/10.1016/j.ygyno.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 36.Hammer A, Lauszus FF, Petersen AC. Ovarian granulosa cell tumor and increased risk of breast cancer. Acta Obstet Gynecol Scand. 2013;92:1422–1425. doi: 10.1111/aogs.12252. http://dx.doi.org/10.1111/aogs.12252. [DOI] [PubMed] [Google Scholar]
- 37.Ohel G, Kaneti H, Schenker JG. Granulosa cell tumors in Israel: a study of 172 cases. Gynecol Onc. 1983;15:278–286. doi: 10.1016/0090-8258(83)90083-5. http://dx.doi.org/10.1016/0090-8258(83)90083-5. [DOI] [PubMed] [Google Scholar]
- 38.Gittleman AM, Price AP, Coren C, Akhtar M, Donovan V, Katz DS. Juvenile granulosa cell tumor. Clin Imaging. 2003;27:221–224. doi: 10.1016/s0899-7071(02)00586-7. http://dx.doi.org/10.1016/S0899-7071(02)00586-7. [DOI] [PubMed] [Google Scholar]
- 39.Malmström H, Högberg T, Risberg B, Simonsen E. Granulosa cell tumors of the ovary: prognostic factors and outcome. Gynecol Oncol. 1994;52:50–55. doi: 10.1006/gyno.1994.1010. http://dx.doi.org/10.1006/gyno.1994.1010. [DOI] [PubMed] [Google Scholar]
- 40.Rosario R, Wilson M, Cheng WT, et al. Adult granulosa cell tumours (GCT): clinicopathological outcomes including FOXL2 mutational status and expression. Gynecol Oncol. 2013;131:325–329. doi: 10.1016/j.ygyno.2013.08.031. http://dx.doi.org/10.1016/j.ygyno.2013.08.031. [DOI] [PubMed] [Google Scholar]
- 41.Caringella A, Loizzi V, Resta L, Ferreri R, Loverro G. A case of Sertoli-Leydig cell tumor in a postmenopausal woman. Int J Gynecol Cancer. 2006;16:435–438. doi: 10.1111/j.1525-1438.2006.00203.x. http://dx.doi.org/10.1111/j.1525-1438.2006.00203.x. [DOI] [PubMed] [Google Scholar]
- 42.Nicoletto MO, Caltarossa E, Donach M, Nardelli GB, Parenti A, Ambrosini A. Sertoli cell tumor: a rare case in an elderly patient. Eur J Gynaecol Oncol. 2006;27:86–87. [PubMed] [Google Scholar]
- 43.El-Bahrawy M. Alpha-fetoprotein-producing non-germ cell tumours of the female genital tract. Eur J Cancer. 2010;46:1317–1322. doi: 10.1016/j.ejca.2010.01.028. http://dx.doi.org/10.1016/j.ejca.2010.01.028. [DOI] [PubMed] [Google Scholar]
- 44.Jashnani KD, Hegde CV, Munot SP. Alfa-fetoprotein secreting ovarian sex cord-stromal tumor. Indian J Pathol Microbiol. 2013;56:54–56. doi: 10.4103/0377-4929.116152. http://dx.doi.org/10.4103/0377-4929.116152. [DOI] [PubMed] [Google Scholar]
- 45.Outwater EK, Marchetto B, Wagner BJ. Virilising tumors of the ovary: imaging features. Ultrasound Obstet Gynecol. 2000;15:365–371. doi: 10.1046/j.1469-0705.2000.00123.x. http://dx.doi.org/10.1046/j.1469-0705.2000.00123.x. [DOI] [PubMed] [Google Scholar]
- 46.Yanushpolsky EH, Brow DL, Smith BL. Localization of small ovarian Sertoli-Leydig cell tumors by transvaginal sonography with color Doppler. Ultrasound Obstet Gynecol. 1995;5:133–135. doi: 10.1046/j.1469-0705.1995.05020133.x. http://dx.doi.org/10.1046/j.1469-0705.1995.05020133.x. [DOI] [PubMed] [Google Scholar]
- 47.Hamm B, Forstner R, Baert AL, Knauth M, Sartor K. MRI and CT of the female pelvis. New York: Springer; 2007. CT and MRI in ovarian carcinoma; p. 258. [Google Scholar]
- 48.Bhat RA, Lim YK, Chia YN, Yam KL. Sertoli-Leydig cell tumor of the ovary: analysis of a single institution database. J Obstet Gynaecol Res. 2013;39:305–310. doi: 10.1111/j.1447-0756.2012.01928.x. http://dx.doi.org/10.1111/j.1447-0756.2012.01928.x. [DOI] [PubMed] [Google Scholar]
- 49.Sigismondi C, Gadducci A, Lorusso D, et al. Ovarian Sertoli-Leydig cell tumors. a retrospective MITO study. Gynecol Oncol. 2012;125:673–676. doi: 10.1016/j.ygyno.2012.03.024. http://dx.doi.org/10.1016/j.ygyno.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 50.Martins I, Félix A, Cunha TM. Steroid cell tumour of the ovary - a case report {Online} Available at: http://www.eurorad.org/case.php?id=2798.0.