

Abstract
To study the sequence determinants governing protein fold evolution, we generated hybrid sequences from two homologous proteins with 40% identity but different folds: Pfl 6 Cro, which has a mixed α + β structure, and Xfaso 1 Cro, which has an all α-helical structure. First, we first examined eight chimeric hybrids in which the more structurally conserved N-terminal half of one protein was fused to the more structurally divergent C-terminal half of the other. None of these chimeras folded, as judged by circular dichroism spectra and thermal melts, suggesting that both halves have strong intrinsic preferences for the native global fold pattern, and/or that the interfaces between the halves are not readily interchangeable. Second, we examined 10 hybrids in which blocks of the structurally divergent C-terminal region were exchanged. These hybrids showed varying levels of thermal stability and suggested that the key residues in the Xfaso 1 C terminus specifying the all-α fold were concentrated near the end of helix 4 in Xfaso 1, which aligns to the end of strand 2 in Pfl 6. Finally, we generated hybrid substitutions for each individual residue in this critical region and measured thermal stabilities. The results suggested that R47 and V48 were the strongest factors that excluded formation of the α + β fold in the C-terminal region of Xfaso 1. In support of this idea, we found that the folding stability of one of the original eight chimeras could be rescued by back-substituting these two residues. Overall, the results show not only that the key factors for Cro fold specificity and evolution are global and multifarious, but also that some all-α Cro proteins have a C-terminal subdomain sequence within a few substitutions of switching to the α + β fold.
Keywords: chimera, folding specificity, hybrid sequence, structural evolution
Introduction
Numerous design, engineering and selection studies have used hybrids of two differently folded protein sequences to elucidate sequence determinants of folding specificity (Lattman and Rose, 1993; Rose and Creamer, 1994), and to assess the potential for evolutionary pathways between folds. Most early hybrid domains with many residues from each parent protein showed low stability, aggregation and/or poorly defined tertiary structure (Yuan and Clarke, 1998; Blanco et al., 1999; Dalal and Regan, 2000), although at least one early quasi-hybrid protein, Janus, appeared stable and well-folded (Dalal et al., 1997). In later efforts, Orban and coworkers obtained hybrid proteins with well-defined structures and sufficient solubility for nuclear magnetic resonance (NMR) structure determination (Alexander et al., 2005; He et al., 2005). Eventually, they carefully designed nearly identical pairs of hybrid sequences with distinct well-defined folds and even distinct binding functions (Alexander et al., 2007, 2009; He et al., 2008, 2012).
These hybrid approaches demonstrated that the specificity of a sequence for its native fold could reside in very few residues, and that gradual sequence mutation could switch a protein's topology while preserving the ability to fold and function in the intermediate steps. They also suggested, however, that fold-switching pathways require careful design to identify. The studies cited earlier also utilized pairs of protein sequences without any apparent evolutionary relationship, perhaps due to a paucity of known homologous proteins with different folds. The results speak to the general potential for mutationally induced switching between folds, but not necessarily to mechanisms and specificity determinants in the natural evolution of new folds.
Meanwhile, examples of natural pairs of homologous protein sequences with different folds were gradually being discovered (Grishin, 2001; Kinch and Grishin, 2002; Andreeva and Murzin, 2006; Murzin, 2008; Bryan and Orban, 2010), opening new avenues for studies of folding specificity and evolution using hybrid sequences. Some successful early studies of this kind involved switches in topology induced by simple hybrid substitutions in very small, highly disulfide-bonded protein domains (Meier et al., 2007; Yeates, 2007) or domains with secondary structure changes localized to a short stretch of sequence (Tidow et al., 2004).
Our first efforts involved the Cro family of bacteriophage transcription factors, a ∼65-residue DNA-binding domain. Cro proteins can fold as monomers in solution (Jana et al., 1997; Newlove et al., 2004; Roessler et al., 2008) and dimerize to varying extents (Jana et al., 1997; Darling et al., 2000; LeFevre and Cordes, 2003; Newlove et al., 2004; Dubrava et al., 2008; Roessler et al., 2008), but bind DNA in the dimeric form (Albright and Matthews, 1998). Some Cro proteins have an all α-helical fold while others have a mixed α + β fold (Fig. 1). The two folds conserve a three-helix DNA-binding subdomain in the N-terminal half of the sequence, while the dimerization subdomain, consisting mostly of the C-terminal half, is α-helical in some family members and β-sheet in others. The α-helical fold is ancestral, while the α + β fold is a descendant. Differently folded Cro proteins share global sequence similarity, suggesting that the α + β fold evolved from the α-helical fold by accumulation of small sequence mutations (Newlove et al., 2004; Roessler et al., 2008).
Fig. 1.
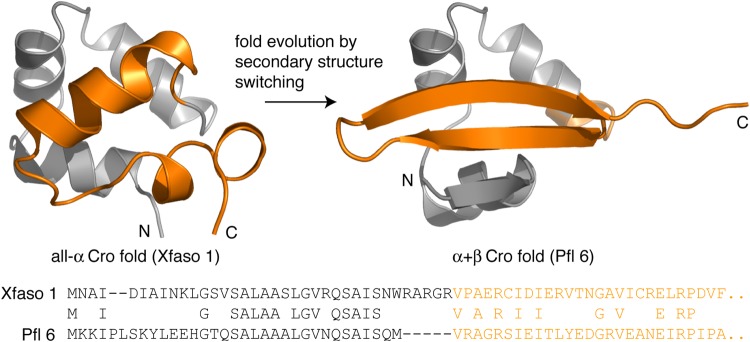
Evolution from an all-α to an α + β fold in the Cro protein family. The N-terminal half of the sequence (gray) contains the helix-turn-helix DNA-binding motif and is mostly structurally conserved, while the structurally divergent C-terminal half (orange) contains much of the homodimer interface. Xfaso 1 (3BD1, chain A) and Pfl 6 (2PIJ, chain A), respectively, represent the ancestral and descendant folds and have 40% sequence identity across this 65-residue alignment.
In an early study, we generated a set of hybrid point substitutions based on a sequence alignment of two distant Cro homologs with 25% sequence identity: P22 Cro, which represented the all α-helical fold, and λ Cro, which represented the mixed α + β fold (Van Dorn et al., 2006). The results suggested that limited mutations could change the Cro fold, but not by any trivial mechanism. We found that the C-terminal portion of each protein contained at least five or six residues that strongly destabilized the other protein, suggesting that both P22 Cro and λ Cro contained multiple specificity determinants that ruled out the alternate fold. However, we also designed chameleon sequences (largely hybrids) that could be substituted for the C-terminal region of either P22 Cro or λ Cro (Van Dorn et al., 2006; Anderson et al., 2011). These sequences folded as α-helix when grafted onto the N-terminal half of P22 Cro, but as β-sheet when introduced into λ Cro. The successful design of Cro chameleon sequences suggested not only that the different C-terminal folding patterns could be encoded in a single sequence, but also that the N-terminal half held specificity factors that could govern the folding pattern of the C terminus.
We later discovered a pair of differently folded Cro proteins with 40% sequence identity, Xfaso 1 and Pfl 6 (Fig. 1) (Roessler et al., 2008). We reasoned that moving to hybrid studies of Xfaso 1 and Pfl 6, a sequence pair with closer homology than P22 and λ, might yield a simpler picture of specificity determinants and potential mechanisms for fold switching. In our first study of Xfaso 1 and Pfl 6 hybrids, we generated a set of hybrid insertion/deletion mutations based on two alignment gaps (Fig. 1), one at the N terminus and one in a central linker connecting the N- and C-terminal halves (Stewart et al., 2013b). All N-terminal deletions were highly destabilizing to Pfl 6, and the central linker deletions completely unfolded Xfaso 1, showing that sequence length in these regions was clearly an important specificity determinant for their respective structures. However, none of the insertions or deletions switched either protein to the other fold, indicating that other sequence differences must also be important.
We now present an extensive study of simple block hybrids of Xfaso 1 and Pfl 6. First, we generated eight chimeric hybrids containing approximately the N-terminal half of one sequence and the C-terminal half of the other. Second, we generated 10 additional hybrid sequences containing mostly one sequence but with block substitutions of fragments of the C-terminal region of the other protein. We did not discover any simple block substitutions that switch the Cro fold; however, the C-terminal sequence of Xfaso 1 does appear to be within a few substitutions of switching from α-helix to β-sheet, when grafted to an N-terminal sequence that supports such a switch.
Materials and methods
Construction of hybrid sequences
Gene sequences encoding Pfl 6 and Xfaso 1 Cro were previously introduced into pET21b expression vectors, yielding constructs for expression of the wild-type sequences with C-terminal LEHHHHHH tags to enable nickel affinity purification (Roessler et al., 2008). The central RARGR sequence was introduced into the wild-type Pfl 6 construct by insertion mutation using the QuikChange method (Stratagene) (Stewart et al., 2013b). The initial set of half-and-half chimeras was then constructed as follows. To construct chimera PX1, the coding sequences for wild-type Xfaso 1 and Pfl6 (RARGR insertion) were amplified, including flanking NdeI and PaeR7I sites, and digested with Bme1580I, which cut both within the central RARGR sequence. The appropriate purified fragments (5′ half of Pfl 6 coding sequence and 3′ half of Xfaso 1 coding sequence) were religated to yield the coding sequence for PX1 flanked by NdeI and PaeR7I sites. This sequence was then digested with NdeI and PaeR7I and ligated back into a pET21b NdeI/PaeR7I fragment. Chimera XP1 was constructed by introduction of silent SacII restriction sites into the constructs for wild-type Xfaso 1 and Pfl 6 (RARGR insertion), digestion of both with SacII and PaeR7I, and religation of the large fragment from the Xfaso 1 digestion with the small fragment from the Pfl 6 digestion. Chimeras PX2-PX4 and XP2-XP4 were then constructed by introducing substitution, insertion or deletion mutations into PX1 or XP1 using the QuikChange method (Stratagene): PX2 and XP2 by deletion of the RARGR sequence from PX1 and XP1, respectively; XP3 by deletion of the VRAGR sequence from XP1; PX3 by introduction of P40R and E42G substitutions into PX1, followed by deletion of the RARGR sequence; PX4 by insertion of the VPAER sequence into PX3; and XP4 by introduction of R38P and G40E substitutions into XP1. The remaining hybrids (XP5-XP10 and PX5-PX8) were constructed by a variety of approaches, including introduction of synthetic double-stranded oligonucleotide cassettes by restriction digestion and ligation (e.g. for XP5 and XP6) and/or successive substitution mutagenesis to the sequence of wild-type or a previously constructed hybrid using QuikChange (Stratagene).
Protein purification
Proteins were expressed and purified by denaturing Ni-NTA affinity chromatography essentially as described (LeFevre and Cordes, 2003) or, in the case of small-scale cultures, by a spin-column version of the same procedure (Qiagen). Purification of cysteine-containing variants also included 15 mM β-mercaptoethanol in the lysis and wash buffers and 3–5 mM β-mercaptoethanol in the elution buffer to prevent disulfide bond formation. For uniform 15N-labeled samples, proteins were expressed in M9T minimal medium containing 0.8 g/l 15NH4Cl as the sole nitrogen source, and purified in the same manner as unlabeled proteins. Purified proteins were refolded by dialysis into SB250 buffer [50 mM Tris (pH 7.5), 250 mM KCl and 0.2 mM EDTA], and dialysates were centrifuged to remove precipitates prior to measurement of soluble protein concentrations. For cysteine-containing variants, 1 mM DTT was included to prevent disulfide bond formation. Protein concentrations were estimated by ultraviolet absorbance as described previously (Stewart et al., 2013b).
Circular dichroism spectroscopy
Circular dichroism (CD) spectra and thermal denaturation curves were obtained on an OLIS DSM-20 CD spectropolarimeter. Wavelength scans were obtained at 20°C at a protein concentration of 25 μM in a 1 mm pathlength cylindrical cell, from 260 to 205 nm in 1 nm steps with an integration time of 5–30 s. Signals were averaged from three to five scans, and spectra were corrected for buffer baseline signals. In most of the figures, signals are reported as raw ellipticity rather than mean residue ellipticity, because Xfaso 1 and Pfl 6 contain disordered C-terminal tails of different lengths that contribute little signal across most of the spectrum. Thermal denaturation curves were obtained at a protein concentration of 25 μM in a 2 mm pathlength cylindrical cell, from a low temperature of 14–20°C to a high temperature of 76–90°C in 2°C steps, with 2 min equilibration time and 25–55 s signal integration time for each temperature point. Tm values were obtained by fitting to the following relationship (Becktel and Schellman, 1987):
The free energy of unfolding ΔGu relates directly to the fraction of molecules in the unfolded state, and this fraction in turn relates to the position of the measured ellipticity value relative to the unfolded and folded baselines. Baseline slopes and intercepts were allowed to vary in fits. The heat capacity of unfolding (ΔCp) was fixed based on an estimate of 14 cal mol−1 K−1 per residue (Myers et al., 1995).
NMR spectroscopy
15N–1H NMR correlation spectra were recorded at 293 K on a Varian Inova 600 MHz spectrometer equipped with a triple-resonance cryogenic probe. Spectra were processed using NMRPipe/NMRDraw (Delaglio et al., 1995) and analyzed using Sparky (T.D. Goddard and D.G. Kneller, SPARKY 3, University of California, San Francisco) or NMRView (Johnson and Blevins, 1994). Pfl 6 M33W/I58D backbone resonance assignments were obtained by strip plot analysis of 3D NOESY-HSQC and 3D TOCSY-HSQC experiments in samples containing 2 mM protein in 50 mM phosphate (pH 6) at 293 K.
Results
Half-and-half chimeras
We expressed and purified eight hybrids containing approximately the N-terminal half of Xfaso 1 or Pfl 6 fused to the C-terminal half of the other protein (XP1–XP4 and PX1–PX4; Table I). We subjected these chimeric hybrids to far ultraviolet CD wavelength scans and thermal denaturation to gauge secondary structure content and stability (Fig. 2). At 20°C, all eight chimeras showed far ultraviolet CD spectra with weak ellipticity at 222 nm and relatively strong negative ellipticity near 205 nm (Fig. 2A). This spectral shape and intensity resembles that observed for other unfolded variants of Pfl 6 and Xfaso 1 (Stewart et al., 2013b). All eight chimeras showed essentially flat profiles for thermal denaturation monitored by CD (Fig. 2B), further suggesting that they are unfolded at ambient temperature; only XP2 and XP3 showed more than 10% ellipticity decrease when heated to 76°C. XP2 and XP3 also have the strongest ellipticity at 222 nm at 20°C (Fig. 2A). These chimeras may have a weakly populated native-like state or a denatured state with significant residual structure, but it is unclear at present whether such structure more closely resembles Xfaso 1 or Pfl 6 (Morrone et al., 2011). By and large, despite the sequence homology between Xfaso 1 and Pfl 6, these simple combinations of the two sequences do not fold, and the two halves of the two sequences are not structurally compatible.
Table I.
Hybrids of Xfaso1 and Pfl6 used in this study
 |
Fig. 2.
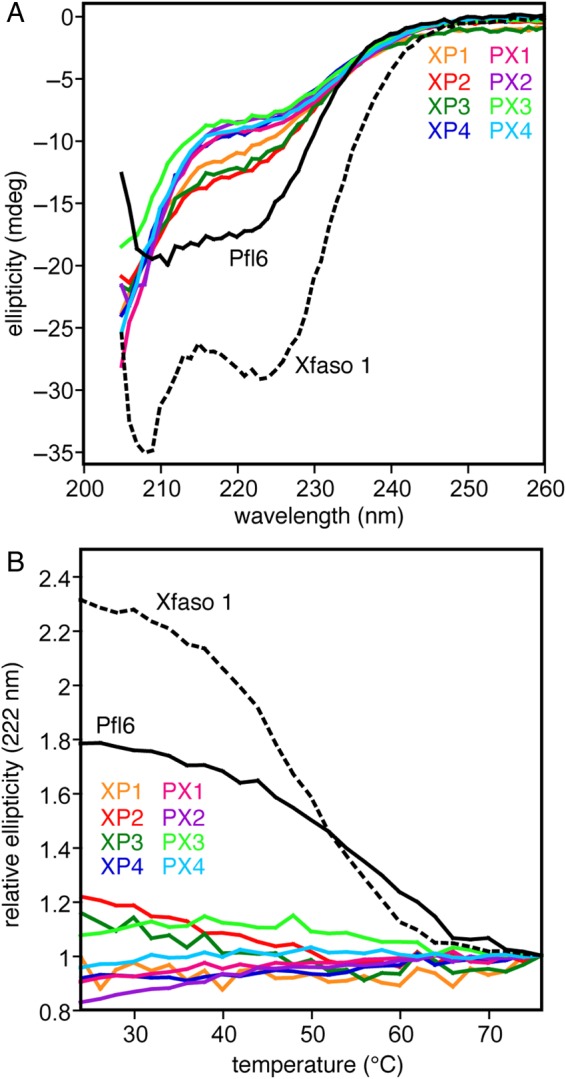
Half-and-half chimeras of Xfaso 1 and Pfl 6 are largely unfolded: (A) far ultraviolet CD spectra (25 μM protein at 1 mm pathlength, 20°C) of eight chimeras compared with wild-type Pfl 6 and Xfaso 1, (B) thermal denaturation of chimeras monitored at 222 nm, shown as ellipticity relative to the baseline signal from denatured protein at 75°C, to illustrate that chimeras show much smaller changes upon heating than the wild-type proteins. See Table I for sequences of the chimeras.
This incompatibility is explainable if the N-terminal half and central linker exert a strong bias on the conformation of the C terminal half (Anderson et al., 2011), but each of the structurally divergent C-terminal halves also strongly resists switching to the alternate fold. One mechanism by which the N-terminal half and the central linker region sequence bias the global fold is the different length of the two sequences at the very N-terminal end and in the central linker (see Fig. 1). In a previous study, variants of Xfaso 1 and Pfl 6 with indels corresponding to both of these gap positions in the alignment did not fold at all (Stewart et al., 2013b). Four of the eight chimeras studied here (XP1, XP4, PX2 and PX3) should be strongly biased by these factors toward one fold or the other. For XP1 and XP4, the combination of short N-terminal sequence and long central linker region sequence should permit the all-α form while effectively prohibiting the α + β topology; PX2 and PX3 have the opposite pattern and should be restricted to the α + β pattern. The failure of these chimeras to fold suggests that the C-terminal regions of both proteins also have a strong folding bias that opposes the influence of the N-terminal/central region.
Block substitutions within the C-terminal region
We next examined which elements within the C-terminal halves were least and most compatible with an alternate conformation. We thus generated and characterized several hybrid sequences in which fragments/blocks of each C terminus were substituted into the other (Table I; Figs 3 and 4). For the first round of experiments, we divided the C terminus into two blocks, approximately at the turn connecting the major C-terminal secondary structure elements. The smaller of the two swapped fragments (giving chimeras XP5 and PX5) corresponded to residues 42–49 of Xfaso 1 (CIDIERVT) and 39–46 of Pfl 6 (SIEITLYE), representing most of the fourth helix of Xfaso 1 and the entire second strand of Pfl 6. A second, much larger swapped fragment (giving chimeras XP6 and PX6) consisted of residues 50–79 of Xfaso 1 or 47–67 of Pfl 6, corresponding to helices 5 and 6 of Xfaso 1 or strand 3 of Pfl 6, along with a turn/loop region connecting them to the preceding secondary structure element. This region also includes stretches at the C-terminal end (residues 66–79 of Xfaso 1 and residues 61–67 of Pfl 6) that do not appear in the crystal structures and are probably disordered (Roessler et al., 2008).
Fig. 3.
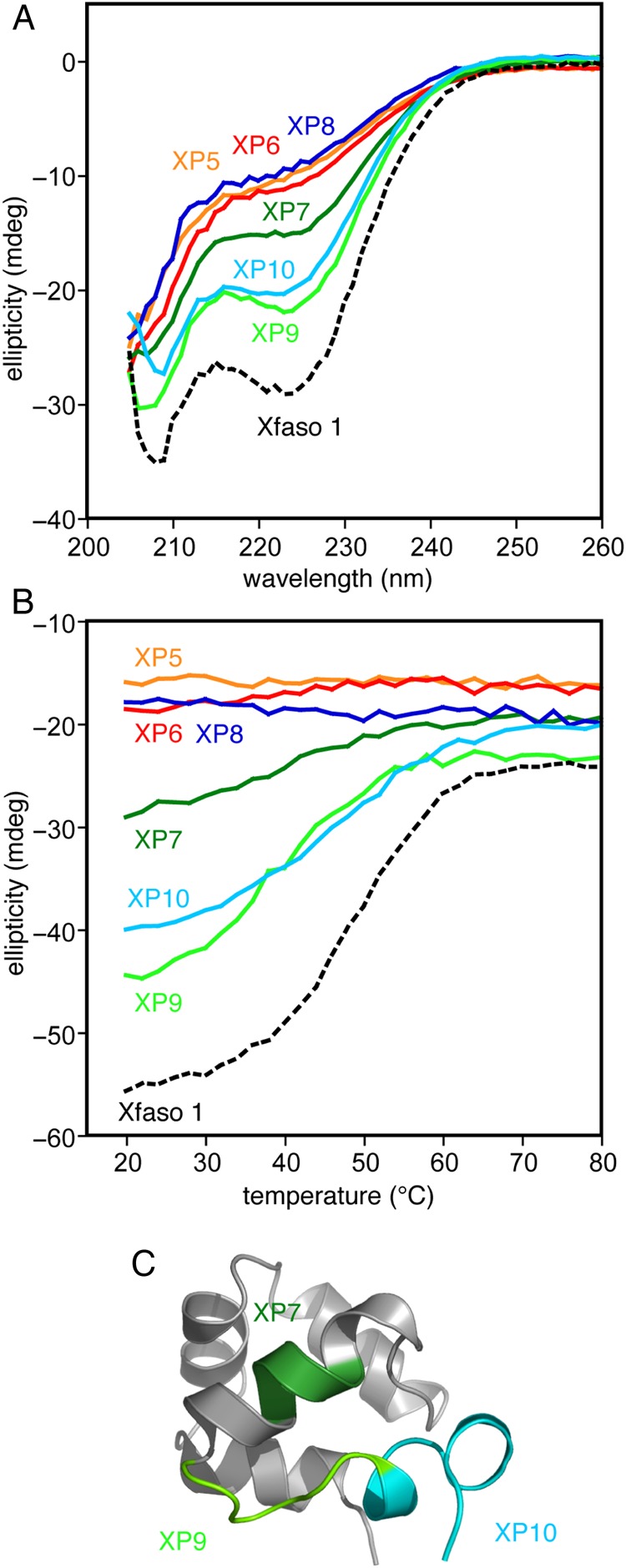
Block substitutions of Pfl 6 C-terminal sequence into Xfaso 1 show varying abilities to fold: (A) far ultraviolet CD spectra (25 μM protein at 1 mm pathlength, 20°C) of eight block hybrids compared with the parent sequence, wild-type Xfaso 1, (B) thermal denaturation of the block hybrids monitored by CD at 222 nm (25 μM protein at 1 mm pathlength, 20°C), (C) regions for which the block substitutions are reasonably well tolerated, mapped onto the subunit structure of Xfaso 1. See Table I for sequences.
Fig. 4.
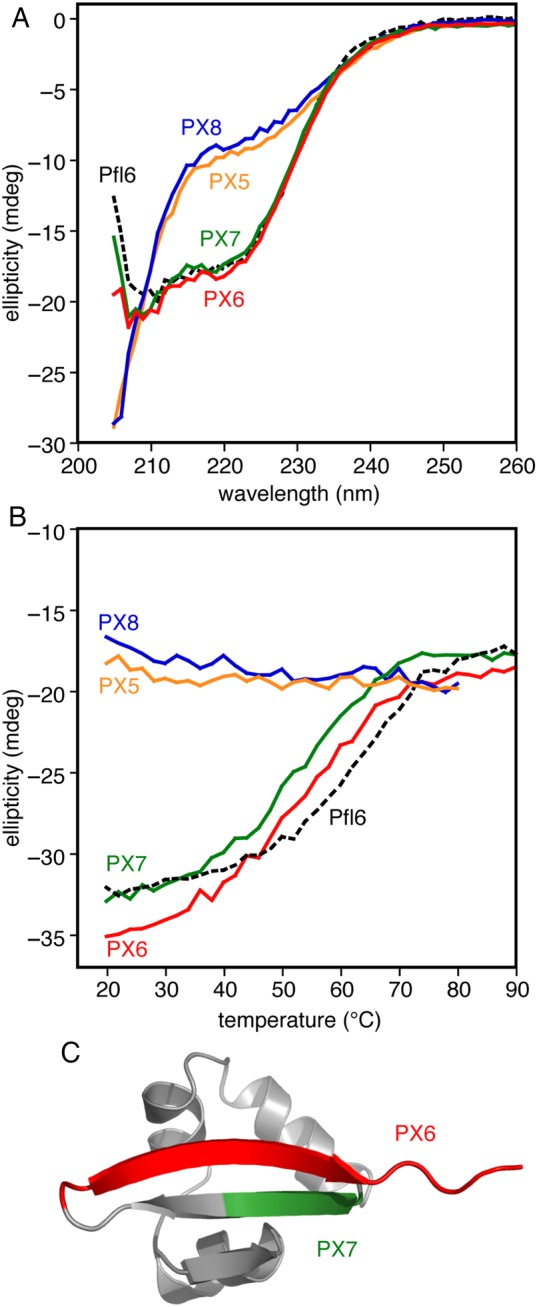
Block substitutions of Xfaso 1 C-terminal sequence into Pfl 6 show varying abilities to fold: (A) far ultraviolet CD spectra (25 μM protein at 1 mm pathlength, 20°C) of eight block hybrids compared with the parent sequence, wild-type Pfl 6, (B) thermal denaturation of the block hybrids monitored by CD at 222 nm (25 μM protein at 1 mm pathlength, 20°C), (C) regions for which the block substitutions are reasonably well tolerated, mapped onto the subunit structure of Pfl 6. See Table I for sequences.
Of these four swapped constructs, only PX6, containing the helix 5/6 region of Xfaso 1 swapped into Pfl 6, showed any folding at all, but it was relatively stable, with a Tm of 54°C (ΔTm = −10°C; see also Fig. 7 for a curve fit). The far ultraviolet CD spectrum of PX6 suggests that it adopts the β-sheet fold of Pfl 6. An 15N–1H correlation NMR spectrum of PX6 (Fig. 5) shows patterns of dispersed peaks similar to those belonging to the central strand of the three-stranded β-sheet Pfl 6, further suggesting that the β-sheet framework is largely intact. We conclude that a significant contiguous portion of the C-terminal half of Xfaso 1 is compatible with the β-sheet fold: the region encoding the short helices 5 and 6, and the preceding loop/turn, can substitute for strand 3 of Pfl 6. In contrast, neither of the two C-terminal fragments of Pfl 6 was compatible with the α-helical fold.
Fig. 7.

Folding of PX3 is rescued by R44L and V45Y substitutions. (A) far ultraviolet CD spectra (25 μM protein at 1 mm pathlength, 20°C) and C-terminal subdomain sequences of PX3-R44L/V45Y compared with PX6, showing that both are likely to have the α + β fold of Pfl 6 despite introduction of large amounts of Xfaso 1 sequence (underlined residues), (B) thermal denaturation monitored by CD at 222 nm (25 μM protein at 1 mm pathlength, 20°C), including curve fits (see ‘Materials and methods’ section).
Fig. 5.
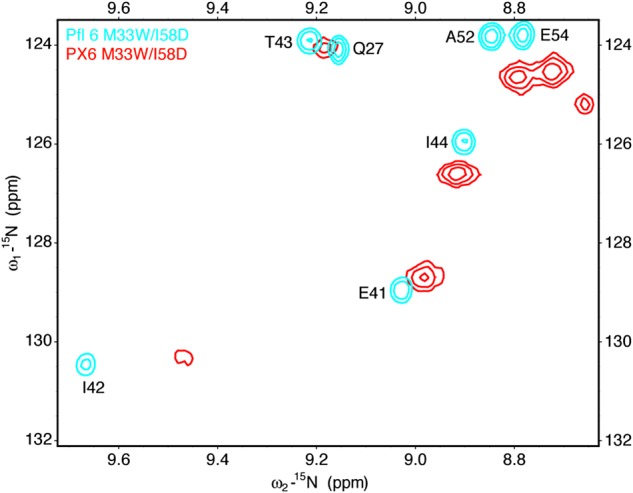
Limited comparison of Pfl 6 (cyan) and PX6 (red) 15N–1H correlation spectra suggests retention of the Pfl 6 β-sheet in PX6. Both proteins contain M33W/I58D mutations, which render Pfl 6 monomeric and slightly more stable, to improve spectral quality. Pfl 6 M33W/I58D is at 2 mM protein concentration in 50 mM sodium phosphate (pH 6), with 10% D2O; PX6 M33W/I58D sample is at 1 mM protein concentration in 50 mM sodium phosphate (pH 7.2), 200 mM NaCl with 10% D2O. Only the Pfl 6 M33W/I58D spectrum could be assigned by strip analysis of three-dimensional spectra.
Since the C-terminal fragments from the helix 4/strand 2 region of both proteins were incompatible with the other sequence, we investigated this region further by dividing it into two equal four-residue blocks of sequence. The corresponding block substitutions yielded chimeras XP7, XP8, PX7 and PX8 (Table I; Figs 3 and 4). One of the two blocks, comprising residues 39–42 of Pfl 6 (SIEI) and 42–45 of Xfaso 1 (CIDI), could be swapped in both directions with at least some retention of folding, perhaps because these two sequences differ by a pair of conservative substitutions. The substitution of the Xfaso 1 sequence into Pfl 6 was much better tolerated, giving a Tm of 53°C (ΔTm = −11°C), while the inverse substitution showed only a weak denaturation curve and much less ellipticity than native Xfaso 1 at 20°C. This variant probably has a Tm < 20°C (ΔTm > −30°C). One possible explanation for this destabilization is the substitution of serine for cysteine at a buried position. Swapping of residues 43–46 of Pfl 6 (TLYE) for 46–49 of Xfaso 1 (ERVT) and vice versa was poorly tolerated with no significant folding observed. These subsequences are quite dissimilar, being related by four non-conservative substitutions, including replacements of polar for hydrophobic residues. These differences might explain their poor interchangeability.
Since residues 47–67 of Pfl 6 were incompatible with substitution for residues 50–79 of Xfaso 1, we also divided this region into two smaller blocks, yielding chimeras XP9 and XP10. Residues 47–52 (DGRVEA) of Pfl 6 could be substituted into Xfaso 1 (NGAVIC) with some preservation of folding (XP9; Tm of 39°C and ΔTm = −12°C). This region corresponds essentially to the loop connecting helices 5 and 6 of Xfaso 1. Substitution of 53–67 of Pfl 6, which corresponds to replacing the sequence encoding the short fifth and sixth helices 5 and 6 of Xfaso 1, showed a similar level of folding stability (XP10). For both XP9 and XP10, the lack of a clear baseline in the thermal melts makes it somewhat difficult to distinguish whether the variant has a higher native helicity and very low thermal stability, or a lowered native helicity but moderate thermal stability.
Overall, the block substitution analysis of the C-terminal halves suggests that Pfl 6 has higher tolerance for replacement with Xfaso 1 C-terminal residues than Xfaso 1 does for Pfl 6 replacements. Multiple regions of the Pfl 6 C-terminus strongly destabilize Xfaso 1, while only one of the fragments of Xfaso 1's C-terminus strongly destabilizes Pfl 6. An important caveat is that the thermal stability of wild-type Xfaso 1 is ∼13°C lower than that of Pfl 6, meaning that less thermal destabilization is required to unfold Xfaso 1. This caveat aside, we suggest that (i) the Xfaso 1 C-terminal sequence is more nearly capable of switching to the β-sheet fold than the Pfl 6 C-terminal sequence is capable of switching to the α-helical fold, and/or (ii) the key C-terminal determinants favoring the helix fold in Xfaso 1 are more centralized in a single region than the determinants specifying the β-sheet fold for Pfl 6.
Point substitution analysis of critical region in helix 4/strand 2
One feature shared by the two proteins is that the sequence block corresponding to residues 43–46 in Pfl 6 and 46–49 in Xfaso 1 cannot be exchanged in either direction without complete unfolding. To further investigate this apparently critical region for fold specificity, we exchanged each aligned residue individually (Fig. 6). The Pfl 6 variants L44R and Y45V showed severe destabilization (Tm ≤ ∼25°C), while T43E and E46T were mildly to moderately destabilizing with Tm values of 48°C (ΔTm = −16°C) and 54°C (ΔTm = −10°C), respectively. The Xfaso 1 variant T49E is completely unfolded, while R47L is rather stable. V48Y and E46T substitutions in Xfaso 1 both destabilize it, with Tm values estimated at <15°C assuming that the native ellipticity is similar to that of the wild type. The only single-residue swap that is strongly destabilizing in both directions is Y45V/V48Y: each residue appears to strongly favor one fold or the other. At the other three positions, at least one of the two wild-type residues can coexist with both structures.
Fig. 6.

Single hybrid substitutions exchanging aligned residues in the most critical region of the C-terminal sequence. (A) thermal denaturation curves of Xfaso 1 variants monitored by CD at 222 nm (25 μM protein at 1 mm pathlength, 20°C), (B) thermal denaturation curves of Pfl 6 variants, (C) interactions made between residues in this region in the Xfaso 1 structure (cyan) and other side chain or backbone groups (yellow), with hydrogen bonds shown as dashed lines, (D) equivalent analysis for this region of Pfl 6.
The importance of this four-residue block and the effects of mutations can be rationalized by examination of three-dimensional structures (Fig. 6C and D). This region displays strongly different patterns of interaction and solvent exposure in the two folds. L44 in Pfl 6 is a hydrophobic core residue, while the aligned residue R47 in Xfaso 1 is on the outer surface of helix 4. Y45 is on the outer face of strand 2 in Pfl 6, while V48 is partially buried on the interior face of helix 4. E46 in Pfl 6 is on the surface in the turn between strand 2 and strand 3; T49 in Xfaso 1 is highly buried and makes hydrogen bonds to interior-facing backbone amide groups. The first residue in the block is somewhat different: here, both residues are on solvent-exposed faces of secondary structure elements but make different interactions: E46 in Xfaso 1 makes side chain-to-main chain hydrogen bonds, while T43 in Pfl 6 is packed mostly against methylene groups of other side chains in the β-sheet. The least destabilizing mutations involve replacement of solvent-facing residues: Xfaso 1 R47L, Pfl 6 E46T and Pfl 6 T43E; the more destabilizing mutations often but not always involve replacement of hydrophobic core residues.
Rescue of half-and-half chimera folding
The above results suggest that the inability of Xfaso 1's C terminus to conform to the β-sheet fold is mostly due to two highly destabilizing residues: R47 and V48. To test this idea, we examined whether folding of the half-and-half chimera PX3 could be rescued by reversing the swap at these two positions. Indeed, while PX3 is completely unfolded (Fig. 2), PX3-R44L/V45Y (Pfl 6 numbering) shows a folded CD spectrum similar to that of Pfl 6 and almost superimposable on that of the chimera PX6 (Fig. 7A). Recall that prior to PX3-R44L/V45Y, PX6 was the chimera that contained the largest portion of the Xfaso 1 C terminus while folding and retaining the β-sheet (Fig. 5). Not surprisingly, since it contains four additional Xfaso 1 residues in strand 2, the thermal stability of PX3-R44L/V45Y (44°C) is lower than that of PX6 (54°C; Fig. 7B). However, the decrement of 10°C is less than expected based on the combined ΔTm values of the Pfl 6 variants containing the four additional Xfaso 1 residues: specifically, the SIEI → CIDI fragment substitution has ΔTm = −11°C and the T43E and E46T substitutions have ΔTm = −16 and −10°C, respectively. The non-additive behavior of the hybrid mutations in strands 2 and 3 may derive from cross-strand interactions between side chains.
We obtained NMR spectra of PX3-R44L/V45Y with reasonable dispersion but insufficient spectral quality for resonance assignment, and the sequence differences in strand 2 hindered even a limited overlay comparison like that shown in Fig. 5. Regardless, the CD data strongly suggest that the Xfaso 1 C-terminal region (from Cys 42 onward) can be rendered basically compatible with the Pfl 6 N-terminal region (and apparently with the β-sheet fold) by two substitutions. The Xfaso 1 C terminal ‘half’ may also be defined as beginning at residue 37 and including the VPAER linker sequence, which can be aligned to the VRAGR sequence in Pfl 6 (Fig. 1 and Table I). Based on this larger definition, as many as four substitutions may be required, but in any case it is only a few sequence changes from having chameleon properties that could facilitate switching from the ancestral to the descendant fold. Interestingly, the overall sequence identity of PX3-R44L/V45Y with Xfaso 1 (61% over the span shown in Table I) is nearly as high as its sequence identity with Pfl 6 (70%).
Discussion
Lessons about specifying the Cro folds from this and previous studies
In previous studies of the P22/λ model system, we employed alanine scanning and hybrid scanning mutagenesis to identify individual residues important for stability and specificity, respectively (Van Dorn et al., 2006). We also designed C-terminal chameleon sequences based on insights gained from the mutagenesis studies. In our current studies of the Xfaso 1/Pfl 6 pair, we are focusing on the effects of swapping blocks of sequence between the two proteins, generating either insertion/deletion mutation events at alignment gaps or block substitutions in other regions. Together, these mutagenesis and design studies support several conclusions about the determinants of fold specificity in Cro proteins.
The stability effects of swapping residues in the C-terminal regions suggest that, in most Cro proteins, this part of the sequence strongly prefers the native fold to the alternative Cro fold. In the P22/λ comparison, five or six individual residues from each C-terminal sequence were found to destabilize the other protein by more than 10°C (Van Dorn et al., 2006). Similarly, in the Pfl 6/Xfaso 1 system, we identified one or more sequence blocks within the C terminus of each protein that caused complete unfolding when swapped into the other protein. Even so, the features of the C-terminal region of Xfaso 1 that critically destabilize the α + β fold appear confined to a few positions, most particularly R47 and V48. This finding suggests that some α-helical Cro proteins, such as Xfaso 1, could be comparatively susceptible to mutationally induced fold switching.
The specific features of the C-termini that exclude the alternate fold are not always shared between homologs with the same fold. This conclusion is evident from a comparison between hybrid point mutations for the four-residue high-specificity region identified for Xfaso 1/Pfl 6 and the same region in the P22/λ sequence alignment. In the P22/λ comparison, the respective sequences for this region are EIVT and TINA, while in the Xfaso 1/Pfl 6 comparison, the sequences are ERVT and TLYE. There are some notable similarities in the effects observed in the hybrid point variants: at the fourth position, a buried Thr residue that makes hydrogen bond interactions is critical for the all-α fold, and both α + β homologs lack it (λ has an Ala residue and Pfl 6 has a Glu residue); at the third position, the small hydrophobic residue Val is important for forming the hydrophobic core of the all-α fold, and substitutions of larger or more polar residues from the α + β homologs (Asn from λ or Tyr from Pfl 6) cause moderate to severe destabilization.
There are also important and telling differences, however. At the second position, a hydrophobic residue (Ile in λ and Leu in Pfl 6) is probably required for the core of the α + β fold, but the all-α homologs differ qualitatively in sequence at this position: P22 has an Ile residue which is identical with the residue in λ, while Xfaso 1 has an Arg which is highly incompatible with the α + β fold. This difference illustrates how evolutionary drift, often at surface positions, can cause variation in the pattern of specificity determinants that rule out an alternate fold. Two homologous proteins with the same fold might both be three substitutions away from an alternate fold, but these substitutions could be at different positions. At the first position, the sequence difference of E/T is identical between the two pairs, but surprisingly the effects are different. In the Xfaso 1/Pfl 6 pair, the Glu residue in Xfaso 1 is less tolerant of the swap than the Thr residue in Pfl 6, while in the P22/λ pair the opposite is the case. This difference illustrates how identical equivalent residues in homologs with the same fold may not contribute equally to stability and specificity: contextual and combinatorial effects lead to complexities in how the folds are determined, and these complexities cannot be fully understood by examining single-site mutations in a limited number of family members.
Switching sequences with the ancestral all-α fold to the α + β fold may be easier than doing the reverse. Recent computational work suggests that the α + β Cro fold has a higher sequence capacity, or number of sequences that fold into it, than the all-α fold (Cao and Elber, 2010). Our work is consistent with this view. In the P22/λ chameleon study, we designed C-terminal chameleon constructs that adopted both folds, albeit with a thermal stability decrement of ∼20°C compared with each parent sequence (Van Dorn et al., 2006). However, our best chameleon design was 63% identical to the C terminus of P22 Cro (residues 34–57) and only 42% identical to the same region from λ, suggesting that the α + β conformation supports greater sequence variation than the all-α conformation. In our Xfaso 1/Pfl 6 study, we find that every sizeable block of the Pfl 6 C-terminal sequence led to a strong drop in the ellipticity of Xfaso 1 at 20°C, probably due to destabilization effects of more than ∼15°C in each case; in contrast, only a few specific residues in the C terminus of Xfaso 1 destabilize the α + β fold.
Last but not least, the N-terminal regions, including the flanking sequence gaps, are clearly important determinants of the Cro fold along with the structurally divergent C-terminal regions. In the P22/λ comparison, the folding pattern of the designed chameleons is fully controlled by the N-terminal sequence to which it is attached (Anderson et al., 2011). In the Xfaso 1/Pfl 6 comparison, the failure of the half-and-half chimeras to fold further demonstrates the importance of both halves of the sequence and the interface and linkage between them. In these chimeras, the preference of the C-terminal region for one fold is incompatible with the preference of the N-terminal region for the other fold. This incompatibility is probably partly a function of intrinsic topological preferences in each region and partly a function of the interaction interface between them. Regardless, the determinants specifying different Cro folds are not confined to the structurally divergent C-terminal region, and are not simple.
Relationship to other work
As noted in the ‘Introduction’ section, our study is one of many to examine sequence hybrids of two differently folded proteins; it is distinct from most other studies of this kind in examining hybrids of homologous rather than unrelated proteins. Most hybrid sequence approaches have offered insights into the general potential for evolutionary switching between any two folds of a given size, and their connectedness in sequence space; ours, on the other hand, may be seen as an attempt to probe possible mechanisms and determinants for a particular fold switch that actually occurred during evolution.
Previous studies involving unrelated, completely different folds generally suggested that only very careful design studies, especially those informed by mutagenesis data, could reveal viable mutational paths for fold switching (Blanco et al., 1999; Alexander et al., 2007, 2009; He et al., 2008, 2012). It might be expected that this search would be easier, perhaps even trivial, for folds related by natural evolution, especially when some aspects of the sequence and structure are conserved, as is the case for Xfaso 1 and Pfl 6. Perhaps in such cases, careful design would not be necessary, and naive hybrid construction would reveal simple determinants of fold specificity and switching. Indeed, for some very small disulfide-bonded domains, or domains with very localized topological changes, evolutionary fold switching may be understandable in terms of one or two key substitutions (Tidow et al., 2004; Meier and Ozbek, 2007).
Our results on block hybrids of Xfaso 1 and Pfl 6, however, suggest that fold specificity determinants can be multifarious and globally distributed, even among proteins with fairly similar sequences and partly conserved folds. Simple block substitutions, insertions and deletions between Xfaso 1 and Pfl 6 Cro do not switch the fold and in many cases lead to drastic reductions in stability or complete unfolding. Our studies do suggest that the fold of Xfaso 1 might be switched by a combination of a few key substitutions in the C-terminal region, coupled with several appropriately designed indels and substitutions in the rest of the protein. This prediction is a subject of ongoing investigation.
In general, more sophisticated approaches to Cro hybrid construction may yield additional lessons and may lead to different conclusions. For example, our initial division of the proteins into N- and C-terminal halves does not perfectly coincide with conserved and divergent regions of backbone topology; in particular, the N-terminal half of Pfl 6 contains a β-strand that interacts with the C terminus and is not present in Xfaso 1. Examination of hybrids based more strictly on regions of structural divergence and conservation may clarify our picture of the sequence determinants of fold. Additionally, moving beyond simple hybrids of two sequences to incorporate sequence conservation information will surely be useful and informative, as it has been for the design of conformational switches (Hori and Sugiura, 2002; Cerasoli et al., 2005; Ambroggio and Kuhlman, 2006). For the Cro family, this depends on being able to classify Cro sequences a priori by fold, and we are progressing on this front.
An increasingly favorable view is emerging of the potential for evolutionary flow between protein folds. For example, very recent hybrid sequence studies (Porter et al., 2015), as well as other mutational and computational work, point toward the existence of networks and supernetworks of multiple folds that may be connected directly or indirectly by mutational pathways that conserve stability (Babajide et al., 2001; Burke and Elber, 2012; Stewart et al., 2013a). To what extent does evolution actually traverse routes between folds? The lesson from the Cro family so far is that evolution can find pathways between folds even when hybrid studies suggest that the determinants of fold switching are complex and multifarious.
Funding
This work was supported by the National Institute for General Medical Sciences at the National Institutes of Health (grant number R01 GM066806 to M.H.J.C.).
Acknowledgements
This paper is dedicated to the memory of Matthew Dubrava, who first came up with the idea for this study.
References
- Albright R.A., Matthews B.W. (1998) J. Mol. Biol., 280, 137–151. [DOI] [PubMed] [Google Scholar]
- Alexander P.A., Rozak D.A., Orban J., Bryan P.N. (2005) Biochemistry, 44, 14045–14054. [DOI] [PubMed] [Google Scholar]
- Alexander P.A., He Y., Chen Y., Orban J., Bryan P.N. (2007) Proc. Natl Acad. Sci. U.S.A., 104, 11963–11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander P.A., He Y., Chen Y., Orban J., Bryan P.N. (2009) Proc. Natl Acad. Sci. U.S.A., 106, 21149–21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambroggio X.I., Kuhlman B. (2006) Curr. Opin. Struct. Biol., 16, 525–530. [DOI] [PubMed] [Google Scholar]
- Anderson W.J., Van Dorn L.O., Ingram W.M., Cordes M.H.J. (2011) Protein Eng. Des. Sel., 24, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva A., Murzin A.G. (2006) Curr. Opin. Struct. Biol., 16, 399–408. [DOI] [PubMed] [Google Scholar]
- Babajide A., Farber R., Hofacker I.L., Inman J., Lapedes A.S., Stadler P.F. (2001) J. Theor. Biol., 212, 35–46. [DOI] [PubMed] [Google Scholar]
- Becktel W.J., Schellman J.A. (1987) Biopolymers, 26, 1859–1877. [DOI] [PubMed] [Google Scholar]
- Blanco F.J., Angrand I., Serrano L. (1999) J. Mol. Biol., 285, 741–753. [DOI] [PubMed] [Google Scholar]
- Bryan P.N., Orban J. (2010) Curr. Opin. Struct. Biol., 20, 482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke S., Elber R. (2012) Proteins Struct Funct Bioinformatics, 80, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B.Q., Elber R. (2010) Proteins Struct Funct Bioinformatics, 78, 985–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerasoli E., Sharpe B.K., Woolfson D.N. (2005) J. Am. Chem. Soc., 127, 15008–15009. [DOI] [PubMed] [Google Scholar]
- Dalal S., Regan L. (2000) Protein Sci., 9, 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal S., Balasubramanian S., Regan L. (1997) Nat. Struct. Biol., 4, 548–552. [DOI] [PubMed] [Google Scholar]
- Darling P.J., Holt J.M., Ackers G.K. (2000) Biochemistry, 39, 11500–11507. [DOI] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Dubrava M.S., Ingram W.M., Roberts S.A., Weichsel A., Montfort W.R., Cordes M.H. (2008) Protein Sci., 17, 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin N.V. (2001) J. Struct. Biol., 134, 167–185. [DOI] [PubMed] [Google Scholar]
- He Y., Yeh D.C., Alexander P., Bryan P.N., Orban J. (2005) Biochemistry, 44, 14055–14061. [DOI] [PubMed] [Google Scholar]
- He Y., Chen Y., Alexander P., Bryan P.N., Orban J. (2008) Proc. Natl Acad. Sci. U.S.A., 105, 14412–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Chen Y., Alexander P., Bryan P., Orban J. (2012) Structure, 20, 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y., Sugiura Y. (2002) J. Am. Chem. Soc., 124, 9362–9363. [DOI] [PubMed] [Google Scholar]
- Jana R., Hazbun T.R., Mollah A.K., Mossing M.C. (1997) J. Mol. Biol., 273, 402–416. [DOI] [PubMed] [Google Scholar]
- Johnson B.A., Blevins R.A. (1994) J. Biomol. NMR, 4, 603–614. [DOI] [PubMed] [Google Scholar]
- Kinch L.N., Grishin N.V. (2002) Curr. Opin. Struct. Biol., 12, 400–408. [DOI] [PubMed] [Google Scholar]
- Lattman E.E., Rose G.D. (1993) Proc. Natl Acad. Sci. U.S.A., 90, 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeFevre K.R., Cordes M.H. (2003) Proc. Natl Acad. Sci. U.S.A., 100, 2345–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S., Ozbek S. (2007) Bioessays, 29, 1095–1104. [DOI] [PubMed] [Google Scholar]
- Meier S., Jensen P.R., David C.N., Chapman J., Holstein T.W., Grzesiek S., Ozbek S. (2007) Curr. Biol., 17, 173–178. [DOI] [PubMed] [Google Scholar]
- Morrone A., McCully M.E., Bryan P.N., Brunori M., Daggett V., Gianni S., Travaglini-Allocatelli C. (2011) J. Biol. Chem., 286, 3863–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin A.G. (2008) Science, 320, 1725–1726. [DOI] [PubMed] [Google Scholar]
- Myers J.K., Pace C.N., Scholtz J.M. (1995) Protein Sci., 4, 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlove T., Konieczka J.H., Cordes M.H. (2004) Structure, 12, 569–581. [DOI] [PubMed] [Google Scholar]
- Porter L.L., He Y., Chen Y., Orban J., Bryan P.N. (2015) Biophys. J., 108, 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler C.G., Hall B.M., Anderson W.J., Ingram W.M., Roberts S.A., Montfort W.R., Cordes M.H. (2008) Proc. Natl Acad. Sci. U.S.A., 105, 2343–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose G.D., Creamer T.P. (1994) Proteins, 19, 1–3. [DOI] [PubMed] [Google Scholar]
- Stewart K.L., Dodds E.D., Wysocki V.H., Cordes M.H. (2013a) Protein Sci., 22, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart K.L., Nelson M.R., Eaton K.V., Anderson W.J., Cordes M.H. (2013b) Proteins, 81, 1988–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidow H., Lauber T., Vitzithum K., Sommerhoff C.P., Rosch P., Marx U.C. (2004) Biochemistry, 43, 11238–11247. [DOI] [PubMed] [Google Scholar]
- Van Dorn L.O., Newlove T., Chang S., Ingram W.M., Cordes M.H. (2006) Biochemistry, 45, 10542–10553. [DOI] [PubMed] [Google Scholar]
- Yeates T.O. (2007) Curr. Biol., 17, R48–R50. [DOI] [PubMed] [Google Scholar]
- Yuan S.M., Clarke N.D. (1998) Proteins, 30, 136–143. [DOI] [PubMed] [Google Scholar]