

 This work is licensed under a
This work is licensed under a Abstract
Although the role of estrogen signaling in breast cancer development has been extensively studied, the mechanisms that regulate the indispensable role of estrogen in normal mammary gland development have not been well studied. Because of the unavailability of culture system to maintain estrogen-receptor-positive (ERα+) cells in vitro, the molecular mechanisms that regulate estrogen/ERα signaling in the normal human breast are unknown. In the present study, we examined the effects of estrogen signaling on ERα+ human luminal progenitors using a modified matrigel assay and found that estrogen signaling increased the expansion potential of these progenitors. Furthermore, we found that blocking ERα attenuated luminal progenitor expansion and decreased the luminal colony-forming potential of these progenitors. Additionally, blocking ERα decreased H19 expression in the luminal progenitors and led to the development of smaller luminal colonies. We further showed that knocking down the H19 gene in the luminal progenitors significantly decreased the colony-forming potential of the luminal progenitors, and this phenotype could not be rescued by the addition of estrogen. Lastly, we explored the clinical relevance of the estrogen–H19 signaling axis in breast tumors and found that ERα+ tumors exhibited a higher expression of H19 as compared with ERα− tumors and that H19 expression showed a positive correlation with ERα expression in those tumors. Taken together, the present results indicate that the estrogen–ERα–H19 signaling axis plays a role in regulating the proliferation and differentiation potentials of the normal luminal progenitors and that this signaling network may also be important in the development of ER+ breast cancer tumors.
Keywords: ERα, H19, luminal progenitors, ER+ breast cancer cells
Introduction
Estrogen signaling through estrogen receptor α (ERα) is pivotal to the survival and maintenance of ERα+ breast cancer tumors. Furthermore, the estrogen–ERα signaling axis has been shown to be indispensable to mammary gland development, seeing as ERα knockout or ovariectomized mice only develop rudimentary breast structures (Daniel et al. 1987, Bocchinfuso & Korach 1997a ). The results of previous investigations have indicated that the nature of the estrogen signaling network changes dramatically during human breast tumorigenesis. This is evidenced by the fact that increased ERα expression in the normal breast increases the risk of developing breast cancer and the fact that elevated ERα expression is an early event that occurs in premalignant lesions (Clarke et al. 1997, Clemons & Goss 2001, Lee et al. 2006). These observations indicate that alterations to estrogen signaling and its molecular targets may serve as early, premalignant changes in breast epithelial cells. However, the exact mechanisms by which estrogen signaling regulates the proliferation and differentiation of normal human breast cells are unknown.
Current results indicate that estrogen acts as an indirect mitogen in normal breast epithelial cells (Lippman et al. 1977, Clarke et al. 1997, Shoker et al. 1999). This concept is based on the observation that although increased ERα expression and an increased number of ERα+ cells can be observed during the menstrual cycle in humans (Soderqvist et al. 1997), few, if any, ERα+ cells also show staining for proliferation markers (Clarke et al. 1997, Anderson et al. 1998). Also, the progesterone receptor (PR, a classical ERα target in breast cancer cells) and ERα mostly show divergent expression profiles in the normal breast (Hilton et al. 2012). On the basis of these observations, it can be postulated that ERα+ cells act in a paracrine manner to stimulate the proliferation of ERα− cells (Mallepell et al. 2006). However, no functional studies have been conducted to examine the direct effects of estrogen signaling on normal ERα+ human breast cells. Therefore, our understanding of the molecular mechanisms that are regulated by estrogen signaling in the breast is primarily based on ERα+ human breast cancer cell lines or xenografts in athymic mice. Because of the lack of ERα+ non-malignant cell lines (Anderson et al. 1998), a functional analysis of primary ERα+ cells obtained from human breast tissue is required in order to study the mechanisms by which the estrogen–ERα signaling network regulates mammary gland development.
Human breast tissue is made up of luminal and myoepithelial cells that are continuously produced from bipotent and lineage-restricted progenitors, which are themselves generated from a rare population of breast stem cells (Eirew et al. 2008, 2010, Raouf et al. 2008). The transcriptome profiling of these different progenitor subtypes has revealed that the undifferentiated bipotent progenitors showed high enrichment for PR transcripts, whereas the luminal progenitors showed enrichment for the ERα transcripts (Raouf et al. 2008). The results of recent studies in human and mouse mammary glands have provided some insights into the potential role of progesterone in the regulation of bipotent progenitors and stem cells (Joshi et al. 2010, Hilton et al. 2014). More recently, it has been demonstrated that ERα+ human breast cells are able to grow and generate colonies in vitro and mammary structures in vivo that are restricted to the luminal lineage (Honeth et al. 2014). However, there is a gap in our knowledge about the molecular mechanisms by which estrogen signaling regulates the proliferation and differentiation potentials of these ERα+ luminal progenitors. Such knowledge would be important for understanding how known breast cancer oncogenes function in healthy ERα+ cells and how alterations to their functions can lead to breast tumor development. In this context, H19 is an estrogen-regulated gene (Adriaenssens et al. 1999) that was previously identified as a breast tumor oncogene (Berteaux et al. 2005). H19 is a long non-coding RNA (lncRNA) that harbors the microRNA-675 in its first exon and has been shown to regulate estrogen-induced proliferation of ERα+ breast cancer cells (Lottin et al. 2002, Cai & Cullen 2007, Keniry et al. 2012). However, it is not clear if the H19 gene plays a similar role in regulating the proliferation of healthy ERα+ cells.
In the present study, we therefore used a modified in vitro matrigel assay to culture primary human ERα+ luminal progenitors under estrogen-depleted growth conditions in order to directly examine the effect of estrogen signaling on these cells at the molecular and cellular levels. Using this assay and the colony-forming cell (CFC) assay, we found that estrogen–ERα signaling enhances the proliferation and differentiation potentials of the luminal progenitors through increased H19 gene expression. Furthermore, we provide evidence that H19 is highly expressed in ERα+ breast cancer tumors and that its expression positively correlates with ERα expression in these tumors. These findings are significant because increased estrogen signaling has been demonstrated to be a risk factor for developing ERα+ breast tumors; therefore, understanding the molecular mechanisms that govern ERα and estrogen signaling in healthy breast cells provides insights into how alterations to this signaling axis may lead to the development of premalignant lesions.
Materials and methods
For more detailed descriptions, see the Supplementary Materials and methods (see section on supplementary data given at the end of this article).
Isolation of luminal and bipotent progenitors
Luminal and bipotent progenitors were isolated from reduction mammoplasty samples that were obtained through informed, written patient consent in accordance with the University of Manitoba's Health Research Ethics Board (REB no. H2010:292) policies. Breast reduction samples were enzymatically dissociated, and the organoid-enriched fractions were obtained as described previously (Raouf & Sun 2013). Single-cell suspensions were prepared from these organoid-enriched fractions and were placed in co-cultures with irradiated 3T3 mouse fibroblasts (ATCC, X-irradiated at 30 Grey, i3T3) in SF7 media (Stingl et al. 1998, Raouf et al. 2008, Raouf & Sun 2013) supplemented with 5% fetal bovine serum (FBS). After 3 days, adherent cells were made into single-cell suspensions as described previously (Raouf & Sun 2013) and were blocked with 10% human serum. Cells were then stained with anti-epithelial cell adhesion molecule (EpCAM, 1:100 dilution; StemCell Technologies, Vancouver, BC, Canada) conjugated to phycoerythrin (PE) and anti-α6 integrin (CD49f) conjugated to Alexa Fluor 647 (1:100 dilution; Biolegend, San Diego, CA, USA). For isotype controls, IgG antibodies directly conjugated to PE or allophycocyanin (APC) were used (both from BD Biosciences, San Jose, CA, USA). Propidium iodide (PI, 1 mg/ml) was added to each sample to distinguish live cells. PI−EpCAMhighCD49flow (luminal progenitors) or PI−EpCAMlowCD49bright (bipotent progenitors) cells were sorted via fluorescence-activated cell sorting (FACS).
Matrigel cultures
For gene expression studies, the luminal or bipotent progenitors (2.0–3.5×104 cells/well) were combined with i3T3 cells (2.0×105 cells/well) and placed in 96-well plates containing 50 μl of polymerized, growth-factor-reduced, and phenol-red-free (PRF) matrigel (BD Biosciences). Cells were grown in complete media (PRF-SF7 supplemented with bovine pituitary extract (BPE) at 100 μg/ml) for 7 days and then cultured in estrogen-depleted growth media (PRF-DMEM supplemented with 5% charcoal-stripped serum (v/v) (Weitsman et al. 2006)) for 48 h. Thereafter, the cultures were treated with 17β-estradiol (E2, 10 nM) or ethanol (EtOH) for an additional 24 h, at which point the gels were dissociated and RNA was extracted from the cells using TRIzol Reagent (Life Technologies). For some of the experiments, two individual reduction mammoplasty samples were pooled to yield sufficient numbers of luminal progenitors for carrying out the experiments.
To quantify the effects of E2 on luminal progenitor expansion and differentiation, isolated progenitors were placed in matrigel cultures with 5.0×104 cells/well or in co-cultures with i3T3 (2.0–3.5×104 luminal progenitors combined with 2.0×105 i3T3 cells per well in a 96-well plate). After 7 days, the growth media was replaced with PRF-SF7 supplemented with E2 (10 nM) or E2 (10 nM) plus ICI (500 nM) or with ethanol. After an additional 7 days, gels were dissociated and cells were placed in CFC assays as described below. The frequencies of luminal progenitors before and after matrigel cultures were ascertained by CFC assays as described in the next section.
CFC assays
Luminal and bipotent progenitors were examined using CFC assays essentially as described previously (Raouf et al. 2008) with the following modifications: 1000 progenitors were combined with 8.0×104 i3T3 cells and were cultured in collagen-treated six-well tissue culture plates using SF7 growth medium supplemented with 2% FBS for 7–10 days. Colonies were fixed with an acetone:methanol (1:1 ratio) solution and stained with a crystal violet solution (0.05% w/v). Some CFC assays were set up as described above, except that PRF SF7 medium supplemented with 2% FBS was used. After 18 h, growth media were replaced with PRF SF7 media supplemented with E2 (10 nM) or E2 plus ICI 182,780 (ICI, at the indicated concentrations), EtOH, or ICI only (at the indicated concentrations) for 6 days. Subsequently, the colonies were fixed and stained as described above. The colony numbers were obtained using an inverted microscope (Evos XL Core), and colony size (number of cells/colony) was determined (analyzed using Adobe Acrobat X Pro, San Jose, CA, USA).
H19-deficient luminal progenitors
Single-cell suspensions prepared from the organoid-enriched fractions (1×106 cells) were placed in co-cultures with 1.6×105 i3T3 cells in SF7 media supplemented with 5% FBS. After 24 h, cells were infected separately with 1×107 lentiviral particles prepared from a pool of three different pGIPZ-puro-GFP plasmids containing short hairpin (sh)-RNA that target the H19 RNA or a pGIPZ-puro-GFP vector that expresses a scrambled sh-RNA fragment (Thermo Fisher, Waltham, MA, USA; for details on lentivirus production and infection, see Raouf et al. (2008)). After 4 days, the GFP+ luminal progenitors were isolated via FACS and placed in CFC assays. For some of the experiments, the growth media were supplemented with either 10 nM E2, E2 plus ICI (500 nM), or EtOH after 18 h, cultures were grown for 6 additional days, and colonies were fixed and analyzed as described above.
Overexpression or knockdown of ERα and ERβ in breast cancer cells
ERα was expressed in the MDA-MB231 by transfecting the pCDNA 3.1-ERα vector into the cells using the conventional calcium chloride method (Wigler et al. 1977, Chen & Okayama 1987). The transfected cells were selected using geneticin (2.5 μg/ml, Sigma–Aldrich) and then grown in estrogen-depleted growth media for 48 h, at which point cells were treated with E2 (10 nM) or ethanol-supplemented growth media for 24 h. ERα expression in the MCF7 cells was knocked down by infection with lentivirus prepared from pGIPZ-sh-ERα or pGIPZ-scrambled control (Thermo Fisher) for 4 h. Three separate pGIPZ-sh-ERα constructs (1, 2, and 3) were used. After 48 h, the transduced cells were puromycin selected (2 μg/ml, Sigma–Aldrich) for 72 h. The MDA-MB231-ERβ cells (Murphy et al. 2005) were placed in estrogen-depleted growth media for 48 h and then grown in PRF-DMEM supplemented with 5% CS-FBS, doxycycline (2 μg/ml; Sigma), and E2 (10 nM) or EtOH for 24 h.
Chromatin immunoprecipitation assay
MCF7 cells were grown in estrogen-depleted growth media for 48 h and treated with either E2 (10 nM) at 1 h, 3 h, 6 h, and 24 h or with ethanol for 24 h. ERα chromatin immunoprecipitation (ChIP) was carried out as described previously (Schmidt et al. 2009, Ross-Innes et al. 2012). Equal amounts of ChIP DNA were analyzed by qPCR for the estrogen responsive elements (EREs) in the H19 promoter using the oligonucleotides indicated in Supplementary Table 1, see section on supplementary data given at the end of this article. Melting temperatures were examined to ensure that a singular amplicon was obtained from each primer set. The binding enrichment to the H19 promoter regions was obtained using the percentage of ChIP DNA (relative to input DNA) normalized to a nonspecific control region of the genome, as described previously (Holmes et al. 2008).
Selection of archival human breast tumors samples
All of the human breast cancers tumors were obtained from the Manitoba Breast Tumour Bank (MBTB) (Watson et al. 1996). All of the samples were obtained and used in accordance with the University of Manitoba's Health Research Ethics Board (REB no. H2001:083). ERα and PR protein levels in each tumor were determined using the ligand-binding assay, where >3 fmol/mg of protein was considered positive (Watson et al. 1996, Barnes et al. 2008). A total of 70 tumor samples were selected, of which 39 were deemed to be ER+ and 31 were categorized as ER− (i.e., <3 fmol/mg protein). RNA was extracted from frozen tumor sections using TRIzol Reagent, treated with DNase, and turned into cDNA.
Statistical analysis
The differential expression of genes was determined using the two-tailed Student's t-test, and the differential expression of H19 in the breast tumor samples was determined using the two-sided Mann–Whitney rank test. The statistical significance of an increased dose of ICI on H19 transcript levels was assessed by ANOVA. All of the tests were conducted with the GraphPad Prism 4.02 program (GraphPad, La Jolla, CA, USA). The correlation between the ERα transcript levels and H19 expression was determined using Fisher's exact test. For this purpose, the best-fit line (Microsoft Excel) was forced through the origin, because more than half of the data points fell close to the origin of the scatter plot.
Results
EpCAM+CD49flow subset of human breast epithelial cells are enriched for ERα+ luminal progenitors
Luminal and bipotent progenitors were isolated from reduction mammoplasty samples by FACS (Fig. 1A). With this strategy, EpCAM+CD49flow cells were enriched for the luminal progenitors (21.6% purity), whereas EpCAMlowCD49f+ cells were enriched for the bipotent progenitors (16.7% purity) (Fig. 1A and B). The frequency of the luminal colonies was determined by CFC assay, an in vitro clonal assay that provides a prospective measure of luminal progenitor numbers in the initial sample and their differentiation potential to form mature luminal colonies (i.e., through the quantification of colony size). Next, we confirmed the high expression of ERα in the freshly isolated luminal progenitors as compared with the bipotent progenitors through immunofluorescence staining (Supplementary Figure 1A, see section on supplementary data given at the end of this article). The luminal phenotype of the cells in the colonies was determined by measuring cytokeratin 18 expression (Supplementary Figure 1B).
Figure 1.
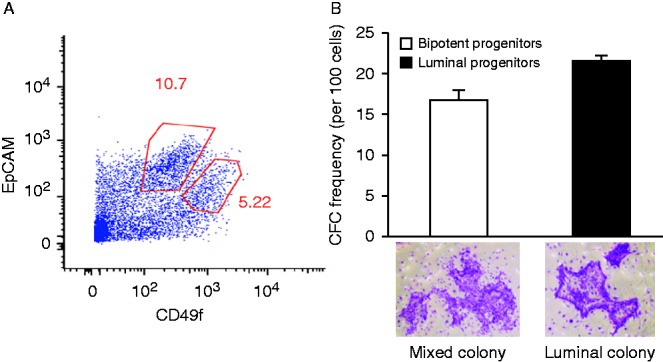
ERα is strongly expressed in luminal progenitors. (A) Luminal (EpCAMbrig ht CD49flow) and bipotent (EpCAMlowCD49fbright) progenitors were isolated from reduction mammoplasty samples using FACS. (B) CFC assays were used to determine the purity of different progenitor subtypes. As shown, 21.6% of the EpCAMbrightCD49flow cells formed pure luminal colonies, whereas 16.7% of the EpCAMlow CD49fbright cells formed mixed colonies (n=3). Representative luminal and mixed colonies are shown in the photographs.
Estrogen–ERα signaling enhances luminal progenitor expansion and differentiation
Although estrogen signaling is indispensable for mammary gland development, the precise mechanisms that underlie its action are not clear (Bocchinfuso & Korach 1997b , Bocchinfuso et al. 2000). To address this issue, we examined whether estrogen signaling plays a role in regulating the proliferation and differentiation of ERα+ human luminal progenitors. For this purpose, CFC assays were set up either in the presence of E2 or increasing concentrations of fulvestrant (ICI 182,780), a specific estrogen receptor antagonist (Fig. 2A and Supplementary Figure 2A, see section on supplementary data given at the end of this article). The addition of E2 caused a modest yet statistically significant (1.48-fold) increase in the luminal colony numbers. However, the addition of 250 nM or 500 nM ICI in the presence of E2 decreased colony-forming potential of the luminal progenitors by 3.2- and 5.3-fold respectively (Fig. 2A). Because CFC assays were unamenable to estrogen-deprived culture conditions, these experiments were performed in complete media, which would contain sources of estrogens. It is therefore not surprising that only a modest increase in the number of colonies was observed in the presence of additional E2. Interestingly, we also observed that increased concentrations of ICI led to significantly decreased luminal numbers (Supplementary Figure 2A) and that no colonies were observed in the presence of higher concentrations of ICI. Colony size is a measure of the potential of luminal progenitors to generate mature luminal colonies (Stingl et al. 1998). We therefore quantified the number of cells per colony as a prospective measure of the differentiation potential of luminal progenitors. Interestingly, in the presence of ICI, we observed a significant reduction in luminal colony size (Fig. 2B). It is noteworthy that the addition of E2 did not increase colony size. We posit that this was because of the existing levels of estrogens in the growth media, and the addition of E2 to these cultures was therefore ineffective in enhancing the luminal colony size. These results indicate that estrogen–ERα signaling is essential to the proliferation and differentiation of luminal progenitors.
Figure 2.
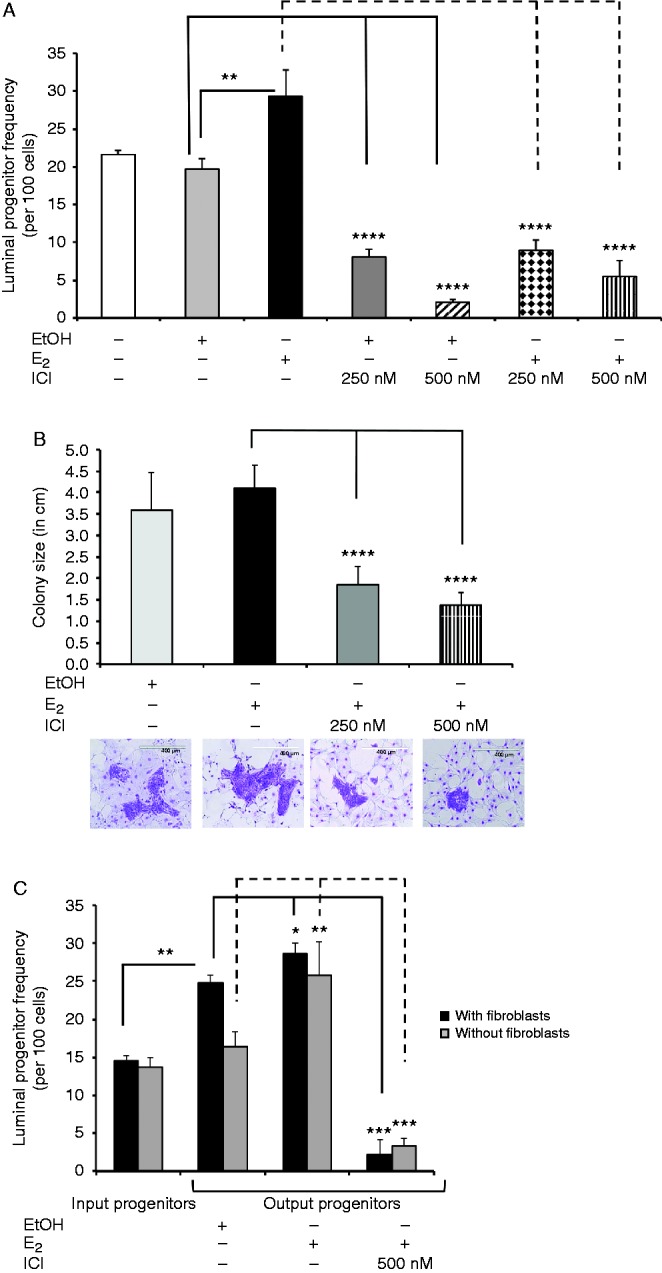
Estrogen signaling enhances luminal progenitor proliferation. (A) Luminal progenitors were examined using CFC assays and were either treated with E2, EtOH, E2 plus ICI, or EtOH plus ICI at the indicated concentrations. The CFC cultures were fixed and stained, and the colony numbers were quantified (n=3). As shown, in the presence of E2, the colony-forming capacity of luminal progenitors was increased (1.48-fold as compared to EtOH), and blocking ERα significantly attenuated this induction. (B) Luminal progenitors were treated as in (A), and colony size (i.e., the ability of the luminal progenitors to form mature luminal cells) was determined by counting the number of cells in each colony. As shown, ICI treatment significantly reduced colony size in a dose-dependent manner. Images show representative colonies from each treatment. (C) Luminal progenitors were placed in matrigel cultures with or without irradiated fibroblasts (i3T3) in complete medium for 7 days and were then treated with E2, E2 plus ICI, or EtOH for an additional 7 days. Thereafter, the frequency of progenitors was assessed via CFC assay. In matrigels without i3T3, E2 significantly increased luminal progenitor frequency, which was circumvented with ICI. Interestingly, the addition of i3T3 alone increased progenitor cell numbers, and the addition of E2 had a small but statistically significant effect. Aliquots from the same samples were placed in the CFC assays before the matrigel cultures to give the starting luminal progenitor frequency. *P<0.05, **P<0.005, ***P<0.0001, ****P<0.00001.
The examination of estrogen signaling at the molecular level in luminal progenitors requires the luminal progenitors to be placed into estrogen-depleted growthconditions. This was accomplished by placing isolated luminal progenitors into matrigel cultures along with irradiated mouse embryonic fibroblasts (i3T3) for 7 days, after which the cultures were treated with E2. Using immunofluorescence staining and FACS, we determined that cells in the E2-treated gels showed a 3.2-fold increased expression of ERα (Supplementary Figure 1C and D). Next, we examined the frequency of luminal progenitors in the matrigel cultures that had been treated with E2 or E2 plus ICI. Using CFC assays, we found that compared with the EtOH-treated controls, E2 stimulation increased the frequency of luminal progenitors by 13%, which was completely attenuated in the presence of ICI (Fig. 2C, black bars, input luminal progenitors as compared with output progenitors) and exhibited a twofold increase as compared with the input controls. Also, we observed that placing the luminal progenitors in matrigel cultures with i3T3 cells plus EtOH significantly increased the frequency of the progenitors (Fig. 2C, black bars, input as compared with the ethanol controls), which indicates that either fibroblasts or ethanol alone could increase the frequency of luminal progenitors and that estrogen may not act directly on luminal progenitors. It is noteworthy that in the presence of ICI, the colony-forming potential of the luminal progenitors was less than that of the cultures treated with ethanol as controls. This observation was most probably a result of the residual estradiol in the matrigel preparation that was present in the ethanol-treated cultures but was completely neutralized in presence of ICI. To examine whether the estrogen-induced expansion of the numbers of luminal progenitors in the matrigel cultures required fibroblasts, matrigel cultures initiated with luminal progenitors without fibroblasts were treated with EtOH, E2, or E2 plus ICI (Fig. 2C). Luminal progenitor frequency remained the same in the absence of i3T3 plus ethanol, which indicates that fibroblasts alone can increase the frequency of luminal progenitors. However, upon stimulation with E2 and in the absence of fibroblasts, luminal progenitor frequency increased by twofold, but this increase was abrogated by ICI. We examined 7-day matrigel cultures that had been initiated with luminal progenitors and determined that as many as 60% of the cells in these gels expressed ERα+ (Supplementary Figure 2B and C). We also found that the matrigel treated with E2 contained significantly more Ki67+ cells as compared with the EtOH-treated gels (Supplementary Figure 2D). To ensure that placing luminal progenitors in matrigel cultures does not alter their phenotype, the expression of CD49f, EpCAM, cytokeratin 18, and cytokeratin 14 was examined in cells obtained from these gels after 7 days of treatment with E2 or EtOH. We found no appreciable difference in the expression of these markers in cells grown in EtOH- and E2-treated gels (Supplementary Figure 2E). These observations indicate that placing primary human luminal progenitors in matrigel cultures for 14 days does not alter their phenotype.
The results of these experiments indicate that estrogen signaling could increase the proliferation of luminal progenitors and that fibroblasts play a major role in this signaling network. It is interesting that ICI was able to diminish the colony-forming ability of the luminal progenitors in the absence of fibroblasts, which indicates that E2 could directly act on ER+ luminal progenitors to regulate their proliferation.
H19 but not PR expression is regulated by estrogen signaling in luminal progenitors
A number of estrogen target genes, such as PR, pS2, and H19, have been shown to be involved in regulating the proliferation of ER+ breast cancer cells (Brown et al. 1984, Adriaenssens et al. 1999, Cunliffe et al. 2003, Carroll et al. 2006). However, the nature of the estrogen regulation of these genes in normal ERα+ human breast cells has not been explored. Using matrigel cultures, we observed that the transcript expression of PR and pS2 genes showed no significant change in E2-stimulated luminal progenitor cultures as compared with the controls, whereas the transcript expression of ERα and H19 was significantly increased (5.3- and 8.5-fold respectively, Fig. 3A). No significant change in the expression of H19 or ERα resulting from estrogen treatment could be detected in the bipotent progenitor-initiated matrigel cultures (Supplementary Figure 3A and B, see section on supplementary data given at the end of this article). To the best of our knowledge, this was the first direct measurement of estrogen signaling on isolated normal, non-transformed human ERα+ breast cells at the molecular level. Moreover, the addition of E2 to the CFC cultures increased H19 transcript levels significantly, whereas ICI blocked this increase, which indicates that H19 might be involved in the estrogen–ERα induced expansion of luminal progenitors (Fig. 3B).
Figure 3.

Estrogen signaling enhances luminal progenitor proliferation through H19. (A) Luminal progenitors were placed in matrigel assays for 7 days and were then grown in estrogen-depleted growth media for 48 h and were subsequently treated with E2 (10 nM) or EtOH for an additional 24 h. qPCR was used to examine the expression of the estrogen target genes (PR, pS2, ERα, and H19) in cells obtained from the gels. Interestingly, no changes in the transcript levels of PR or pS2 could be detected. However, ERα and H19 expression levels were increased 5.3- and 8.5-fold respectively in E2-exposed luminal progenitors. For comparison, ER+ breast cancer cells (MCF7 and T47D) were exposed to EtOH or E2 for 24 h, and the expression of estrogen target genes was assessed using qPCR. Solid bars represent transcript expression in EtOH-treated cells, and crossed bars represent E2-treated cells. All of the transcript expression levels were normalized to the GAPDH levels, and the means and s.d.s are shown (n=3). (B) Luminal progenitors were isolated and placed in CFC assays; they were then treated with E2 (10 nM), E2 plus ICI, or EtOH for 7 days. H19 expression was examined via qPCR. As shown, in E2-stimulated colonies, H19 expression was increased 1.6-fold, whereas the addition of ICI significantly decreased H19 expression. (C) Lenti sh-H19 or lenti sh-scrambled-infected luminal progenitors (GFP+) were isolated via FACS and placed in CFC assays. Scale bars represent 400 μm. Representative pictures show the GFP+ colonies that developed from the transduced luminal progenitors. (D) Average colony counts were used as a prospective measure of the transduced luminal progenitor frequency (n=3). Compared with the scrambled control, the sh-H19-infected progenitors formed threefold fewer colonies, and the addition of E2 had no effect on these transduced progenitors. *P<0.05, **P<0.005, ***P<0.0005.
The colony-forming potential of the luminal progenitors requires H19
The H19 gene encodes a lncRNA whose expression in adults is restricted to only a few tissues (e.g., mammary glands) (Pachnis et al. 1984, Poirier et al. 1991, Doucrasy et al. 1993, Dugimont et al. 1995, Ariel et al. 1997). The biological role of H19 has been mostly described in relation to IGF2 signaling; however, its function in breast epithelial cells remains puzzling. Interestingly, H19 is highly expressed in breast cancer cells (Qiu et al. 2013) and may play a role as an oncogene (Lottin et al. 2002, Berteaux et al. 2005). In addition to autocrine factors (hepatocyte growth factor or fibroblast growth factor 2 (Adriaenssens et al. 2002)), H19 expression is also regulated by estrogen signaling in ERα+ breast cancer cells (Adriaenssens et al. 1999). We therefore proposed the hypothesis that H19 might be involved in regulating the estrogen-induced expansion of luminal progenitors. To test this hypothesis, we infected single-cell suspensions prepared from reduction mammoplasty samples with lentivirus that expressed a shRNA against the H19 RNA (sh-H19) or a scrambled control RNA sequence. The transduced luminal progenitors were isolated via FACS (Fig. 3C) and placed in CFC assays in the presence E2 or ethanol as a vehicle control. The level of H19 knockdown in the luminal colonies was assessed using qPCR (Supplementary Figure 4, see section on supplementary data given at the end of this article). Interestingly, the sh-H19-transduced human breast cells contained significantly reduced luminal progenitors, which were not rescued with the addition of E2 (Fig. 3D). Notably, the loss of H19 expression led to a threefold decrease in the frequency of luminal progenitors (Fig. 3D). In contrast, the ICI treatment of the luminal progenitors led to a 5.3-fold decrease in the colony-forming ability of the luminal progenitors (Fig. 2A), which indicates that H19 is an important mediator of estrogen signaling in the luminal progenitors but that other genes may be involved as well. It is interesting that the colonies that developed from the sh-H19-infected luminal progenitors contained the same number of cells per colony as the scrambled control-infected progenitors (data not shown). This indicates that H19 does not play an essential role in the differentiation potential of luminal progenitors.
Estrogen-induced expression of H19 is primarily regulated through ERα
As has been shown previously (Adriaenssens et al. 1999), we also found that estrogen signaling increases the expression of H19 in MCF7 and T47D cells (Supplementary Figure 5A, see section on supplementary data given at the end of this article). To ascertain the kinetics of regulation of H19 expression by estrogen, MCF7 cells were cultured in estrogen-depleted media, and H19 expression was determined using RT-PCR at various times post-E2 stimulation (Supplementary Figure 5B). Interestingly, we found that E2 treatment increased H19 expression as early as 6 h after stimulation and reached its apex within 24 h; this increase was followed by a marked decrease after 48 h. This results indicate that estrogen-induced H19 expression shows a dynamic pattern of activity that is typical of ERα-regulated gene promoters. Furthermore, increasing doses of ICI led to a sequential decrease in H19 expression in the MCF7 and T47D cells, which indicates that ERα is directly involved in the regulation of H19 gene expression (Supplementary Figure 5C and D).
Estrogen signaling can be mediated through ERα and ERβ receptors (Murphy & Watson 2002, Clarke et al. 2004). To examine the ERα requirement for the estrogen regulation of H19 expression, lenti shRNA was used to knock down the expression of ERα in MCF7 cells. Compared with the scramble-control-infected cells, loss of ERα expression led to a marked decrease in the expression of H19 (Fig. 4A). Also, the expression and activation of ERα in MDA-MB231, an ERα− breast cancer cell line, increased H19 expression by 2.9-fold (Supplementary Figure 6A and B, see section on supplementary data given at the end of this article).
Figure 4.
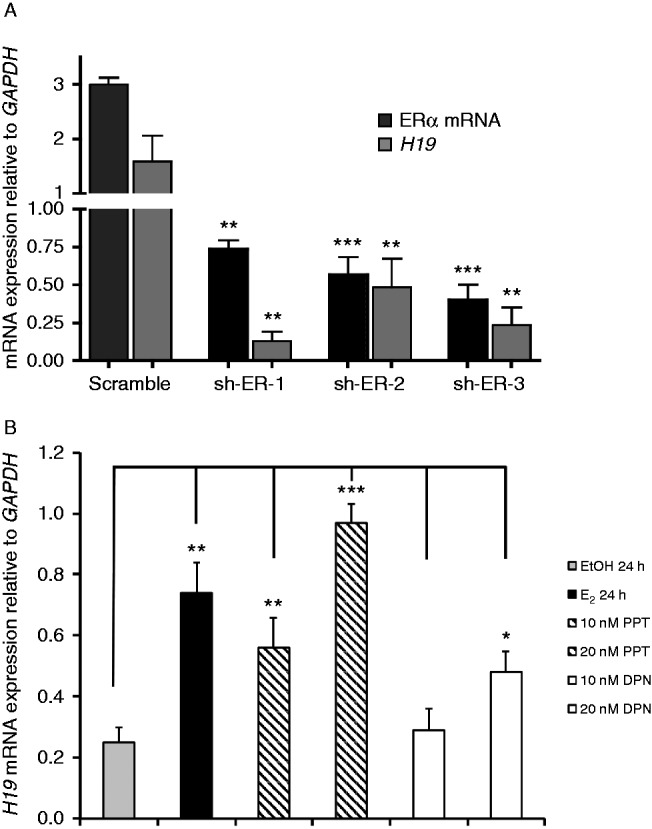
Estrogen-regulated expression of H19 is mainly regulated through ERα. (A) ERα transcript levels were decreased in MCF7 cells with three different sh-RNA fragments (sh-ER-1, -2, and -3) using lentiviral transduction. ERα and H19 transcript levels were examined in the transduced cells by qPCR. All of the data are normalized to the GAPDH transcript levels. (B) MCF7 cells were grown under estrogen-deprived growth conditions and treated with either EtOH, E2, PPT (a selective ERα agonist), or DPN (a selective ERβ agonist) for 24 h. H19 expression was ascertained using qPCR and was normalized with respect to GAPDH expression (n=3). Compared with EtOH, as little as 10 nm PPT increased H19 transcript levels, whereas DPN at the 10 nM concentration level had no effect. However, at 20 nM concentration, both PPT and DPN increased H19 expression (3.9- and 1.9-fold respectively). *P<0.05, **P<0.005, ***P<0.0005.
To investigate if ERβ is also involved in estrogen-induced H19 expression, we used tet-on MDA-MB-231-ERβ cells and found that E2 and doxycycline treatment did not increase H19 expression in these cells (Supplementary Figure 6C and D). Furthermore, we observed that a specific agonist for ERα, PPT (Kraichely et al. 2000), was a more potent inducer of H19 expression (3.9-fold) as compared with an ERβ agonist, DPN (Meyers et al. 2001), which provides further support the hypothesis that ERα plays a more dominant role in regulating H19 expression in MCF7 (Fig. 4B) and in T47D (Supplementary Figure 6E).
ERα binds to estrogen-responsive elements in the H19 promoter
We first used inhibitors of transcription and protein synthesis to validate that estrogen induction of H19 gene expression requires de novo transcription without the need for de novo protein synthesis (Supplementary Figure 7A, B, C, D and E, see section on supplementary data given at the end of this article).
The analysis of the H19 proximal promoter and enhancer region revealed seven ERα response elements half-sites (ERE, AGGTCA) (Carroll et al. 2006) within 1500 base pairs (bp) of the transcription start site (TSS) (Fig. 5A). To ascertain if ERα is able to bind to any or all of these seven ERE half-sites, we performed ChIP assays. For this purpose, DNA-bound ERα fragments were precipitated after 3, 6, and 24 h of exposure to estradiol or ethanol as a vehicle control. Because of the close proximity of the EREs located at 1483–1503 bp from the TSS, these sites were measured as one site. As early as 1 h after E2 treatment, a significant enrichment for each of the six ERE sites was observed (Fig. 5B). After 3 and 6 h of E2 stimulation, all of the sites showed a significant decrease in ERα occupancy as compared with 1 h of exposure to E2. However, after 24 h of exposure to E2, all of the ERE sites appeared to be once again occupied by ERα. Interestingly, the ERE located at 1004 bp from the TSS appears to be bound to ERα at all of the times tested and showed the highest level of occupancy. This pattern of ERα occupancy on the H19 promoter is reminiscent of the cycling, on-and-off binding of ER to other ER target promoters in breast cancer cell lines (Shang et al. 2000, Reid et al. 2003).
Figure 5.
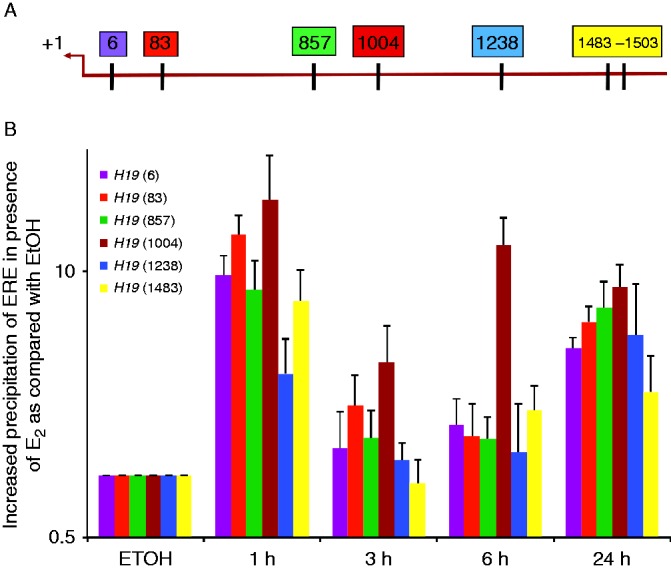
ERα binds to H19 promoter through EREs. (A) This figure shows the location of EREs within 1500 base pairs of the transcription start site (TSS, +1) of the H19 proximal promoter. Each box is representative of one ERE half-site, and the number within each box shows its location away from the TSS. (B) Chromatin immunoprecipitation was performed to examine the ERα occupancy of each ERE. MCF7 cells were grown in estrogen-depleted media and treated either with EtOH or E2 for the indicated times. ERα bound to each ERE was quantified using qPCR. As shown, ERα binding to the H19 promoter was detectable after 1 h of exposure to E2.
H19 expression correlates with ERα expression in breast cancer tumors
To assess the clinical relevance of our findings, we examined the expression of H19 in 70 primary breast tumors that were obtained from postmenopausal women. Only samples that contained more than 70% tumor tissue and known ERα and PR expression levels were included. The comparative analysis of H19 expression in these tumors using qPCR revealed a significantly higher expression of H19 in the ER+ tumors (39 samples) as compared with the ER− tumors (31 samples, P=0.0030, Fig. 6A). This observation was further validated using a larger TCGA data set that was analyzed using the UCSC Cancer Genome Browser (Supplementary Figure 8A, see section on supplementary data given at the end of this article). Next, we observed that nearly 40% of the changes in the expression of H19 in our dataset were related to changes in ERα expression (Fig. 6B, r=0.73). Also, when the expression of H19 was examined in the ERα+ breast tumors, a strong positive correlation was obtained (r=0.763, Supplementary Figure 8B), whereas no such correlation was observed when H19 expression was examined against the ERαlow/− tumors (r=0.38, Supplementary Figure 8C). These results indicate that H19 is expressed at a higher level in ER+ breast tumors as compared with ER− tumors.
Figure 6.
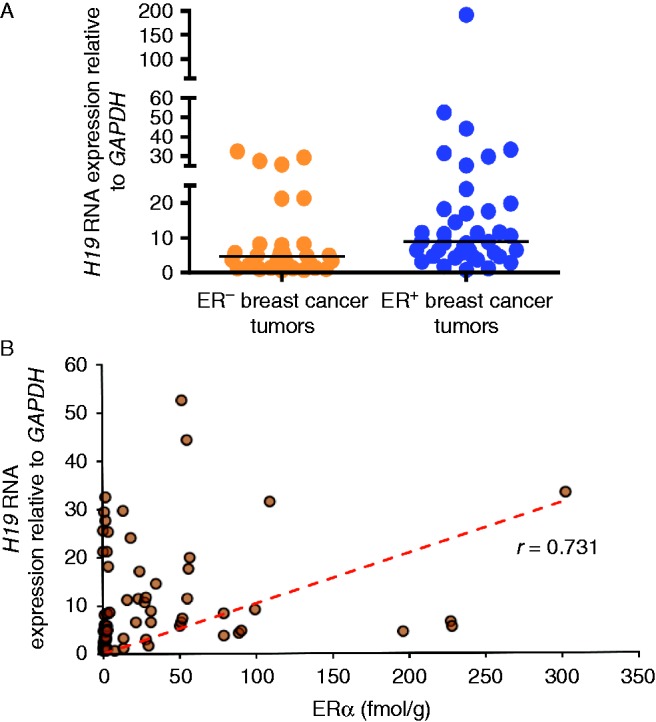
H19 expression is associated with ER+ breast tumors. (A) Expression of the H19 gene was quantified in 39 ER+ and 31 ER− primary breast tumors using qPCR. On average, the ER+ breast tumors showed higher expression of H19 (mean±s.d.=17.65±31.04) as compared with the ER− tumors (8.11±9.4) as per the Mann–Whitney test (P=0.0030). (B) Based on a ligand binding assay, the expression of ERα was found to range from 0 to 302 fmol/mg protein. Pearson correlation revealed a positive correlation between ERα and H19 expression in the breast tumors (r=0.731).
Discussion
The present evidence indicates that increased estrogen signaling and increased ERα expression are significant risk factors for the development of breast cancer (Khan et al. 1994). Recent results have indicated that ERα+ cells represent a luminal-restricted progenitor cell population in healthy human breast tissue. Therefore, understanding the molecular mechanisms by which estrogen signaling regulates the proliferation and differentiation of these ERα+ luminal progenitors and how enhanced activation of estrogen–ERα signaling can lead to breast cancer is likely to be of value in the early detection of ER+ breast cancers or even in the development of preventative measures. Because of the unavailability of ERα+ non-malignant human breast epithelial cell lines, our current understanding of estrogen signaling at the molecular level is based on ER+ breast cancer cell lines. Therefore, little information is available about the targets of estrogen–ERα signaling axis in normal human breast epithelial cells. In the present report, we examined the effects of estrogen signaling on the biological functions of primary human ERα+ luminal progenitors and found that estrogen directly increased luminal progenitor cell proliferation and that blocking ERα strongly attenuated this effect of estrogen. Moreover, we also observed that co-culturing fibroblasts with luminal progenitors significantly increased progenitor numbers and that the addition of estrogen to these co-cultures had a small synergistic effect on luminal progenitor cell expansion. The present observations are in agreement with results described in previous reports that indicated that estrogen could act through paracrine effects to regulate ductal elongation in mammary glands (Mallepell et al. 2006, Brisken & Duss 2007, Fillmore et al. 2010). Our results further indicate that estrogen could act directly on ERα+ luminal progenitors and that stromal fibroblasts and the estrogen–ERα signaling axis cooperate to regulate ductal elongation during puberty. Interestingly, we found that estrogen signaling increased the expression of H19 and ERα but not the well-identified breast cancer targets of estrogen signaling PR and pS2 in matrigel cultures that had been initiated with luminal progenitors. We further showed that H19 expression is important for estrogen-induced luminal progenitor expansion. This observation could be important to the pathogenesis of ER+ breast tumors, seeing as we found that H19 is more significantly expressed in ER+ breast cancer tumors and that its expression is correlated with ER expression in these tumors. This finding is interesting because results of previous studies have indicated that H19 could act as an oncogene in breast cancer cells (Berteaux et al. 2005). Previously, it was reported that c-MYC, an estrogen-regulated gene in ER+ breast cancer cells (Dubik & Shiu 1988), was also able to bind to and activate the transcription of the H19 gene (Barsyte-Lovejoy et al. 2006), which indicates that estrogen signaling might in fact indirectly regulate the expression of H19 through the activation of c-MYC. In the present report, we provide evidence that ligand-activated ERα binds to six ERE sites within the H19 promoter and could directly activate the transcription of this gene in breast cancer cells. Although the ChIP data indicated that ERα occupies the EREs in the H19 promoter as early as 1 h after induction with E2, the increase in H19 transcript levels could only be detected after 10 h of exposure to E2, which indicates that the ERE half-sites may not be as transcriptionally active as full ERE sites are. Although we can potentially extrapolate to suggest that this is also the case in normal ERα+ luminal progenitors, because ChIP studies are only possible in ER+ breast cancer cells, we cannot exclude the possibility that estrogen signaling could indirectly regulate H19 transcription and expression in healthy ER+ luminal progenitors. It would be interesting to investigate if H19 and MIR675 regulate the expression of the GATA3 gene, which is known to play a role in breast cancer development as well as the development and maturation of luminal cells in the mammary gland. Although H19 expression is increased in ERα+ luminal progenitors, the H19 gene is expressed in undifferentiated bipotent progenitors, which indicates that H19 may regulate the expression of a set of genes that are associated with proliferation and differentiation in both progenitor subtypes. Perhaps such a commonly expressed progenitor-associated gene set would help explain the plasticity that has been exhibited by luminal progenitors in vitro and in vivo (Visvader & Stingl 2014).
Because H19 has been shown to have oncogenic effects in breast cancer cells, its expression has been studied in ER+ and triple-negative breast tumors. In these efforts, the in situ hybridization technique was used to examine H19 expression in 97 breast cancer tumors (Adriaenssens et al. 1998). That study, however, did not yield any informative data, because H19 expression could only be detected in three of the tumor samples, whereas all of the other samples showed only a strong stromal expression of H19. Most probably this was a result of the limited resolution of the in situ hybridization technique that was employed. In the present study, we used the sensitive and quantifiable qPCR assay to examine the expression of H19 in ER+ and ER− breast tumor samples that contained at least 70% tumor cells and found that H19 expression was highly correlated with ERα expression in these tumors. Lastly, our observation that estrogen signaling in the luminal progenitors does not increase the expression of PR and pS2, which are classical targets of estrogen signaling in breast cancer cells, indicates that in healthy luminal progenitors, estrogen signaling could activate a different network of molecular targets to that it activates in breast cancer cells.
Conclusion
The present results provide new insights into the molecular mechanism through which estrogen regulates human mammary gland development, and they identify H19, a lncRNA, as a regulator of luminal progenitor cell expansion. Our observations also lay the foundation for new studies aimed at understanding the differences in the molecular targets of estrogen signaling in healthy versus transformed primary human cell types and breast cancer cells. Based on these findings, it is possible to propose the hypothesis that some of the breast cancer-specific targets of estrogen signaling may function as early premalignant changes in the process that leads to breast cancer initiation and development. Furthermore, some of these breast-cancer-specific targets may prove useful in ascertaining the risk of developing ER+ breast cancer in the high-risk patient population.
Supplementary data
This is linked to the online version of the paper at http://dx.doi.org/10.1530/ERC-15-0105.
Author contribution statement
P Basak, L C Murphy, and A Raouf conceived of and designed the experiments. The experimental procedures were performed by P Basak, S Chatterjee, and S Weger and were analyzed by P Basak, S Chatterjee, M C Bruce, A Raouf, and L C Murphy. P Basak, L C Murphy, and A Raouf prepared the manuscript. L C Murphy and A Raouf contributed reagents, materials, and analysis tools.
Supplementary Material
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was made possible through generous funding support from the CancerCare Manitoba Foundation (grant number 761099745, CancerCare.org), the University of Manitoba (grant number 125972, umanitoba.ca), and the Manitoba Health Research Council (grant number 125348, mhrc.mb.ca), and Canadian Breast Cancer Foundation (CBCF-Prairies/NWT chapter, grant number 76-109-9750, cbcf.org) to A Raouf. L C Murphy was supported by grants from the Canadian Breast Cancer Foundation (CBCF-Prairies/NWT chapter, grant number 1213, cbcf.org) and the Canadian Cancer Society Research Institute (cancer.ca). M C Bruce was supported by a Postdoctoral Fellowship from the Canadian Breast Cancer Foundation (CBCF-Prairies/NWT chapter, grant number 1301, cbcf.org), and P Basak was funded by a Postdoctoral Fellowship from the Manitoba Health Research Institute (MHRC.mb.ca).
References
- Adriaenssens E, Dumont L, Lottin S, Bolle D, Lepretre A, Delobelle A, Bouali F, Dugimont T, Coll J, Curgy JJ. H19 overexpression in breast adenocarcinoma stromal cells is associated with tumor values and steroid receptor status but independent of p53 and Ki-67 expression. American Journal of Pathology. 1998;153:1597–1607. doi: 10.1016/S0002-9440(10)65748-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriaenssens E, Lottin S, Dugimont T, Fauquette W, Coll J, Dupouy JP, Boilly B, Curgy JJ. Steroid hormones modulate H19 gene expression in both mammary gland and uterus. Oncogene. 1999;18:4460–4473. doi: 10.1038/sj.onc.1202819. [DOI] [PubMed] [Google Scholar]
- Adriaenssens E, Lottin S, Berteaux N, Hornez L, Fauquette W, Fafeur V, Peyrat JP, Le Bourhis X, Hondermarck H, Coll J, et al. Cross-talk between mesenchyme and epithelium increases H19 gene expression during scattering and morphogenesis of epithelial cells. Experimental Cell Research. 2002;275:215–229. doi: 10.1006/excr.2002.5500. [DOI] [PubMed] [Google Scholar]
- Anderson E, Clarke RB, Howell A. Estrogen responsiveness and control of normal human breast proliferation. Journal of Mammary Gland Biology and Neoplasia. 1998;3:23–35. doi: 10.1023/A:1018718117113. [DOI] [PubMed] [Google Scholar]
- Ariel I, Ayesh S, Perlman EJ, Pizov G, Tanos V, Schneider T, Erdmann VA, Podeh D, Komitowski D, Quasem AS, et al. The product of the imprinted H19 gene is an oncofetal RNA. Molecular Pathology. 1997;50:34–44. doi: 10.1136/mp.50.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes RO, Parisien M, Murphy LC, Watson PH. Influence of evolution in tumor biobanking on the interpretation of translational research. Cancer Epidemiology, Biomarkers & Prevention. 2008;17:3344–3350. doi: 10.1158/1055-9965.EPI-08-0622. [DOI] [PubMed] [Google Scholar]
- Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL, Tsao MS, Penn LZ. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Research. 2006;66:5330–5337. doi: 10.1158/0008-5472.CAN-06-0037. [DOI] [PubMed] [Google Scholar]
- Berteaux N, Lottin S, Monte D, Pinte S, Quatannens B, Coll J, Hondermarck H, Curgy JJ, Dugimont T, Adriaenssens E. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. Journal of Biological Chemistry. 2005;280:29625–29636. doi: 10.1074/jbc.M504033200. [DOI] [PubMed] [Google Scholar]
- Bocchinfuso WP, Korach KS. Mammary gland development and tumorigenesis in estrogen receptor knockout mice. Journal of Mammary Gland Biology and Neoplasia. 1997a;2:323–334. doi: 10.1023/A:1026339111278. [DOI] [PubMed] [Google Scholar]
- Bocchinfuso WP, Korach KS. Estrogen receptor residues required for stereospecific ligand recognition and activation. Molecular Endocrinology. 1997b;11:587–594. doi: 10.1210/mend.11.5.9931. [DOI] [PubMed] [Google Scholar]
- Bocchinfuso WP, Lindzey JK, Hewitt SC, Clark JA, Myers PH, Cooper R, Korach KS. Induction of mammary gland development in estrogen receptor-α knockout mice. Endocrinology. 2000;141:2982–2994. doi: 10.1210/endo.141.8.7609. [DOI] [PubMed] [Google Scholar]
- Brisken C, Duss S. Stem cells and the stem cell niche in the breast: an integrated hormonal and developmental perspective. Stem Cell Reviews. 2007;3:147–156. doi: 10.1007/s12015-007-0019-1. [DOI] [PubMed] [Google Scholar]
- Brown AM, Jeltsch JM, Roberts M, Chambon P. Activation of pS2 gene transcription is a primary response to estrogen in the human breast cancer cell line MCF-7. PNAS. 1984;81:6344–6348. doi: 10.1073/pnas.81.20.6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Cullen BR. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA. 2007;13:313–316. doi: 10.1261/rna.351707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. Genome-wide analysis of estrogen receptor binding sites. Nature Genetics. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Molecular and Cellular Biology. 1987;7:2745–2752. doi: 10.1128/MCB.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RB, Howell A, Potten CS, Anderson E. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Research. 1997;57:4987–4991. [PubMed] [Google Scholar]
- Clarke RB, Anderson E, Howell A. Steroid receptors in human breast cancer. Trends in Endocrinology and Metabolism. 2004;15:316–323. doi: 10.1016/j.tem.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Clemons M, Goss P. Estrogen and the risk of breast cancer. New England Journal of Medicine. 2001;344:276–285. doi: 10.1056/NEJM200101253440407. [DOI] [PubMed] [Google Scholar]
- Cunliffe HE, Ringner M, Bilke S, Walker RL, Cheung JM, Chen Y, Meltzer PS. The gene expression response of breast cancer to growth regulators: patterns and correlation with tumor expression profiles. Cancer Research. 2003;63:7158–7166. [PubMed] [Google Scholar]
- Daniel CW, Silberstein GB, Strickland P. Direct action of 17β-estradiol on mouse mammary ducts analyzed by sustained release implants and steroid autoradiography. Cancer Research. 1987;47:6052–6057. [PubMed] [Google Scholar]
- Doucrasy S, Coll J, Barrois M, Joubel A, Prost S, Dozier C, Stehelin D, Riou G. Expression of the human fetal BAC H19 gene in invasive cancers. International Journal of Oncology. 1993;2:753–758. doi: 10.3892/ijo.2.5.753. [DOI] [PubMed] [Google Scholar]
- Dubik D, Shiu RP. Transcriptional regulation of c-myc oncogene expression by estrogen in hormone-responsive human breast cancer cells. Journal of Biological Chemistry. 1988;263:12705–12708. [PubMed] [Google Scholar]
- Dugimont T, Curgy JJ, Wernert N, Delobelle A, Raes MB, Joubel A, Stehelin D, Coll J. The H19 gene is expressed within both epithelial and stromal components of human invasive adenocarcinomas. Biology of the Cell. 1995;85:117–124. doi: 10.1016/0248-4900(96)85272-5. [DOI] [PubMed] [Google Scholar]
- Eirew P, Stingl J, Raouf A, Turashvili G, Aparicio S, Emerman JT, Eaves CJ. A method for quantifying normal human mammary epithelial stem cells with in vivo regenerative ability. Nature Medicine. 2008;14:1384–1389. doi: 10.1038/nm.1791. [DOI] [PubMed] [Google Scholar]
- Eirew P, Stingl J, Eaves CJ. Quantitation of human mammary epithelial stem cells with in vivo regenerative properties using a subrenal capsule xenotransplantation assay. Nature Protocols. 2010;5:1945–1956. doi: 10.1038/nprot.2010.148. [DOI] [PubMed] [Google Scholar]
- Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, Kuperwasser C. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. PNAS. 2010;107:21737–21742. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton HN, Graham JD, Kantimm S, Santucci N, Cloosterman D, Huschtscha LI, Mote PA, Clarke CL. Progesterone and estrogen receptors segregate into different cell subpopulations in the normal human breast. Molecular and Cellular Endocrinology. 2012;361:191–201. doi: 10.1016/j.mce.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Hilton HN, Santucci N, Silvestri A, Kantimm S, Huschtscha LI, Graham JD, Clarke CL. Progesterone stimulates progenitor cells in normal human breast and breast cancer cells. Breast Cancer Research and Treatment. 2014;143:423–433. doi: 10.1007/s10549-013-2817-2. [DOI] [PubMed] [Google Scholar]
- Holmes KA, Song JS, Liu XS, Brown M, Carroll JS. Nkx3-1 and LEF-1 function as transcriptional inhibitors of estrogen receptor activity. Cancer Research. 2008;68:7380–7385. doi: 10.1158/0008-5472.CAN-08-0133. [DOI] [PubMed] [Google Scholar]
- Honeth G, Lombardi S, Ginestier C, Hur M, Marlow R, Buchupalli B, Shinomiya I, Gazinska P, Bombelli S, Ramalingam V, et al. Aldehyde dehydrogenase and estrogen receptor define a hierarchy of cellular differentiation in the normal human mammary epithelium. Breast Cancer Research. 2014;16:R52. doi: 10.1186/bcr3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, Stingl J, Waterhouse PD, Khokha R. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465:803–807. doi: 10.1038/nature09091. [DOI] [PubMed] [Google Scholar]
- Keniry A, Oxley D, Monnier P, Kyba M, Dandolo L, Smits G, Reik W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r . Nature Cell Biology. 2012;14:659–665. doi: 10.1038/ncb2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SA, Rogers MA, Obando JA, Tamsen A. Estrogen receptor expression of benign breast epithelium and its association with breast cancer. Cancer Research. 1994;54:993–997. [PubMed] [Google Scholar]
- Kraichely DM, Sun J, Katzenellenbogen JA, Katzenellenbogen BS. Conformational changes and coactivator recruitment by novel ligands for estrogen receptor-α and estrogen receptor-β: correlations with biological character and distinct differences among SRC coactivator family members. Endocrinology. 2000;141:3534–3545. doi: 10.1210/endo.141.10.7698. [DOI] [PubMed] [Google Scholar]
- Lee S, Mohsin SK, Mao S, Hilsenbeck SG, Medina D, Allred DC. Hormones, receptors, and growth in hyperplastic enlarged lobular units: early potential precursors of breast cancer. Breast Cancer Research. 2006;8:R6. doi: 10.1186/bcr1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman ME, Osborne CK, Knazek R, Young N. In vitro model systems for the study of hormone-dependent human breast cancer. New England Journal of Medicine. 1977;296:154–159. doi: 10.1056/NEJM197701202960307. [DOI] [PubMed] [Google Scholar]
- Lottin S, Adriaenssens E, Dupressoir T, Berteaux N, Montpellier C, Coll J, Dugimont T, Curgy JJ. Overexpression of an ectopic H19 gene enhances the tumorigenic properties of breast cancer cells. Carcinogenesis. 2002;23:1885–1895. doi: 10.1093/carcin/23.11.1885. [DOI] [PubMed] [Google Scholar]
- Mallepell S, Krust A, Chambon P, Brisken C. Paracrine signaling through the epithelial estrogen receptor α is required for proliferation and morphogenesis in the mammary gland. PNAS. 2006;103:2196–2201. doi: 10.1073/pnas.0510974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-β potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. Journal of Medicinal Chemistry. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- Murphy LC, Watson P. Steroid receptors in human breast tumorigenesis and breast cancer progression. Biomedicine & Pharmacotherapy. 2002;56:65–77. doi: 10.1016/S0753-3322(01)00157-3. [DOI] [PubMed] [Google Scholar]
- Murphy LC, Peng B, Lewis A, Davie JR, Leygue E, Kemp A, Ung K, Vendetti M, Shiu R. Inducible upregulation of oestrogen receptor-β1 affects oestrogen and tamoxifen responsiveness in MCF7 human breast cancer cells. Journal of Molecular Endocrinology. 2005;34:553–566. doi: 10.1677/jme.1.01688. [DOI] [PubMed] [Google Scholar]
- Pachnis V, Belayew A, Tilghman SM. Locus unlinked to α-fetoprotein under the control of the murine raf and Rif genes. PNAS. 1984;81:5523–5527. doi: 10.1073/pnas.81.17.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier F, Chan CT, Timmons PM, Robertson EJ, Evans MJ, Rigby PW. The murine H19 gene is activated during embryonic stem cell differentiation in vitro and at the time of implantation in the developing embryo. Development. 1991;113:1105–1114. doi: 10.1242/dev.113.4.1105. [DOI] [PubMed] [Google Scholar]
- Qiu MT, Hu JW, Yin R, Xu L. Long noncoding RNA: an emerging paradigm of cancer research. Tumour Biology. 2013;34:613–620. doi: 10.1007/s13277-013-0658-6. [DOI] [PubMed] [Google Scholar]
- Raouf A, Sun YJ. In vitro methods to culture primary human breast epithelial cells. Methods in Molecular Biology. 2013;946:363–381. doi: 10.1007/978-1-62703-128-8_23. [DOI] [PubMed] [Google Scholar]
- Raouf A, Zhao Y, To K, Stingl J, Delaney A, Barbara M, Iscove N, Jones S, McKinney S, Emerman J, et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell. 2008;3:109–118. doi: 10.1016/j.stem.2008.05.018. [DOI] [PubMed] [Google Scholar]
- Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Molecular Cell. 2003;11:695–707. doi: 10.1016/S1097-2765(03)00090-X. [DOI] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–393. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Spyrou C, Brown GD, Hadfield J, Odom DT. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods. 2009;48:240–248. doi: 10.1016/j.ymeth.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/S0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- Shoker BS, Jarvis C, Sibson DR, Walker C, Sloane JP. Oestrogen receptor expression in the normal and pre-cancerous breast. Journal of Pathology. 1999;188:237–244. doi: 10.1002/(SICI)1096-9896(199907)188:3%3c237::AID-PATH343%3e3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Soderqvist G, Isaksson E, von Schoultz B, Carlstrom K, Tani E, Skoog L. Proliferation of breast epithelial cells in healthy women during the menstrual cycle. American Journal of Obstetrics and Gynecology. 1997;176:123–128. doi: 10.1016/S0002-9378(97)80024-5. [DOI] [PubMed] [Google Scholar]
- Stingl J, Eaves CJ, Kuusk U, Emerman JT. Phenotypic and functional characterization in vitro of a multipotent epithelial cell present in the normal adult human breast. Differentiation. 1998;63:201–213. doi: 10.1111/j.1432-0436.1998.00201.x. [DOI] [PubMed] [Google Scholar]
- Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes and Development. 2014;28:1143–1158. doi: 10.1101/gad.242511.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PH, Snell L, Parisien M. The NCIC-Manitoba Breast Tumor Bank: a resource for applied cancer research. Canadian Medical Association Journal. 1996;155:281–283. [PMC free article] [PubMed] [Google Scholar]
- Weitsman GE, Li L, Skliris GP, Davie JR, Ung K, Niu Y, Curtis-Snell L, Tomes L, Watson PH, Murphy LC. Estrogen receptor-α phosphorylated at Ser118 is present at the promoters of estrogen-regulated genes and is not altered due to HER-2 overexpression. Cancer Research. 2006;66:10162–10170. doi: 10.1158/0008-5472.CAN-05-4111. [DOI] [PubMed] [Google Scholar]
- Wigler M, Silverstein S, Lee LS, Pellicer A, Cheng Y, Axel R. Transfer of purified herpes virus thymidine kinase gene to cultured mouse cells. Cell. 1977;11:223–232. doi: 10.1016/0092-8674(77)90333-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.