

Abstract
We report on a ‘green’ method for the utilization of carbon dioxide as C1 unit for the regioselective synthesis of (E)‐cinnamic acids via regioselective enzymatic carboxylation of para‐hydroxystyrenes. Phenolic acid decarboxylases from bacterial sources catalyzed the β‐carboxylation of para‐hydroxystyrene derivatives with excellent regio‐ and (E/Z)‐stereoselectivity by exclusively acting at the β‐carbon atom of the C=C side chain to furnish the corresponding (E)‐cinnamic acid derivatives in up to 40% conversion at the expense of bicarbonate as carbon dioxide source. Studies on the substrate scope of this strategy are presented and a catalytic mechanism is proposed based on molecular modelling studies supported by mutagenesis of amino acid residues in the active site.
Keywords: biotransformations, enzyme catalysis, para‐hydroxystyrenes, phenolic acid decarboxylases, reaction mechanism, regioselective carboxylation
Introduction
The use of CO2 as a C1 carbon source for the synthesis of valuable organic compounds and polymeric materials has become very attractive,1 although the high demand of energy required for the activation of carbon dioxide represents a tough challenge for its synthetic utilization. Nature has solved the disfavored energetic balance of CO2 functionalization through the utilization of light.2 In recent years, numerous chemical carboxylation strategies based on (transition) metal catalysis have been developed, predominantly replacing harsh reaction conditions by eco‐friendly catalytic processes. A large variety of substrates were successfully applied to (transition) metal‐catalyzed C—C bond forming reactions utilizing CO2 as C1 unit. Overall, CO2 activation proceeds via four mechanistic pathways depending on its coordination to the metal centre:3
-
i
The Kolbe–Schmitt reaction represents the first pioneering example for CO2 fixation onto electron‐rich aromatics, such as phenols, which is exemplified by the production of salicylic acid on an industrial scale.4
-
ii
Carbon dioxide fixation onto isolated and conjugated (di)enes, (di)ynes and allenes proceeds via oxidative cycloaddition catalyzed by low‐valent transition metal [Ni(0)] complexes and yields the corresponding carboxylate derivatives via a Hoberg‐type oxanickelacycle species.5 Depending on the substrate type, products were obtained in moderate to high yields with acceptable to excellent regio‐ and stereoselectivities.3a,b
-
iii
The carboxylation of arylalkenes proceeds via the corresponding boronic esters catalyzed by Cu(I) complexes bearing bis‐oxazoline6 or N‐heterocyclic carbene ligands.7 Beside a large number of arylboronic esters, which underwent carboxylation at the aromatic system, a few (para‐substituted) α‐ and β‐styrylboronic esters were carboxylated at the respective side‐chain position in up to 92% yield. Furthermore, the unsubstituted β‐styrylboronic ester was carboxylated by a Rh catalyst with 69% yield.3a,b,6 The carboxylation of organozinc reagents with both palladium and nickel catalysts has so far only been reported for aromatic or aliphatic substrates.3b,d
-
iv
In the reductive (hydro)carboxylation a Ni(II)8 or Fe(II)9 complex acts as catalyst in combination with Et2Zn (for the Ni‐catalyzed process) or EtMgBr (for the Fe‐catalyzed process) as hydride/reductant source, to furnish the reductive carboxylation of styrene‐type alkenes at the α‐position of the C=C bond yielding the corresponding saturated α‐arylcarboxylic acids as major product, whereas β‐carboxylation products were detected only in traces.10 The Ni‐catalyzed process has been successfully applied to various electron‐deficient and electron‐neutral styrene derivatives, whereas the Fe‐catalyzed variant showed excellent results for electron‐rich styrenes. Overall, the reductive carboxylation has also been applied to different substrate classes3b,8,11 including aryl halides12 and allyl acetate derivatives13 which are rare examples for originally electrophilic compounds entirely converted to nucleophiles during the transformation with carbon dioxide.
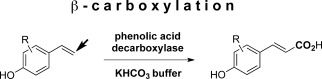
In contrast, the biocatalytic approach described herein is complementary and exclusively yields (E)‐cinnamic acid derivatives through a regioselective redox‐neutral carboxylation, which does not have a direct counterpart in traditional chemical methodology.14 The corresponding products of the β‐carboxylation, such as para‐coumaric or ferulic acid, exhibit diverse roles in nature, particularly in plants (cross‐linkers in the cell walls, signalling and defence molecules).15 Furthermore, ferulic acid has therapeutic functions as antioxidant, antimicrobial, anticancer and anti‐inflammatory agent and is used in food and cosmetic applications.16 The enzymatic strategy makes use of (de)carboxylases acting in reverse.17 These cofactor‐independent, oxygen‐stable enzymes are involved in the detoxification of phenolic species and act under mild reaction conditions at the expense of bicarbonate as CO2 source.14,18 Whereas ortho‐benzoic acid decarboxylases catalyze the highly regioselective ortho‐carboxylation of a broad range of phenol‐type substrates as a biocatalytic equivalent to the Kolbe–Schmitt reaction,14,17,18a,b,19 the regiocomplementary β‐carboxylation of styrene derivatives is achieved by phenolic acid decarboxylases (PADs) (Scheme 1).14,20
Scheme 1.
Regioselective β‐carboxylation of para‐hydroxystyrenes catalyzed by phenolic acid decarboxylases yielding (E)‐cinnamic acids.
Results and Discussion
Enzyme Candidates
In nature, phenolic acid decarboxylases (PADs) catalyze the decarboxylation of cinnamic acids, such as ferulic, caffeic, coumaric and sinapinic acids, which are derived via oxidative biodegradation of lignin through the action of Mn‐peroxidase and related enzymes.21 The reverse β‐carboxylation of para‐hydroxystyrenes was discovered only recently by employing PADs from Lactobacillus plantarum (PAD_Lp) and Bacillus amyloliquefaciens (PAD_Ba).14 Both enzymes were able to catalyze the energetically ‘uphill’ carboxylation in the presence of elevated concentrations of bicarbonate (3 M) using the ‘natural’ substrates para‐vinylphenol (1a) and 2‐methoxy‐4‐vinylphenol (2a) yielding para‐coumaric and ferulic acid, respectively, with low to moderate conversion (2–30%). Encouraged by these results a sequence similarity search was initiated in the NCBI gene bank taking PAD_Lp as a lead.22 The relationship of PADs/FDC was set within a range of 40–80% identity to ensure a reasonably broad coverage of enzyme candidates with complementary substrate tolerance. Both too closely related (iso)enzymes with identical substrate preference and too distant relatives lacking the desired activity were excluded. Based on these constraints, the following phenolic/ferulic acid decarboxylases were chosen: phenolic acid decarboxylases from Mycobacterium colombiense (PAD_Mc), Methylobacterium sp. (PAD_Ms), Pantoea sp. (PAD_Ps), Lactoccocus lactis (PAD_Ll) and ferulic acid decarboxylase (FDC) from Enterobacter sp. (FDC_Es) (Table 1, for sequence alignment see Figure S6 in the Supporting Information). In order to analyze the evolutionary relationship of PADs/FDC (acting on the β‐C atom of styrenes) with ortho‐benzoic acid decarboxylases (acting on the ortho‐position of phenols), a phylogenetic tree was calculated based on primary amino acid sequences using 2,6‐dihydroxybenzoic acid decarboxylase from Rhizobium sp. (2,6‐DHBD_Rs) as representative (Figure 1). As Figure 1 shows, the sequence relationship of 2,6‐DHBD_Rs lies far off (identity <10%) which indicates different folds and mechanism of action.19a
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Figure 1.
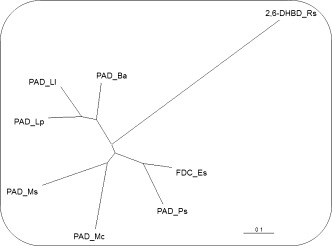
Phylogenetic tree of selected PADs/FDC related to 2,6‐dihydroxybenzoic acid decarboxylase from Rhizobium sp.
The genes encoding for the selected enzyme candidates were synthesized and subcloned into a pET vector. A standard E. coli BL21(DE3) host was used for overexpression which was induced by addition of IPTG. SDS‐PAGE analysis showed successful overexpression of all enzymes, which were applied as lyophilized whole cells for simplified handling. Negative controls confirmed the absence of carboxylation activities in empty host cells.14 In order to verify the expected activities, all biocatalysts were tested in the ‘downhill’ decarboxylation mode using para‐coumaric acid (1b) and ferulic acid (2b) as substrates. After verification of excellent decarboxylation activities of all enzymes, detailed studies in the ‘uphill’ carboxylation direction were performed.
Substrate Scope
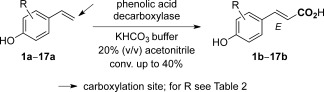
In order to evaluate the substrate scope of PADs/FDC, structurally diverse compounds differing in the electronic and steric properties were investigated (Table 2). In general, all PADs/FDC tolerated a remarkably broad substitution pattern on the aromatic system (1a–8a, entries 1–8), whereas modification of the styrene side chain in the α‐ or β‐position was detrimental (9a, 10a, 16a and 17a, entry 9). With few exceptions, the enzyme candidates showed similar conversions for a given substrate. While PAD_Lp, PAD_Ba and PAD_Mc were rather flexible, the remaining four candidates were somewhat restricted. A common feature of all enzymes is their strict requirement for a para‐hydroxy group, which appears to be a mechanistic prerequisite (see below). Replacing the p‐OH group by a chloro, methoxy or amino group led to complete loss of activity (11a–15a, entry 9). The variety of substituents tolerated in the meta‐position is rather broad and encompasses alkyl and alkoxy groups as well as halogens (entries 2–6). Steric restrictions do not appear with mono‐ and dimethyl analogues (entries 4 and 7), but are apparent with larger methoxy derivatives (entries 2 and 8) and upon increasing the size of the halogens (entries 5 and 6).
Table 2.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
On the other hand, electronic effects played only a minor role, since substrates bearing electron‐donating (entries 2 and 4) and electron‐withdrawing meta‐substituents of approximately the same size (entry 5) showed comparable results. In summary, best results were obtained with substrate 3a bearing an ethoxy group in the meta‐position, where four out of seven decarboxylases showed more than 30% conversion (entry 3). Almost equally good results were achieved with substrate 2a (m‐MeO group), for which all decarboxylases (except PAD_Ll) showed conversions ≥20% (entry 2).
Furthermore, numerous microbial wild‐type organisms from our in house strain collection were screened for β‐carboxylation activity when applied as whole lyophilized cells. Three candidates showed the desired activity: Mycobacterium paraffinicum NCIMB 10420, Fusarium solani DSM 62418 and Bacillus subtilis DSM 10. Surprisingly, the activities observed with substrates 1a and 2a were comparable to those of cloned and overexpressed PADs, and were even superior with substrate 4a. However, they were much lower for the remaining test substrates. This unexpected observation indicates that screening of wild‐type cells might be a promising alternative to the sequence relation‐based approach for the identification of novel carboxylation activities.
Proposed Mechanism
In contrast to Zn2+‐dependent ortho‐benzoic acid decarboxylases,19a PADs/FDC neither contain a metal nor a cofactor, but act through an acid‐base mechanism. Based on the crystal structure of ferulic acid decarboxylase from Enterobacter sp. (2.1 and 2.4 Å resolution, respectively),24 a catalytic mechanism was proposed for the decarboxylation. However, the proposal was later questioned, because the electron density (believed to represent the substrate) was identified as a HEPES buffer molecule.25 An alternative substrate binding mode deeper inside the binding pocked was suggested along with an unprecedented asymmetric hydration of the C=C‐bond as catalytic promiscuity of some PADs/FDC.23 Based on these, we developed a mechanistic proposal for the carboxylation (Scheme 2): Carboxylation is initiated by deprotonation of the essential para‐hydroxy group of the substrate through Tyr19, which is activated through a hydrogen‐bonding network by Tyr21. This induces a flow of electron density through the aromatic core to C‐β of the alkene moiety. In the next step, C‐β launches a nucleophilic attack on bicarbonate, which is tightly coordinated through a salt bridge and H‐bonds onto Arg49. The latter leads to the formation of a charge‐stabilized quinone‐methide oxyanion intermediate.26,27 Elimination of water through the aid of Glu72 initiates tautomerization which forms the product by regaining aromaticity.
Scheme 2.
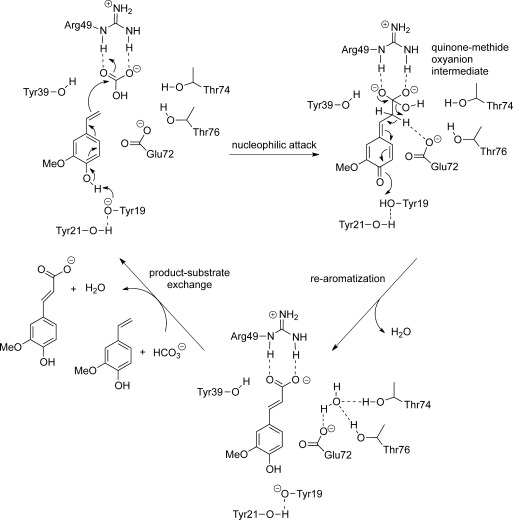
Proposed catalytic mechanism for the β‐carboxylation of para‐hydroxystyrene (2a) based on docking experiments in the crystal structure of ferulic acid decarboxylase from Enterobacter sp. [PDB entry 3NX2]24 and mutant experiments (Table 3).
Mechanistic proposals for the decarboxylation of para‐coumaric acid by para‐coumaric acid decarboxylase from Lactobacillus plantarum (PDB entry 2W2A)26 and phenolic acid decarboxylase from Bacillus subtilis (PDB entry 4ALB)28 suggested the same catalytic residues, but a 180° flipped substrate orientation. However, the substrate orientation proposed in Figure 2 is analogous to that in hydroxycinnamoyl‐CoA hydratase‐lyase (HCHL) which involves the same oxyanion quinine‐methide intermediate.23,29 Furthermore, Arg49 is a much better activator for bicarbonate than Tyr19 and/or Tyr21.
Figure 2.
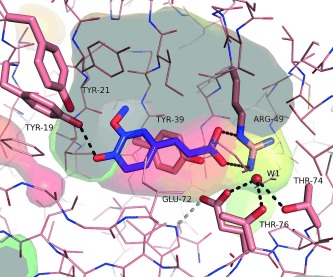
Ferulic acid (blue) docked into the active site of ferulic acid decarboxylase (PDB entry 3NX2). The transparent surface of the active site cavity is coloured to indicate hydrophobicity (hydrophobic=red, hydrophilic=green‐blue).31 Gray areas indicate the interior of the cavity (cut open by plane), catalytically important amino acid residues are shown in stick representation and are labelled. A coordinated water molecule (W1) is shown as a ball. The picture was generated using PyMOL[32].
To confirm the substrate orientation, co‐crystallization and soaking experiments with 2‐methoxy‐4‐vinylphenol (2a) and ferulic acid (2b), respectively, were performed but no substrate‐complex structure could be obtained. However, we determined two high‐resolution structures of wild‐type FDC_Es and the E72A variant (Supporting Information, Figure S4) with a resolution of 1.15 Å and 1.5 Å, respectively. The enzyme exhibits the lipocalin‐like fold, common for this enzyme family.24,26,28,30 An additional electron density within the active site was detected in both structures and only [SO4]2− or [PO4]3− could be fitted with certainty into the cavity of the E72A variant (Supporting Information, Figure S5). Both phosphate and sulfate were present in the protein storage buffer and in the crystallization conditions, respectively. The phosphate or sulfate group in the active site is situated at the same position as the carboxylate anion of the docked ferulic acid, which supports the binding of the ferulic acid in the proposed orientation (Figure 2, Supporting Information, Figure S5). In the same fashion, a sulfate ion was bound in the active site of Bacillus pumilus phenolic acid decarboxylase (PDB entry 3NAD)30 and para‐coumaric acid decarboxylase from Lactobacillus plantarum (PDB entry 2GC9, Joint Center for Structural Genomics).
In order to support the proposed catalytic mechanism, amino acid residues in the active site of ferulic acid decarboxylase from Enterobacter sp. were mutated (Table 3) and the carboxylation activity of enzyme variants was verified using substrate 2a. The replacement of both Tyr19 and Tyr21 by Phe led to a sharp drop in conversion indicating their importance in catalysis. Remarkably, Tyr39, which is close to C‐β of the substrate does not take part in catalysis, since mutation Y39F had no effect on the carboxylation activity. In contrast, the presence of Glu72 is mandatory, because it provides an appropriate environment for the elimination of water (assisted by Thr76 and Thr74), which goes in hand with re‐aromatization. After product release, another substrate molecule and bicarbonate can enter the active site to initiate the next cycle.
Table 3.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Conclusions
The enzymatic carboxylation of styrene‐type substrates catalyzed by phenolic acid decarboxylases at the expense of bicarbonate as CO2 source exclusively proceeded at the C‐β atom of the alkene side chain to yield the corresponding (E)‐cinnamic acids. The substrate tolerance is remarkably broad and encompasses derivatives bearing alkyl, alkoxy and halogen groups on the aromatic moiety. On the other hand, a para‐hydroxy group was mandatory and substitution of the vinyl side chain was not allowed. Incomplete conversions in the energetically unfavoured uphill direction may be improved through reaction engineering.33 Based on data obtained with single‐point mutants, which are supported by molecular modelling studies and crystal structure determination, a mechanism for the β‐carboxylation of para‐hydroxystyrenes catalyzed by phenolic acid decarboxylases has been proposed. It is noteworthy that no counterpart in traditional chemical methodology exists for this biocatalytic method.
Experimental Section
General
2‐Methoxy‐4‐vinylphenol (2a, >98%), para‐coumaric acid (1b, >98%), isoeugenol (9a, 98%), ferulic acid (2b, 99%) and sinapic acid (8b, >98%) were purchased from Sigma Aldrich, para‐vinylphenol (1a, ∼10% solution in propylene glycol) from Alfa Aesar, (E)‐3‐(3‐ethoxy‐4‐hydroxyphenyl)acrylic acid (3b, >98.5%) from ChemCollect GmbH and petroleum ether (boiling range 60–95 °C) from VWR. THF was freshly distilled from potassium.
TLCs were run on silica plates (Merck, silica gel 60, F254), for column chromatography silica gel 60 from Merck was used, compounds were visualized using UV (254 nm) and cerium ammonium molybdate [5 g (CeSO4)2, 25 g (NH4)6Mo7O24⋅4 H2O, 50 mL conc. H2SO4, 450 mL H2O]. NMR spectra were acquired on a Bruker Avance III 300 MHz spectrometer using a 5 mm BBO probe with z‐axis gradients at 300 K. Chemical shifts (δ) are reported in ppm and coupling constants (J) are given in Hz. Protein concentrations were determined spectrophotometrically at λ=280 nm with a NanoDrop apparatus from ThermoScientific. Crystallizations were set up with an Oryx 8 Douglas Instruments robot using the sitting drop method.
Phenolic acid decarboxylase from Lactobacillus plantarum (PAD_Lp, GI: 300769086) and from Bacillus amyloliquefaciens (PAD_Ba, GI: 308175189), subcloned in a pET 28a (+) vector, were kindly provided by Byung‐Gee Kim (Seoul, South Korea). Phenolic acid decarboxylase from Mycobacterium colombiense (PAD_Mc), Methylobacterium sp. (PAD_Ms), Pantoea sp. (PAD_Ps), Lactoccocus lactis (PAD_Ll) and ferulic acid decarboxylase from Enterobacter sp. (FDC_Es) were synthesized at Life Technologies (Germany). DNA sequences were allocated in the NCBI Genebank (GI: 212525355 for FDC_Es, GI: 304396594 for PAD_Ps, GI: 15673912 for PAD_Ll, GI: 342860341 for PAD_Mc, GI: 168197631 for PAD_Ms). The overexpression of PADs/FDC was performed as previously reported.23
General Screening Procedure for β‐Carboxylation
Carboxylation reactions were performed in glass vials capped with septums. Lyophilized whole cells (30 mg E. coli host cells containing the corresponding overexpressed enzyme or whole wild‐type microbial cells) were resuspended in phosphate buffer (800 μL or 700 μL, pH 5.5, 100 mM) and were rehydrated by shaking at 120 rpm at 30 °C for 30 min. The substrate was added either directly or from a stock solution (100 μL of stock solution) to yield a final concentration of 10 mM, followed by addition of acetonitrile (20% v/v, 200 μL) and KHCO3 (3 M, 300 mg). The vials were tightly closed and the mixture was shaken at 30 °C and 120 rpm for 24 h. Thereafter the mixture was centrifuged (13000 rpm, 15 min), an aliquot of 100 μL was diluted with 1 mL of an H2O/acetonitrile mixture (v/v 1:1) supplemented with trifluoroacetic acid (3% v/v, 30 μL). After incubation at room temperature for 5 min, the samples were recentrifuged (13000 rpm, 15 min) and the conversions were analyzed on a reverse‐phase HPLC. Products were identified by comparison with authentic reference material.
Synthesis of Substrates and Reference Material: 2‐Bromo‐4‐vinylphenol (6a), 2,6‐Dimethyl‐4‐vinyl‐ phenol (7a), 4‐(Prop‐1‐en‐2‐yl)phenol (10a) and 2‐Methoxy‐5‐vinylphenol (11a)34
Sodium bis(trimethylsilyl)amide (1.05 g, 5.7 mmol, Sigma Aldrich, 95 %) was added under cooling (ice bath) to a stirred solution of methyl(triphenyl)phosphonium bromide (1.85 g, 5.2 mmol, Sigma Aldrich, 98 %) in freshly distilled THF (8 mL), which caused a yellow colour change. After 1.5 h of stirring at room temperature the corresponding solid aldehyde (2.54 mmol, Sigma Aldrich, >97%) was added to the ylide solution and stirring was continued for 4 h. The mixture was acidified using H2SO4 (0.1 M, 5 mL) and extracted with CH2Cl2. The combined organic phases were dried over Na2SO4, evaporated and purified by flash chromatography on silica.
2‐Bromo‐4‐vinylphenol (6a): Eluent for flash chromatography: petroleum ether/EtOAc 2/1; isolated yield: 80 mg (0.40 mmol, 16%); TLC: R f=0.75 (petroleum ether/EtOAc 2/1); GC‐MS: m/z=198; 1H NMR (acetone‐d 6): δ=4.99 (1 H, d, J=10.95 Hz), 5.54 (1 H, dd, J=0.67 and 17.61 Hz), 6.50 (1 H, dd, J=10.95 and 17.62 Hz), 6.84 (1 H, d, J=8.37 H), 7.19 (1 H, dd, J=2.03 and 8.37 Hz), 7.48 (1 H, d, J=2.03 Hz), 8.80 (1 H, s); 13C NMR (acetone‐d 6): δ=109.7, 111.8, 116.4, 126.5, 130.7, 131.2, 135.2, 153.7.
2,6‐Dimethyl‐4‐vinylphenol (7a): Eluent for flash chromatography: petroleum ether/EtOAc 10/1; isolated yield: 243 mg (1.64 mmol, 66%); TLC: R f=0.51 (petroleum ether/EtOAc 10/1); GC‐MS: m/z=148; 1H NMR (acetonitrile‐d 3): δ=2.21 (6 H, s), 5.06 (1 H, d, J=10.92 Hz), 5.61 (1 H, d, J=17.64 Hz), 6.14 (1 H, s), 6.61 (1 H, dd, J=10.93 and 17.64 Hz), 7.07 (2 H, s); 13C NMR (acetonitrile‐d 3): δ=15.6, 110.2, 123.8, 126.4, 129.4, 136.7, 152.9.
4‐(Prop‐1‐en‐2‐yl)phenol (10a): Eluent for flash chromatography: petroleum ether/EtOAc 2/1; isolated yield: 48 mg (0.35 mmol, 14%); TLC: R f=0.71 (petroleum ether/EtOAc 2/1); GC‐MS: m/z=134; 1H NMR (acetonitrile‐d 3): δ=2.03 (3 H, m), 4.89–4.90 (1 H, m), 5.21 (1 H, m), 6.69–6.71 (2 H, m), 7.29–7.32 (2 H, m); 13C NMR (acetonitrile‐d 3): δ=20.7, 109.3, 114.6, 126.3, 132.3, 142.5, 156.2.
2‐Methoxy‐5‐vinylphenol (11a): Eluent for flash chromatography: petroleum ether/EtOAC 2/1; isolated yield: 264 mg (1.76 mmol, 71%); TLC: R f=0.64 (petroleum ether/EtOAC); GC‐MS: m/z=150; 1H NMR (CDCl3): δ=3.91 (3 H, s), 5.15 (1 H, dd, J=0.83 and 10.84 Hz), 5.61 (1 H, s), 5.62 (1 H, dd, J=0.87 and 17.55 Hz), 6.64 (1 H, dd, J=10.87 and 17.55 Hz), 6.82 (1 H, d, J=8.28 Hz), 6.90 (1 H, dd, J=2.01 and 8.28 Hz), 7.07 (1 H, d, J=2.03 Hz); 13C NMR (CDCl3): δ=5.60, 110.5, 111.6, 112.1, 118.8, 131.5, 136.3, 145.7, 146.5.
(E)‐3‐(4‐Hydroxy‐3‐methylphenyl)acrylic Acid (4b) and (E)‐3‐(4‐Hydroxy‐3,5‐dimethylphenyl)acrylic Acid (7b)35
For the protection of the phenolic OH group, imidazole (328 mg, 4.8 mmol, Sigma Aldrich,>99.5 %) and TBDMS (361 mg, 2.4 mmol) were added to a stirred solution of the corresponding aldehyde (2 mmol, Sigma Aldrich, >97%) in dry THF (4 mL). The reaction mixture turned milky and stirring was continued for 2 h at room temperature. Thereafter the TBDMS protected benzaldehyde was filtered, washed with THF (2 mL) and purified by flash chromatography. The purified compound (1.0 mmol) was mixed with malonic acid (229 mg, 2.2 mmol, Sigma Aldrich, 99%), pyridine (450 μL, Sigma–Aldrich, 99.8%) and piperidine (18 μL, Fluka, p.a.) followed by stirring under reflux at 130 °C for 3 h. The reaction mixture was cooled to room temperature and HCl (9 mL, 2 M) was added. The resulting precipitate was filtered, dried overnight over P2O5 and purified by flash chromatography.
(E)‐3‐(4‐Hydroxy‐3‐methylphenyl)acrylic acid (4b): Eluent for flash chromatography: petroleum ether/EtOAc 1/1; isolated yield: 65 mg (0.37 mmol, 34%); TLC: R f=0.36 (petroleum ether/EtOAc 1/1); 1H NMR (acetonitrile‐d 3): δ=2.20 (3 H, s), 6.31 (1 H, d, J=15.95 Hz), 6.83 (1 H, d, J=8.92 Hz), 7.33 (1 H, dd, J=1.69 and 8.29 Hz), 7.43 (1 H, s), 7.60 (1 H, d, J=15.93 Hz); 13C NMR (acetonitrile‐d 3): δ=15.1, 114.2, 115.0, 125.1, 126.2, 127.8, 131.0, 145.5, 157.4, 168.2. The aromatic substitution pattern was confirmed by a 2D HMBC experiment (Supporting Information).
(E)‐3‐(4‐Hydroxy‐3,5‐dimethylphenyl)acrylic acid (7b): Eluent for flash chromatography: petroleum ether/EtOAc 2/1; isolated yield: 124 mg (0.65 mmol, 65%); TLC: R f=0.43 (petroleum ether/EtOAc 2/1); 1H NMR (acetone‐d 6): δ=2.13 (6 H, s), 6.19 (1 H, d, J=15.93 Hz), 7.17 (2 H, s), 7.42 (1 H, d, J=15.94 Hz); 13C NMR (acetone‐d 6): δ=15.6, 114.6, 124.4, 126.0, 128.8, 145.1, 155.7, 167.4. The aromatic substitution pattern was confirmed by a 2D HMBC experiment (Supporting Information).
(E)‐3‐(3‐Chloro‐4‐hydroxyphenyl)acrylic Acid (5b) and (E)‐3‐(3‐Bromo‐4‐hydroxyphenyl)acrylic Acid (6b)
The corresponding aldehyde (1.0 mmol, Sigma Aldrich) was mixed with malonic acid (229 mg, 2.2 mmol, Sigma Aldrich, 99%), pyridine (450 μL, Sigma–Aldrich, 99.8%) and piperidine (18 μL, Fluka) at room temperature and stirred under reflux at 80 °C. After 5 h the reaction mixture was cooled to room temperature and HCl (9 mL, 2 M) was added. The resulting precipitate was filtered, dried overnight over P2O5 and purified by flash chromatography if necessary.
(E)‐3‐(3‐Chloro‐4‐hydroxyphenyl)acrylic acid (5b): flash chromatography was not required; isolated yield; 101 mg (0.55 mmol, 55%); TLC: R f=0.46 (petroleum ether/EtOAc 2/1); 1H NMR (acetonitrile‐d 3): δ=6.28 (1 H, d, J=15.98 Hz), 6.94 (1 H, d, J=8.45 Hz), 7.37 (1 H, dd, J=2.14 and 8.48 Hz), 7.49 (1 H, d, J=16.00 Hz), 7.57 (1 H, d, J=2.13 Hz); 13C NMR (acetonitrile‐d 3): δ=116.2, 116.9, 120.7, 127.7, 128.4, 129.8, 143.6, 154.3, 167.4. The aromatic substitution pattern was confirmed by a 2D HMBC (Supporting Information).
(E)‐3‐(3‐Bromo‐4‐hydroxyphenyl)acrylic acid (6b): Eluent for flash chromatography: petroleum ether/EtOAc 2/1; isolated yield: 101 mg (0.41 mmol, 41%), TLC: R f=0.15 (petroleum ether/EtOAc 2/1); 1H NMR (acetone‐d 6): δ=6.29 (1 H, d, J=15.79 Hz), 6.93 (1 H, d, J=8.44 Hz); 7.41–7.47 (2 H, m), 7.74 (1 H, d, J=2.04 Hz); 13C NMR (acetone‐d 6): δ=110.1, 116.7, 116.7, 128.0, 128.8, 133.0, 143.0, 155.9, 167.1. The aromatic substitution pattern was confirmed by a 2D HMBC (Supporting Information).
Analytical Procedures
All GC‐MS measurements were carried out on an Agilent 7890 A GC system, equipped with an Agilent 5975C mass‐selective detector (electron impact, 70 eV) and a HP‐5‐MS column (30 m×0.25 mm×0.25 μm film) using He as carrier gas at a flow of 0.55 mL min−1. The following temperature program was used for all GC‐MS measurements: initial temperature 100 °C, hold for 0.5 min, 10 °C min−1 to 300 °C.
Determination of Conversion
All analyses were carried out on a Shimadzu HPLC system equipped with a diode array detector (SPD‐M20 A) and a reversed‐phase Phenomenex Luna column C18 (2) 100 A (250 mm×4.6 mm×5 μm, column temperature 24 °C). Conversions were determined by comparison with calibration curves for products and substrates prepared with authentic reference material. The method was run over 17 min with H2O/TFA (0.1%) as the mobile phase at a flow rate of 1 mL min−1 and an MeCN/TFA (0.1%) gradient (0–2 min 0%, 2–15 min 0–100%, 15–17 min 100%). The column temperature was 24 °C and compounds were spectrophotometrically detected at 270 and 280 nm, respectively. Retention times: 1a 13.6 min, 1b 11.3 min, 2a 14.2 min, 2b 11.5 min, 3a 15.4 min, 3b 13.0 min, 4a 15.0 min, 4b 12.0 min, 5a 15.2 min, 5b 12.3 min, 6a 15.4 min, 6b 12.5 min, 7a 16.1 min, 7b 12.8 min, 8a 13.8 min, 8b 11.5 min.
Protein Mutagenesis
All FDC_Es mutants were constructed using the ‘QuickChange Site‐Directed Mutagenesis PCR Kit’ from Stratagene. The mutagenic primers used to introduce the amino acid changes were the following (FP=forward primer, RP=reverse primer):
FDC_Es Y19F: FP‐Y19F (GTTGGTAAACATCTGGTGTTTACCTATGATAATGGTGG) and RP‐Y19F (CCAGCCATTATCATAGGTAAACACCAGATGTTTACCAAC);
FDC_Es Y21F: FP‐Y21F (CATCTGGTGTATACCTTTGATAATGGCTGGGAG) and RP‐Y21F (CTCCCAGCCATTATCAAAGGTATACACCAGATG);
FDC_Es Y39F: FP‐Y39F (AATGAAAACACCCTGGATTTTCGCATTCATAGCGGTCTG) and RP‐Y39F (CAGACCGCTATGAATGCGAAAATCCAGGGTGTTTTCATT);
FDC_E72A: FP‐E72A (AATCAGCTGGACCGCGCCGACCGGCACCG) and RP‐E72 A (CGGTGCCGGTCGGCGCGGTCCAGCTGATT).
Structure Determination
Purification and crystallization: Expressed wild‐type protein (FDC_Es WT) and the FDC_Es E72A variant were purified by a two‐step procedure via His‐tag affinity chromatography and anion exchange chromatography (MonoQ, GE Healthcare), respectively, followed by size‐exclusion chromatography (Superdex200 HiLoad 1660 column, GE Healthcare) using a multicomponent buffer containing MES, l‐malic acid, Tris (100 mM, pH 7.0) and NaCl (250 mM).36 For the crystallization of FDC_Es E72A variant, the buffer was changed to succinic acid, sodium dihydrogen phosphate monohydrate and glycine (100 mM, pH 7.0).36 Concentration of the proteins was determined spectrophotometrically at λ=280 nm.
Crystallizations were set up with an Oryx 8 robot (Douglas Instruments) using the sitting drop method. A drop (0.5 μL) of enzyme solution (∼20 mg mL−1) was mixed with reservoir solution (0.5 μL) using standard screens (Index and Morpheus) and incubated at 20 °C. First crystal clusters were obtained for the wild‐type protein and were observable after 24 h. Crystallization conditions were optimized and good diffracting wild‐type crystals were obtained with potassium bromide (0.15 M) and PEG monomethyl ether 2000 (30% w/v). Good diffracting crystals of the FDC_Es E72A variant were obtained after approximately two months in the presence of ammonium sulfate (0.2 M), Bis‐Tris (0.1 M, pH 5.5) and PEG 3350 (25% w/v).
Data collection and structure determination: Complete data sets for the wild‐type protein (1.15 Å resolution) and the FDC_Ec E72A variant (1.50 Å resolution) were collected at the beamline BM30A37 and ID2938 at the ESRF Grenoble, France.
Data were processed using XDS39 and software from the CCP4 suite.40 The structures were solved by molecular replacement with the program Phaser41 using PDB 3NX224 as a search template. Structure rebuilding and refinement was done in Coot42 and phenix.refine.43 The stereochemistry and geometry were analyzed using Molprobity.44 The atomic coordinates and structure factors (PDB codes: 4UU3 for the wild‐type protein and 4UU2 for the FDC_Es E72A variant) have been deposited in the Protein Data Bank. All structure‐related pictures were generated using PyMOL.32 For the analysis of the hydrophobicity of the cavities the hydrophobic calculation part of the program VASCo31 was used. Cavities were calculated using a LIGSITE algorithm.45
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
This work has been supported by the Austrian BMWFW, BMVIT, SFG, Standortagentur Tirol and ZIT through the Austrian FFG‐COMET‐Funding Program. M. Fuchs is thankful to the Austrian Science Fund (FWF) for an Erwin‐Schrödinger fellowship (J3466_N28), Klaus Zangger and his co‐workers (Graz) are thanked for their great support in NMR spectroscopy and Byung‐Gee Kim (Seoul, South Korea) for providing vectors for PAD_Lp and PAD_Ba.
Contributor Information
Silvia M. Glueck, Email: Si.Glueck@Uni-Graz.at
Kurt Faber, Email: Kurt.Faber@Uni-Graz.at.
References
- 1.
- 1a. Aresta M., Dibenedetto A., Angelini A., Chem. Rev. 2014, 114, 1709–1742; [DOI] [PubMed] [Google Scholar]
- 1b. Sakakura T., Choi J.‐C., Yasuda H., Chem. Rev. 2007, 107, 2365–2387; [DOI] [PubMed] [Google Scholar]
- 1c. Quadrelli E. A., Centi G., Duplan J.‐L., Perathoner S., ChemSusChem 2011, 4, 1194–1215. [DOI] [PubMed] [Google Scholar]
- 2. Ishida N., Shimamoto Y., Murakami M., Angew. Chem. 2012, 124, 11920–11922; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012, 51, 11750–11752. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Tsuji Y., Fujihara T., Chem. Commun. 2012, 48, 9956–9964; [DOI] [PubMed] [Google Scholar]
- 3b. Huang K., Sun C.‐L., Shi Z.‐J., Chem. Soc. Rev. 2011, 40, 2435–2452; [DOI] [PubMed] [Google Scholar]
- 3c. Nurhanna Riduan S., Zhang Y., Dalton Trans. 2010, 39, 3347–3357; [DOI] [PubMed] [Google Scholar]
- 3d. Correa A., Martin R., Angew. Chem. 2009, 121, 6317–6320; [Google Scholar]; Angew. Chem. Int. Ed. 2009, 48, 6201–6204. [DOI] [PubMed] [Google Scholar]
- 4. Lindsey A. S., Jeskey H., Chem. Rev. 1957, 57, 583–620. [Google Scholar]
- 5.
- 5a. Burkhart G., Hoberg H., Angew. Chem. 1982, 94, 75; [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1982, 21, 76; [Google Scholar]
- 5b. Hoberg H., Peres Y., Krüger C., Tsay Y.‐H., Angew. Chem. 1987, 99, 799–800; [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1987, 26, 771–772. [Google Scholar]
- 6. Takaya J., Tadami S., Ukai K., Iwasawa N., Org. Lett. 2008, 10, 2697–2700. [DOI] [PubMed] [Google Scholar]
- 7. Ohishi T., Nishiura M., Hou Z., Angew. Chem. 2008, 120, 5876–5879; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008, 47, 5792–5795. [DOI] [PubMed] [Google Scholar]
- 8. Williams C. M., Johnson J. B., Rois T., J. Am. Chem. Soc. 2008, 130, 14936–14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greenhalgh M. D., Thomas S. P., J. Am. Chem. Soc. 2012, 134, 11900–11903. [DOI] [PubMed] [Google Scholar]
- 10. Zhang Y., Riduan S. N., Angew. Chem. 2011, 123, 6334–6336; [Google Scholar]; Angew. Chem. Int. Ed. 2011, 50, 6210–6212. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Shirakawa E., Ikeda D., Masui S., Yoshida M., Hayshi T., J. Am. Chem. Soc. 2012, 134, 272–279; [DOI] [PubMed] [Google Scholar]
- 11b. Li S., Yuan W., Ma S., Angw. Chem. Int. Ed. 2011, 50, 2578–2582; [DOI] [PubMed] [Google Scholar]
- 11c. Fujihara T., Xu T., Semba K., Terao J., Tsuji Y., Angew. Chem. 2011, 123, 543–547; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011, 50, 523–527. [Google Scholar]
- 12.
- 12a. Correa A., Martin R., J. Am. Chem. Soc. 2009, 131, 15974–15975; [DOI] [PubMed] [Google Scholar]
- 12b. Fujihara T., Nogi K., Xu T., Terao J., Tsuji Y., J. Am. Chem. Soc. 2012, 134, 9106–9109. [DOI] [PubMed] [Google Scholar]
- 13. Greco G. E., Gleason B. L., Lowery T. A., Kier M. J., Hollander L. B., Gibbs S. A., Worthy A. D., Org. Lett. 2007, 9, 3817–3818. [DOI] [PubMed] [Google Scholar]
- 14. Wuensch C., Glueck S. M., Gross J., Koszelewski D., Schober M., Faber K., Org. Lett. 2012, 14, 1974–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mandal S. M., Chakraborty D., Dey S., Plant Signal. Behav. 2010, 5, 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ou S., Kwok K.‐C., J. Sci. Food Agric. 2004, 84, 1261–1269. [Google Scholar]
- 17. Glueck S. M., Gümüs S., Fabian W. M. F., Faber K., Chem. Soc. Rev. 2010, 39, 313–328. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Ishii Y., Narimatsu Y., Iwasaki Y., Arai N., Kino K., Kirimura K., Biochem. Biophys. Res. Commun. 2004, 324, 611–620; [DOI] [PubMed] [Google Scholar]
- 18b. Matsui T., Yoshida T., Yoshimura T., Nagasawa T., Appl. Microbiol. Biotechnol. 2006, 73, 95–102; [DOI] [PubMed] [Google Scholar]
- 18c. Wieser M, Fujii N., Yoshida T., Nagasawa T., Eur. J. Biochem. 1998, 257, 495–499. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Wuensch C., Gross J., Steinkellner G., Lyskowski A., Gruber K., Glueck S. M., Faber K., RSC Adv. 2014, 4, 9673–9679; [Google Scholar]
- 19b. Kamath A. V., Dasgupta D., Vaidyanathan C. S., Biochem. Biophys. Res. Commun. 1987, 145, 586–595; [DOI] [PubMed] [Google Scholar]
- 19c. Santha R., Appaji Rao N., Vaidyanathan C. S., Biochim. Biophys. Acta 1996, 1293, 191–200; [DOI] [PubMed] [Google Scholar]
- 19d. Yoshida T., Hayakawa Y., Matsui T., Nagasawa T., Arch. Microbiol. 2004, 181, 391–397; [DOI] [PubMed] [Google Scholar]
- 19e. Yoshida M., Fukuhara N., Oikawa T., J. Bacteriol. 2004, 186, 6855–6863; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19f. Iwasaki Y., Kino K., Nishide H., Kirimura K., Biotechnol. Lett. 2007, 29, 819–822. [DOI] [PubMed] [Google Scholar]
- 20.For decarboxylation activities of PADs see: Jung D.‐H., Choi W., Choi K.‐Y., Jung E., Yun H., Kazlauskas R. J., Kim B.‐G., Appl. Microbiol. Biotechnol. 2013, 97, 1501–1511. [DOI] [PubMed] [Google Scholar]
- 21. Bugg T. D. H., Ahmad M., Hardiman E. M., Singh R., Curr. Opin. Biotechnol. 2011, 22, 394–400. [DOI] [PubMed] [Google Scholar]
- 22. Rodriguez H., de La Rivas B., Munoz R., Mancheno J. M., Acta Crystallogr. 2007, F63, 300–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wuensch C., Gross J., Steinkellner G., Gruber K., Glueck S. M., Faber K., Angew. Chem. 2013, 125, 2349–2353; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2013, 52, 2293–2297. [DOI] [PubMed] [Google Scholar]
- 24. Gu W., Yang J., Lou Z., Liang L., Sun Y., Huang J., Li X., Cao Y., Meng Z., Zhang K.‐Q., PLoS ONE 2011, 6, e16262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0016262.
- 26. Rodriguez H., Angulo I., de Las Rivas B., Campillo N., Paez J. A., Munoz R., Mancheno J. M., Proteins 2010, 78, 1662–1676. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. van den Heuvel R. H. H., Fraaije M. W., van Berkel W. J. H., FEBS Lett. 2000, 481, 109–112; [DOI] [PubMed] [Google Scholar]
- 27b. Mattevi A., Fraaije M. W., Mozzarelli A., Olivi L., Coda A., Van Berkel W. J. H., Structure 1997, 5, 907–920; [DOI] [PubMed] [Google Scholar]
- 27c. Hopper D. J., Cottrell L., Appl. Environ. Microbiol. 2003, 69, 3650–3652; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27d. Reeve C. D., Carver M. A., Hopper D. J., Biochem. J. 1989, 263, 431–437; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27e. Gargouri M., Chaudiere J., Manigand C., Mauge C., Bathany K., Schmitter J.‐M., Gallois B., Biol. Chem. 2010, 391, 219–227; [DOI] [PubMed] [Google Scholar]
- 27f. Sugumaran M., Dali H., Sementi V., Biochem. J. 1991, 277, 849–853; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27g. Saul S. J., Dali H., Sugumaran M., Arch. Insect Biochem. Physiol. 1991, 16, 123–138; [DOI] [PubMed] [Google Scholar]
- 27h. Reilly C. A., Henion F., Bugni T. S., Ethirajan M., Stockmann C., Pramanik K. C., Srivastava S. K., Yost G. S., Chem. Res. Toxicol. 2013, 26, 55–66; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27i. Annan N., Paris R., Jordan F., J. Am. Chem. Soc. 1989, 111, 8895–8889; [Google Scholar]
- 27j. Calisti C., Ficca A. G., Barghini P., Ruzzi M., Appl. Microbiol. Biotechnol. 2008, 80, 475–483. [DOI] [PubMed] [Google Scholar]
- 28. Frank A., Eborall W., Hyde R., Hart S., Turkenburg J. P., Grogan G., Catal. Sci. Technol. 2012, 2, 1568–1574. [Google Scholar]
- 29. Bennett J. P., Bertin L., Moulton B., Fairlamb I. J. S., Brzozowski A. M., Walton N. J., Grogan G., Biochem. J. 2008, 414, 281–289. [DOI] [PubMed] [Google Scholar]
- 30. Matte A., Grosse S., Bergeron H., Abokitse K., Lau P. C. K., Acta Crystallogr. 2010, F66, 1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steinkellner G., Rader R., Thallinger G. G., Kratky C., Gruber K., BMC Bioinform. 2009, 10, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.
- 33. Wuensch C., Schmidt N., Gross J., Grischek B., Glueck S. M., Faber K., J. Biotechnol. 2013, 168, 264–270. [DOI] [PubMed] [Google Scholar]
- 34. Mure M., Wang S. X., Klinman J. P., J. Am. Chem. Soc. 2003, 125, 6113–6125. [DOI] [PubMed] [Google Scholar]
- 35. Szymanski W., Wu B., Weiner B., De Wildeman S., Feringa B. L., Janssen D. B., J. Org. Chem. 2009, 74, 9152–9157. [DOI] [PubMed] [Google Scholar]
- 36. Newman J., Acta Crystallogr. 2004, D60, 610–612. [DOI] [PubMed] [Google Scholar]
- 37. Ferrer J. L., Acta Crystallogr. 2001, D57, 1752–1753. [DOI] [PubMed] [Google Scholar]
- 38. de Sanctis D., Beteva A., Caserotto H., Dobias F., Gabadinho J., Giraud T., Gobbo A., Guijarro M., Lentini M., Lavault B., Mairs T., McSweeney S., Petitdemange S., Rey‐Bakaikoa V., Surr J., Theveneau P., Leonard G. A., Mueller‐Dieckmann C., J. Synchrotron Rad. 2012, 19, 455–461. [DOI] [PubMed] [Google Scholar]
- 39. Kabsch W., Acta Crystallogr. 2010, D66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Acta Crystallogr. 2011, D67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCoy A. J., Grosse‐Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. 2010, D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse‐Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., Acta Crystallogr. 2010, D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen V. B., Arendall 3rd W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., Acta Crystallogr. 2010, D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hendlich M., Rippmann F., Barnickel G., J. Mol. Graph. Model. 1997, 15, 359–363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information