

Abstract
From a series of gold complexes of the type [t-BuXPhosAu(MeCN)]X (X=anion), the best results in intermolecular gold(I)-catalyzed reactions are obtained with the complex with the bulky and soft anion BAr4F− [BAr4F−=3,5-bis(trifluoromethyl)phenylborate] improving the original protocols by 10–30% yield. A kinetic study on the [2+2] cycloaddition reaction of alkynes with alkenes is consistent with an scenario in which the rate-determining step is the ligand exchange to generate the (η2-phenylacetylene)gold(I) complex. We have studied in detail the subtle differences that can be attributed to the anion in this formation, which result in a substantial decrease in the formation of unproductive σ,π-(alkyne)digold(I) complexes by destabilizing the conjugated acid formed.
Keywords: cycloaddition, cyclobutenes, gold catalysis, mechanistic study
Introduction
Gold(I)-catalyzed intramolecular cycloisomerization reactions have been widely studied during the last decade.[1] Gold(I) complexes have been found to be powerful homogeneous catalysts for carbon-carbon, carbon-oxygen or carbon-nitrogen bond formation proceeding by nucleophilic additions to alkynes, allenes and alkenes, giving access to new carbo- and heterocyclic compounds. Despite these major advances, the development of intermolecular cycloadditions using alkynes as the substrates has been shown to be challenging.[2]
By using cationic gold(I) complexes with bulky ligands, we developed the intermolecular reaction of alkynes with alkenes to form regioselectively cyclobutenes of type 3a (Scheme 1).[3,4] More recently, we have developed a synthesis of phenols 5 by the intermolecular reaction of alkynes with furans such as 4,[5,6] as well as a synthesis of oxabicyclo[3.2.1]oct-3-enes of type 7a by cycloaddition between oxoalkene 6 and 1a.[7]
Scheme 1.
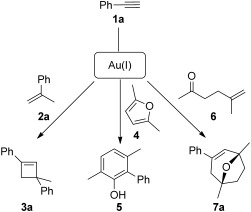
Gold(I)-catalyzed intermolecular reactions.
During our studies on the [2+2+2] cycloaddition of alkynes with oxoalkenes,[7] we discovered that formation of the active gold species was more complex than expected. Although it was possible to observe the phenylacetylene gold(I) complex (8a) at −60 °C, the resting state under the reaction conditions was the unreactive σ,π-(phenylacetylene)digold(I) complex 9a, which decreases the reaction efficiency (Scheme 2).
Scheme 2.
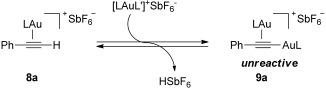
Formation of σ,π-digold(I) complexes from (η2-alkyne)gold(I) complexes (L=t-BuXPhos).
Different research groups reported the formation of very similar digold(I) complexes and their influence on the reactivity in catalytic transformations.[8] In this context, we have focused on tuning the catalyst structure to minimize the formation of digold(I) complexes. In intermolecular reactions involving alkynes, we reasoned that the use of a more bulky, non-coordinating, and less basic counterion could slow down the deprotonation of terminal alkynes to form the σ-acetylide gold(I) intermediates. Hence, we have pre- pared the new gold(I) complexes [t-BuXPhosAu(MeCN)]BAr4F A2 and [IPrAu(PhCN)]BAr4F B2 with the BAr4F− anion [BAr4F−=3,5-bis(trifluoromethyl)phenylborate] (Figure 1).[9] These are close relatives of the corresponding complexes A1[10] and B1[11] with hexafluoroantimonate anion, which have been used as the catalysts of choice in gold(I)-catalyzed intermolecular reactions.[3,4,5,7] Since complex B2 showed slightly better performance than B1 in the intermolecular synthesis of phenols of type 5,[5] we decided to study in detail the effect of the anion on the corresponding t-BuXPhos complexes A1 and A2. Analogous complexes with BF4− and PF6− anions A3 and A4 have also been studied. Herein we present a mechanistic study of the intermolecular [2+2] cycloaddition of alkynes with alkenes in order to understand the influence of the counterion on the reactivity of these processes. This work shows that A2 is the catalyst of choice for intermolecular reactions of terminal alkynes.
Figure 1.
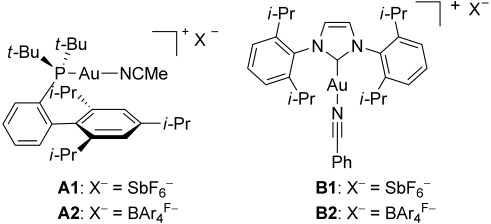
Cationic gold(I) complexes with SbF6− and BAr4F− anions (BAr4F−=3,5-bis(trifluoromethyl)phenylborate).
Results and Discussion
The [2+2] cycloaddition of alkynes with alkenes was developed using complex [t-BuXPhosAu(MeCN)] SbF6 (A1) as catalyst, furnishing regioselectively cyclobutenes 3 in moderate to good yields.[3] This cycloaddition proceeded under mild conditions in dichloromethane at room temperature. Although, as expected, the ligand had a strong influence on the selectivity, we were surprised by the notable difference observed when changing the counterion (Table 1). Thus, replacing SbF6− in A1 by BAr4F− (A2) leads to an increase in the yield of the cycloaddition of 1a with 2a from 80 to 95% (Table 1, entries 1 and 2). The use of BF4−, PF6−, NTf2− or OTf− as counterions led to 3a in lower yields (Table 1, entries 3–6).
Table 1.
Intermolecular gold(I)-catalyzed [2+2] cycloaddition between phenylacetylene (1a) and α-methylstyrene (2a) with different gold(I) catalysts A.[a]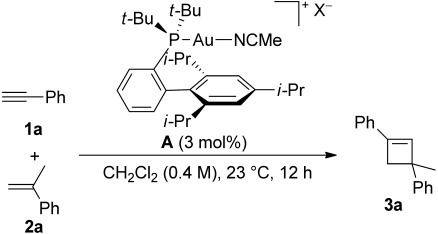
| Entry | X− | 3a(Yield [%])[b] |
|---|---|---|
| 1 | SbF6− | 80 |
| 2 | BAr4F− | 95 |
| 3 | BF4− | 62 |
| 4 | PF6− | 19 |
| 5[c] | NTf2− | 26 |
| 6[c] | OTf− | 18 |
[a] 2a/1a=2:1.
[b] Yield determined by 1H NMR using 1,4-diacetylbenzene as internal standard.
[c] Catalysts generated in situ with [LAuCl] and the corresponding silver salts.
The cycloaddition between different terminal alkynes and 2a using catalysts A1 and A2 is shown in Table 2. In most cases, yields using A2 were 10–30% higher (Table 2), with the exception of MeO-substituted alkynes 1c, 1g, and 1l, which afforded the corresponding cyclobutenes in very similar yields (Table 2, entries 6, 14, and 24). In the case of 1n, a lower yield was obtained (Table 2, entry 28). Cyclobutene 3a was also obtained in 95% yield by performing the reaction on a larger scale (2.0 mmol). Generating in situ A2 by simple mixing of (t-BuXPhos)gold(I) chloride and NaBAr4F did not mean any drop in the yield.
Table 2.
Intermolecular gold(I)-catalyzed [2+2] cycloaddition between alkynes (1a–n) and α-methylstyrene (2a)[a]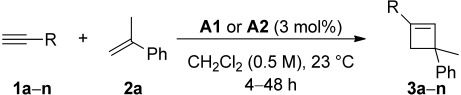
| Entry | R | Catalyst | Product (Yield [%])[b] |
|---|---|---|---|
| 1 | Ph (1a) | A1 | 3a (80)[c] |
| 2 | A2 | 3a (95) | |
| 3 | p-Tol (1b) | A1 | 3b (74)[c] |
| 4 | A2 | 3b (86) | |
| 5 | p-MeOC6H4 (1c) | A1 | 3c (68)[c] |
| 6 | A2 | 3c (64) | |
| 7 | p-FC6H4 (1d) | A1 | 3d (75)[c] |
| 8 | A2 | 3 d (84) | |
| 9 | p-ClC6H4 (1e) | A1 | 3e (61)[c] |
| 10 | A2 | 3 e (91) | |
| 11 | p-BrC6H4 (1f) | A1 | 3f (74)[c] |
| 12 | A2 | 3 f (97) | |
| 13 | m-MeOC6H4 (1g) | A1 | 3g (80) |
| 14 | A2 | 3g (78) | |
| 15 | m-Tol (1h) | A1 | 3h (78)[c] |
| 16 | A2 | 3h (91) | |
| 17 | m-HOC6H4 (1i) | A1 | 3i (74)[c] |
| 18 | A2 | 3i (98) | |
| 19 | m-FC6H4 (1j) | A1 | 3j (67) |
| 20 | A2 | 3j (77) | |
| 21 | m-ClC6H4 (1k) | A1 | 3k (60) |
| 22 | A2 | 3k (83) | |
| 23 | o-MeOC6H4 (1l) | A1 | 3l (30) |
| 24 | A2 | 3l (24) | |
| 25 | 3-thienyl (1m) | A1 | 3m (84) |
| 26 | A2 | 3m (86) | |
| 27 | cyclopropyl (1n) | A1 | 3n (46)[c] |
| 28 | A2 | 3n (35) |
Improved yields were also obtained in general when phenylacetylene (1a) was used with different alkenes (Table 3). The reaction can also be extended to allylsilane 2d, allyl ether 2e and allyl silyl ether 2f, although yields were modest due to the lower nucleophilicity of these alkenes.
Table 3.
Intermolecular gold(I)-catalyzed [2+2] cycloaddition between phenylacetylene (1a) and alkenes 2b–f[a]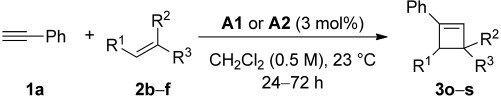
| Entry |  |
Catalyst | Product (Yield [%]) |
|---|---|---|---|
| 1 |  |
A1 | 3o (74)[b,c] |
| 2 | A2 | 3o (79)[b] | |
| 3 | 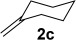 |
A1 | 3p (53)[b,c] |
| 4 | A2 | 3p (69)[b] | |
| 5 | 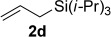 |
A1 | 3q (48)[d] |
| 6 | A2 | 3q (71)[d] | |
| 7 | 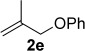 |
A1 | 3r (26)[d] |
| 8 | A2 | 3r (31)[d] | |
| 9 | 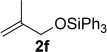 |
A1 | 3s (21)[d] |
| 10 | A2 | 3s (31)[d] |
[a] 2b–f/1a=2:1.
[b] Isolated yields.
[c] Ref.[3]
[d] Yield determined by 1H NMR using 1,4-diacetylbenzene as internal standard.
The yield in the macrocyclization of 1,14-enyne 10 to form 13-membered derivative 11 was also improved from 57% using A1 to 82% with A2 (Scheme 3).[7]
Scheme 3.
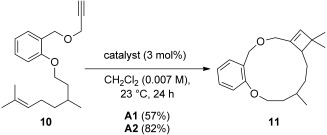
Gold(I)-catalyzed macrocyclization of a 1,14-enyne.
We also explored the influence of the counterion in the intermolecular [2+2+2] cycloaddition of alkynes with oxoalkene 6 to furnish 8-oxabicyclo[3.2.1]oct-3-enes 7a–d using A1 and A2. For this more challenging cascade reaction, we could also observe a moderate improvement of the yields using catalyst A2 (Table 4, entries 2, 4, 6, and 8).
Table 4.
Intermolecular gold(I)-catalyzed cyclization of 5-methylhex-5-en-2-one (6) with terminal alkynes (1a–h)[a]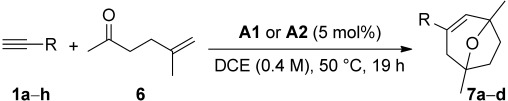
| Entry | R | Catalyst | Product (Yield [%])[b] |
|---|---|---|---|
| 1 | Ph (1a) | A1 | 7a (68)[c] |
| 2 | A2 | 7a (72) | |
| 3 | p-ClC6H4 (1e) | A1 | 7b (51)[c] |
| 4 | A2 | 7b (62) | |
| 5 | m-HOC6H4 (1i) | A1 | 7c (65)[c] |
| 6 | A2 | 7c (81) | |
| 7 | m-Tol (1h) | A1 | 7d (70)[c] |
| 8 | A2 | 7d (72) |
To define the role of the anion in intermolecular reactions, we studied experimentally the mechanism of the [2+2] cycloaddition between alkynes and alkenes. According to previous theoretical work,[7,12] the catalytic cycle for the [2+2] cycloaddition of alkynes with alkenes was expected to proceed by a rate-determining attack of the electron-rich alkene to the (η2-alkyne)gold(I) complex 8 forming the cyclopropyl gold(I) carbene 12 (Scheme 4). Then, the ring expansion occurs to form benzylic carbocation 13, which forms cyclobutene 3a after demetallation. An associative ligand exchange between (η2-cyclobutene)gold(I) complex 14 and the starting alkyne closes the catalytic cycle, regenerating 8.
Scheme 4.
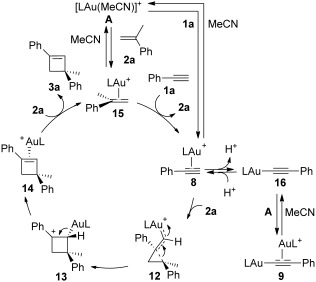
Mechanism of the [2+2] cycloaddition between alkynes and alkenes considering all the gold(I) species in equilibrium.
However, in our previous study on the [2+2+2] cycloaddition of alkynes with oxoalkenes,[7] we had observed that the formation of the (η2-alkyne)gold(I) complex 8a is complicated by the competitive formation of σ,π-digold(I) alkyne complex 9a (Scheme 2). Therefore, we determined the order of the reagents in the rate equation to gain further insight in the mechanism. Initial rates were calculated for each component by 1H NMR using diphenylmethane as internal standard (Figure 2). First order was observed for both the alkyne 1a and the gold(I) catalyst A2, whereas the reaction showed zero order dependence for the alkene 2a.
Figure 2.
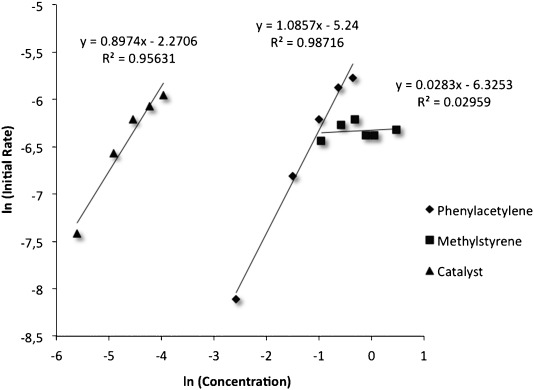
Order of the reagents in the [2+2] cycloaddition between phenylacetylene (1a) and α-methylstyrene (2a) with complex A2.
These results are consistent with a scenario in which the actual rate-determining step is the ligand exchange to generate the active species 8 (Scheme 4). Complex A undergoes ligand exchange with 2a forming 15. Generation of 8 is the slowest step due to its unstability and rapid evolution to 9 or 3a.
Monitoring the [2+2] cycloaddition reaction by 1H NMR showed a significant dependence on the anion (Figure 3). Besides the difference in the final yields, the reaction rate increases with the bulkiness and the softness of the counterion: BAr4F−>SbF6−>BF4−.
Figure 3.
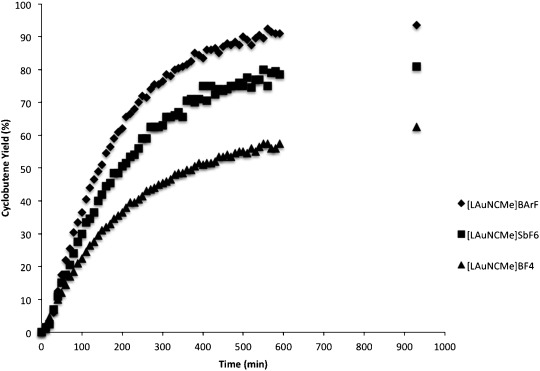
Kinetics of the [2+2] cycloaddition between phenylacetylene (1a) and α-methylstyrene (2a) with different gold(I) complexes (L=t-BuXPhos).
Analysis of the reaction mixture by 31P NMR showed only the (alkene)gold(I) complex 15 and the digold complex 9. The ratio between these species [15]/[9] increased following the same trend: BAr4F− (b)>SbF6− (a)>BF4− (c). Thus, [15]/[9] drops from 115 (BArF4F−) to 30 (SbF6−) and finally to 4 for BF4− resulting in a smaller reservoir of the cationic gold(I) species.
The equilibrium constants for the formation of 9 and 15 from A1 and A2 were also determined, endothermic and exothermic, respectively (Scheme 5). Formation of digold(I) complex with SbF6− (9a) anion is more favored than with BAr4F− (9b), probably due to the minor stability of the bulkier conjugated acid. We also checked that A2 binds stronger to 2a than A1.
Scheme 5.
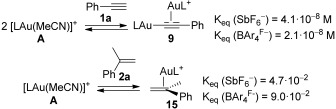
Determination of the equilibrium constants from A to 9 and 15.
These results suggest that when BAr4F− is used, the concentration of catalytically active species 8b (BAr4F− as counteranion) is higher than with SbF6− (8a). We also studied the evolution of the gold(I) species formed with A1 or A2 and 1a from −60 to 20 °C by 31P NMR. With A1, the digold(I) complex 9a was observed from −60 °C, becoming the only species at −20 °C (Scheme 6). However, in the case of A2, the corresponding digold(I) complex 9b was not observed until 0 °C. Furthermore, the catalytically active species 8b was clearly observed up to the same temperature.
Scheme 6.
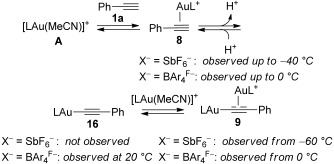
Gold(I) species formed between phenylacetylene (1a) and A1 or A2 from 213 to 293 K.
Complexes 15b and 9b (Figure 4) could be isolated and were fully characterized by X-ray crystallography.[13] The main divergence between 9a and 9b in the solid state (Figure 4b and c, respectively) is the radically different position of the counterions. Whereas BAr4F− is located alongside the phenylacetylene moiety in the same plane, SbF6− is placed between both gold atoms bending slightly the cation entity. Thus, the angle of the π-coordinated gold(I), the alkyne and the counterion is 130.3° for BAr4F− (Au–B 10.22 Å) and 77.3° for SbF6− (Au–Sb 8.23 Å) and the angle of the σ-gold is 210.0° for BAr4F− (Au–B 11.52 Å) and 60.6° for SbF6− (Au–Sb 7.34 Å). Complex 16 was independently prepared by reaction of the neutral gold(I) complex with lithium phenylacetylide and its structure was determined by X-ray diffraction.[13,14]
Figure 4.
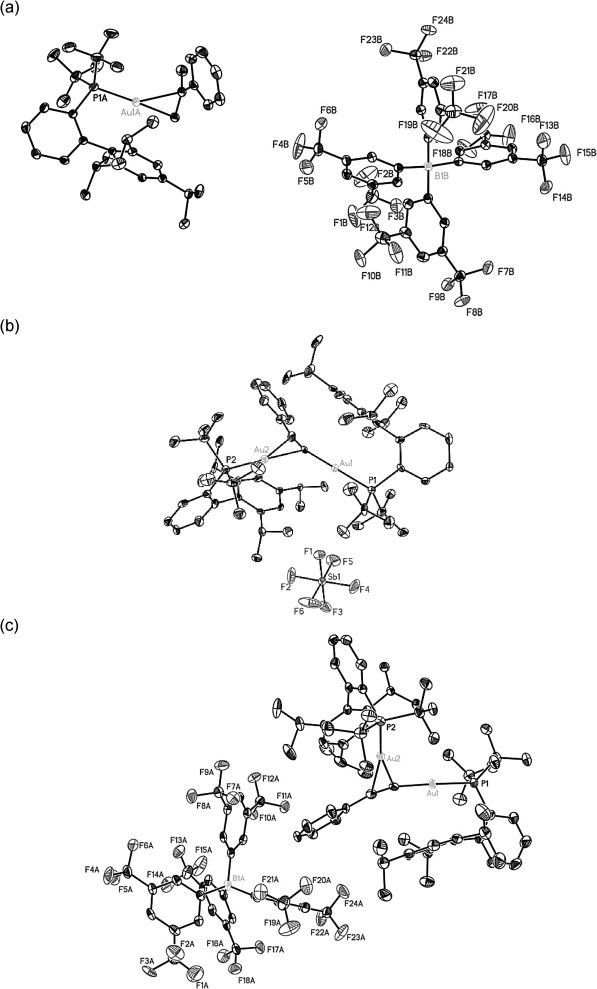
X-Ray crystal structures: (a) α-methylstyrene gold(I) complex 15b, (b) [(t-BuXPhosAu)2C≡CPh]+[SbF6]− 9a (taken from ref.[7]) and (c) [(t-BuXPhosAu)2C≡CPh]+[BAr4F]− 9b. ORTEP plot (50% thermal ellipsoids). Hydrogens are omitted for clarity.
We performed DFT calculations of the key complexes [t-BuXPhosAu(η2-phenylacetylene)]X 8 (X=BF4−, SbF6−, BAr4F−) [M06, 6-31G(d) (C, H, P, B, F) and SDD (Au, Sb), CH2Cl2]. First, we evidenced the steric congestion around the substrate hampering its deprotonation depending on the counterion. We analyzed the charge distribution by electron density from total SCF density mapped with ESP (ρ=0.03 e Å3) and the positive charge is widely distributed around the ligand instead of being concentrated in the metal center (Figure 5). We also checked the pattern between the bulkiness of the counterion and the acidity of phenylacetylene by determining the Mulliken atomic charges. The electron density decreases with the anion size: BF4−<SbF6−<BAr4F−, although the differences are modest: 0.250 for BF4− (8c), 0.243 for SbF6− (8a) and 0.237 for BAr4F− (8 b). Presumably, the large cation forms a more stable complex 8 with a softer counterion as BAr4F−.
Figure 5.
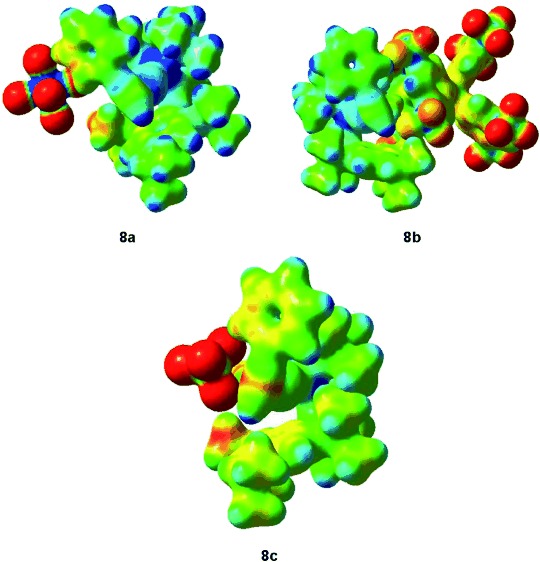
Electron density from total SCF density mapped with ESP (ρ=0.03 e Å3) for complexes 8 [t-BuXPhosAu(η2-phenylacetylene)X, X=SbF6− (8a), BAr4F− (8b), and BF4− (8c)].
Finally, we performed some additional experiments to exclude other mechanistic pathways.[14] We started by reacting the isolable intermediates under stoichiometric conditions with α-methylstyrene (2a). Neither complex 9 nor 16 reacted with 2a in CH2Cl2 at 23 °C for 8 h in the absence or presence of A2 as a catalyst. On the other hand, complex 15b (Figure 4) reacts with 1a to form cyclobutene 3a in 72% (CH2Cl2, 23 °C, 8 h) therefore that equilibrium is not inhibiting the process.
Complex 16 is not catalytically active for the formation of 3a by [2+2] cycloaddition, although the activity is restored upon addition of HSbF6, which cleaves the Au–C bond generating the gold(I) catalyst (Table 5, entries 1 and 2). More significantly, as we observed before in another context,[7] digold complexes 9a and 9b are very poor catalysts for the [2+2] cycloaddition between 1a with 2a (Table 5, entries 3 and 4), although the reaction proceeds smoothly after addition HSbF6 (Table 5, entry 5).
Table 5.
Regeneration of the catalytic activity in the intermolecular gold(I)-catalyzed [2+2] cycloaddition between 1a and 2a in the presence of a Brønsted acid[a]
| Entry | [Au] (mol%) | Additive (mol%) | Yield [%][b] |
|---|---|---|---|
| 1 | 16 (3) | – | – |
| 2 | 16 (3) | HSbF6⋅6 H2O (3) | 75 |
| 3 | 9a (1.5) | – | 13 |
| 4 | 9b (1.5) | – | 13 |
| 5 | 9a (1.5) | HSbF6⋅6 H2O (1.5%) | 70 |
[a] 23 °C, 8 h.
[b] Yield determined by 1H NMR using 1,4-diacetylbenzene as internal standard.
Conclusions
We have designed a new generation of gold(I) complexes bearing BAr4F− as counterion [BAr4F−=3,5-bis(trifluoromethyl)phenylborate]. These have proven to be more efficient in different intermolecular gold(I)-catalyzed reactions, improving the yields between 10 and 30%. We have then studied in detail the subtle anion effects in the gold(I)-catalyzed [2+2] cycloaddition of terminal alkynes with alkenes, which sum up in a substantial decrease in the formation of unproductive σ,π-(alkyne)digold(I) complex when the BAr4F− anion is used.
Our kinetic study of the gold(I)-catalyzed [2+2] cycloaddition reaction of terminal alkynes with alkenes is consistent with a scenario in which the rate- determining step is the first ligand exchange of [LAu(MeCN)]X to generate the active (η2-phenylacetylene)gold(I) complex, which is more stable with softer counterions according to DFT calculations. From a general practical perspective, we have found that the best results in intermolecular gold(I)-catalyzed reactions are obtained using [t-BuXPhosAu(MeCN)]BAr4F as catalyst.
Experimental Section
Procedure for the Synthesis of Gold(I) Complex A2
Chloro[(2′,4′,6′-triisopropyl-1,1′-biphenyl-2-yl)di-tert-butylphosphine]gold(I) (100.0 mg, 0.152 mmol) and acetonitrile (9.5 μL, 0.183 mmol) were dissolved in dichloromethane (6.6 mL). Then, sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (135.0 mg, 0.152 mmol) was added and the reaction mixture was stirred at room temperature for 30 min. The crude was filtered through Celite, then Teflon 0.22 and concentrated to obtain a white powder; yield: 224 mg (97%).
Procedure for the Synthesis of Cyclobutenes (3)
Alkyne (1 equiv.) and alkene (2 equiv.) were dissolved in dichloromethane (0.48 M) and the cationic gold(I) catalyst (3 mol%) was added. The reaction mixture was stirred at room temperature until no alkyne was observed by TLC. Then, it was quenched by adding a drop of a solution of Et3N in cyclohexane (1 M) and the solvent was removed. Preparative TLC was used to purify the resulting cyclobutenes.
Acknowledgments
We thank the MICINN (CTQ2010-16088/BQU and FPI fellowship to A. H.), the AGAUR (project 2009 SGR 47 and Beatriu de Pinós postdoctoral fellowship to D. L.), the MEC (FPU fellowship to C. O.), the European Research Council (Advanced Grant No. 321066), and the ICIQ Foundation for financial support. We also thank the ICIQ X-ray diffraction, the Nuclear Magnetic Resonance and the Spectroscopic and Kinetics units.
Supporting Information
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201300704.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Zhang L, Sun J, Kozmin SA. Adv. Synth. Catal. 2006;348:2271–2296. [Google Scholar]
- 1b.Hashmi ASK, Hutchings GJ. Angew. Chem. 2006;118:8064–8105. [Google Scholar]
- Angew. Chem. Int. Ed. 2006;45:7896–7936. doi: 10.1002/anie.200602454. [DOI] [PubMed] [Google Scholar]
- 1c.Fürstner A, Davies PW. Angew. Chem. 2007;119:3478–3519. [Google Scholar]
- Angew. Chem. Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]
- 1d.Hashmi ASK. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x. [DOI] [PubMed] [Google Scholar]
- 1e.Li Z, Brouwer C, He C. Chem. Rev. 2008;108:3239–3265. doi: 10.1021/cr068434l. [DOI] [PubMed] [Google Scholar]
- 1f.Arcadi A. Chem. Rev. 2008;108:3266–3325. doi: 10.1021/cr068435d. [DOI] [PubMed] [Google Scholar]
- 1g.Jiménez-Núñez E, Echavarren AM. Chem. Rev. 2008;108:3326–3350. doi: 10.1021/cr0684319. [DOI] [PubMed] [Google Scholar]
- 1h.Gorin DJ, Sherry BD, Toste FD. Chem. Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1i.Michelet V, Toullec PY, Genêt J-P. Angew. Chem. 2008;120:4338–4386. doi: 10.1002/anie.200701589. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2008;47:4268–4315. doi: 10.1002/anie.200701589. [DOI] [PubMed] [Google Scholar]
- 1j.Fürstner A. Chem. Soc. Rev. 2009;38:3208–3221. doi: 10.1039/b816696j. [DOI] [PubMed] [Google Scholar]
- 1k.Aubert C, Fensterbank L, Garcia P, Malacria M, Simonneau A. Chem. Rev. 2011;111:1954–1993. doi: 10.1021/cr100376w. [DOI] [PubMed] [Google Scholar]
- 1l.Krause N, Winter C. Chem. Rev. 2011;111:1994–2009. doi: 10.1021/cr1004088. [DOI] [PubMed] [Google Scholar]
- 2a.Ferrer C, Amijs CHM, Echavarren AM. Chem. Eur. J. 2007;13:1358–1373. doi: 10.1002/chem.200601324. For selected examples, see. [DOI] [PubMed] [Google Scholar]
- 2b.Dateer## RB, Shaibu BS, Liu RS. Angew. Chem. 2012;124:117–121. doi: 10.1002/anie.201105921. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:113–117. [Google Scholar]
- 2c.Yeom H-S, Koo J, Park H-S, Wang Y, Liang Y, Yu Z-X, Shin S. J. Am. Chem. Soc. 2012;134:208–211. doi: 10.1021/ja210792e. [DOI] [PubMed] [Google Scholar]
- 2d.Kramer S, Skrydstrup T. Angew. Chem. 2012;124:4759–4762. doi: 10.1002/anie.201200307. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:4681–4684. doi: 10.1002/anie.201200307. [DOI] [PubMed] [Google Scholar]
- 2e.Luo Y, Ji K, Li Y, Zhang L. J. Am. Chem. Soc. 2012;134:17412–17415. doi: 10.1021/ja307948m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.López-Carrillo V, Echavarren AM. J. Am. Chem. Soc. 2010;132:9292–9294. doi: 10.1021/ja104177w. [DOI] [PubMed] [Google Scholar]
- 4.Obradors C, Lebœuf D, Aydin J, Echavarren AM. Org. Lett. 2013;15:1576–1579. doi: 10.1021/ol400358f. Conceptually analogous macrocyclization by cycloaddition of alkynes with alkenes, see. [DOI] [PubMed] [Google Scholar]
- 5.Huguet N, Lebœuf D, Echavarren AM. Chem. Eur. J. 2013;19:6581–6585. doi: 10.1002/chem.201300646. [DOI] [PubMed] [Google Scholar]
- 6a.Hashmi ASK, Kurpejovic E, Wölfle M, Frey W, Bats JW. Adv. Synth. Catal. 2007;349:1743–1750. First example of an intramolecular gold-catalyzed formation of phenol with a furan and an alkyne, see. [Google Scholar]
- 6b.Hashmi ASK, Blanco MC, Kurpejovic E, Frey W, Bats JW. Adv. Synth. Catal. 2006;348:709–713. [Google Scholar]
- 6c.Hashmi ASK, Blanco MC. Eur. J. Org. Chem. 2006:4340–4342. [Google Scholar]
- 7.Obradors C, Echavarren AM. Chem. Eur. J. 2013;19:3547–3551. doi: 10.1002/chem.201300131. [DOI] [PubMed] [Google Scholar]
- 8a.Wei C, Li CJ. J. Am. Chem. Soc. 2003;125:9584–9585. doi: 10.1021/ja0359299. [DOI] [PubMed] [Google Scholar]
- 8b.Lavallo V, Frey GD, Kousar S, Donnadieu B, Bertrand G. Proc. Natl. Acad. Sci. USA. 2007;104:13569–13573. doi: 10.1073/pnas.0705809104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c.Cheong PHY, Morganelli P, Luzung MR, Houk KN, Toste FD. J. Am. Chem. Soc. 2008;130:4517–4526. doi: 10.1021/ja711058f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d.Hooper TN, Green M, Russell CA. Chem. Commun. 2010;46:2313–2315. doi: 10.1039/b923900f. [DOI] [PubMed] [Google Scholar]
- 8e.Simonneau A, Jaroschik F, Lesage D, Karanik M, Guillot R, Malacria M, Tabet J-C, Goddard J-P, Fensterbank L, Gandon V, Gimbert Y. Chem. Sci. 2011;2:2417–2422. [Google Scholar]
- 8f.Grirrane A, Garcia H, Corma A, Álvarez E. ACS Catal. 2011;1:1647–1653. [Google Scholar]
- 8g.Brown TJ, Widenhoefer RA. Organometallics. 2011;30:6003–6009. [Google Scholar]
- 8h.Raducan M, Moreno M, Bour C, Echavarren AM. Chem. Commun. 2012;48:52–54. doi: 10.1039/c1cc15739f. [DOI] [PubMed] [Google Scholar]
- 8i.Hashmi ASK, Lauterbach T, Nösel P, Vilhelmsen MH, Rudolph M, Rominger F. Chem. Eur. J. 2013;19:1058–1065. doi: 10.1002/chem.201203010. [DOI] [PubMed] [Google Scholar]
- 8j.Gómez-Suárez A, Dupuis S, Slawin AMZ, Nolan SP. Angew. Chem. 2013;125:972–976. [Google Scholar]
- Angew. Chem. Int. Ed. 2013;52:938–942. [Google Scholar]
- 8k.Zhdanko A, Ströbele M, Maier ME. Chem. Eur. J. Chem. -Eur. J. 2012;18:14732–14744. doi: 10.1002/chem.201201215. [DOI] [PubMed] [Google Scholar]
- 8l.Hashmi ASK, Braun I, Nösel P, Schädlich J, Wieteck M, Rudolph M, Rominger F. Angew. Chem. 2012;124:4532–4536. doi: 10.1002/anie.201109183. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:4456–4460. doi: 10.1002/anie.201109183. [DOI] [PubMed] [Google Scholar]
- 8m.Rubial B, Ballesteros A, González JM. Adv. Synth. Catal. 2013;355:3337–3343. [Google Scholar]
- 9.Weber SG, Zahner D, Rominger F, Straub BF. Chem. Commun. 2012;48:11325–11327. doi: 10.1039/c2cc36171j. A gold(I) carbene with tetrakis[3,5-bis(trifluoromethyl)phenyl]borate as the counterion has been reported to undergo C–B cleavage to from an aryl-gold(I) complex due to the high electrophilic of the metal center. However, we never observed this behavior with complex A2. [DOI] [PubMed] [Google Scholar]
- 10.Pérez-Galán P, Delpont N, Herrero-Gómez E, Maseras F, Echavarren AM. Chem. Eur. J. 2010;16:5324–5332. doi: 10.1002/chem.200903507. [DOI] [PubMed] [Google Scholar]
- 11.Amijs CHM, López-Carrillo V, Raducan M, Pérez-Galán P, Ferrer C, Echavarren AM. J. Org. Chem. 2008;73:7721–7730. doi: 10.1021/jo8014769. [DOI] [PubMed] [Google Scholar]
- 12a.Nieto-Oberhuber C, López S, Muñoz MP, Cárdenas DJ, Buñuel E, Nevado C, Echavarren AM. Angew. Chem. 2005;117:6302–6304. doi: 10.1002/anie.200501937. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2005;44:6146–6148. doi: 10.1002/anie.200501937. [DOI] [PubMed] [Google Scholar]
- 12b.Escribano-Cuesta A, Pérez-Galán P, Herrero-Gómez E, Sekine M, Braga AAC, Maseras F, Echavarren AM. Org. Biomol. Chem. 2012;10:6105–6111. doi: 10.1039/c2ob25419k. [DOI] [PubMed] [Google Scholar]
- 13. CCDC 953708 (12b) CCDC 953710 (9b), and CCDC 953709 (16) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
- 14. See the Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information